
Abstract
Although neoadjuvant chemotherapy with docetaxel + cisplatin + 5-fluorouracil (CF) has been the standard treatment for stage II and III esophageal cancers, it is associated with severe adverse events caused by docetaxel. Consequently, this study aimed to construct a prognostic system for CF regimens, especially for locally advanced esophageal cancers. Biopsy specimens from 82 patients treated with the CF regimen plus radical surgery were analyzed. Variants in 56 autophagy- and esophageal cancer-related genes were identified using targeted enrichment sequencing. Overall, 13 single-nucleotide variants, including 8 non-synonymous single-nucleotide variants, were identified as significantly associated with esophageal cancer recurrence (p < 0.05). Particularly, variants of ATG2A p.R478C and ULK2 splice-site also showed significant differences in recurrence-free and overall survival. Subsequently, machine learning was used to construct a model for predicting esophageal cancer recurrence based on 21 features, including eight patient characteristics. A Naive Bayes machine-learning model was shown to be highly reliable for predicting esophageal cancer recurrence with an accuracy of 0.88 and an area under the curve of 0.9. We believe that our results provide useful guidance in the selection of neoadjuvant adjuvant chemotherapy, including avoidance of docetaxel.
Similar content being viewed by others

Introduction
Esophageal cancer ranks seventh in terms of incidence and sixth in terms of mortality globally1. The conventional standard treatment for stage II and III esophageal cancer in Japan was neoadjuvant chemotherapy with the cisplatin + 5-fluorouracil (5-FU) (CF) regimen followed by radical surgery2. According to previous reports, the 5-year survival rate for stage II cancers after neoadjuvant chemotherapy with the CF regimen is good at 69%. In contrast, the 5-year survival rate for stage III cancer is poor at 52%, indicating that neoadjuvant chemotherapy with the CF regimen has a limited effect in locally advanced cases3,4. Therefore, more potent neoadjuvant chemotherapy with the docetaxel + cisplatin + 5-FU (DCF) regimen has attracted attention in recent years. A phase III trial (JCOG1109) that compared the superiority of neoadjuvant chemotherapy with DCF regimens versus CF regimens demonstrated an overall survival (OS) advantage in the neoadjuvant DCF arm5. Based on these results, neoadjuvant DCF therapy became the standard treatment in Japan in February 20226. However, exacerbations of adverse events with docetaxel, specifically grade 3 or higher leukopenia (6.7% to 63.8%), neutropenia (23.4% to 85.2%), and hyponatremia (6.2% to 26.0%), have also been reported simultaneously5. This has raised concerns that a high level of chemotherapy-related adverse events may force a series of treatment interruptions, prevent the maintenance of ideal chemotherapy dose intensity, and make it difficult to complete treatment, including subsequent surgery. Additionally, although chemotherapy-induced leukopenia is a known prognostic factor for chemotherapy in some malignancies, it is not currently evident in esophageal cancer7,8,9. Therefore, we believe that the establishment of prognostic markers, particularly for CF regimens for cT3 resectable advanced esophageal cancers will provide useful guidance in the selection of neoadjuvant chemotherapy, including avoidance of docetaxel administration.
Autophagy is a highly regulated process of degradation and recycling of cellular components. The most important feature of autophagy is that it degrades intracellular proteins and organelles and recycles them as a new source of nutrients10. Recently, autophagy has been shown to contribute to the acquisition of chemotherapy resistance in established cancers via intracellular recycling, providing a substrate for metabolism, and maintaining a functional pool of mitochondria11. A high expression of PINK1, an initiator of mitophagy, was associated with poor prognosis in patients with esophageal cancer receiving CF and DCF regimens, suggesting that PINK1-mediated mitophagy contributes to resistance to neoadjuvant therapy12. However, it was not established as a biomarker because high PINK1 protein expression did not correlate with the response to neoadjuvant chemotherapy in biopsy specimens taken before neoadjuvant adjuvant chemotherapy. In contrast, we reported that single nucleotide variants (SNVs) in the PINK1 gene may be a biomarker for non-recurrence in colorectal cancer patients treated with postoperative adjuvant chemotherapy13.
Therefore, this study aimed to construct a prognostic system for CF regimens, particularly for locally advanced cancers. This system can aid in the selection of neoadjuvant chemotherapy, including avoidance of docetaxel administration.
Results
Original target enrichment sequencing
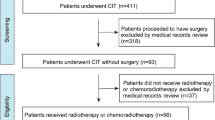
Biopsy specimens from patients with esophageal cancer undergoing radical surgery after treatment with the CF regimen were used to identify SNVs and insertions/deletions (INDELs) for 56 genes related to autophagy and esophageal cancer using targeted enrichment sequencing to construct a prognostic system for the CF regimen. Between May 2012 and June 2020, 91 patients underwent the CF regimen + radical surgery at Saitama Medical University International Medical Center, of which 82 patients were eligible for the study after the required amount of DNA was obtained. The clinical characteristics of the 82 patients are shown in Table 1. Among the 82 patients, the total number of recurrent cases was 45, representing a recurrence rate of 55%. Next-generation sequencing yielded a median of 2,252,009 reads per sample (range: 714,042–6,247,424 reads per sample). Among the designed target bases, 87.1% (range: 40.2%–98.4% per sample) had at least a 15-fold coverage, with a mean coverage of 660-fold (range: 156.11–1,963 fold) per nucleotide in the coding region of the target gene (Fig. 1a,b).

Results of the original target enrichment sequencing in our ESCC clinical cases. (a) The violin plot depicts the distribution of the coverage ratio for each of the 82 multiplexed samples. Percentage of regions with a depth of coverage greater than 15x. (b) The violin plot depicts the distribution of the mean depth for each of the 82 multiplexed samples. (c) Variant filtering thresholds (AltSeq: Alt, any other allele found at that locus). (d) The number of SNVs or INDELs identified by the original target enrichment sequencing is shown. The classification was performed by variant type.
Breakdown of SNVs and INDELs
The next-generation sequencing data analysis only reports the presence of AltSeq (Alt, any other allele found at that locus); therefore, if there are no sequence reads, AltSeq is considered absent. Variant filtering based on criteria such as depth of coverage, variant allele frequency, and AltSeq counts reduces false positive results and ensures confidence in the variants detected. Therefore, by setting thresholds for "Depth of coverage ≥ 15, Variant allele frequency ≥ 5%, and AltSeq ≥ 2," we aimed to ensure that detected variants are supported by a sufficient number of sequencing reads and are present at a significant level to be considered genuine (Fig. 1c). The original target enrichment sequencing for cases of neoadjuvant chemotherapy showed that a total of 12,562 SNVs or INDELs were detected within the target region (Fig. 1d). Among these variants, 7,962 were non-synonymous SNVs, meaning they resulted in amino acid changes in the protein sequences. Additionally, 88 frameshift deletions and 17 frameshift insertions were detected, indicating alterations that cause a shift in the reading frame of the gene. SNVs associated with stop-gain variants were identified in 596 locations (Fig. 1d). These variants result in the premature termination of protein synthesis.
Variants correlated with recurrence
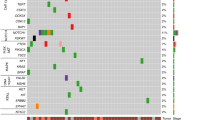
We examined the association between SNVs or INDELs and recurrence. Variants found in samples from 82 patients and recurrence were treated as binary events and subjected to Fisher’s exact test. A total of 13 variants were found to have significant differences with a p < 0.05. Among these variants, eight were nonsynonymous SNVs, four were synonymous SNVs, and one was a splicing site variant (Table 2).
Survival analysis of 13 candidate SNVs in recurrence-free and overall survival
Recurrence-free survival (RFS) analysis was conducted for the 13 identified variants, with significant differences revealed for 6 of the 13 variants (Fig. 2a,b; Supplementary Table S2). OS analysis was also performed for the 13 identified variants, with significant differences observed for two variants: ATG2A p.R478C (p < 0.005) and ULK2 splice-site (p = 0.05) (Fig. 2c,d).

Relationship between variants of ATG2A p.R478C and ULK2 splice-site and ESCC prognosis with CF neoadjuvant chemotherapy. Recurrence-free survival with (a) ATG2A p.R478C or (b) ULK2 1442–1 G > T. Overall survival with or without (c) ATG2A p.R478C or (d) ULK2 1442–1 G > T.
Variant ATG2A p.R478C, extracted as a candidate prognostic factor, showed an association not only with RFS but also with OS in the univariate analysis (p = 0.025 and 0.002, respectively). Variant ULK2 splice-site showed an association with RFS but not with OS (p = 0.016 and 0.054, respectively). Additionally, the multivariate Cox regression analysis revealed that either the presence of variant ATG2A_R478C or the absence of variant ULK2_1442-2G > T (p = 0.046, hazard ratio = 2.076) and conventional open thoracotomy (p = 0.018, hazard ratio = 2.096) were independent prognostic factors for RFS. Likewise, the multivariate Cox regression analysis of OS revealed that either the presence of variant ATG2A_R478C or the absence of variant ULK2_1442-2G > T (p = 0.029, hazard ratio = 2.764) and clinical lymph node metastasis (p = 0.040, hazard ratio = 2.604) were independent prognostic factors for OS (Tables 3, 4). The clinical background characteristics of all 78 patients were compared, and the differences in patient backgrounds between the two identified SNV statuses are shown in Supplementary Table S3. There were no statistically significant differences in patient background factors for either SNV status.
Correlation between pathogenic/likely pathogenic SNVs and recurrence rate
SNVs and INDELs classified as pathogenic or likely pathogenic mutations in ClinVar were found at 212 locations in 22 genes, including 17 esophageal cancer-related genes. Among these variants, 11 frameshift deletions, 15 splicing variants, 95 nonsynonymous SNVs, and 87 stop-gain variants were identified. Fisher’s exact test was conducted for each gene to assess their association with recurrence, but none of the genes showed statistically significant differences (Supplementary Fig. S1). Notably, pathogenic/likely pathogenic variants were observed in 81 of 82 specimens analyzed. This suggests that while these variants were prevalent among the patient samples, they did not appear to influence the recurrence or prognosis significantly.
Machine learning model to predict recurrence
In this study, 13 SNVs were found to be candidate predictors of the recurrence of neoadjuvant chemotherapy with CF regimens for esophageal cancer. However, some specimens had multiple types of SNVs, making it unclear which SNVs should be trusted for prediction (Supplementary Fig. S2a). Therefore, we constructed a recurrence prediction model using machine learning, considering 21 factors including the SNVs found in this study and patient background (Supplementary Fig. S2b).
Fifteen algorithms were trained using the Pycaret classification module, and 21 features, including patient background and SNVs, were used to construct a model with recurrence as the correct answer. The accuracy level of the entire model was compared based on the value of Accuracy, and the result showed the highest value of 0.8467 in Naive Bayes (Supplementary Tables S4 and S5). Furthermore, when tune_model was performed for Accuracy, the value became 0.883, which was defined as the final_model (Fig. 3a). When eight types of patient backgrounds were used as features, the value of Accuracy was 0.546 in Naive Bayes (Fig. 3a). Additionally, other evaluation metrics such as area under the curve (AUC), Recall, Precision, F1 Score, Kappa, and Matthews Correlation Coefficient also demonstrated the superiority of the model using all 21 factors, including SNVs (Fig. 3a). Based on the learning curve results, it is unlikely that overlearning occurred (Fig. S2C). These results showed that incorporating SNVs along with patient background information significantly improves the predictive performance of the model.

Machine learning model to predict recurrence. (a) An indicator to evaluate the prediction of recurrence by Naive Bayes with patient background or patient background + SNVs as features. (Prec.: precision). (b) The ROC (receiver operating characteristic) curve for fine-tuned Naive Bayes. Class 0 means non-recurrence. Class 1 means recurrence. (c) Confusion matrix for fine-tuned Naive Bayes.
A receiver operating characteristic curve for the Naive Bayes algorithm was generated, showing an AUC value of 0.9 for both class 0 (no recurrence) and class 1 (recurrence) (Fig. 3b). This AUC value indicates that this model has excellent discriminative ability to distinguish possible recurrences.
The confusion matrix is one of the representations used in machine learning to evaluate the performance of classification models. From the confusion table, the true positive value was 7, which is higher than the false negative value of 4 (Fig. 3c). Furthermore, it is noteworthy that the number of true negatives was 14, and the number of false positives was 0 (Fig. 3c). These results suggest that the Naive Bayes classification model has high performance in terms of both sensitivity (true positive rate) and specificity (true negative rate) in predicting recurrence in patients with esophageal cancer.
Comparison of the expression of coding RNAs between recurrence and non-recurrence groups
In this study, several prediction systems for the ineffectiveness group with CF regimens were demonstrated. Therefore, we performed a comprehensive expression analysis to understand the biological characteristics of the poor response group to propose a selective treatment. In the analysis of differential gene expression between recurrence and non-recurrence specimens among the 19,972 coding genes, 241 genes were found to have higher expression levels in the recurrence group compared to the non-recurrence group, while 428 genes showed lower expression levels in the recurrence group compared to the non-recurrence group, based on the criteria of fold change ≥ 2 and p < 0.05 (Fig. 4a).

Expression analysis between recurrence and non-recurrence groups. (a) Volcano plot of differentially expressed genes between recurrence (n = 7) and non-recurrence (n = 8) groups. (b, c) Summary of biological processes in gene ontology (GO-BP) and KEGG-pathway analyses of genes with elevated expression in recurrence and (d, e) non-recurrence groups. (f) Principal component analysis performed on 19 genes involved in glutathione metabolism and keratinization. Red specimen names indicate non-recurrence group; blue specimen names indicate recurrence group.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using the DAVID database revealed enrichment of specific biological processes associated with highly expressed genes in the recurrence and non-recurrence groups (Fig. 4b–e; Supplementary Tables S6 and S7). In the recurrence group, eight genes with high expression were enriched in processes related to “glutathione metabolism” (Fig. 4c). In contrast, in the non-recurrence group, 11 genes with high expression were enriched in processes related to “keratinization” (Fig. 4d). These genes, especially those contributing to "glutathione metabolism," may be potential additional therapeutic targets for patients with poor response to the CF regimen.
Possibility of predicting recurrence by analysis of gene set expression levels
Prediction of recurrence was examined by analyzing the expression levels of a set of 19 genes associated with GO “keratinization” and KEGG analysis “glutathione metabolism” terms. Principal component analysis (PCA) was performed on the gene expression data for the 19 genes. Plotting the data showed that the first PCA effectively distinguished between recurrent and non-recurrent outcomes (Fig. 4f). In particular, the non-recurrence group appeared to have less variance.
The expression levels of 37 autophagy-related genes were also examined; however, none of them showed significant differences (Supplementary Table S8). A PCA analysis of the 37 autophagy-related genes was also performed (Supplementary Fig. S3), but no clear distinction of recurrence could be made. These observations suggest that the 37 genes associated with autophagy are not compatible with the prediction of recurrence by expression level analysis.
Discussion
In this study, we aimed to construct a prognostic system for CF regimens for locally advanced cancers. We identified variants of autophagy and esophageal cancer-related genes as biomarkers that could predict the efficacy of CF therapy prior to treatment. Based on these results, we constructed a machine-learning model that can predict postoperative recurrence based on 21 factors consisting of clinical factors and SNVs. Additionally, we established a highly heterogeneous treatment selection system using a machine-learning model.
High expression of PINK1 protein, an initiator of mitophagy, correlated with poor response to neoadjuvant chemotherapy with CF or DCF regimens in surgical specimens from patients with esophageal cancer receiving neoadjuvant chemotherapy; however, no correlation was observed in biopsy specimens taken prior to chemotherapy. Thus, PINK1 protein expression is not considered a predictive biomarker for response to neoadjuvant chemotherapy with CF or DCF regimens in patients with esophageal cancer. In contrast, we identified 13 SNVs in this study as prognostic predictors of neoadjuvant chemotherapy with CF regimen in patients with esophageal cancer. Specifically, SNVs such as p.R478C in ATG2A and the splice site of ULK2 were found to be significant. To the best of our knowledge, this is the first study to report these SNVs as prognostic predictors of esophageal cancer. However, SNVs in the PINK1 gene (c.1018G > A and c.1562A > C), previously suggested to be prognostic factors for 5-FU-based adjuvant chemotherapy in colon cancer, showed no significance in esophageal cancer. Additionally, SNVs identified in esophageal cancer did not show significant differences in colorectal cancer. The fact that prognosis-related SNVs differ between colorectal cancer and esophageal cancer suggests that genetic characteristics affecting treatment response and outcome may differ significantly between different cancer types. This underscores the importance of considering organ-specific genetic profiles when developing personalized medicine approaches.
ATG2A plays an important role in autophagosome formation, an early step in autophagy, and promotes lipid translocation required for autophagosome membrane expansion14. ATG2A is involved in promoting colony formation and cell migration in glioblastoma cell lines by activating autophagy. This suggests its involvement in cancer progression and therapeutic response15. In this study, an ATG2A p.C478 minor variant was found to be significantly associated with worse RFS and OS compared to p.R478 major variant. This suggests that the p.C478 variant may contribute to a worse response to CF regimens by activating autophagy. Additionally, in silico analysis with PolyPhen2 showed that the p.C478 variant is “probably damaging” with a score of 1.000 indicating that the high score is functionally significant. Nevertheless, further biochemical characterization is required to better understand the functional impact and role of the p.C478 mutation in ATG2A on neoadjuvant chemotherapy for esophageal cancer. Such characterization efforts may pave the way for the development of targeted therapies aimed at modulating the activity of this mutant and the identification of biomarkers to guide treatment decisions, especially in relation to CF regimens.
Thirteen variants showed significant differences in this study, predicting recurrence after treatment with CF regimens for esophageal cancer. Therefore, we constructed a machine learning model to predict recurrence using 21 features, including eight patient backgrounds and 13 SNVs, which showed high values of Accuracy = 0.88 and AUC = 0.9. However, we have recognized some limiting factors. First, although the difference in the importance of the features of the 13 SNVs was examined, the Naive Bayes algorithm is difficult to compute directly and has not been shown. Second, in this study, the hyperparameters were optimized collectively using the tune_model function; however, it is also possible to effectively tune individual parameters. Third, the number of samples was limited due to the fact that there were 25 test samples and a single cohort. Consequently, in the future, we aim to improve the decision rate when SNV analysis and pathological image results are added to the machine learning features.
Prognostic prediction of CF regimens by SNVs showed that the ATG2A p.R478C (p < 0.005) and ULK2 splice-site (p = 0.05) variants were candidates. Furthermore, when machine learning was performed considering the information from the 13 SNVs as features, the AUC = 0.9 was high. On the other hand, the confusion matrix in Fig. 3c had four cases of non-recurrence where recurrence was expected. PCA performed on 19 genes that were shown to be differentially expressed in the expression analysis between the recurrence and non-recurrence groups suggested the possibility of predicting the likelihood of recurrence. Since the purpose of RNAseq analysis in this study was to determine the biological characteristics of the recurrence group, the number of samples was limited to 15. However, in the future, the number of specimens should be 82, the same as that in the SNV analysis, to examine whether prognosis can also be predicted by expression analysis. We would also like to consider improving the decision rate when the results of the expression analysis are added to the machine learning features. This study involved an analysis of a small number of cases at a single institution, and external validation using a larger cohort may be warranted in the future.
The KEGG pathway analysis showed that genes contributing to glutathione and drug metabolism were enriched in the recurrence group. The G6PD gene, included in the glutathione metabolism term, encodes a glucose-6-phosphate dehydrogenase, whose contribution to the generation of NADPH (the reduced equivalent) is essential for redox homeostasis and reductive biosynthesis in the cell. Rapidly proliferating cancer cells support cellular requirements for NADPH production and fatty acid and nucleic acid synthesis. As a result, aberrant activation of G6PD leads to increased cell proliferation and adaptation in many types of cancer16,17. One study evaluated cells of 94 patients with lymph node-positive invasive breast cancer using tissue microarrays and two public databases. The results indicated that high G6PD mRNA expression was correlated with significantly poorer OS18. A panel of patient-derived spheroids was established from clinical material of ovarian cancer, and an integrated analysis combining chemotherapy resistance data with gene expression profiling was conducted. The results revealed that cisplatin resistance was significantly associated with increased levels of glucose-6-phosphate dehydrogenase (G6PD) and glutathione-producing oxidoreductase19. Furthermore, the combination of a G6PD inhibitor and cisplatin suppressed spheroid growth in vitro and almost eliminated peritoneal metastases in a mouse xenograft model19. These reports suggest that significantly higher G6PD gene expression in the recurrent group may correlate with a lower efficacy of cisplatin in CF therapy. Therefore, the combination of CF therapy with a G6PD inhibitor may increase the efficacy of neoadjuvant chemotherapy.
However, we believe that our model for predicting the efficacy of CF therapy is a significant finding because it is expected to avoid excessive drug toxicity caused by DCF therapy while simultaneously providing a non-inferior therapeutic effect. Even in cases where CF therapy is deemed ineffective, the therapeutic effect of standard DCF therapy can be expected. In future studies, it is necessary to examine whether our model for predicting the efficacy of CF therapy has any relevance for the efficacy of DCF therapy, especially whether patients who are expected to respond poorly to CF therapy can be rescued by DCF therapy. While our results represent an important step toward clinical application, further validation is needed implementation in clinical practice is possible. If the usefulness of this biomarker is demonstrated by other studies in the future, it will be possible to determine the optimal treatment strategy for individual patients before treatment is initiated through routine next-generation sequencing analysis.
In conclusion, we identified candidate genes that can predict the prognosis of CF regimens and constructed a machine-learning model to further predict recurrence in patients with esophageal cancer treated with neoadjuvant chemotherapy using CF regimens. We believe this information is useful for the selection of neoadjuvant chemotherapy, including the avoidance of docetaxel. Avoidance of unnecessary drugs may be useful not only for patients but also for health economics.
Methods
Tissue samples
This study was approved by the Institutional Review Board of the Saitama Medical University International Medical Center (2022–113 and 2024–055), and the requirement for obtaining informed patient consent was waived due to the retrospective nature of the study.
Ninety-one patients with esophageal cancer who underwent a neoadjuvant CF regimen plus radical surgery at Saitama Medical University International Medical Center between May 2012 and June 2020 were enrolled in the study. Of these, tissue samples from 82 with sufficient DNA content, obtained during biopsy, were included in the analysis (Table 1). The tissue specimens were processed and embedded into paraffin blocks, which were used for further analysis. Tumor cells in the tissue specimen were determined visually and microscopically by a pathologist using hematoxylin and eosin-stained sections.
Target sequencing in our clinical ESCC cases
Fifty-six autophagy- and ESCC-related genes were selected for target-enrichment sequencing. Among them, 37 autophagy-related genes have been reported previously, with some additions or omissions13,20. Whole-genome analysis of 552 ESCC cases identified cancer driver genes, among which 19 genes detected at a high frequency were selected21. The target regions were designed to enrich the exonic regions and exon–intron junctions of the 56 genes (Supplementary Table S1). The mean percentile of the target regions covered was 99.57%.
DNA extraction, library preparation, and data analysis for targeted capture sequencing
Biopsy specimens from 82 patients were analyzed. The assessment and recovery of cancerous tissue regions were performed using previously reported methods13,22. From the extracted DNA, a library of all exonic sequences of the 56 genes was prepared using the HaloPlex Target Enrichment kit (Agilent Technologies, Santa Clara, CA), according to the manufacturer’s instructions. The libraries were high-throughput sequenced on a NextSeq platform (Illumina, San Diego, CA) with 150-bp paired-end reads, according to the manufacturer’s protocol. Identification of SNVs and small INDELs was based on Mutect2 of the Genome Analysis Toolkit, as previously reported13,23. SNVs that showed multiallelic sites were excluded. The violin plot figure was generated using R package (https://bioconductor.org/packages/release/-bioc/html/edgeR.html).
Study design and statistical analyses
A 2 × 2 cross-tabulation table was created with and without variants, and with and without recurrence. A Fisher’s exact test was performed using R, based on the cross-tabulation table, to examine the association between gene variants and recurrence.
The variants with a preliminarily inferred association with postoperative recurrence were examined for their associations with OS and RFS. OS was defined as the period from the date of surgery to the date of death. RFS was defined as the period from the date of surgery to the date of first evidence of recurrence. RFS or OS was censored at the last confirmed date of no recurrence for patients who did not show recurrence or die. OS and RFS were analyzed using the Kaplan–Meier method, and significance was determined by the log-rank test using the open-source Python software package. The median follow-up period for patients surviving without death was 51.2 (range: 7.0–107.8) months. Survival analysis was performed for 78 samples that had sufficient sample volume to allow the analysis of all variants that were candidates for prognostic factors. Univariate and multivariate survival analyses were performed using a stratified Cox proportional hazard model. In the multivariate analyses, covariates were selected using backward elimination. All statistical tests were two-sided, and p < 0.05 was considered statistically significant.
RNA extraction, library preparation, and data analysis for RNA sequence
Total RNA was isolated from formalin-fixed paraffin-embedded (FFPE) biopsy specimens (n = 15) from patients with esophageal cancer treated between 2012 and 2019. Libraries for RNA sequencing were prepared from total RNA as described previously24. Of the 15 specimens, 8 and 7 specimens showed non-recurrence and recurrence, respectively.
The resulting library was sequenced on an Illumina HiSeqX platform (2 × 150-bp read length). Data analysis was based on previously reported methods with some modifications24. Differentially expressed genes were defined as genes that showed a two-fold or greater difference in the expression level of transcripts per million values between the recurrence and non-recurrence groups and a significant difference of p < 0.05. The significance estimate of the differences in gene expression, such as the p-value, was calculated from RSEM’s expected counts using edgeR package in R, and the volcano plot figure was generated using R. For the enrichment analysis, DAVID (https://david.ncifcrf.gov/) was used for GO and KEGG analyses25.
Raw counts from the gene expression data were normalized to log counts per million (log-CPM) and further transformed into z-scores. PCA was performed using R, based on log-CPM (z-score) values.
Machine learning model construction
The model development was performed using the Google Collab platform, and Pycaret was the first package used for machine learning, which required the installation of packages containing pandas, NumPy, warnings, and Pycaret (version: 3.3.2, Moez A. PyCaret: an open-source, low-code machine learning library in Python. https://www.pycaret.org). Feature sets from 8 different patient backgrounds and 21 different patient backgrounds + SNVs were entered separately into Pycaret to build a recurrence prediction model. Pycaret divided each set into training (70%) and independent test cohorts (30%). Each feature set was trained on 15 machine learning models, and the stability of the models was evaluated by performing tenfold cross-validation of the performance of each model and the genomic features that contributed the most to automatic generation in the training cohort. The most accurate models were subjected to hyperparameter tuning, and the tuned models were assembled using the blending method. The missing values in the SNVs were filled using Pandas df. fillna (data.mean()).
Data availability
The datasets extracted and/or analysed during the current study are available in the DDBJ (https://www.ddbj.nig.ac.jp/dra/index-e.html), and the accession numbers were PRJDB19608 and PRJDB19609. However, the above correspondence table linking patient identification codes to personal information is not publicly available due to privacy and ethical constraints. When an application for secondary use of sequence data is submitted, we will ask the applicant to present the purpose of use and review the pros and cons of granting access before making a decision. The person who handles applications for use is Masataka Hirasaki (hirasaki@saitama-med.ac.jp).
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71(3), 209–249. https://doi.org/10.3322/CAAC.21660 (2021).
Kitagawa, Y. et al. Esophageal cancer practice guidelines 2017 edited by the Japan Esophageal Society: part 1. Esophagus 16(1), 1–24. https://doi.org/10.1007/s10388-018-0641-9 (2019).
Yokota, T. et al. Prognostic factors in patients receiving neoadjuvant 5-fluorouracil plus cisplatin for advanced esophageal cancer (JCOG9907). Oncology https://doi.org/10.1159/000381065 (2015).
Ando, N. et al. A randomized trial comparing postoperative adjuvant chemotherapy with cisplatin and 5-fluorouracil versus preoperative chemotherapy for localized advanced squamous cell carcinoma of the thoracic esophagus (JCOG9907). Ann. Surg. Oncol. https://doi.org/10.1245/S10434-011-2049-9/FIGURES/5 (2012).
Kato, K. et al. A randomized controlled phase III trial comparing two chemotherapy regimen and chemoradiotherapy regimen as neoadjuvant treatment for locally advanced esophageal cancer, JCOG1109 NExT study. (2022). https://doi.org/10.1200/JCO.2022.40.4_SUPPL.238.
Kitagawa, Y. et al. Esophageal cancer practice guidelines 2022 edited by the Japan esophageal society: Part 1. Esophagus https://doi.org/10.1007/s10388-023-00993-2 (2023).
Shitara, K. et al. Neutropenia as a prognostic factor in advanced gastric cancer patients undergoing second-line chemotherapy with weekly paclitaxel. Ann. Oncol. https://doi.org/10.1093/ANNONC/MDQ248 (2010).
Hara, H. et al. Influence of preoperative chemotherapy-induced leukopenia on survival in patients with esophageal squamous cell carcinoma: exploratory analysis of JCOG9907. Esophagus https://doi.org/10.1007/s10388-020-00752-7 (2021).
Miyoshi, N. et al. Myelotoxicity of preoperative chemoradiotherapy is a significant determinant of poor prognosis in patients with T4 esophageal cancer. J. Surg. Oncol. https://doi.org/10.1002/JSO.21235 (2009).
Devenport, S. N. & Shah, Y. M. Functions and implications of autophagy in colon cancer. Cells https://doi.org/10.3390/CELLS8111349 (2019).
Wang, Y. et al. The role of mitochondrial dynamics and mitophagy in carcinogenesis, metastasis and therapy. Front. Cell Dev. Biol. https://doi.org/10.3389/FCELL.2020.00413 (2020).
Yamashita, K. et al. High expression of the mitophagy-related protein Pink1 is associated with a poor response to chemotherapy and a poor prognosis for patients treated with neoadjuvant chemotherapy for esophageal squamous cell carcinoma. Ann. Surg. Oncol. https://doi.org/10.1245/S10434-017-6096-8 (2017).
Mihara, Y. et al. PTEN-induced kinase 1 gene single-nucleotide variants as biomarkers in adjuvant chemotherapy for colorectal cancer: A retrospective study. BMC Gastroenterol. https://doi.org/10.1186/s12876-023-02975-1 (2023).
van Vliet, A. R. et al. ATG9A and ATG2A form a heteromeric complex essential for autophagosome formation. Mol. Cell https://doi.org/10.1016/j.molcel.2022.10.017 (2022).
Chu, F. et al. MGCG regulates glioblastoma tumorigenicity via hnRNPK/ATG2A and promotes autophagy. Cell Death Dis. https://doi.org/10.1038/s41419-023-05959-x (2023).
Yang, H. C. et al. The redox role of g6pd in cell growth, cell death, and cancer. Cells 2019(8), 1055. https://doi.org/10.3390/CELLS8091055 (2019).
Yang, H. C. et al. What has passed is prolog: New cellular and physiological roles of G6PD. Free Radical Res. https://doi.org/10.1080/10715762.2016.1223296 (2016).
Debeb, B. G. et al. Histone deacetylase inhibitor-induced cancer stem cells exhibit high pentose phosphate pathway metabolism. Oncotarget https://doi.org/10.18632/ONCOTARGET.8631 (2016).
Yamawaki, K. et al. Integrative analyses of gene expression and chemosensitivity of patient-derived ovarian cancer spheroids link G6PD-driven redox metabolism to cisplatin chemoresistance. Cancer Lett. https://doi.org/10.1016/J.CANLET.2021.08.018 (2021).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy https://doi.org/10.1080/15548627.2015.1100356 (2016).
Moody, S. et al. Mutational signatures in esophageal squamous cell carcinoma from eight countries with varying incidence. Nat. Genet. https://doi.org/10.1038/s41588-021-00928-6 (2021).
Inoue, H. et al. Predicting the radiosensitivity of HPV-negative oropharyngeal squamous cell carcinoma using miR-130b. Acta Otolaryngol. https://doi.org/10.1080/00016489.2021.1897160 (2021).
McKenna, A. et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. https://doi.org/10.1101/GR.107524.110 (2010).
Ichinose, Y. et al. Vimentin-positive invasive breast carcinoma of no special type: A breast carcinoma with lethal biological characteristics. Pathol. Int. https://doi.org/10.1111/pin.13350 (2023).
Kanehisa, M. et al. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. https://doi.org/10.1093/NAR/GKAE909 (2025).
Acknowledgements
We thank the staff of the Division of Analytical Science, Hidaka Branch of Biomedical Research Center, Saitama Medical University for providing research equipment and offering important advice. We would like to thank Editage (www.editage.jp) for English language editing.
Funding
This work was supported by the Hidaka Research Project (4-D-1-04) and the Takeda Science Foundation (MH). MH is a recipient of grants from the Japan Society for the Promotion of Science (JSPS) KAKENHI, grant number 21K06825.
Author information
Authors and Affiliations
Contributions
YM collected patient data, performed statistical analysis, and prepared the manuscript. MH performed chromosomal DNA and total RNA extraction, analyzed and interpreted the data from the next-generation sequencer and prepared the manuscript. YK made sections of FFPE samples. TK determined the tumor area and collected and managed patient FFPE samples. YB is an advisor for machine learning. TS and SY are advisors for bioinformatics analysis. HF, KU, YM, HS and TH designed and supervised the study and helped revise the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
The entire study followed the ethical standards of Declaration of Helsinki and its later amendments. It was approved by the Institutional Review Board of Saitama Medical University International Medical Center (2022–113 and 2024–055). The requirement for informed consent was waived by the Institutional Review Board of Saitama Medical University International Medical Center (2022–113 and 2024–055) in view of the retrospective nature of this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Miyawaki, Y., Hirasaki, M., Kamakura, Y. et al. Developing a machine-learning model to enable treatment selection for neoadjuvant chemotherapy for esophageal cancer. Sci Rep 15, 25669 (2025). https://doi.org/10.1038/s41598-025-11252-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-11252-y
