
Abstract
Extracellular vesicles (EVs) play a crucial role in cell-to-cell communication and serve as a source of biomarkers in several pathologies. In this study, we aimed to characterize plasma-derived EVs isolated by ultracentrifugation (UC) or size exclusion chromatography (SEC) to define the best method for proteomic and functional studies. EVs characterization included nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM), detection of biomarkers by western blotting, and quantitative proteomic analysis. SEC-EVs samples had higher particle and protein concentration, particle-to-protein ratio, and smaller size compared to UC-EVs. A total of 171 proteins were identified through Sequential Window Acquisition of All Theoretical Mass Spectra (SWATH-MS) analysis, with 11 increased in SEC-EVs and 5 in UC-EVs. The proteins increased in UC-EVs are complement proteins and immunoglobulins, while proteins increased in SEC-EVs are apolipoproteins and proteins present in the extracellular space. Functional studies with EVs from activated platelets confirmed that EVs isolated by SEC exacerbate platelet aggregation, whereas there was no effect induced by UC-EVs. The latter suggests that EVs obtained by SEC seem more suitable for platelet-related functional studies compared to those obtained by UC. The results presented pave the way for future clinical orientated studies involving plasma-derived EVs.
Similar content being viewed by others


Introduction
Extracellular vesicles (EVs) are a heterogeneous group of 30 to 1000 nm spherical nanoparticles secreted by cells containing proteins, lipids, RNA, and DNA and coated by a lipid bilayer. In 1946, Chargaff and West were the first to describe the procoagulant activity of what they called “lipoproteins of a very high particle weight” that were obtained from the sediment after a strong centrifugal field (31,000 g)1. Some years later, in 1967, Peter Wolf referred to those lipid-rich particles that can be separated by ultracentrifugation as “platelet dust” due to their coagulant properties and their release from platelets2. In 2012, the International Society for Extracellular Vesicles (ISEV) was created3 and in 2014 the first MISEV guidelines were published4. Many terms have been used to refer to EVs according to their origin, nature, and function such as exosomes, microvesicles, ectosomes, microparticles, or apoptotic bodies5. However, due to the inefficacy to obtain a pure population of each type of EVs, the 2018 MISEV guidelines endorsed “extracellular vesicles” as the generic term for particles released from the cells that are limited by a lipid bilayer and cannot replicate. The guidelines propose terms for EVs subtypes that refer to size, density, molecular composition and/or cellular origin6,7. According to size, EVs can be classified as small (sEVs) and medium/large EVs (mEVs, lEVs); according to density, as low, medium or high density; EVs can also be classified according to their molecular composition (the presence of a specific molecule); and finally, according to the cellular origin, EVs can be classified as exosomes if they are originated via the multivesicular bodies (MVB) or ectosomes (known as microparticles, microvesicles) if they originate from the cell membrane by evagination5,7. However, there is no established cut-off of size or density for vesicles, and most EVs techniques do not isolate EVs according to their biogenesis pathway since there are no universal biomarkers for microvesicles, microparticles, exosomes or other EV subtypes7.
EVs can be isolated from different biofluids such as blood, urine, breast milk, saliva, amniotic fluid, semen, cerebrospinal fluid, ascites, synovial fluid, bile, nasal fluid, uterine fluid, and bronchoalveolar fluid8,9. Almost every cell can release EVs by two main mechanisms: exocytosis of the MVB and budding/blebbing of the plasma membrane10,11. The release of EVs enables cells to communicate with each other in physiological and pathological contexts since the EVs cargo is highly context-dependent12,13 and their ability to reach cells and tissues inaccessible for the donor cells make them a key player in cell-to-cell communication13,14. Once EVs are released, they bind to the plasma membrane of the recipient cell, activate surface receptors and signaling, and ends with vesicle internalization by endocytosis or fusion with the target cell12. Endocytosis mechanisms include clathrin-mediated endocytosis, caveolin-dependent endocytosis, macropinocytosis, phagocytosis, and lipid raft-mediated endocytosis12,15.
EVs play an important role in the development and progression of several diseases such as hematologic and cardiovascular diseases16,17, cancer14,18, autoimmune diseases19, metabolic syndrome20,21, neurodegenerative diseases22, and infectious diseases23. However, they are also involved in the maintenance of homeostasis and regulation of physiological functions such as immune response, hemostasis, wound healing, cell proliferation and differentiation, liver repair, synaptic plasticity and so on9. Due to their importance in genesis, progression, and ending of several pathophysiological processes, EVs are an interesting source of disease biomarkers. For instance, an EV-based surface protein assay of three different subpopulations of EVs present in plasma can predict early stages of hepatocellular carcinoma24 and the presence of microvesicles from plasma containing miRNA-126 and miRNA-199a are used for the prognosis of cardiovascular events in patients with stable coronary artery disease25. Moreover, EVs analysis following liquid biopsies have emerged as a promising non-invasive approach to identify and validate biomarkers that allow a better stratification and diagnosis of cancer26,27.
Blood is a very complex biofluid with high abundance of soluble proteins, cells, lipoproteins and EVs; which makes EVs isolation very challenging28. There are several methods of EVs isolation from human plasma based on several physical and biochemical properties such as (i) size (ultracentrifugation (UC), size exclusion chromatography (SEC), and ultrafiltration); (ii) density (density gradient ultracentrifugation (dUC) made with iodixanol or sucrose); (iii) charge (precipitation with polyethylene glycol, protamine or sodium acetate); or (iv) molecular composition (immunoaffinity chromatography with antibodies that bind to EVs surface proteins)29,30. Nowadays, key isolation methods are UC, dUC, and SEC. However, these methods are not completely able to avoid the presence of blood contaminants and the presence of EVs generated during the isolation method mainly due to platelet activation31. Moreover, the EVs isolation method influences drastically on the type of EVs obtained and their protein cargo32. In conclusion, the need to develop a reproducible isolation method of pure and functional EVs remains crucial.
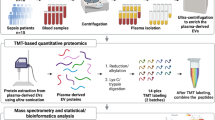
In the present study we aim to propose the best plasma-derived EVs isolation method for platelet-related functional studies comparing the classical UC-based method (improved with an additional washing step in potassium bromide to ensure maximum removal of soluble proteins from plasma), and SEC. Indeed, there is a need for functional studies to investigate the involvement of EVs in intercellular communication (e.g. platelet-platelet, platelet-endothelium, platelet-leukocytes), or for future therapeutic interventions based on EVs. In the present study, EVs isolated by both UC and SEC methods were characterized by nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM) and biomarkers expression; analyzed by proteomics and immunoblotting; and functionally evaluated in platelet aggregation assays (Fig. 1).

Schematic overview of the general workflow depicting the steps followed in the study. Created with Biorender.com.
Results
EVs samples isolated using SEC have a higher yield of EVs, smaller in size, and a greater particle-to-protein ratio compared to UC-EV samples
In order to evaluate the differences in the characteristics of EVs due to the isolation method, we isolated them from each donor by two methods, UC and SEC.
To confirm the EVs integrity and the rounded morphology, EVs isolated by UC and SEC were visualized by TEM (Fig. 2a and b). Moreover, EVs concentration and size were analyzed by NTA (Fig. 2c and d). The particle concentration of SEC-EVs was 6.91 × 109 (4.61 × 109 − 1.14 × 1010) particles per mL of platelet-free plasma (PFP), which was significantly higher than the particle concentration of the UC-EVs (3.75 × 108 (2.83 × 108 – 6.94 × 108) particles/PFP mL) (p = 0.0005) (Fig. 2e). This is also consistent with TEM images (Fig. 2a and b). SEC-EVs were smaller with a mean diameter of 154.24 ± 18.37 nm and a mode of 116.65 (105.70 – 122.31) nm compared to the UC-EVs with a mean of 202.93 ± 18.42 nm and a mode of 157.10 (136.08 – 180.66) nm (p < 0.0001 and p = 0.0024, respectively) (Fig. 2f and g). Protein concentration was higher in SEC-EVs (1.06 ± 0.66 mg/mL) compared to UC-EVs samples (0.46 ± 0.22 mg/mL) (p = 0.0448) (Fig. 2h); while particle-to-protein ratio was also higher in SEC-EVs samples (1.13 × 109 ± 8.01 × 108 particles/µg) compared to UC-EVs (7.83 × 107 ± 3.29 × 107 particle/µg) (p = 0.0224) (Fig. 2i). These differences in particle-to-protein ratio suggest a lower presence of soluble serum proteins in SEC-EVs samples compared to UC-EVs33; however, it could also indicate a greater number of particles without high content of proteins such as lipoproteins34. A 1D SDS-PAGE was performed to determine the protein profile of SEC-EVs (fractions 2–3 and fractions 2–4) and UC-EVs compared to PFP and platelet lysate samples. The band profile was highly similar between SEC-EVs and UC-EVs samples, whereas when compared to PFP and platelet lysate samples, there were marked differences. A significant band was observed at approximately 66 kDa in PFP samples, which likely corresponds to albumin. This band was much less intense in platelet lysates and both vesicles’ samples suggesting low level of plasma contamination. Moreover, the abundant band present in platelet lysates at 42 kDa corresponds to actin, and it was less pronounced in the other samples. However, the bands above 180 kDa in platelet lysates and vesicle samples may correspond to structural proteins such as myosin heavy chain (~ 200–230 kDa), von Willebrand Factor (~ 250 kDa), and spectrin (~ 220–240 kDa) (Fig. 2j). These proteins, present in platelets, have been also reported in EVs35,36,37. Finally, the presence of EVs biomarkers such as integrin αIIb, CD81 and CD9 was assessed by western blotting (Fig. 2k). Moreover, the absence of certain proteins considered contaminants by ISEV guidelines7such as histone H3, was also confirmed by western blotting in EVs samples (see Supplementary Fig. S1 online). Interestingly, a protein marker of large EVs, such as calnexin, was found absent from SEC-EVs samples by western blotting, with a very faint band detected in UC samples (see Supplementary Fig. S1 online); nevertheless, the latter is in line with the subtle differences detected in the size of the population of EVs depending on the isolation method.

EVs characterization by TEM, NTA, protein quantification, SDS-PAGE and western blot. Representative images of (a) SEC-EVs and (b) UC-EVs taken by transmission electron microscopy (TEM). Representative image of an analysis by NTA of (c) SEC-EVs and (d) UC-EVs. Results of analysis by NTA of (e) particle concentration (particles/mL of PFP), (f) mean particle size (nm), and (g) mode particle size (nm) comparing both isolation methods (N = 12). (h) Protein concentration (mg/mL) and (i) particle-to-protein ratio (particle/µg) of SEC-EVs compared to UC-EVs (N = 6). (j) SDS-PAGE representative image comparing the protein profile of PFP, platelets (PLT), UC-EVs (UC), SEC-EVs fractions 2–3 (SEC 2–3), and SEC-EVs fractions 2–4 (SEC 2–4). The protein ladder (PL) indicates molecular weight markers (kDa). (k) Representative western blot images of EVs biomarkers (integrin αIIb, CD81 and CD9). * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Analysis by SWATH-MS identified 16 differentially abundant proteins between UC and SEC isolation methods, while DDA analysis allowed the functional characterization of all proteins identified
To analyze proteome differences between the two EVs isolation methods, two different and complementary Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) approaches were used, a qualitative analysis through data-dependent acquisition (DDA) mode, and a high-throughput and label free quantitative analysis by Sequential Window Acquisition of All Theoretical Mass Spectra (SWATH-MS).
To have a general view of the two types of samples analyzed, an unsupervised multivariate statistical analysis was performed using a principal component analysis (PCA) and the two groups clustered perfectly with an 83.9% (Fig. 3a). A total of 171 proteins were identified and quantified through SWATH-MS analysis with a ppm of 30 and a false discovery rate (FDR) of 1%. The comparison with the Volcano plot analysis revealed the presence of 16 proteins differentially expressed (p < 0.05) between SEC-EVs and UC-EVs with a minimum fold change (FC) of 2 (Fig. 3b). Among these proteins, 11 showed higher abundance in the SEC-EVs group and 5 were more abundant in the UC-EVs group (Table 1). In addition to the Volcano plot analysis, we performed a heatmap cluster analysis of the 16 differential proteins depicting the Z-score of the normalized abundances with proteins arranged in rows and samples in columns. The analysis identified two main clusters that matched with the two isolation methods (Fig. 3c). Among the proteins increased in SEC-EV samples, there was a predominance of apolipoproteins (i.e. APOC4, APOA1, and APOL1), collagen, and proteins secreted and/or present in the extracellular space, while in the UC-EV samples immunoglobulins and complement proteins were more abundant (Table 1).

Proteomic results by SWATH-MS and DDA analysis of SEC-EVs and UC-EVs samples. (a) Principal Component Analysis (PCA) bi-plot depicting the separation of both groups (dark green: SEC-EVs; light green: UC-EVs). (b) Volcano plot representing the proteins identified by SWATH-MS. Proteins with a fold change < 0.5 and p < 0.05 were upregulated in UC-EVs (light green); and proteins with fold change > 2 and p < 0.05 were upregulated in SEC-EVs (dark green). (c) Heatmap illustrates the 16 proteins differentially expressed among EVs samples. Each row represents a protein, and each column represents an EV sample. Protein expression values were log2-normalized, and cluster analysis was performed using Z-score. Color intensity indicates the relative protein abundance across samples, with green colors indicating more abundant levels, while red colors indicate reduced levels. (d) Venn diagram representing the number of protein clusters identified by DDA in the SEC-EVs samples (dark green) compared to the UC-EVs samples (light green). (e) Venn diagram indicating the number of proteins clusters previously described in Vesiclepedia database. (f–k) Bioinformatic Gene Ontology analysis of biological process, molecular function, and cellular components of the proteins identified by DDA in the SEC-EVs samples (f, h, and j; dark green) and in the UC-EVs samples (g, i, and k; light green). Each row represents a process, function or cellular component and they are sorted by the corresponding p value.
A qualitative analysis by DDA showed the presence of 280 proteins identified in total with a minimum requirement of two peptides per protein and identified with less than 1% FDR. Among these proteins, 156 were present in both groups, 50 only in the SEC-EVs group, and 74 only in the UC-EVs group (Fig. 3d). Nearly 80% of the total proteins identified in UC-EVs (78.6%) and SEC-EVs (79.6%) overlapped with the Vesiclepedia database (Fig. 3e). Analyzing in depth the proteins not described in Vesiclepedia, most of them were specific variants of the variable region of certain immunoglobulins and are involved in adaptative immune response and immunoglobulin mediated immune response. The three main biological processes in which all EVs proteins identified are involved are platelet aggregation, negative regulation of blood coagulation, and homotypic cell-cell adhesion. In addition, SEC-EVs proteins are also involved in cholesterol transport (Fig. 3f) whereas UC-EV proteins are also involved in zymogen activation and fibrinolysis (Fig. 3g). Furthermore, according to molecular functions, endopeptidase inhibitor activity is the main molecular function in which SEC-EVs and UC-EVs proteins are implicated (Fig. 3h and i respectively). Moreover, SEC-EVs proteins are involved in lipoprotein particle receptor binding, phosphatidylcholine-Sterol O-acyltransferase activator activity, protease binding and serine-type endopeptidase activity (Fig. 3h); whereas UC-EVs are involved in serine-type endopeptidase and peptidase activity, lipoprotein particle receptor binding and peptidase inhibitor activity (Fig. 3i). Finally, SEC-EVs and UC-EVs proteins are present in collagen-containing extracellular matrix, different secretory and platelet granules and endoplasmic reticulum lumen (Fig. 3j and k).
Validation studies confirm proteomic data: C3 is upregulated in UC-EVs samples whereas APOC4 is downregulated
SWATH-MS results were validated by western blotting in an independent cohort of 6 donors. We focused on the validation of two proteins that were prevalent in each of the groups compared in the proteomic analysis: complement C3 (CO3), upregulated in UC-EVs, and apolipoprotein C4 (APOC4), upregulated in SEC-EVs. In that way, it was confirmed that CO3 and APOC4 are more abundant in UC-EVs and in SEC-EVs, respectively (p = 0.0312 in both cases), in agreement with the SWATH-MS data (Fig. 4).

Western blot validations confirm the upregulation of complement C3 and apolipoprotein C4 in UC and SEC samples, respectively. (a) Representative western blot images of complement C3 and apolipoprotein C4 in an independent cohort of 6 donors from whom EVs were isolated by UC and SEC. Densitometry data for (b) complement C3 and (c) apolipoprotein C4. Western blot analysis was performed in N = 12; * p < 0.05.
SEC-EVs enhance platelet aggregation at different conditions whereas UC-EVs have no effect
To evaluate the functional effects of EVs on platelets, we performed platelet aggregation assays incubating platelets with activated platelet-rich plasma (PRPa)-derived EVs isolated by UC and SEC. Platelet activation induces the release of EVs, which is consistent with the observed increase in the number of EVs isolated from PRPa compared to PRP without previous platelet activation (p = 0.0068; EVs isolated by SEC) (see Supplementary Fig. S2 online).
Due to the low yield of EVs isolated by UC from PRPa from a single donor, we first established the experimental conditions for the functional assays using SEC-EVs. Platelets (2.5 × 108 platelets/mL) were incubated with SEC-EVs in a 1:100 ratio for 15–25 min at 37 °C before platelet activation. Following the addition of platelet agonists, platelet aggregation was measured for 5 min. Thrombin (0.05 U/mL and 0.1 U/mL) and collagen (10 µg/mL and 40 µg/mL) were used to induce platelet aggregation via PAR-1/4 receptor and GPVI receptor signaling pathways, respectively. While SEC-EVs had no effect on platelet aggregation induced by 0.05 U/mL thrombin with a previous EVs pre-incubation time of 15 min (Fig. 5a), when incubating for 25 min, EVs enhanced platelet aggregation (86.92 ± 4.72% compared to 71.75 ± 7.99% with no EVs; p < 0.0001) (Fig. 5b). Moreover, when platelet aggregation was induced with 0.1 U/mL thrombin, the treatment with EVs increased the aggregation levels with EVs incubation times of 15 and 25 min (p = 0.0019 and p = 0.0042, respectively) (Fig. 5c and d). Regarding platelet aggregation induced by collagen, EVs increased aggregation only with the lower concentration of collagen (10 µg/mL). In that case, EVs increased platelet aggregation by 12.78 ± 11.73% (p = 0.0178) after 15 min of incubation and by 14.88 ± 8.82% (p = 0.0020) after 25 min of incubation compared to collagen alone (Fig. 5e and f). However, at a higher concentration of collagen (40 µg/mL), EVs were not able to increase platelet aggregation compared to the control group (Fig. 5g and h).

Effect of SEC-EVs and UC-EVs on platelet aggregation. Aggregation profiles upon platelet activation at the following conditions: platelet-SEC-EVs incubation for (a) 15 min or (b) 25 min and activation with 0.05 U/mL thrombin; platelet-SEC-EVs incubation for (c) 15 min or (d) 25 min and activation with 0.1 U/mL thrombin; platelet-SEC-EVs incubation for (e) 15 min or (f) 25 min and activation with 10 µg/mL collagen; and platelet-SEC-EVs incubation for 15 min (g) or 25 min (h) and activation with 40 µg/mL collagen. (i) Comparison of the effect of SEC-EVs and UC-EVs on platelet aggregation with 0.05 U/mL thrombin and 25 min incubation time at the endpoint of 6 min. (j) Representative aggregation curves of platelets incubated for 25 min with PBS (control, blue), SEC-EVs (dark green), and UC-EVs (light green) and then activated with 0.05 U/mL thrombin. (k) Aggregation levels of platelets incubated for 25 min with PBS (control, blue), SEC-EVs (dark green), and UC-EVs (light green) and then activated with 0.05 U/mL thrombin. Values were annotated every minute; agonist addition was at minute 1. The effect of the SEC-EVs addition on thrombin and collagen-induced platelet aggregation was evaluated in N = 8, except for thrombin 0.05 U/mL and 25 min of incubation, which was evaluated in N = 12. Comparison of SEC-EVs and UC-EVs was performed in N = 6. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
To achieve a platelet-to-EV ratio of 1:100 with EVs isolated by UC, we had to create pools of UC-EVs from PRPa from 4 donors due to the low recovery of EVs with the UC protocol. According to the preliminary results obtained with SEC-EVs, we decided to test platelet aggregation with 0.05 U/mL thrombin and an incubation time of 25 min for the comparison. Thrombin was used instead of collagen because SEC-EVs increased platelet aggregation in a more reproducible manner with that agonist. Moreover, an incubation time of 25 min was established because, independently of the thrombin concentration, SEC-EVs were able to enhance platelet aggregation at that incubation time.
After the incubation of UC-EVs with platelets for 25 min and the addition of 0.05 U/mL thrombin, EVs isolated by UC had no effect on platelet aggregation compared to control (PBS) and SEC-EVs, while SEC-EVs remained increasing platelet aggregation by 16.67 ± 4.20% compared to the control group at the same conditions (p = 0.0005) (Fig. 5j and k).
Analyzing in depth the aggregation curve induced by 0.05 U/mL thrombin after 25 min of platelet-EVs incubation, it is evident that SEC-EVs induced a stronger and faster platelet aggregation since the addition of the agonist compared to the control group without EVs. One minute after thrombin addition, the mean difference between the control and the SEC-EVs group was 25.83 ± 8.70% (p = 0.0001); whereas at the endpoint of the aggregation curve, the difference was of 16.67 ± 4.20% (p = 0.0005). This effect was not observed with UC-EVs (Fig. 5j and k).
Discussion
The present study provides guidelines for platelet functional studies with plasma-derived EVs and compares data from two major EVs isolation methods: UC and SEC. A comparative proteomic study of EVs isolated by both methods is also provided. In recent years, new EVs roles have emerged in several pathophysiological processes given the EVs role in cell-to-cell communication within one tissue and between them. This EVs function made them an important source of biomarkers for different stages in several pathologies. Also, their presence in biofluids such as blood make them available in a non-invasive way for the diagnosis and prognosis of several diseases24. However, these biofluids contain more than EVs, such as soluble proteins, cells, or lipoproteins, so the isolation method is a critical step. Several parameters need to be considered for the latter, such as isolation efficiency, EVs purity, reproducibility, scalability, and overall cost. The complexity of the task led to the development of several protocols for EVs isolation, each of them with different characteristics29. In the present study, we performed a comparison by NTA, TEM, western blotting, proteomics, and functional platelet aggregation assays of plasma-derived EVs isolated by two of the most commonly used methods: SEC and UC.
Plasma preparation before EVs isolation is a critical step to avoid contaminations with platelets and the release of platelet-derived EVs during the procedure, which would interfere in the results obtained28,38. In our study, we performed several low-speed centrifugations in the presence of ACD and prostacyclin to remove all platelets preventing their artificial activation. The EVs variability between donors and within the same donor at different times is very high because EVs are highly context-dependent, and their half-life might last a few minutes13. Therefore, we decided to isolate EVs from the same donor by both methods. Blood is a complex biofluid and the separation of a pure EVs sample from the other particles is a challenging issue due to similar physical properties39. In the UC protocol for EVs isolation, we included an additional step for removal of plasma proteins, such as lipoproteins, by addition of potassium bromide40. Moreover, as it is possible to decide which fractions to collect during EVs isolation by SEC, it was possible to establish a protocol to isolate highly pure EVs collecting fractions 2 and 3 (proteomic analysis) and increasing their yield by adding fraction 4 (functional analysis). Nevertheless, as can be seen in Fig. 2j, the protein profile of EVs isolated by both methods is very similar, in agreement with the proteomic analysis, and even the SEC method seems to have a diminishing of some protein bands present in the plasma lane. The overall profile of EVs proteins differs from platelets and PFP, indicating the success of the EVs enrichment with both methods.
The variability in the characterization of isolated EVs samples from human plasma between different studies and isolation techniques is very high41,42,43,44,45. In our study, we observed that the EVs yield in SEC-EV samples was higher compared to UC-EV samples, in contrast to other studies that suggested that these two methods lead to a similar number of particles44,45; however, other studies reported a higher particle concentration in UC compared to SEC41 or inversely42. NTA is not specific for EVs quantification and there might be some lipoproteins as contaminants that would be quantified too39. However, all studies agreed that in the size distribution profile of SEC-EVs, EVs smaller in size predominate compared to UC-EVs41,42,43,44,45. A wide range of EVs’ size was observed in the NTA analysis; nevertheless, the NTA profiles and TEM images suggest a higher concentration of sEVs (< 200 nm) compared to other larger EVs subtypes, such as apoptotic bodies (ApoBD)46. Moreover, the apparent larger size of EVs in UC samples may be the result of the aggregation of vesicles due to high-speed centrifugations47which can be observed in TEM images (Fig. 2b). According to protein concentration, we determined that UC-EV samples had higher protein concentration compared to SEC-EVs in agreement with other comparison studies16,42,45. Although the bands of some EVs markers, such as integrin αIIb, were weak (Fig. 2k), its presence was confirmed by proteomic analysis in both EVs samples. Moreover, we performed a western blot analysis of the endoplasmic reticulum (ER) protein calnexin and a faint band was only detected in UC-EVs samples compared to SEC-EVs (see Supplementary Fig. S1 online). Nevertheless, calnexin was not detected in the proteomic analyses of EVs samples. ER proteins are not enriched in the sEVs (< 200 nm), however, may be present in larger EVs5 which is consistent with the differences in size observed in the NTA analyses of both isolation methods.
The EVs proteome is highly dependent on the isolation method48,49. After the characterization of EVs, we performed a proteomic analysis using two different LC-MS/MS approaches. SWATH analysis revealed the presence of 16 proteins differentially present between both isolation methods (with a strict 2-fold change cutoff). SEC-EVs samples had a higher concentration of apolipoproteins (APOC4, APOL1, and APOA1), suggesting the presence of lipoproteins as potential particle contaminants, which might correlate with the increased number of particles identified by NTA in these samples compared to UC-EVs. In contrast, in UC-EVs samples predominated the presence of complement proteins (CO3 and C1QC) and immunoglobulin heavy chains (IGHG2 and IGG1). In both cases, some of the above proteins might be considered contaminants. However, recent studies suggest the formation of a protein corona on the surface of EVs isolated from blood plasma, which influences EVs function49,50. One of these studies49 identified the presence of nine EVs corona proteins, where some apolipoproteins such as APOA1, some complement proteins such as CO3, and some immunoglobulin heavy constant chains such as IGHG2 were included. Therefore, we cannot discard that the identification of these proteins is because they are part of the vesicles’ corona. Moreover, EVs proteins identified using the DDA method (comprising samples isolated by both UC and SEC) overlapped in nearly 80% with the Vesiclepedia database (http://microvesicles.org/index.html ). These results correlate with previous studies in which frozen plasma-derived EVs proteins isolated by UC overlapped in a 79% with the Vesiclepedia51.
Complement C3 and apolipoprotein C4 were chosen for validation studies due to their role in different pathologies. A previous study from our laboratory described CO3 as a potential biomarker for cardiovascular disease in patients with obesity40. Regarding APOC4, it is an exchangeable apolipoprotein transferred among high-density lipoproteins and APOB-containing lipoproteins52. The role of APOCs in plasma triglyceride and cholesterol levels regulation has been extensively studied, however, APOC4 mechanism of action is poorly understood52,53. In this context, APOC1, APOC3 and APOC4 are thought to increase plasma triglycerides by interfering in the binding of APOE to the hepatic receptors52. Furthermore, APOC4 has been described as a potential prognostic biomarker for hepatitis B virus-related hepatocellular carcinoma54.
The development of an EVs isolation protocol that maintains them functional is crucial to study EVs functions in vitro and in vivo; and to use them as therapeutic mediators16. Bearing in mind that platelets are the main contributors to circulating EVs in the blood55and that the number of these vesicles is increased in pathologies linked to platelet hyper-reactivity such as obesity or acute myocardial infarction, we decided to carry out the platelet aggregation-based functional assays. Considering 2 × 1010 EVs/mL as the estimated concentration of EVs in plasma56and assuming 2.5 × 108 platelets/mL an average platelet concentration in healthy individuals, the physiological ratio platelets: EVs should be 1:80. However, the activation of platelets (e.g. in pathological circumstances) increases the release of EVs, so we decided to increase the ratio to 1:100 for functional assays. Regarding the platelet-EVs incubation time prior to the addition of agonists, we chose 15 and 25 min because those were the conditions in which we observed better results. These times agree with previous data checking interactions between cells and platelet-derived EVs57.
After setting up the conditions, we proceed with the studies and, interestingly, the functional effect of EVs obtained from PRPa on platelet aggregation was only detected with EVs obtained by SEC. Indeed, SEC-EVs from PRPa were able to enhance platelet aggregation inducing a more intense and faster response, while UC-EVs had no effect at the same conditions. This can be explained by the fact that UC may lead to the formation of EVs aggregates thereby decreasing their activity44; moreover, the EVs protein corona might be also affected by the UC isolation method49. In addition to the above, it is important to investigate the potential presence of soluble protein aggregates in SEC fractions that could impact functional assays; however, the most relevant proteins upregulated in SEC samples (e.g. COL6A1, COL6A3, SVEP1, APOC4, FCGBP) have been identified in plasma-derived EVs and are reported in the Vesiclepedia. Finally, to improve the efficiency of functional analyses in future studies, since EVs samples were frozen in PBS at -80 °C following their isolation, the incorporation of a cryoprotectant might be considered to minimize EVs population changes in size, concentration, membrane proteins, and molecular surface extensions58. Although all frozen EVs aliquots in the present study were treated in the same way, and thawed only once, the above recommendation is of relevance, especially in clinical studies involving freeze-thaw cycles.
Conclusions
In conclusion, the present study demonstrates that the characteristics of plasma-derived EVs differ depending on the isolation method, with SEC-EVs samples having higher particle and protein concentration and particle-to-protein ratio, and smaller size compared to UC-EVs. In line with the above, both SEC and UC protocols do not allow an efficient separation of different EVs subtypes, although there is an enrichment in sEVs, especially in the case of SEC samples. Regarding the proteome profile, although similar, it presents some relevant differences with an increased presence of complement proteins and immunoglobulins in UC-EVs samples, and apolipoproteins in SEC-EVs. Platelet aggregation assays with plasma-derived EVs and different agonists, such as thrombin or collagen, demonstrate that PRPa-derived EVs obtained by SEC can increase platelet reactivity, which is not the case when EVs are obtained by UC. Our data pave the way for future clinically orientated functional studies involving EVs.
Methods
Plasma and platelet isolation
Fresh whole blood was obtained from healthy volunteers who gave their informed consent and were not under chronic medication, or antiplatelet drugs for the previous 10 days. The study was approved by the Galician Clinical Investigation Ethics Committee (Reg. Code No. 209/270) and developed according to the principles outlined in the Declaration of Helsinki.
Blood from cubital vein was collected in 3.2% sodium citrated tubes of 3.5 mL (Vacuette®, Greiner Bio-One International GmbH, Kremsmünster, Austria) and processed in less than one hour after extraction. Fifteen tubes were collected per donor. Whole blood was supplemented with 10% acid-citrate-dextrose (ACD; 78 mM citric acid, 96.6 mM trisodium citrate, and 111 mM glucose) and centrifuged at 200 × g for 20 min. PRP was collected, supplemented with prostaglandin I2 (Cayman Chemical, Ann Arbor, MI, USA), and centrifuged at 1,000 × g for 10 min to obtain the platelet-poor plasma (PPP) and the pellet of platelets. On the one hand, PPP was centrifuged twice at 1,500 × g for 10 min and 15,000 × g for 2 min to obtain the PFP. PFP was frozen at -80 °C until EVs isolation. On the other hand, washed human platelets were obtained by centrifugation of the platelet pellet resuspended in modified Tyrode’s buffer (MTB; 134 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20 mM HEPES, 1 mM MgCl2, 5 mM glucose, pH 7.3) and supplemented with prostaglandin I2 and 10% ACD. Finally, platelets were resuspended in MTB at 2.5 × 108 platelets/mL followed by a resting step of 30 min.
EVs isolation by ultracentrifugation
EV isolation by UC was performed as previously described40. Briefly, 7.2 mL PFP (6 UC tubes) was mixed with HEPES buffer (10 mM HEPES, 5 mM KCl, 1 mM MgCl2, 136 mM NaCl, pH 7.4) and ultracentrifuged at 200,000 × g for 90 min at 4 °C. That centrifugation speed was to fully recover small EVs7. The EVs pellet was resuspended in 0.25 M KBr and incubated for 20 min on ice to remove soluble serum proteins. Samples were ultracentrifuged at 200,000 × g for 90 min at 4 °C and after washing the EVs with 0.22 μm filtered PBS, samples were ultracentrifuged at the same conditions. The pellet of EVs was resuspended in PBS, and frozen at -80 °C until further analysis. In the case of proteomic samples, one of the tubes was for EVs characterization and the other 5 were pooled for proteomic analysis.
EVs isolation by size exclusion chromatography
To remove any cells and large particles, PFP was centrifuged at 10,000 × g for 8 min and supernatant was collected. The qEV10 35 nm Gen 2 Izon columns (Izon Science, Lyon, France) were used for EV isolation. Firstly, the column was flushed with 0.22 μm freshly filtered PBS, and 10 mL of sample (PFP for functional analysis or 6 mL PFP diluted with 4 mL PBS for proteomic analysis) was loaded into the reservoir of the column. After discarding the default buffer volume (DBV), two or three EVs-enriched fractions of 5 mL each (fractions 2–3 or 2–4) were pooled, concentrated using Amicon Ultra 3 kDa (Merck, Darmstadt, Germany), and frozen at -80 °C until further analysis. A small sample of unconcentrated EVs was used for NTA analysis (see below). Two EVs-enriched fractions were collected for proteomic analysis (fractions 2 and 3) and three for functional analysis (fractions 2, 3 and 4).
Nanoparticle tracking analysis
Particle size and concentration of EVs isolated by both methods were evaluated using Nanoparticle Tracking Analysis (Nanosight NS300, Malvern Instruments Ltd, Malvern, UK). EVs samples were diluted (dilution range from 1:2 − 1:1000 in PBS, with a measurement concentration range between 106 particles/mL and 109 particles/mL) and automatically injected into the laser module sample chamber of the NTA system with a continuous syringe pump flow of 40. Particles under Brownian motion were visualized as small points of light moving rapidly using the sCMO8 camera and the Blue488 laser. Five videos of 60 s of the particles were recorded and analyzed using the NTA 3.4 software (Malvern Instruments Ltd). Software settings were as follows: camera level, 12–14; and detection threshold, 7–12.
Transmission Electron microscopy
EVs samples were adsorbed onto a carbon-coated 400-mesh 240 formvar grid for 5 min at RT, stained with 2% uranyl acetate for 1 min at RT, and visualized in the JEOL JEM-1011 transmission electron microscope (JEOL, Ltd) operating at 100 kV.
EVs Lysis and protein quantitation
EVs were lysed with sodium dodecyl sulfate (SDS) (2% final concentration). Protein lysates were quantified by Pierce™ 660 nm protein assay after mixing the ionic detergent compatibility reagent (IDCR, ThermoFisher Scientific, Rockford, IL, USA) with the Pierce 660 nm protein assay reagent (ThermoFisher Scientific). Samples were mixed with the Pierce reagent, incubated for 5 min at RT, and absorbance was measured at 660 nm using a Genesys 20 (4001/4) Spectrophotometer (ThermoFisher Scientific). Protein concentration was assessed from a BSA standard curve.
SDS-PAGE
Five micrograms of SEC-EVs, UC-EVs, PFP (diluted 1:5 in PBS), and platelet protein lysates were supplemented with Laemmli sample buffer (2% w/v SDS, 5% v/v 2-mercaptoethanol, 10% v/v glycerol, 25 mM Tris, pH 6.8, final concentration), boiled at 98 °C for 5 min, centrifuged at 10,000 × g for 2 min, and loaded into a 11% Tris-Glycine SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE). After protein separation, the gel was stained with Sypro® Ruby protein gel stain (Lonza, Rockland, ME, USA) overnight at RT, and visualized using the ChemiDoc MP instrument (Bio-Rad Laboratories, California, USA).
Western blot
EVs, PFP and platelet protein lysates (15 µg) were loaded into 12% SDS-PAGE gels and transferred to a polyvinylidene difluoride (PVDF) membrane at 200 mA at 4 °C for 120 min. The membranes were blocked with 5% bovine serum albumin (BSA) (Merck) in TBS-T (20 mM Tris-HCl (pH 7.6), 150 mM NaCl and 0.1% Tween 20) overnight at 4 °C. Immunodetection was following membrane incubation, with the corresponding primary antibodies for 90 min at RT, and then for 60 min with the secondary antibody conjugated to horseradish peroxidase (HRP). Finally, the Pierce™ ECL Western Blotting Substrate (ThermoFisher Scientific) was used for protein visualization with the ChemiDoc MP instrument (Bio-Rad Laboratories).
The primary antibodies used for EVs characterization were rabbit polyclonal anti-integrin αIIb (sc-15328, Santa Cruz Biotechnology, Dallas, TX, USA), dilution 1:500; mouse anti-CD81 (sc-166029, Santa Cruz Biotechnology), dilution 1:500; and mouse monoclonal anti-CD9 (sc-13118, Santa Cruz Biotechnology), dilution 1:200. The primary antibodies for proteomic validations were mouse monoclonal anti-APOC4 (7-RE36) (sc-134263, Santa Cruz Biotechnology), dilution 1:200; and mouse monoclonal anti-C3 (B-9) (sc-28294, Santa Cruz Biotechnology), dilution 1:200. Secondary antibodies were Pierce® goat anti-mouse IgG HRP conjugated (31430, ThermoFisher Scientific), dilution 1:5,000; and Pierce® goat anti-rabbit IgG HRP conjugated (31460, ThermoFisher Scientific), dilution 1:5,000.
Proteomic analyses by liquid chromatography tandem mass spectrometry (LC-MS/MS)
Sample Preparation for proteomic analysis
Sample preparation for LC-MS/MS was performed as previously described59. Briefly, 25 µg of EVs protein lysates from 6 different donors isolated by both methods (12 samples) were loaded into a 10% SDS-PAGE gel. After concentrating the proteins in a band, the gel was fixed for 30 min with 40% ethanol and 10% (v/v) acetic acid, stained for one hour with Coomassie Blue (ThermoFisher Scientific), and destained in the same fixing solution overnight. Next, protein bands were cut, washed three times with washing solution (50 nM ammonium bicarbonate and 50% methanol) for 10 min at RT at 600 rpm, dehydrated with acetonitrile for 10 min at RT at 600 rpm, and vacuum dried with SpeedVac. Proteins were reduced with 10 mM dithiothreitol (DTT) for 30 min at 56 °C, alkylated with 55mM iodoacetamide for 20 min at RT, and in-gel digested with 0.025 µg/µL sequencing-grade trypsin (Merck) overnight at 37 °C at 450 rpm. Peptides were extracted from the gel using an extraction buffer (60% acetonitrile and 0.5% HCOOH), and vacuum dried with SpeedVac. Peptides were dried and stored at -20 °C until LC-MS/MS analysis. To perform global protein identification and quantification, samples were analyzed using two methods: DDA to increase protein identification, and SWATH-MS for protein quantification. Both analyses were performed using a micro LC system coupled to a high-speed TripleTOF 6600 mass spectrometer (Sciex, Redwood City, CA, USA) following an established procedure60,61,62,63. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE64 partner repository with the dataset identifier PXD056613. Further information is available in the Supplementary Methods.
Systems biology analysis
Volcano plot was created using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA). Heatmap was generated using http://www.heatmapper.ca/expression/ (accessed on 3 February 2024). Venn diagrams were generated using https://www.statskingdom.com/venn-diagram-maker.html (accessed on 8 August 2024) based on the analysis provided by FunRich Analysis software (version 3.1.3) when comparing UC-EVs, SEC-EVs and Vesiclepedia database65. The enrichment analysis was performed using EnrichR software with the following ontology sources: gene ontology (GO) biological processes, molecular functions, and cellular components (accessed on 17 August 2024). A p value < 0.05 was set for significant enrichment.
PRP activation for functional studies
For PRP activation, firstly, whole blood was centrifuged in sodium citrate tubes without the addition of ACD, so the PRP was collected. To achieve effectively platelet aggregation and the release of the greatest number of EVs, platelet aggregation was induced by a cocktail of agonists: thrombin receptor activating peptide 6 (TRAP-6) (Tocris Bioscience, Bristol, UK) and Horm® collagen (Takeda Austria GmbH, Linz, Austria), that activates PAR-1/4 and GPVI receptors, respectively. Agonists concentrations were established to ensure at least a 70% of platelet aggregation, typically 10–20 µM TRAP-6 and 10 µg/mL collagen. PRP aggregation levels were checked in a Chrono-Log® 490-X aggregometer (Chrono-log Corporation, Havertown, PA, USA) to establish the concentration of each agonist (see below Light Transmission Platelet Aggregation). PRP aggregation was performed after the addition of the agonists cocktail, and incubation at 37 °C for 10 min under stirring. Activated PFP (PFPa) was obtained from PRPa after serial centrifugations: 1,000 × g for 10 min at 4 °C to obtain the PPPa, and 1,500 × g for 10 min at RT and 15,000 × g for 2 min at RT to obtain the PFPa. Samples were frozen at -80 °C until further analyses.
Light transmission platelet aggregation
Platelet aggregation levels of PRP after agonist addition were checked before whole PRP activation. For that, 300 µL of PRP were incubated at 37 °C for 4 min, and one additional minute under constant stirring at 1,200 rpm. Platelet aggregation was then measured for 5 min after the addition of the cocktail of agonists (10–20 µM TRAP-6 and 10 µg/mL collagen) in the Chrono-Log® 490-X aggregometer (Chrono-log Corporation). The conditions were established to ensure a PRP aggregation above 70%. PPP was used as a blank.
To assess the EVs effect on platelet aggregation, platelets were incubated with EVs from PRPa isolated by both methods, UC and SEC. 300 µL of washed platelets (2.5 × 108 platelets/mL) were pre-incubated with EVs at a 1:100 ratio for 15–25 min at 37 °C in the aggregometer, and for 1 additional minute at 37 °C with constant stirring at 1,200 rpm. Platelet aggregation was measured for 5 min after the addition of platelet agonists. collagen (10–40 µg/mL) or thrombin (0.05–0.1 U/mL) (Merck).
Statistical analysis
Data are expressed as the mean ± standard deviation (SD) or median (interquartile range) when appropriate. Normality of data was checked using the Shapiro-Wilk normality test. Non-parametric tests were used when the data did not pass the normality tests. Differences between groups were analyzed by paired or unpaired t-test as well as ANOVA as appropriate. P values of < 0.05 were considered significant. Statistical analyses were performed using GraphPad Prism 10.3.0 (GraphPad Software).
Data availability
The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD056613 (https://www.ebi.ac.uk/pride/archive/projects/PXD056613). Unprocessed original blot images are included in the Supplementary Information file. The remaining raw data is available from the corresponding author (angel.garcia@usc.es) on reasonable request.
References
Chargaff, E. & West, R. The biological significance of the thromboplastic protein of blood. J. Biol. Chem. 166, 189–197 (1946).
Wolf, P. The Nature and Significance of Platelet Products in Human Plasma (1967).
Kowal, J., Tkach, M. & Théry, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell. Biol. 29, 116–125 (2014).
Lötvall, J. et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the international society for extracellular vesicles. J. Extracell. Vesicles 3, 26913 (2014).
Théry, C. et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7 (2018).
Maas, S. L. N., Breakefield, X. O. & Weaver, A. M. Extracellular vesicles: Unique intercellular delivery vehicles. Trends Cell. Biol. 27, 172–188 (2017).
Welsh, J. A. et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J. Extracell. Vesicles 13, e12404 (2024).
Puhm, F., Boilard, E. & Machlus, K. R. Platelet extracellular vesicles: beyond the blood. Arterioscler. Thromb. Vasc. Biol. 41, 87–96 (2021).
Yáñez-Mó, M. et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 4, 27066 (2015).
Colombo, M., Raposo, G. & Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell. Dev. Biol. 30, 255–289 (2014).
Akers, J. C., Gonda, D., Kim, R., Carter, B. S. & Chen, C. C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 113, 1–11 (2013).
van Niel, G., D’Angelo, G. & Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell. Biol. 19, 213–228 (2018).
Dixson, A. C., Dawson, T. R., Di Vizio, D. & Weaver, A. M. Context-specific regulation of extracellular vesicle biogenesis and cargo selection. Nat. Rev. Mol. Cell. Biol. 24, 454–476 (2023).
Marar, C., Starich, B. & Wirtz, D. Extracellular vesicles in Immunomodulation and tumor progression. Nat. Immunol. 22, 560–570 (2021).
Mulcahy, L. A., Pink, R. C. & Carter, D. R. F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 3 (2014).
Han, C., Yang, J., Sun, J. & Qin, G. Extracellular vesicles in cardiovascular disease: Biological functions and therapeutic implications. Pharmacol. Ther. 233, 108025 (2022).
Konkoth, A. et al. Multifaceted role of extracellular vesicles in atherosclerosis. Atherosclerosis 319, 121–131 (2021).
Cocks, A. et al. Diverse roles of EV-RNA in cancer progression. Semin. Cancer Biol. 75, 127–135 (2021).
Withrow, J. et al. Extracellular vesicles in the pathogenesis of rheumatoid arthritis and osteoarthritis. Arthritis Res. Ther. 18, 286 (2016).
Pardo, F., Villalobos-Labra, R., Sobrevia, B., Toledo, F. & Sobrevia, L. Extracellular vesicles in obesity and diabetes mellitus. Mol. Aspects Med. 60, 81–91 (2018).
Kumar, V., Kiran, S., Kumar, S. & Singh, U. P. Extracellular vesicles in obesity and its associated inflammation. Int. Rev. Immunol. 41, 30–44 (2022).
Jiang, Y. L. et al. The protective effects of osteocyte-derived extracellular vesicles against Alzheimer’s disease diminished with aging. Adv. Sci. 9, e2105316 (2022).
Xia, B. et al. Extracellular vesicles mediate antibody-resistant transmission of SARS-CoV-2. Cell. Discov. 9, 2 (2023).
Sun, N. et al. HCC EV ECG score: An extracellular vesicle-based protein assay for detection of early-stage hepatocellular carcinoma. Hepatology 77, 774–788 (2023).
Jansen, F. et al. MicroRNA expression in circulating microvesicles predicts cardiovascular events in patients with coronary artery disease. J. Am. Heart Assoc. 3 (2014).
Brokāne, A. et al. Validation of potential RNA biomarkers for prostate cancer diagnosis and monitoring in plasma and urinary extracellular vesicles. Front. Mol. Biosci. 10 (2023).
Whiteside, T. L. Validation of plasma-derived small extracellular vesicles as cancer biomarkers. Nat. Rev. Clin. Oncol. 17, 719–720 (2020).
Nieuwland, R. & Siljander, P. R. M. A beginner’s guide to study extracellular vesicles in human blood plasma and serum. J. Extracell. Vesicles 13, e12400 (2024).
Monguió-Tortajada, M., Gálvez-Montón, C., Bayes-Genis, A., Roura, S. & Borràs, F. E. Extracellular vesicle isolation methods: Rising impact of size-exclusion chromatography. Cell. Mol. Life Sci. 76, 2369–2382 (2019).
Konoshenko, M. Y., Lekchnov, E. A., Vlassov, A. V. & Laktionov, P. P. Isolation of extracellular vesicles: General methodologies and latest trends. Biomed. Res. Int. 2018, 1–27 (2018).
Jia, Y. et al. Small extracellular vesicles isolation and separation: Current techniques, pending questions and clinical applications. Theranostics 12, 6548–6575 (2022).
Abyadeh, M. et al. Proteomic profiling of mesenchymal stem cell-derived extracellular vesicles: Impact of isolation methods on protein cargo. J. Extracell. Biol. 3, e159 (2024).
Webber, J. & Clayton, A. How pure are your vesicles? J. Extracell. Vesicles 2 (2013).
Takov, K., Yellon, D. M. & Davidson, S. M. Comparison of small extracellular vesicles isolated from plasma by ultracentrifugation or size-exclusion chromatography: yield, purity and functional potential. J Extracell. Vesicles 8 (2019).
El-Shennawy, L. et al. Circulating ACE2-expressing extracellular vesicles block broad strains of SARS-CoV-2. Nat. Commun. 13, 405 (2022).
Capriotti, A. L. et al. Proteomic characterization of human platelet-derived microparticles. Anal. Chim. Acta 776, 57–63 (2013).
Yamada, N. et al. Glycosylation changes of vWF in circulating extracellular vesicles to predict depression. Sci. Rep. 14, 29066 (2024).
Lucien, F. et al. MIBlood-EV: Minimal information to enhance the quality and reproducibility of blood extracellular vesicle research. J. Extracell. Vesicles 12 (2023).
Simonsen, J. B. What are we looking at? Extracellular vesicles, lipoproteins, or both? Circ. Res. 121, 920–922 (2017).
Barrachina, M. N. et al. A combination of proteomic approaches identifies A panel of circulating extracellular vesicle proteins related to the risk of suffering cardiovascular disease in obese patients. Proteomics 19, e1800248 (2019).
Williams, S. et al. Comparison of extracellular vesicle isolation processes for therapeutic applications. J. Tissue Eng. 14, 204173142311746 (2023).
Arntz, O. J. et al. An optimized method for plasma extracellular vesicles isolation to exclude the copresence of biological drugs and plasma proteins which impairs their biological characterization. PLoS One 15, e0236508 (2020).
Veerman, R. E. et al. Molecular evaluation of five different isolation methods for extracellular vesicles reveals different clinical applicability and subcellular origin. J. Extracell. Vesicles 10 (2021).
Mol, E. A., Goumans, M. J., Doevendans, P. A., Sluijter, J. P. G. & Vader, P. Higher functionality of extracellular vesicles isolated using size-exclusion chromatography compared to ultracentrifugation. Nanomedicine 13, 2061–2065 (2017).
Brennan, K. et al. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 10 (2020).
Shi, B., Phan, T. K. & Poon, I. K. H. Extracellular vesicles from the dead: The final message. Trends Cell. Biol. 35, 439–452 (2025).
Linares, R., Tan, S., Gounou, C., Arraud, N. & Brisson, A. R. High-speed centrifugation induces aggregation of extracellular vesicles. J. Extracell. Vesicles 4, 29509 (2015).
Zhang, Z. et al. Comprehensive characterization of human brain-derived extracellular vesicles using multiple isolation methods: Implications for diagnostic and therapeutic applications. J. Extracell. Vesicles 12, e12358 (2023).
Tóth, E. et al. Formation of a protein Corona on the surface of extracellular vesicles in blood plasma. J. Extracell. Vesicles 10 (2021).
Wolf, M. et al. A functional Corona around extracellular vesicles enhances angiogenesis, skin regeneration and Immunomodulation. J. Extracell. Vesicles 11, e12207 (2022).
Tsamchoe, M., Petrillo, S., Lazaris, A. & Metrakos, P. Isolation of extracellular vesicles from human plasma samples: The importance of controls. Biotechnol. J. 18, (2023).
Hsu, C. C., Kanter, J. E., Kothari, V. & Bornfeldt, K. E. Quartet of APOCs and the different roles they play in diabetes. Arterioscler. Thromb. Vasc. Biol. 43, 1124–1133 (2023).
Mehta, A. & Shapiro, M. D. Apolipoproteins in vascular biology and atherosclerotic disease. Nat. Rev. Cardiol. 19, 168–179 (2022).
Wang, X. et al. Diagnostic and prognostic significance of mRNA expressions of apolipoprotein A and C family genes in hepatitis B virus-related hepatocellular carcinoma. J. Cell. Biochem. 120, 18246–18265 (2019).
Mezouar, S. et al. Involvement of platelet-derived microparticles in tumor progression and thrombosis. Semin. Oncol. 41, 346–358 (2014).
Johnsen, K. B., Gudbergsson, J. M., Andresen, T. L. & Simonsen, J. B. What is the blood concentration of extracellular vesicles? Implications for the use of extracellular vesicles as blood-borne biomarkers of cancer. Biochim. Biophys. Acta Rev. Cancer 1871, 109–116 (2019).
Koponen, A. et al. Label-free characterization and real-time monitoring of cell uptake of extracellular vesicles. Biosens. Bioelectron. 168, 112510 (2020).
Walker, S. A. et al. Sucrose-based cryoprotective storage of extracellular vesicles. Extracell. Vesicle 1 (2022).
Chantada-Vázquez, M. P., García Vence, M., Serna, A., Núñez, C. & Bravo, S. B. SWATH-MS protocols in human diseases. In Methods in Molecular Biology Vol. 2259, 105–141 (Humana Press Inc., 2021).
Hermida-Nogueira, L. et al. Proteomic analysis of extracellular vesicles derived from platelet concentrates treated with Mirasol® identifies biomarkers of platelet storage lesion. J. Proteom. 210, 103529 (2020).
Hermida-Nogueira, L. et al. Deciphering the secretome of leukocyte-platelet rich fibrin: Towards a better understanding of its wound healing properties. Sci. Rep. 10, 14571 (2020).
López-Valverde, L. et al. Characterization of the plasma proteomic profile of Fabry disease: Potential sex- and clinical phenotype-specific biomarkers. Transl. Res. 269, 47–63 (2024).
Álvarez, J. V. et al. Morquio A syndrome: Identification of differential patterns of molecular pathway interactions in bone lesions. Int. J. Mol. Sci. 25 (2024).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Chitti, S. V. et al. Vesiclepedia 2024: An extracellular vesicles and extracellular particles repository. Nucleic Acids Res. 52, D1694–D1698 (2024).
Acknowledgements
We would like to thank the personnel from the Servizo Vixilancia da Saúde, Universidade de Santiago de Compostela, for their assistance on blood collection from healthy volunteers. We would also like to thank the technical staff from the Center for Research in Biological Chemistry and Molecular Materials (CiQUS) for their support with EVs isolation and NTA analysis. Finally, we would like to thank Dr. Albina Román-Castro, from the Electronic and Confocal Microscopy Unit, Universidade Santiago de Compostela Lugo Campus, for performing the TEM analyses.We gratefully acknowledge grant support from Xunta de Galicia (ED431C 2022/20 and ED431G 2023/02) and European Regional Development Fund (ERDF). E.M.F-S is supported by a predoctoral fellowship from Fundación Instituto Investigación Sanitaria de Santiago de Compostela (FIDIS).
Author information
Authors and Affiliations
Contributions
E.M.F.-S. conceived the experiments, conducted the experiments, analyzed the results and wrote the manuscript; S.B.B. conducted the mass spectrometry experiments, analyzed the results and wrote the manuscript; C.P. conducted the mass spectrometry experiments; A.G. designed research, conceived the experiments, contributed with key equipment and analytical tools, and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fernández-Sáez, E.M., Bravo, S.B., Pena, C. et al. The proteome of circulating extracellular vesicles and their functional effect on platelets vary with the isolation method. Sci Rep 15, 20490 (2025). https://doi.org/10.1038/s41598-025-05374-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-05374-6
