
Abstract
The fall armyworm, Spodoptera frugiperda, is an invasive, polyphagous pest that threatens approximately 353 plant species across 72 families worldwide. Due to morphological similarities with other noctuid pests during the early larval, pupal, and adult stages, traditional identification methods are labour-intensive and require specialist expertise. Rapid, reliable detection is essential given the pest’s potential for widespread destruction. Through genome-wide in-silico analysis, this study identified a unique region within a signal peptide gene of S. frugiperda, which served as the basis for developing PCR, LAMP, and RPA-based assays for detection. The PCR assay produced a specific 550 bp amplicon for S. frugiperda, showing no cross-reactivity with negative controls. In the LAMP assay, positive samples exhibited a sky-blue colour, while negative samples turned violet when hydroxynaphthol blue dye was used. The RPA assay, with SYBR green dye, displayed bright green in positive samples and brick-red in negatives. Sensitivity tests demonstrated that PCR detected as low as 1 pg/µL, while LAMP and RPA achieved a higher sensitivity of 100 fg/µL. This study introduces the first RPA colorimetric assay for S. frugiperda, providing a time-efficient, cost-effective option that requires minimal equipment, ideal for field detection, thereby supporting timely pest monitoring and management.
Similar content being viewed by others


Introduction
Fall armyworm (FAW), Spodoptera frugiperda (J. E. Smith, 1797), is an invasive polyphagous lepidopteran pest that infests about 353 species of crop plants from 72 families worldwide1. Fast growth, rapid reproduction, ability to migrate long distances, phenotypic plasticity, tolerance of a wide range of environmental conditions, a diverse range of food habits, and a strong potential to compete with native species are the characteristics that facilitate its rapid dispersal and establishment in exotic regions. FAW has emerged as a serious threat to cereal crop productivity, particularly maize and sorghum, two of the major staple food crops of Asia and Africa’s smallholder farmers, jeopardizing regional food security2. The pest feeds on economically important crops such as maize, rice, wheat, millet, barley, and cotton3. Maize is a major economic crop cultivated across 180.63 million hectares in 165 countries with an estimated production of 1,134 million tonnes (https://apeda.gov.in). In India, FAW has emerged as a significant pest of maize, causing production losses up to 34%4. In the early stages of the crop, the young larvae feed on leaves by scraping the chlorophyll content, subsequently, they devour the entire leaf, as well as crop tassels and cobs5. Morphologically, FAW closely resembles other Spodoptera species, especially in their early larval instars, making it difficult to distinguish them6. Female moths are typically tough to identify, especially if they are preserved in poor condition with missing wing scales. Adult specimens are normally identified by dissecting male genital structures and examining them under a microscope. In the maize ecosystem, the life stages of this insect, viz. egg, larva, and pupa, often overlap with those of other lepidopteran pests, adding complexity to its identification of the insect species. The morphological similarity between species makes it challenging to distinguish and necessitates the expertise of a taxonomist or the use of a laboratory-based PCR assay7. As FAW has become a serious pest globally, fast identification, particularly in the early stages, is critical so that management strategies can be planned properly, by utilizing biocontrol agents, semiochemicals, and insecticides4.
To date, several molecular approaches have been developed for species identification, including PCR, species-specific multiplex PCR, and real-time PCR assays7. These techniques, while successful, are time-consuming and often expensive, necessitating highly specialized laboratories and trained staff8. PCR is the most commonly utilized nucleic acid-based molecular diagnostic technology. However, the PCR-based diagnostic method requires an expensive thermal cycler and takes three hours to achieve complete species identification9. The success of PCR as a diagnostic technique relies on the quality and quantity of DNA for amplification, while polysaccharides, ethanol, phenol, proteins, and proteinases can act as inhibitors of the PCR process10. To address these limitations, a specialized technique called loop-mediated isothermal amplification (LAMP) assay was invented11, which is a simple and accurate technique used for pest detection12. In comparison to traditional PCR, LAMP reactions are performed by isothermal incubation. As a result, LAMP assays can be effectively used in field conditions utilizing a simple heat block, with no further procedure necessary for confirmation13. Numerous LAMP-based colorimetric assays have been developed to date for the detection of S. frugiperda4,14,15,16,17,18. LAMP, though simpler than PCR, operates at a higher temperature and thus may not be feasible in the field19,20. Consequently, an alternative, isothermal detection assay was needed for the diagnosis of S. frugiperda that avoids both high temperature and specialized laboratory equipment. Although LAMP assays are simpler than PCR, they require a high operating temperature of 65 °C for optimal DNA amplification. This temperature requires a heating block with a 225 V power supply, which may not be practical for field application.
The Recombinase polymerase amplification assay, which consists of three main components (recombinase, single-stranded DNA binding protein, and strand-displacing polymerase), is an isothermal amplification method that has shown promise for developing diagnostic tests, particularly effective in the detection of insects, plant viruses, and zoonotic diseases19,21. RPA has the same advantages as LAMP, including quick reaction times, high specificity, and sensitivity. However, RPA has a simpler design and more multiplexing capability. Unlike LAMP tests, which require high temperatures (60–65 °C), RPA operates at a consistently lower temperature. With an optimal range of 37–42 °C, RPA can often be performed at ambient room temperatures in tropical countries, eliminating the need for a water bath or heating block and making it a more practical approach for field-based applications19,21. Several recent investigations have demonstrated that incubation-free, body-heat-mediated reverse-transcription RPA assays can be successfully employed in the field19,22.
To develop highly specific RPA detection assays, it is essential to identify distinct, unique regions in the organism’s genome, which can then be used to design specific amplification primers. In this study, a region within a gene coding for signal peptides was employed for this purpose. Signal peptides are short amino-terminal sequences that play a crucial role in directing non-cytoplasmic proteins to their appropriate cellular locations, such as the endoplasmic reticulum (ER) or the plasma membrane. These peptides typically consist of three distinct regions: a positively charged amino-terminal (n-region), a hydrophobic core (h-region), and a polar carboxyl-terminal (c-region) that contains the signal peptidase recognition site. The proper functioning of signal peptides is essential for the translocation of proteins across membranes, as they facilitate the binding of the signal recognition particle (SRP) to the nascent polypeptide, halting translation until the ribosome is targeted to the ER membrane23,24.
Thus, in the current study, an attempt was made to develop a simple, rapid, highly specific, and sensitive RPA assay for detecting S. frugiperda. To the best of our knowledge, this is the first attempt to develop an RPA assay specifically for S. frugiperda detection.
Results
Sample collection and identification based on morphological and molecular characteristics
The species-level identification of the specimens of Spodoptera frugiperda and other lepidopteran species was done by using morphological characteristics of both larvae and adults with the help of catalogues and keys available1,25,26. Based on the characters, insect specimens were confirmed to be S. frugiperda, S. litura, S. exigua, Helicoverpa armigera, Mythimna impura, Sesamia inferens, Chilo sacchariphagus indicus and Chilo infuscatellus. The specimens were photographed using a stereo-zoom microscope equipped with a digital camera (SZX16, Olympus, Japan) (Figs. 1 and 2).

Habitus of different larvae collected in the study; (a) Chilo infuscatellus, (b) Chilo sacchariphagus indicus, (c) Helicoverpa armigera, (d) Mytimna impura, (e) Spodoptera exigua, (f) Spodoptera litura, (g) Spodoptera frugiperda, and (h) Sesamia inferens.

Habitus of adult moths collected in the study; (a) Chilo infuscatellus, (b) Chilo sacchariphagus indicus, (c) Helicoverpa armigera, (d) Mythimna impura, (e) Spodoptera exigua, (f) Spodoptera litura, (g) Spodoptera frugiperda, and (h) Sesamia inferens.
The nucleotide sequences of S. frugiperda and other lepidopteran species obtained after amplification using the COX1 primers were subjected to BLASTn search in NCBI GenBank nucleotide database which showed maximum query cover (100%) with minimum E-value (0.0). The results revealed that the consensus sequences from the current isolates showed more than 99% similarity to corresponding sequences available on the NCBI database. All the sequences were submitted to GenBank and accession numbers were obtained (Supplementary Table S1).
Phylogenetic analysis
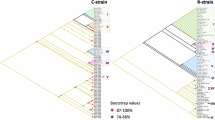
A phylogenetic tree was built utilizing 50 S. frugiperda sequences submitted in GenBank and an additional seven lepidopteran species as an outgroup which were used as negative control for the assays. The current analysis confirmed that significant conservation of amino acid sequences and regional variation in the COX1 is the likely basis for clade split among the S. frugiperda and other lepidopteran species (Fig. 3).

Maximum likelihood unrooted tree representing the phylogeny of Cytochrome c oxidase subunit I (COX1) sequence of the 50 samples of Spodoptera frugiperda and seven other lepidopterans, built with MEGA11 with branch quality evaluated using 1000 bootstrap replicates and visualized with iTOL. Labels correspond to their name in the NCBI genomic database and assembly accession number.
In-silico analysis of Spodoptera frugiperda genome and primer designing
A comprehensive genome-wide in-silico analysis identified a unique target gene in S. frugiperda. The target gene was identified as an uncharacterized protein LOC118262236, located on chromosome number 12, spanning positions 9,582,624 to 9,583,173 bp that encodes a protein containing a signal peptide and a non-cytoplasmic domain. The signal peptide spans amino acids 1 to 25, and the presence of a predicted non-cytoplasmic domain showed its extracellular or membrane-bound localization. However, this protein currently lacks detailed functional annotation in available databases. Based on this sequence, primers for the PCR, LAMP, and RPA were designed. The primer sequences and their corresponding annealing temperatures are provided in Table 1.
Specificity assay of PCR, LAMP, and RPA
The concentration of the isolated DNA from S. frugiperda and other lepidopteran species ranged from 800 to 1100 ng/µL, and the absorbance ratio at 260/280 nm and 260/230 for total DNA was between 1.8 and 2.2. The S. frugiperda sample displayed a clear 550 bp band after amplification at 52 °C. Additionally, lower molecular weight bands were observed for H. armigera and C. sacchariphagus indicus, indicating non-specific amplification. No amplification was detected for S. litura, S. exigua, Mythimna impura, S. inferens, Chilo infuscatellus, or the no-template control (NTC) (Fig. 4A).

Specificity assay for Spodoptera frugiperda detection. (A) PCR specificity assay using S. frugiperda specific primers, (B) LAMP specificity assay using S. frugiperda specific primers with HNB dye, (C) RPA specificity assay using S. frugiperda specific primers with SYBR Green I dye. M- 100 bp marker, Lane 1: Spodoptera frugiperda, Lane 2: Spodoptera litura, Lane 3: Spodoptera exigua, Lane 4: Helicoverpa armigera, Lane 5: Mythimna impura, Lane 6: Sesamia inferens, Lane 7: C. sacchariphagus indicus, Lane 8: Chilo infuscatellus and Lane 9: non-template control (NTC). Original blots/gels are presented in Supplementary Fig. 1.
The LAMP assay was optimized using designed primers at 60 °C for 60 min, amplifying only S. frugiperda genomic DNA. The amplification was visualized using an HNB dye-based colorimetric assay, with the positive samples displaying sky blue colour. In contrast, no amplification was observed in S. litura, S. exigua, H. armigera, M. impura, S. inferens, C. sacchariphagus indicus, C. infuscatellus, and also in the NTC, all of which displayed a characteristic violet colour visible to the naked eye (Fig. 4B).
The specificity assay with the RPA primers showed successful amplification in the S. frugiperda samples at 39 °C for 30 min, while no amplification occurred in the other samples. These results were visualized with SYBR Green I dye, where the S. frugiperda sample displayed a bright green colour, whereas the negative samples from lanes two to nine appeared brick-red under the UV transilluminator. (Fig. 4C).
Sensitivity of PCR, LAMP, and RPA assays
The sensitivity of PCR, LAMP, and RPA assays was evaluated by serially diluted DNA samples, viz., 100 ng, 10 ng, 1 ng, 100 pg, 10 pg, 1 pg, 100 fg, and 10 fg based on the initial nanodrop concentration of 1000 ng/µL. The PCR assay demonstrated a sensitivity threshold of 1pg/µL (Fig. 5A). For the LAMP colorimetric sensitivity assay, amplification of S. frugiperda DNA was achieved with as little as 100 fg/µL of DNA, after 60 min of incubation at 60 °C. The results were visualized based on visible colour changes, with lanes one through seven and nine showing a sky-blue colour (Fig. 5B). The results indicated that the lowest concentration of S. frugiperda DNA necessary for successful amplification using the LAMP assay is 100 fg/µL. Hence, LAMP was found to be ten times more sensitive than PCR, detecting lower concentrations of S. frugiperda DNA in less time. In RPA colorimetric sensitivity assay, the colour change to bright green in lanes one to seven confirmed amplification, while lanes 8, 10, and 11 exhibited brick red colour, indicating no amplification (Fig. 5C). This indicates that the RPA assay achieved sensitivity comparable to that of the LAMP assay, successfully amplifying DNA at concentrations down to 100 fg/µL. Thus, both the LAMP and RPA assays were significantly more sensitive and faster than PCR in detecting S. frugiperda.

Sensitivity assay for detection of Spodoptera frugiperda using different concentrations of template DNA. (A) PCR sensitivity assay with agarose gel electrophoresis, (B) LAMP sensitivity assay with HNB dye, (C) RPA sensitivity assay with SYBR Green I dye. M- 100 bp marker, Lane 1: 100 ng, Lane 2: 10 ng, Lane 3: 1 ng, Lane 4: 100 pg, Lane 5: 10 pg, Lane 6: 1 pg, Lane 7: 100 fg, Lane 8: 10 fg, Lane 9: positive control (Spodoptera frugiperda), Lane 10: negative control (Sesamia inferens); Lane 11: non-template control (NTC). Original blots/gels are presented in Supplementary Fig. 2.
Validation of PCR, RPA, and LAMP assays
For the validation of PCR, RPA, and LAMP assays, we used S. frugiperda specimens collected from the different geographical regions of India, along with other lepidopteran species serving as negative controls and NTC. The PCR assay successfully amplified DNA from the 50 field-collected S. frugiperda samples, yielding an amplicon size of 550 bp. Both the LAMP and RPA assays effectively identified S. frugiperda samples, evidenced by a colour change to sky blue and bright green colour when visualized with HNB dye and SYBR Green I dye (Supplementary Fig. 3), respectively. In contrast, the negative control and NTC displayed no banding patterns and exhibited violet and brick red colour, respectively, in the PCR, LAMP, and RPA assays, indicating no amplification.
Discussion
The fall armyworm, S. frugiperda, is a globally significant pest that can cause severe production losses in agricultural and horticultural crops, including maize, sweet corn, sorghum, cotton, peanut, chickpea, soybean, vegetables, and various fruit crops27,28,29. Early larval instars of many noctuid moths, including S. frugiperda, are morphologically similar, making accurate field identification challenging30. Therefore, rapid and precise detection techniques are essential for identifying S. frugiperda in its early stages to make timely decisions to prevent extensive crop damage14. Other lepidopteran pests, such as H. aarmigera, C. partellus, and S. inferens, add to the complexity of field identification due to overlapping symptoms, including cob damage, leaf pinholes, and defoliation. Various diagnostic methods have been developed for species identification, with molecular techniques offering the most sensitivity and accuracy31. However, molecular approaches, including PCR, though accurate, can be time-consuming and require specialized laboratory equipment8,21.
The present study focused on developing colorimetric loop-mediated isothermal amplification (LAMP) and recombinase polymerase amplification (RPA) assays for the rapid and specific detection of S. frugiperda. The LAMP primers were designed based on a unique region distinct to S. frugiperda, coding for a signal peptide with a non-cytoplasmic domain, that is allowing the assay to be performed on any heating block using a colorimetric approach instead of an agarose gel-based method. The HNB dye-based LAMP assay was shown to be species-specific, sensitive, and cost-effective, and could identify S. frugiperda within 60 min at 60 °C. HNB, an azo dye, undergoes a colour change during DNA amplification because the free phosphate molecules bind to the magnesium present in the dye. This interaction leads to magnesium depletion, turning the reaction mixture sky blue in the positive samples due to successful amplification. In contrast, negative controls, where no amplification occurs, retain the violet colour due to the presence of magnesium4,14,15,16. The assay’s specificity was validated across multiple S. frugiperda samples and other lepidopterans such as S. litura, S. exigua, H. armigera, M. impura, S. inferens, C. sacchariphagus indicus, and C. infuscatellus, demonstrating 100% specificity for S. frugiperda and no cross-reactivity. The assay demonstrated excellent success in detecting S. frugiperda from the field-collected populations, producing results within 60 min, faster than PCR. Park et al.17 successfully used LAMP to identify S. frugiperda, achieving amplification within 45 min at an isothermal temperature of 60 °C. LAMP provides faster and more reliable results even with lower DNA concentrations11. The six LAMP primers developed in this study were found to be highly sensitive to S. frugiperda, detecting genomic DNA at 100 fg, whereas standard PCR is less sensitive, requiring at least 1 pg.
While the LAMP assay offers many advantages, it does have limitations, including the need for specific heating equipment, such as a water bath. Additionally, the assay requires six sets of primers, elevated temperatures (60 °C), and longer incubation times, which can limit its practicality for field applications19,21. In contrast, RPA is rapidly gaining popularity as a simpler and more cost-effective diagnostic tool for detecting insects and diseases21. Hence, to overcome the limitations of the LAMP assay, we developed a colorimetric RPA assay that functions at a lower isothermal temperature (39 °C) and requires only two primers, enhancing its suitability for on-site diagnostics. When amplification occurs in positive samples, SYBR Green I dye chelates with DNA, resulting in a stronger signal. The greater the amplification, the more pronounced the chelation and the stronger the signal, enabling the avoidance of gel electrophoresis and the need for associated documentation systems to confirm DNA amplification15,21. The use of SYBR Green I dye in the RPA assay presents numerous advantages over conventional diagnostic methods, including the elimination of the need for reaction tubes and the capability to swiftly detect amplification with a UV transilluminator8. The RPA assay developed in the study showed a bright green colour upon amplification, while negative samples remained brick red, visible under a UV transilluminator. The RPA assay could amplify S. frugiperda at a low isothermal temperature of 39 °C, which is comparable to room temperature and eliminates the need for a heating device or a thermal cycler. The study also demonstrated that the developed RPA assay is ten times more sensitive than the PCR method, achieving detection sensitivity of up to 100 fg/µL compared to 1 pg/µL in PCR. Like the LAMP assay, the colorimetric RPA diagnostic assay was validated using field-collected samples of S. frugiperda and other lepidopterans. The RPA assay demonstrated 100% specificity for S. frugiperda, with no cross-amplification observed in other lepidopteran species. RPA demonstrates substantial improvements over PCR-based thermal amplification previously employed for diagnosing various insects, significantly reducing both manpower and incubation time. The simplicity and speed of the RPA assay position it as an optimal tool with distinct advantages over other molecular diagnostic techniques. The study’s findings are consistent with previous research, highlighting the assay’s reliability and its accessibility in field applications19.
To date, RPA has not been utilized to identify the invasive pest S. frugiperda anywhere in the world. Therefore, this on-site, rapid, and sensitive method can be executed by non-expert personnel without the need for sophisticated equipment. It will be instrumental in quickly developing pest control strategies and minimizing economic losses in various crops.
Conclusion
To our knowledge, this study represents the first effort to develop an RPA-based diagnostic kit for the early detection of the significant invasive pest, S. frugiperda. We have developed colorimetric detection assays that are highly specific, rapid, robust, and sensitive at a low cost. Both the LAMP and RPA assays are straightforward to perform, require less time, and do not necessitate expensive equipment. Compared to PCR and LAMP assays, the RPA colorimetric assay stands out for its simplicity, speed, and affordability, detecting S. frugiperda at 39 °C within 30 min. Since this assay operates at a temperature comparable to room temperature, it eliminates the need for heating devices, making it easy to implement under field conditions. This early detection capability is crucial for effective pest monitoring and the development of appropriate pest management strategies.
Methods
Sample collection and identification based on morphological and molecular characteristics
Larval and adult specimens of S. frugiperda (n = 50 individuals), along with seven other noctuid species (S. litura, S. exigua, H. armigera, M. impura, S. inferens, C. sacchariphagus indicus, and C. infuscatellus) were collected from various agro-climatic zones across India and morphologically identified using available catalogues and identification keys. The larvae of each species were preserved in 100% ethanol at -80 °C for further studies.
Genomic DNA was extracted from a single larva for each species by using a DNeasy blood and tissue kit (Qiagen, Hilden, Germany), following the manufacturer’s protocols with slight modifications32. The quantity and quality of DNA were determined using nanodrop spectrophotometry (7415 Nano, Jenway, UK) in the 260/280 and 260/230 ratios. The integrity of the DNA was evaluated in a 1.5% (w/v) agarose gel electrophoresis, and gel images were documented using the GelDoc Go gel imaging system (Bio-Rad, California). The DNA samples were stored at -20 °C (Celfrost BFS 350 Upright Deep Freezer, USA) until further investigation. PCR amplification was performed on the COI gene, targeting a 658 bp region near the 5′ terminus, using the COX1 gene primers: forward (LCO1490) and reverse (HCO2198)33. The reaction was conducted in a thermal cycler with a 25 µL reaction volume which included 12.5 µL of 2 ×reSource™ Taq Mix (reSourceTaq DNA Polymerase, 6 mM MgCl2, 2 mM dNTPs) (Source Bioscience, UK), 1 µL each of 10 µM forward and reverse primers, 8.5 µL of molecular biology grade water and 2 µL of template DNA. PCR was carried out with an initial denaturation at 94 °C for 5 min, followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 48 °C for 1 min, and extension at 72 °C for 1 min. The reaction was then extended for 5 min at 72 °C before being stored at 4 °C. The PCR products were separated using 1.5% agarose gel electrophoresis, and gel images were documented using the GelDoc Go gel imaging system.
Phylogenetic analysis
The phylogenetic analysis was performed using Molecular Evolutionary Genetics Analysis (MEGA) version 1134 to explain the relationship between S. frugiperda specimens and other closely related lepidopterans. The amplified COX1 nucleotide sequences of all the samples were aligned using the MEGA11 Clustal-W tool and the neighbour-Joining approach to infer evolutionary history. The evolutionary distance was calculated using the Maximum Composite Likelihood technique using the Kimura-two-parameter model with 1000 bootstrap replications35 representing several base substitutions per site. This analysis included 50 nucleotide sequences of S. frugiperda isolates and seven other lepidopterans, viz., S. litura, S. exigua, H. armigera, M. impura, S. inferens, C. sacchariphagus indicus, and C. infuscatellus, as an outgroup. All ambiguities were deleted for each sequence pair (pairwise deletion).
In-silico analysis of Spodoptera frugiperda genome and primer designing
A genome-wide in-silico analysis was performed to identify the S. frugiperda-specific nucleotide sequence. Nucleotide sequences of S. frugiperda and other lepidopteran species (negative controls) were retrieved from the NCBI database. BioEdit software v.7.0 was used to align various sequences and unigenes, reduce gaps, and also to find the variable regions within the gene. This highly variable region was used to generate primers for the detection assay.
PCR primers were designed using primer3plus (http://primer3plus.jp/e) based on the identified unique sequence. Two LAMP outer primers (F3 and B3), two internal primers (FIP and BIP), and loop forward and backward primers (LF and LB), were designed using NEB® Primer Design Tools. The S. frugiperda-specific RPA primers were manually developed by following the unique requirements for amplification. The GC content (%), predicted melting temperature (Tm), and probable secondary structure forms (hairpins or dimers) of all primers were determined using the Integrated DNA Technologies (IDT) online Oligo Analyzer tool (https://sg.idtdna.com/calc/analyser). The chosen primers’ specificity was validated using NCBI-BLAST for additional assurance. The third step was to test the specificity of the sequences by looking for homology using the BLASTn method (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Specificity test of PCR, LAMP, and RPA assays
A gradient PCR was performed from 48 to 60 °C following the protocol described previously with specific primers designed using the variable region of S. frugiperda. The amplification was visualized in a 1.5% agarose gel, and the gel images were documented using the GelDoc Go gel imaging system.
The LAMP specificity assay reaction used a 25 µL reaction volume, which comprised 12.5 µL of Warmstart buffer, one µL each of outer, inner, and loop primers (F3, B3, FIP, BIP, LF, LB), 5.5 µL of molecular grade water, and three µL of DNA template. The genomic DNA of S. litura, S. exigua, H. armigera, M. impura, S. inferens, C. sacchariphagus indicus, and C. infuscatellus were used as negative controls. The reaction was also conducted with a non-template control (NTC), using the same reaction mixture but without the DNA template. The reaction reagents were incubated for 60 min at 60 °C in a dry bath (GeNei, Bengaluru, India). To detect the test,1 µL of HNB dye was added to 10 µL of amplified LAMP products which were observed with the naked eye for the colour change.
The RPA reactions were carried out using newly designed primer pairs at 39 °C for 30 min using a dry bath (GeNei, Bengaluru, India). The assay was performed in a 50 µL reaction volume, following the protocol indicated in the TwistAmp® Basic kit (TwistDx, Cambridge, UK) with minor modifications. To start the reaction, 5 µL of DNA template was mixed with 2.5 µL each of forward and reverse primers (10 mM), 30 µL of rehydration buffer, 7.5 µL of molecular biology grade water, and 2.5 µL of magnesium acetate. Genomic DNA from S. litura, S. exigua, H. armigera, M. impura, S. inferens, C. sacchariphagus indicus, and C. infuscatellus served as negative controls. A non-template control, omitting DNA, was also included. After incubation, the RPA-amplified products were mixed with 0.5 µL of SYBR green I dye for colorimetric detection using a UV transilluminator. All the reactions were performed three times to check their accuracy.
Sensitivity test of PCR, LAMP, and RPA assays
To check the sensitivity of the PCR, LAMP and RPA assays, S. frugiperda genomic DNA was serially diluted from the original concentration measured by nanodrop. Dilutions were prepared at concentrations of 100 ng, 10 ng, 1 ng, 100 pg, 10 pg, 1 pg, 100 fg, and 10 fg per microlitre. The amplification of the PCR product was visualized using a 1.5% agarose gel. The LAMP colorimetric assay for different dilutions was performed at 60 °C for 60 min, and the amplification was confirmed by using HNB dye. The RPA sensitivity assay was conducted at 39 °C for 30 min using a dry bath, after which the amplified product was mixed with 0.5 µL of SYBR Green I dye for colorimetric detection. In addition to S. frugiperda, a negative control of S. inferens and an NTC were used for the sensitivity assays. To ensure consistency, the sensitivity test assays were repeated three times.
Validation of PCR, LAMP, and RPA assays
To validate the PCR, LAMP, and RPA detection techniques developed in the study, we utilized 50 field-collected specimens along with the specimens of S. frugiperda, including S. litura, S. exigua, H. armigera, M. impura, S. inferens, C. sacchariphagus indicus and C. infuscatellus. The PCR, LAMP, and RPA reaction mixtures were prepared using the standardized protocol as described previously (Fig. 6).

Overview of the development of isothermal nucleic acid-based assays for the rapid and accurate detection of Spodoptera frugiperda larvae in field conditions.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Shylesha, A. N. et al. Studies on new invasive pest Spodoptera frugiperda (JE Smith) (Lepidoptera: Noctuidae) and its natural enemies. J. Biocontrol. 32 (3), 1–7 (2018).
Keerthi, M. C. et al. Biology and oviposition preference of fall armyworm, Spodoptera frugiperda (J.E. Smith) (Lepidoptera: Noctuidae) on fodder crops and its natural enemies from Central India. Int. J. Pest Manag 69 (3), 215–224 (2021).
Keerthi, M. C. et al. Bio-intensive tactics for the management of invasive fall armyworm for organic maize production. Plants 12 (3), 685 (2023).
Sinha, T. et al. Development of a loop-mediated isothermal amplification assay for accurate and rapid identification of Spodoptera frugiperda in maize from India. Cereal Res. Commun. 52 (3), 1069–1079 (2023).
Sharanabasappa, M. et al. First report of the fall armyworm, Spodoptera frugiperda (JE Smith) (Lepidoptera: Noctuidae), an alien invasive pest on maize in India. Pest Manage. Hortic. Ecosyst. 24 (l), 23–29 (2018).
Pogue, M. G. A world revision of the genus Spodoptera Guenee (Lepidoptera: Noctuidae). Mem. Am. Entomol. Soc. 43, 1–202 (2002).
Tsai, C. L. et al. Rapid identification of the invasive fall armyworm Spodoptera frugiperda (Lepidoptera, Noctuidae) using species-specific primers in multiplex PCR. Sci. Rep. 10 (1), 16508 (2020).
Manjunatha, C. et al. Rapid detection of Puccinia triticina causing leaf rust of wheat by PCR and loop mediated isothermal amplification. PLoS ONE. 13, e0196409 (2018).
Prasannakumar, M. K. et al. Loop-mediated isothermal amplification assay for pre-symptomatic stage detection of Xanthomonas axonopodis Pv. punicae infection in pomegranate. Australasian Plant. Pathol. 49, 467–473 (2020).
Radstrom, P. et al. Pre-PCR processing: strategies to generate PCR-compatible samples. Mol. Biotechnol. 26 (2), 133–146 (2004).
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28 (12), 63–63 (2000).
Blacket, M. J. et al. A LAMP assay for the detection of Bactrocera tryoni Queensland fruit fly (Diptera: Tephritidae). Sci. Rep. 10 (1), 9554 (2020).
Blaser, S. et al. From laboratory to point of entry: development and implementation of a loop-mediated isothermal amplification (LAMP)-based genetic identification system to prevent introduction of quarantine insect species. Pest Manag Sci. 74 (6), 1504–1512 (2018).
Congdon, B. S., Webster, C. G., Severtson, D. & Spafford, H. In-field capable loop-mediated isothermal amplification detection of Spodoptera frugiperda (Lepidoptera: Noctuidae) larvae using a rapid and simple crude extraction technique. J. Econ. Entomol. 114, 2610–2614 (2021).
Agarwal, A. A diagnostic LAMP assay for rapid identification of an invasive plant pest, fall armyworm Spodoptera frugiperda (Lepidoptera: Noctuidae). Sci. Rep. 12 (1), 1116 (2022).
Osabutey, A. (ed, F.) Identification of a fall armyworm (Spodoptera frugiperda)-specific gene and development of a rapid and sensitive loop-mediated isothermal amplification assay. Sci. Rep. 12 874 (2022).
Park, J. Development of a rapid isothermal amplification assay for the fall armyworm, Spodoptera frugiperda (Lepidoptera: Noctuidae), using species-specific genomic sequences. Agron 14 (1), 219 (2024).
Kim, J. Development of a simple and accurate molecular tool for Spodoptera frugiperda species identification using LAMP. Pest Manag Sci. 77, 3145–3153 (2021).
Priti, J. S. et al. A rapid field-based assay using recombinase polymerase amplification for identification of Thrips palmi, a vector of tospoviruses. J. Pes T Sci. 94, 219–229 (2021).
Nanditha, S. C. et al. Development of point-of-need colourimetric, isothermal diagnostic assays for specific detection of Bacillus subtilis using shikimate dehydrogenase gene. Folia microbiol. (2024).
Nanditha, S. et al. Development of recombinase polymerase amplification-based colourimetric detection assay for rapid identification of invasive cassava Mealybug, Phenacoccus Manihoti Matile-Ferrero. Saudi J. Biol. Sci. 31 (6), 104005 (2024).
Kumar, R. et al. Development of reverse transcription recombinase polymerase amplification (RT-RPA): a methodology for quick diagnosis of potato leafroll viral disease in potato. Int. J. Mol. Sci. 24 (3), 2511 (2023).
Freudl, R. Signal peptides for Recombinant protein secretion in bacterial expression systems. Microb. Cell. Fact. 17, 52 (2018).
Bardy, S. L., Eichler, J. & Jarrell, K. F. Archaeal signal peptides-a comparative survey at the genome level. Prot. Sci. 12 (9), 1833–1843 (2003).
Ganiger, P. C. et al. Occurrence of the new invasive pest, fall armyworm, Spodoptera frugiperda (JE Smith) (Lepidoptera: Noctuidae), in the maize fields of Karnataka, India. Curr. Sci. 115 (4), 621–623 (2018).
Vinay, N. et al. First report on the occurrence of fall armyworm, Spodoptera frugiperda (JE Smith) (Lepidoptera: Noctuidae) from North Eastern plain zone of Uttar Pradesh, India. J. Exp. Zool. 25, 1598–1604 (2022).
Montezano, D. G. et al. Host plants of Spodoptera frugiperda (Lepidoptera: Noctuidae) in the Americas. Afr. Entomol. 26, 286–300 (2018).
Maino, J. L. et al. Regional and seasonal activity predictions for fall armyworm in Australia. Curr. Res. Insect Sci. 1, 100010 (2021).
Overton, K. et al. Global crop impacts, yield losses and action thresholds for fall armyworm (Spodoptera frugiperda): a review. Crop Protect. 145, 105641 (2021).
Hong, S. J. et al. Moth detection from pheromone trap images using deep learning object detectors. Agriculture 10 (5), 170 (2020).
Choi, B. H. et al. Development of a highly accurate and sensitive diagnostic tool for pyrethroid-resistant chimeric P450 CYP337B3 of Helicoverpa armigera using loop-mediated isothermal amplification. Arch. Insect Biochem. Physiol. 99, 21504 (2018).
Shivakumara, K. T. et al. First report of Bihar hairy caterpillar, Spilarctia obliqua walker (Lepidoptera: Erebidae), infesting sweet basil in India. Int. J. Pest Manag. 4, 1–2 (2021).
Hebert, P. D. N. et al. DeWaard, Biological identifications through DNA barcodes. Proc. R. Soc. B 2024, 270 (2024).
Tamura, G., Stecher, S. & Kumar MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38 (7), 3022–3027 (2021).
Saitou, N. & Nei, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4 (4), 406–425 (1987).
Acknowledgements
The authors would like to express their sincere gratitude to The Director, ICAR-NBAIR, Bengaluru, for the invaluable support provided during the successful execution of this research. The current research is conducted under the CABIN Project on “Network Project on Agricultural Bioinformatics and Computational Biology” ICAR, New Delhi.
Funding
The current research is funded under the CABIN Project on “Network Project on Agricultural Bioinformatics and Computational Biology” ICAR, New Delhi.
Author information
Authors and Affiliations
Contributions
Conceptualization: MC, VT; Methodology: MC, NS; Validation: AK, CMK, SS; Writing - Original draft: SKT, NN, RRS; Funding acquisition: GKJ, Writing – Review and Editing: GGR, MM; Supervision: JP, SNS.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Channappa, M., Thiruvengadam, V., Shivakumar, N. et al. Recombinase polymerase amplification assay for sensitive and rapid detection of invasive fall armyworm, Spodoptera frugiperda. Sci Rep 15, 18026 (2025). https://doi.org/10.1038/s41598-025-02399-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-02399-9
