
Abstract
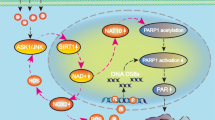
Oxidative stress causes diverse neurological disorders. Parthanatos is a type of programmed cell death, characterised by strong activation of poly (ADP-ribose) (PAR) polymerase-1 (PARP-1), PAR polymer accumulation, and nuclear translocation of apoptosis-inducing factor (AIF), and is involved in cellular oxidative injury. Signalling by c-Jun-N-terminal protein kinase (JNK) is activated by reactive oxygen species (ROS), and this also contributes to ROS production. However, the exact relationship between JNK signalling and parthanatos in neurological disorders triggered by oxidative stress is unclear. In this study, glutamate-treated HT22 neurons were used to investigate whether the signalling by JNK contributes a regulatory role to parthanatos in oxidative stress-related neurological disease. JNK signalling was activated in glutamate-treated HT22 neurons, demonstrated via upregulation of p-JNK levels. Pre-treatment with SP600125 markedly inhibited JNK signalling, increased cell viability, and significantly reversed PARP-1 overproduction, PAR polymer accumulation, and nuclear AIF translocation. In addition, inhibition of JNK signalling severely reduced the production of both intracellular ROS and mitochondria superoxide. This study indicated that parthanatos in glutamate-treated HT22 neurons could be suppressed by JNK signalling inhibition. JNK activation participated in parthanatos via an increase in intracellular ROS levels.
Similar content being viewed by others


Introduction
Preventing neurons from being damaged by stressors provides critical therapy in diverse neurological disorders. Recent research has suggested that cell death is a relatively complex phenomenon that is not only confined to apoptosis and necrosis. Several types of programmed cell death have been defined based on the involved molecules, such as pyroptosis, cuproptosis, and ferroptosis1. Parthanatos is a type of caspase-independent cell death characterized by severe activation of poly (ADP-ribose) (PAR) polymerase-1 (PARP-1)2. PARP-1 overactivation prompts PAR polymer accumulation and is followed by translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus3. Parthanatos can occur in diverse neurological diseases4,5. PARP-1 is involved in initiating parthanatos; it is a nuclear enzyme that both contributes to DNA repair at low concentrations and is harmful to cellular function when it is excessively provoked by heavy DNA injury. Reactive oxygen species (ROS) are well-known genotoxic agents that also contribute to parthanatos. A previous study demonstrated that deoxypodophyllotoxin triggered parthanatos in glioma cells due to excessive ROS induction6. In addition, parthanatos was inhibited in embryonic fibroblasts by reducing ROS levels7.
Glutamate is a well-known neurotransmitter in the central nervous system. However, massive glutamate release leads to excitatory and oxidative toxicity, which play prominent roles in neuronal injury8. Hippocampal HT22 cells are widely used in research on oxidative neuronal damage. HT22 cells lack glutamate ionotropic receptors; therefore, glutamate-induced toxicity in these cells is exclusively ROS-dependent. In a previous study, we demonstrated that ROS accumulated in HT22 neurons with glutamate treatment and in a rat model of epilepsy and that ROS mediated parthanatos9. However, the signalling pathways participating in parthanatos in HT22 neurons in the context of oxidative stress remain unknown, but cellular NAD consumption or a decline in SIRT1 activity was reported to contribute PARP-1 activity10. Signalling by c-Jun-N-terminal protein kinase (JNK) can be provoked by ROS accumulation and plays a role in cell survival11. Interestingly, previous research suggested that JNK promoted apoptosis in hepatoma carcinoma cells via a JNK–NADPH oxidase–ROS self-driven death signal circuit12. We, therefore, speculate that JNK might regulate parthanatos caused by oxidative stress in neurons. In the present study, we examined JNK activation and its roles in ROS production and parthanatos in glutamate-treated HT22 cells. The findings might provide promising strategies for therapies to treat neurological disorders.
Materials and methods
Cell lines and cultures
HT22 neurons were acquired from the Department of Neurology and Neuroscience Center at the First Hospital of Jilin University (Changchun, China). Undifferentiated HT22 neurons were cultured in Dulbecco’s modified Eagle medium (Gibco, Thermo Fisher Scientific, Waltham, MA) containing 10% foetal bovine serum (Gemini, West Sacramento, CA) and 1% penicillin-streptomycin (Gibco). HT22 neurons were incubated in 5% CO2 at 37℃, and the mid-log phase was used in subsequent experiments.
Cell viability assay
HT22 neurons (5,000 per 100 µl of growth medium in each well) were seeded onto 96-well plates and incubated for 24 h. In vitro oxidative toxicity was induced with 5 mM glutamate and the cell counting kit-8 (CCK-8) assay (Dojindo, Kumamoto, Japan) was performed to examine cellular viability at different incubation durations (6, 12, and 24 h), according to the manufacturer’s introductions. To test the effect of JNK inhibition on cellular viability, 20 µM SP600125 was applied to HT22 neurons 2 h prior to glutamate application. Neuronal morphology was assessed with fluorescence microscopy.
Measurement of intracellular oxidative species production
HT22 neurons (5 × 105 per 2 ml growth media) were seeded onto a 35-mm culture dish, then neurons were collected at 12 h after glutamate treatment. Intracellular ROS production in HT22 neurons was detected using DCFH-DA (Meilunbio, Dalian, China) and DHE assays (ATT Bioquest, Pleasanton, CA) according to the manufacturers’ instructions. After incubation (10 µM DCFH-DA or 5 µM DHE) for 20 min at 37℃ in the dark, the medium was removed and washed with phosphate-buffered saline (PBS). Inverted fluorescence microscopy was used to acquire images. Cells were also analysed by flow cytometry.
Measurement of AIF translocation
HT22 neurons (3 × 105 per cell crawling piece) exposed to glutamate for 24 h. Then, neurons were fixed in 4% paraformaldehyde, washed three times with PBS, and incubated with 0.5% Triton X-100. Next, blocking of non-specific antibody binding sites was performed using 5% goat serum (P0216; Beyotime, Shanghai, China). After neurons were incubated with primary antibodies against AIF (#4642; 1:100; Cell Signaling Technology, Danvers, MA) overnight at 4℃, fluorescence-conjugated secondary antibodies (bs-0295G-AF488; 1:100; Bioss, Woburn, MA) was treated for 60 min at room temperature. Subsequently, neurons were washed twice with PBS and incubated with DAPI (C02-04002; Bioss) for 5 min. Images were captured via fluorescence microscopy.
Measurement of mitochondrial superoxide and membrane potentials
Mitochondrial superoxide was tested using MitoSox Red (Thermo Fisher Scientific). Mito-Tracker Green (Beyotime) and Hoechst 33,342 (Solarbio, Beijing, China) were used to mark mitochondria and nuclei, respectively, in HT22 neurons. After treatment with corresponding compounds, 20 nM Mito-Tracker Green and 100× Hoechst 33,342 (diluted to 1×) were added to neurons for 20 min. Subsequently, HT22 neurons were incubated with 5 µM MitoSox Red for 10 min. Then, neurons were washed with Hank’s balanced salt solution (HBSS) three times and captured under a fluorescence microscope.
To measure mitochondrial membrane potentials (MMPs), glutamate-treated HT22 neurons were collected in flow tubes and washed twice in PBS. MMPs were measured using the JC-1 assay (Beyotime). JC-1 staining was prepared according to the manufacturers’ protocols. The fluorescence intensity based on MMPs was measured using flow cytometry. In addition, neurons were also visualised under a fluorescence microscope.
Measurement of the effect of ROS on DNA damage
DNA oxidative damage was detected by 8-OHdG assay (Biofund, China). Pre-treatment with 3 mM N-acetyl-L-cysteine (NAC) was used to inhibit cellular ROS production. After treatment with glutamate for 12 h, 8-OHdG enzyme-linked immunosorbent assay was performed. The results are presented as ratios to the absorbance value at 450 nm of the control cells.
Western blot
Total proteins were extracted by radioimmunoprecipitation lysis buffer (Solarbio) containing phenylmethylsulphonyl fluoride and 1% proteinase inhibitors (Solarbio). Nuclear and cytoplasmic proteins were extracted using commercial reagents (Beyotime).
Proteins were quantified with a BCA kit (Solarbio) following the manufacturer’s instruction. The samples were electrophoresed on 4–20% sodium dodecyl-sulphate polyacrylamide gel (GenScript, Nanjing, China) and transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA). Membranes were blocked with 5% skim milk and incubated with rabbit anti-JNK antibody (#9252 ;1:1000; Cell Signaling Technology), rabbit anti-AIF antibody (#4642; 1:1000; Cell Signaling Technology), rabbit anti-p-JNK antibody (#9251; 1:1000; Cell Signaling Technology), rabbit anti-PARP1 antibody (#9542; 1:1000; Cell Signaling Technology), rabbit anti-β-actin antibody (bs-0061R; 1:3000; Bioss), rabbit anti-GAPDH antibody (bs-10900R; 1:3000; Bioss), mouse anti-PAR antibody (AM80; 1:2000; MilliporeSigma, Burlington, MA), and mouse anti-Histone antibody (bsm-33042 M; 1:3000; Bioss) overnight at 4℃. Western blot bands were quantified using gel densitometry after incubation with horseradish peroxidase-conjugated secondary anti-IgG antibody (bs-0295G-HRP/bs-0296G-HRP; 1:5000; Bioss) for 1 h at room temperature. The relative ratios of proteins to GAPDH/β-actin/Histone were obtained for each sample.
Statistical analysis
Statistical analyses were performed using SPSS 11.0 software (IBM, Armonk, NY) and GraphPad Prism 8.0 (GraphPad Software, San Diego, CA). The Kolmogorov–Smirnoff test was used to judge whether the data conforms to normal distribution, and Levene’s test was used to judge whether the variance is homogeneous. For normal data, one-way analysis of variance (ANOVA) was used for multi-group comparison and Tukey’s test was used for multiple comparison. All data represent six independent experiments (n = 6) and the results are expressed as mean ± standard error of the mean. P-values were corrected using Bonferroni’s test. P-values less than 0.05 were considered statistically significant.
Results
JNK pathway is involved in glutamate-induced HT22 neurons
The activation of JNK signalling in the present study was examined by the level of phosphorylated JNK (p-JNK). Western blot showed that total JNK level was not different between treated and control cells at each timepoint. However, levels of p-JNK were higher at 3, 6, and 12 h of exposure to glutamate, indicating JNK pathway activation in HT22 neurons under oxidative stress (n = 6 per group; Fig. 1a). Next, the JNK inhibitor SP600125 was used to investigate the protective role of JNK in glutamate-induced cell injury. As shown in Fig. 1b, pre-treatment with of 20 µM SP600125 effectively counteracted the upregulation of p-JNK caused by glutamate treatment (n = 6 per group). Subsequently, the CCK-8 assay was used to assess cell viability. As shown in Fig. 2a, HT22 neuron viability decreased after glutamate treatment for different durations (n = 6 per group; 6 h: 88.95 ± 4.90 vs. control [p < 0.05], 12 h: 75.65 ± 3.26 vs. control [p < 0.05], 24 h: 36.80 ± 5.81 vs. control [p < 0.01]). In contrast, JNK inhibition with 20 µM SP600125 significantly promoted neuron survival in glutamate treatment for 24 h (n = 6 per group; 80.12 ± 4.80 vs. 39.67 ± 3.16, p < 0.01) (Fig. 2b, c).

Glutamate alters JNK signalling-related protein expression. (a) Western blot showed JNK activation (presented as p-JNK level) in glutamate-treated HT22 neurons at indicated time points. (b) Western blot showed that SP600125 significantly inhibited JNK signalling (presented as p-JNK level) at 12 h in glutamate-treated cells. GAPDH was used as cytoplasmic loading control. Data are expressed as mean ± standard deviation (SD; *p < 0.05, **p < 0.01).

CCK-8 assay in HT22 neurons after glutamate treatment. (a) Glutamate decreased HT22 neuron viability in a time-dependent manner. (b) JNK inhibition significantly increased HT22 neuron viability for 24 h after glutamate treatment. (c) Disrupted cell morphology was seen in glutamate-treated HT22 neurons; this improved with SP600125 pre-treatment. Data are expressed as mean ± standard deviation (SD; *p < 0.05, **p < 0.01).
JNK inhibition altered parthanatos-related protein expression in HT22 neurons
In a previous study, we demonstrated glutamate-induced parthanatos in HT22 neurons and that pre-treatment with a PARP-1 inhibitor could protect cells from injury. In order to investigate whether JNK signalling has a regulate role on glutamate induced parthanatos in HT22 neurons, we used SP600125 to inhibit JNK pathway and examined the parthanatos-related protein expression levels. As shown in Fig. 3a,b, glutamate induced the upregulation of PARP-1, the formation of PAR polymers, and nuclear accumulation of AIF; these were attenuated when JNK activation was inhibited by 20 µM SP600125 (n = 6 per group). In addition, AIF translocation to the nucleus in glutamate-treated HT22 neurons was visualized using fluorescence immunohistochemistry (Fig. 3c). These results indicated that JNK suppression protected HT22 neurons from parthanatos after oxidative stress.

JNK signalling inhibition altered parthanatos-related protein expression at 12 h in glutamate-treated HT22 neurons. (a) Western blot showed that JNK inhibition decreased PARP-1 and PAR expression levels. (b) Western blot showed that JNK inhibition increased cytoplasmic AIF expression but decreased nuclear AIF expression. Actin and Histone were used as cytoplasmic and nuclear loading controls, respectively. (c) Fluorescence immunohistochemistry of AIF (green) translocation to the nucleus in HT22 neurons incubated with glutamate. Nuclei were stained with DAPI (blue). Data are expressed as mean ± standard deviation (SD; *p < 0.05, **p < 0.01).
ROS contributes to parthanatos in glutamate-treated HT22 neurons
ROS is a well-known genotoxic agent, and we next investigated the relationship between ROS and parthanatos in glutamate-treated HT22 neurons. NAC is an antioxidant that can reduce intracellular ROS. HT22 neurons were incubated with glutamate at each indicated duration time (0, 6, 8, and 12 h) with or without 3 mM NAC pre-treatment, and ROS production was measured using fluorescence spectrometry. As shown in Fig. 4a, ROS production induced by glutamate was severely decreased by the addition of 3 mM NAC at each timepoint (n = 4 per group; 6 h: 183±14.81 vs. 228±11.46 [p < 0.01], 8 h: 189.3±15.67 vs. 271±7.62 [p < 0.01], 12 h: 239±17.57 vs. 380.3±6.02 [p < 0.01]). Moreover, the 8-OHdG assay was performed to detect DNA damage caused by ROS. The results showed that administration of 3 mM NAC significantly decreased the upregulation of 8-OHdG induced by glutamate (n = 4 per group; 33.3 ± 2.01 vs. 41.55 ± 1.99 ng/mg protein, p < 0.01) (Fig. 4b).

Effects of reactive oxygen species (ROS) on parthanatos at 12 h in glutamate-treated HT22 neurons. (a) Fluorescence intensity of DCFH-DA in HT22 neurons was enhanced at the indicated time points (6, 8 and 12 h); this trend was counteracted by NAC pre-treatment. (b) Pre-treatment with NAC decreased glutamate-induced DNA injury in HT22 neurons, as shown by 8-OHdG expression levels. (c) Western blot showed that NAC pre-treatment decreased PARP-1 and PAR expression levels and (d) increased cytoplasmic AIF expression but decreased nuclear AIF expression. Actin and Histone were used as cytoplasmic and nuclear loading controls, respectively. Data are expressed as mean ± standard deviation (SD; *p < 0.05, **p < 0.01).
In order to examine whether the parthanatos-related proteins had a different expression level in HT22 neurons with glutamate treatment, we performed western blotting test. The results showed that the upregulation of PARP-1, formation of PAR polymers, and accumulations of AIF in nuclei by induced by glutamate treatment were all markedly attenuated with the addition of 3 mM NAC (n = 6 per group; Fig. 4c,d). These results indicated that DNA destruction promoted excessive ROS accumulation and further contributed to parthanatos in HT22 neurons under oxidative stress.
JNK activation plays a role in intracellular ROS
As JNK signalling activation could induce parthanatos in glutamate-treated HT22 neurons, we also confirmed that ROS contributed to parthanatos via DNA damage. Further, we conducted DCFH-DA and DHE immunostaining to examine whether JNK activation plays a role in intracellular ROS accumulation. The fluorescence intensities of DCFH-DA and DHE were respectively enhanced by the upregulation of intracellular hydrogen peroxide or superoxide anions. Fluorescence microscopy indicated that the DCF fluorescence (green) and the DHE fluorescence (red) were much brighter in HT22 neurons with glutamate treatment for 12 h than cells in the control group. In contrast, prior administration of 20 µM SP600125 significantly reversed the fluorescence intensity increases (Figs. 5a, 6a). Flow cytometry results (Figs. 5b, 6b) indicated that the mean fluorescence intensity of DCF was obviously lower with 20 µM SP600125 pre-treatment than in control cells (1 × 104 cells per group;1674±126 vs. 4483±341, p < 0.01); this was also true for the DHE fluorescence intensity (1 × 104 cells per group; 543.7±65.04 vs. 1483±166.2, p < 0.01). These findings indicated that JNK activation could lead to increased cellular ROS production.

JNK signalling inhibition altered DCFH-DA fluorescence at 12 h in glutamate-treated HT22 neurons. (a) Fluorescence immunohistochemistry of DCFH-DA staining, with significantly higher green signal in glutamate-treated HT22 neurons; SP600125 decreased green fluorescence intensity. (b) pre-treatment with the JNK inhibitor SP600125 attenuated hydrogen peroxide overproduction in glutamate-treated HT22 neurons, as determined using flow cytometry. Data are expressed as mean ± standard deviation (SD; *p < 0.05, **p < 0.01).

JNK signalling inhibition altered DHE fluorescence at 12 h in glutamate-treated HT22 neurons. (a) Fluorescence immunohistochemistry of DHE, red fluorescence was significantly higher in glutamate-treated HT22 neurons, and SP600125 decreased red fluorescence. (b) pre-treatment with the JNK inhibitor SP600125 attenuated superoxide anion overproduction in glutamate-treated HT22 neurons, as determined using flow cytometry. Data are expressed as mean ± standard deviation (SD; *p < 0.05, **p < 0.01).
JNK activation contributes to mitochondrial dysfunction
Given that mitochondrial superoxide is another main source of intracellular ROS production, we assessed mitochondrial superoxide levels using MitoSox staining. HT22 neurons exposed to glutamate for 12 h at 5 mM had much brighter red fluorescence than control cells. In contrast, administration of the JNK inhibitor markedly reversed the increased fluorescence intensity in glutamate-treated HT22 neurons (Fig. 7). Furthermore, we performed JC-1 staining to detect MMPs. Flow cytometry showed that treatment with 20 µM of SP600125 markedly counteracted the glutamate-induced decreases in MMPs after incubated with glutamate for 12 h in HT22 neurons (1 × 104 cells per group; Fig. 8b). JC-1 immunofluorescence in glutamate-treated HT22 neurons with or without the addition of SP600125 is shown in Fig. 8a.

Fluorescence immunohistochemistry of MitoSox Red (red) was used to measure mitochondrial superoxide levels. JNK inhibition suppressed the mitochondrial superoxide level at 12 h in glutamate-treated HT22 neurons. Mitochondria were stained with Mito-Tracker (green), nuclei were stained with Hoechst 33,342 (blue).

Effects of JNK signalling on mitochondrial membrane potentials (MMPs) at 12 h in glutamate-treated HT22 neurons. (a) JNK inhibition improved MMPs in glutamate induced HT22 neurons, as shown by JC-1 staining. (b) Flow cytometry of MMP in HT22 neurons. JC-1 red labels normal mitochondria, JC-1 green labels abnormal mitochondria. Data are expressed as mean ± standard deviation (SD; *p < 0.05, **p < 0.01).
Discussion
In the present study, we demonstrated that JNK inhibition could promote HT22 neuron survival in a glutamate-treated cell culture model. This was partially related to the attenuation of PARP-1 activation, PAR formation, and AIF translocation, which are characteristic of parthanatos. We further confirmed that ROS contributed to parthanatos. Furthermore, inhibition of the JNK pathway markedly reduced intracellular ROS accumulation and mitochondrial superoxide production. Taken together, this suggests that the JNK pathway can trigger parthanatos in glutamate-treated HT22 neurons via ROS overproduction.
Parthanatos, a relatively newly discovered type of programmed cell death, was first elucidated at Johns Hopkins University in 200813. In this mysterious cellular process, overactivation of PARP-1 is the initiating step, following by excessive synthesis of PAR polymers. PAR polymers are effectors of parthanatos, and they could bind to numerous cellular proteins, leading to malfunction of diverse cellular process. AIF, the molecular locating in mitochondria, works as an executor in parthanatos. When signalling by PAR, AIF is released from mitochondria to cytoplasm, in which they interact with migration inhibitory factor (MIF). In the cooperation with MIF, AIF-MIF complex is recruited to the nucleus, and causes large-scale DNA damage and cell death14. Thus, excessive PARP-1, PAR polymers, and AIF translocation are all necessary for parthanatos, and only the involvement of these three key molecules can be defined as such. In fact, distinctive programmed cell death is not mutually exclusive. Several biochemical processes are shared between parthanatos and other types of cell death. For example, the release of AIF has been reported in caspase-dependent cell death15. Further, parthanatos was found to accompany caspase activation, cytochrome C release, and MMP decreases, which are key characteristics of apoptosis1.
Severe DNA damage is considered to be a direct factor that initiates parthanatos16. ROS has been widely confirmed as a predominant cause of DNA damage. Diverse neurological disorders characterised by oxidative injury are associated with parthanatos, such as epilepsy, ischaemic stroke, and neurodegenerative diseases9,17. In the present study, glutamate treatment significantly induced excessive ROS accumulation in HT22 neurons and subsequently caused DNA damage. In addition, pre-treatment with the ROS inhibitor NAC obviously protected DNA from oxidative injury and prevented parthanatos. In brief, we confirmed that ROS contributed to the process of parthanatos in HT22 neurons through the induction of DNA damage, which was also examined in a previous study. The mechanisms underlying glutamate-induced ROS accumulation in HT22 neurons are relatively complex, including cysteine/glutamate antiporter inhibition and mitochondrial dysfunction, among others18. Therefore, targeting parthanatos could provide a promising strategy in the treatment of oxidative toxicity in neurons.
Although we demonstrated that inhibition of ROS production by NAC could attenuate parthanatos in glutamate-treated HT22 neurons, the regulatory mechanisms involved in parthanatos remain elusive. In a study by Wang et al., supplementation of cellular NAD+ could rescue parthanatos in primary hippocampal neurons after treatment with Mg2+-free medium8. However, energy deficiency seems to not be important in the process of parthanatos. The JNK pathway is a member of the mitogen-activated protein kinase family, and transient JNK activation is beneficial to cell growth; however, constant JNK activation can induce cell death. According to previous studies, the JNK pathway can be activated by ROS, and JNK activation plays an important role in HT22 neuronal injury from oxidative stress19,20. Thus, we speculated that JNK signalling might contribute to parthanatos in glutamate-treated HT22 neurons. In the present study, we showed that JNK signalling was activated in glutamate-treated HT22 neurons, demonstrated by the upregulation of p-JNK. JNK inhibition could significantly increase cell viability and rescue the changes in expression level of parthanatos-related proteins. In conclusion, the inhibition of JNK signalling partially protected HT22 neurons from oxidative injury via reducing parthanatos.
In subsequent experiments, we found that increases in intracellular ROS after glutamate treatment were suppressed by pre-treatment with SP600125, which indicated that JNK signalling can regulate intracellular ROS amplification. Thus, JNK pathway activation induced parthanatos in glutamate-treated HT22 neurons via promotion of intracellular ROS accumulation. The likely mechanism behind this is related to the regulatory role of JNK in cellular NADPH oxidase. NADPH oxidases (NOXs), a kind of transmembrane protein, are the key enzymes of redox signalling in vivo and the main source of ROS in the body. The function of NOX necessarily relies on some regulatory subunits, such as p47phox, p91phox, p22phox. The active form of p47phox translocated from cytoplasm to membrane then interacted with gp91phox and p22phox, which was a critical step in NOX activation21. Zeng et al. suggested that excessive ROS production could be decreased by the inhibition of JNK signalling via reducing the activity of p47phox12.
Mitochondrial superoxide is another main source of intracellular ROS. MitoSox was used to detect superoxide derived from mitochondria in the present study. We showed that mitochondrial superoxide was decreased with the administration of SP600125 in glutamate-treated HT22 neurons. Moreover, we used Mito-Tracker to mark mitochondria, and no differences were seen in glutamate-treated HT22 neurons between treatment with or without SP600125, which suggested that inhibition of JNK signalling influenced the physiological function of mitochondria rather than its content. The possible mechanism could be that the JNK pathway regulates the MMP. As shown in the present study, pre-treatment with SP600125 significantly reversed the decreases in MMP seen in glutamate-induced HT22 neurons, as measured using the JC-1 assay. A previous study also suggested that the JNK pathway regulates MMP in JNK1−/− and JNK2−/−murine embryonic fibroblasts under oxidative stress22. The underlying mechanism might be the interaction of p-JNK with Sab (Sh3bp5), a protein located in the mitochondrial membrane, which could lead to mitochondrial membrane leak currents23.
There were some limitations in the present study. We only tested the regulatory role of JNK on parthanatos using pharmacological inhibitors, and JNK genetic silencing should be conducted to further confirm these results. In addition, investigations on the specific mechanisms relating JNK and NADPH oxidase, as well as how JNK interacts with mitochondria, are needed.
Data availability
Data is available from the corresponding author upon reasonable request.
References
Galluzzi, L. et al. Molecular definitions of cell death subroutines: Recommendations of the nomenclature Committee on Cell Death 2012. Cell. Death Differ.19, 107–120 (2012).
Andrabi, S. A. et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA103, 18308–18313 (2006).
Wang, Y., Dawson, V. L. & Dawson, T. M. Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp. Neurol.218, 193–202 (2009).
Wang, S. J. et al. Poly(ADP-ribose) polymerase inhibitor is neuroprotective in epileptic rat via apoptosis-inducing factor and akt signaling. Neuroreport18, 1285–1289 (2007).
Zhong, H. et al. Propofol inhibits parthanatos via ROS-ER-calcium-mitochondria signal pathway in vivo and vitro. Cell. Death Dis.9, 932 (2018).
Ma, D. et al. Deoxypodophyllotoxin triggers parthanatos in glioma cells via induction of excessive ROS. Cancer Lett.371, 194–204 (2016).
Chiu, L. Y., Ho, F. M., Shiah, S. G., Chang, Y. & Lin, W. W. Oxidative stress initiates DNA damage MNNG-induced poly(ADP-ribose)polymerase-1-dependent parthanatos cell death. Biochem. Pharmacol.81, 459–470 (2011).
Jiang, T. et al. Gastrodin protects against glutamate-induced ferroptosis in HT-22 cells through Nrf2/HO-1 signaling pathway. Toxicol. Vitro62, 104715 (2020).
Wang, X., Zhang, W., Ge, P., Yu, M. & Meng, H. Parthanatos participates in glutamate-mediated HT22 cell injury and hippocampal neuronal death in kainic acid-induced status epilepticus rats. CNS Neurosci. Ther.28, 2032–2043 (2022).
Wang, S. et al. Cellular NAD depletion and decline of SIRT1 activity play critical roles in PARP-1-mediated acute epileptic neuronal death in vitro. Brain Res.1535, 14–23 (2013).
Zhang, W., Wang, X., Yu, M., Li, J. A. & Meng, H. The c-Jun N-terminal kinase signaling pathway in epilepsy: Activation, regulation, and therapeutics. J. Recept Signal. Transduct. Res.38, 492–498 (2018).
Zeng, K. W. et al. Induction of hepatoma carcinoma cell apoptosis through activation of the JNK-nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-ROS self-driven death signal circuit. Cancer Lett.353, 220–231 (2014).
Andrabi, S. A., Dawson, T. M. & Dawson, V. L. Mitochondrial and nuclear cross talk in cell death: Parthanatos. Ann. N. Y. Acad. Sci.1147, 233–241 (2008).
Wang, X. & Ge, P. Parthanatos in the pathogenesis of nervous system diseases. Neuroscience. 449, 241–250 (2020).
Cregan, S. P. et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell. Biol.158, 507–517 (2002).
Caldecott, K. W. Protein ADP-ribosylation and the cellular response to DNA strand breaks. DNA Repair. (Amst)19, 108–113 (2014).
Huang, P. et al. Molecular mechanisms of parthanatos and its role in diverse diseases. Int. J. Mol. Sci.23, 7292 (2022).
Fukui, M., Song, J. H., Choi, J., Choi, H. J. & Zhu, B. T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol.617, 1–11 (2009).
Zhang, L., Wang, H., Xu, J., Zhu, J. & Ding, K. Inhibition of cathepsin S induces autophagy and apoptosis in human glioblastoma cell lines through ROS-mediated PI3K/AKT/mTOR/p70S6K and JNK signaling pathways. Toxicol. Lett.228, 248–259 (2014).
Khan, M., Rutten, B. P. F. & Kim, M. O. MST1 regulates neuronal cell death via JNK/Casp3 signaling pathway in HFD mouse brain and HT22 cells. Int. J. Mol. Sci.20, 2504 (2019).
Ushio-Fukai, M. Compartmentalization of redox signaling through NADPH oxidase-derived ROS. Antioxid. Redox Signal.11, 1289–1299 (2009).
Chambers, J. W. & LoGrasso, P. V. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem.286, 16052–16062 (2011).
Win, S. et al. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol.62, 1367–1374 (2015).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81871008) and the special project for health talent of Jinlin Province (JLSWSRCZX2023-34).
Author information
Authors and Affiliations
Contributions
M.H. designed the experiment. Z.W and S.H conducted the experiment and wrote the main manuscript text. Z.W and L.J prepared figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, W., Sun, H., Zhao, W. et al. Suppression of JNK pathway protects neurons from oxidative injury via attenuating parthanatos in glutamate-treated HT22 neurons. Sci Rep 14, 25793 (2024). https://doi.org/10.1038/s41598-024-76640-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-024-76640-2
