
Abstract
Benzylisoquinoline alkaloids (BIAs) are essential secondary metabolites produced by Papaver somniferum, widely recognized for their pharmaceutical importance. This study employs transcriptome sequencing and weighted gene co-expression network analysis (WGCNA) to investigate the spatiotemporal expression patterns and regulatory networks of BIA-related genes across developmental stages and organs. A total of 23 co-expression modules were identified, revealing stage- and organ-specific dynamics in BIA biosynthesis. Key genes such as TYDC, PPO, and GsSRK demonstrated distinct regulatory roles during flowering and fruit maturation. Functional enrichment analysis uncovered critical pathways and transcription factors involved in alkaloid production. These findings enhance our understanding of the molecular regulation of BIAs and provide valuable insights for improving alkaloid yield through metabolic engineering and molecular breeding strategies.
Similar content being viewed by others

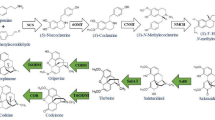

Introduction
Papaver somniferum is a globally significant medicinal plant renowned for producing benzylisoquinoline alkaloids (BIAs), such as morphine, codeine, and papaverine. These alkaloids possess potent analgesic, antitussive, and vasodilatory properties, making P. somniferum an indispensable resource for modern pharmaceuticals1,2,3. BIAs are synthesized through a complex enzymatic cascade, starting from dopamine and 4-hydroxyphenylacetaldehyde, progressing through intermediates such as norcoclaurine and reticuline, and culminating in key metabolites like morphine and sanguinarine4,5,6. The dynamic regulation of these pathways involves a network of biosynthetic genes that exhibit tissue-specific and spatiotemporal expression patterns 7.
While the pharmacological applications of BIAs are well-documented, the regulatory mechanisms underlying their biosynthesis remain incompletely understood, particularly during plant growth and development. As a result, understanding these regulatory networks is essential for optimizing alkaloid yields and advancing metabolic engineering strategies8,9.
Advances in genomics and transcriptomics have provided valuable insights into P. somniferum biology. High-quality genome assemblies have identified key biosynthetic gene clusters controlling alkaloid production10,11. For example, Guo et al. employed 10X genomics technology to assemble the genome of P. somniferum, revealing supergene clusters tightly associated with alkaloid biosynthesis and distinct expression patterns across tissues and developmental stages12. Similarly, Pei et al. demonstrated that long terminal repeat retrotransposons (LTR-RTs) contributed to the expansion of secondary metabolite-related gene families, highlighting the evolutionary significance of genome plasticity in P. somniferum13,14.
Transcriptomic studies have further identified key biosynthetic genes, such as TYDC, PPO, and 7OMT, which are crucial for the production of morphine and sanguinarine. These genes exhibit tissue- and stage-specific expression patterns, with significant upregulation in roots, stems, and fruits during specific developmental phases15,16,17,18. GO and KEGG enrichment analyses have also revealed that these genes are co-regulated with pathways related to stress responses and secondary metabolism, underscoring their multifunctional roles in plant growth and defense19,20,21,22. Previous studies have reported developmental regulation of BIA biosynthesis in opium poppy, highlighting the spatial and temporal variation in the expression of pathway enzymes, such as TYDC and COR23. However, these studies largely focused on individual genes or tissues, and lacked a systems-level perspective integrating developmental and tissue-specific regulation.
Weighted Gene Co-Expression Network Analysis (WGCNA) has emerged as a powerful tool for investigating complex regulatory networks in plants24. By identifying co-expressed gene modules associated with specific traits or biological functions, WGCNA facilitates the discovery of candidate genes for functional validation. Previous studies have successfully applied WGCNA in P. somniferum, linking transcriptional regulation with alkaloid production and uncovering novel regulatory networks25,26,27,28. Despite these advances, a comprehensive understanding of the transcriptional networks regulating BIA biosynthesis across developmental stages and tissues is still lacking, necessitating further exploration.
Understanding the regulatory networks and dynamic expression patterns of genes associated with benzylisoquinoline alkaloid (BIA) biosynthesis is critical for revealing the molecular mechanisms underlying alkaloid production in P. somniferum. This research integrates transcriptomic data and co-expression network analysis to explore the spatial and temporal regulation of key biosynthetic genes. By identifying co-expression modules and candidate regulatory genes, the study provides insights into the transcriptional control of BIA biosynthesis, emphasizing the influence of developmental signals and tissue specificity. Notably, the opium poppy samples used in this study belong to a Chinese Herbal Medicine (CHM)-type cultivar, a traditional and locally preserved germplasm in China. Unlike extensively bred human-selected varieties, such as thebaine-rich or morphine-deficient cultivars, this CHM-type poppy has not undergone modern artificial selection and thus retains more primitive and distinctive genetic features. This unique origin enhances the value of our study by enabling the investigation of BIA biosynthetic regulation in a genetically unmodified background. Such germplasm offers a rare opportunity to uncover native transcriptional patterns and metabolic architectures that may be masked or altered in domesticated lines, thereby contributing to both fundamental plant science and applied pharmaceutical research.
Results
Transcriptome sequencing and gene expression analysis
This study performed 150 bp paired-end transcriptome sequencing on roots, stems, leaves, and fruits (ovaries) of P. somniferum across developmental stages, generating 2.32 billion raw reads. After quality control, 2.26 billion high-quality clean reads were retained, yielding 339.04 Gbp of data suitable for downstream analysis. A total of 56,232 transcripts with FPKM ≥ 1 were identified, confirming high data quality and consistency (Fig. 1A).

Comparative Transcriptome Sequencing and Gene Expression Analysis of Poppy at Different Developmental Stages and Organs. (A) Boxplot of gene expression levels across different organ samples during poppy development. The x-axis represents sample names, while the y-axis denotes log10(FPKM + 1). Each boxplot displays five statistical measures from top to bottom: maximum, upper quartile, median, lower quartile, and minimum. (B) Principal Component Analysis (PCA) results for sample groups. (C) Pearson correlation heatmap of transcriptomic samples across different developmental stages. (D) Hierarchical clustering of samples. Note: R = Root; S = Stem; L = Leaf; C = Ovary; F = Fruit; VP = Vegetative Stage; FLP = Flowering Stage; FRDP = Fruit Development Stage; FRMP = Fruit Maturation Stage.
Principal Component Analysis (PCA) revealed distinct clustering of samples by organ and developmental stage, with root samples showing significant differences between vegetative and reproductive stages. Leaf samples exhibited consistent gene expression during the vegetative stage but increased similarity between fruit development and maturation stages. Stem and fruit samples demonstrated high reproducibility across stages (Fig. 1B).
Pearson correlation analysis highlighted organ-specific expression profiles in roots and leaves, whereas stems and fruits showed less specificity. Root samples exhibited strong correlations within the reproductive stages, while leaves showed the highest correlation during the vegetative stage. Fruit samples demonstrated robust biological reproducibility during fruit development (Fig. 1C).
Hierarchical clustering identified four outlier samples: two root samples from the vegetative stage, one ovary sample from the flowering stage, and one stem sample from the fruit development stage. These samples were excluded, leaving 41 datasets for further analysis (Fig. 1D). This dataset provides a robust foundation for subsequent co-expression network analysis.
Construction and functional module analysis of gene co-expression networks
WGCNA was performed using 41 transcriptome samples, covering 43,557 transcripts. The network was constructed with a correlation coefficient R2 = 0.85 and a soft threshold beta = 12, determined by the pickSoftThreshold function (Fig. 2A). This ensured a scale-free topology with high connectivity. Dynamic tree cutting identified 28 co-expression modules, represented by distinct colors in the dendrogram (Fig. 2B-D).

Co-expression Network Construction and Module-Stage Correlations. (A) Soft threshold selection plot illustrating the relationship between the soft threshold β and network properties, with β = 12 selected as the optimal value to achieve scale-free topology. (B-D) Hierarchical clustering dendrograms of gene modules for blocks 1, 2, and 3, showing the grouping of co-expression modules identified during network construction. The vertical axis represents the proportion of genes, and the horizontal axis displays the gene modules, each distinguished by unique colors. (E) Heatmap of module correlations with developmental stages and samples. The heatmap shows the correlation coefficients between gene co-expression modules and developmental stage samples, with red indicating positive correlation and green indicating negative correlation. The intensity of the color represents the strength of the correlation. Notably, key modules exhibit stage-specific correlations, reflecting the temporal and spatial dynamics of gene regulation during P. somniferum development. VP = Vegetative Stage; FLP = Flowering Stage; FRDP = Fruit Development Stage; FRMP = Fruit Maturation Stage.
Correlation analysis revealed 23 modules with correlation values exceeding 0.5 across developmental stages, comprising 39,224 genes. Each developmental stage and organ were associated with specific modules (Fig. 2E).
During the vegetative stage, organ-specific regulation was evident. The greenyellow module (r = 0.84, 569 genes) in roots was enriched in pathways linked to nutrient uptake and cell wall organization. The yellow module (r = 0.88, 2,861 genes) in stems supported structural integrity and primary metabolism. In leaves, the brown module (r = 0.64, 4,981 genes) regulated photosynthesis and energy production.
In the flowering stage, regulatory complexity increased. Roots were dominated by the turquoise module (r = 0.63, 187 genes), linked to hormonal signaling and root elongation. Stems showed strong associations with the black module (r = 0.85, 1,250 genes), supporting structural development and metabolite transport. Leaves were regulated by the lightyellow module (r = 0.61, 189 genes), associated with photosynthetic activity, and the grey60 module (r = 0.66, 211 genes), enriched in stress responses. Ovaries exhibited regulatory input from the magenta module (r = 0.79, 580 genes) and the midnightblue module (r = 0.75, 320 genes), both critical for reproductive processes.
During fruit development, module interactions became more intricate. The royalblue module (r = 0.54, 13,777 genes) dominated root-related pathways, focusing on nutrient acquisition and stress adaptation. Stems were regulated by the cyan module (r = 0.84, 326 genes) and the skyblue module (r = 0.50, 61 genes), which supported vascular transport and structural reinforcement. In leaves, the orange module (r = 0.78, 107 genes) enhanced photosynthetic efficiency, while the green module (r = 0.56, 583 genes) contributed to secondary metabolism. Fruits were regulated by the blue module (r = 0.86, 2,583 genes), critical for biosynthetic activity, with supplementary input from the darkgrey module (r = 0.53, 293 genes).
In the fruit maturation stage, regulatory networks exhibited high specialization. The pink module (r = 0.72, 653 genes) in roots supported nutrient remobilization and stress responses. The red module (r = 0.54, 1,744 genes) in stems maintained structural integrity and metabolite transport. Leaves were regulated by the lightgreen module (r = 0.55, 209 genes), maintaining photosynthetic activity. In fruits, the purple module (r = 0.89, 577 genes) dominated alkaloid biosynthesis, with additional input from the lightcyan (r = 0.80, 293 genes), darkgrey (r = 0.73, 107 genes), and darkred (r = 0.54, 175 genes) modules.
These results provide a comprehensive framework of stage- and organ-specific regulatory mechanisms in P. somniferum, highlighting the transcriptional coordination underlying growth, development, and alkaloid biosynthesis.
GO and KEGG enrichment analysis of developmentally relevant gene modules
To explore the functional roles of genes in P. somniferum, GO and KEGG enrichment analyses were performed on co-expression modules, revealing dynamic, organ- and stage-specific regulatory mechanisms underlying growth, development, and secondary metabolism (Fig. 3, Supplementary Figure S1-S8).

Expression of genes related to the growth and development of P. somniferum as analyzed by WGCNA. Note: R: Root; S: Stem; L: Leaf; C: Ovary; F: Fruit.
In the vegetative stage, the enriched pathways reflected the specialized roles of roots, stems, and leaves in supporting early growth. Root-associated modules showed significant enrichment in processes related to cell division, elongation, and oxidative stress responses, indicating their role in promoting cellular expansion and environmental adaptation. Stems were primarily involved in resource distribution and structural growth, with enrichment observed in pathways related to carbohydrate and lipid metabolism as well as substance transport. Leaves exhibited robust photosynthetic activity, as evidenced by enrichment in chlorophyll metabolism, thylakoid formation, and photosystem-related pathways, underscoring their critical function as energy producers during this phase.
The transition to the flowering stage marked a shift in regulatory networks to accommodate reproductive and metabolic demands. Roots displayed increased activity in DNA synthesis and repair pathways, reflecting their ongoing cell proliferation to support heightened metabolic requirements. Stems exhibited a dual regulatory role, contributing to amino acid biosynthesis and flowering-related hormone production, such as flavonoids and indole compounds, while also engaging in secondary metabolic pathways associated with BIA biosynthesis. Leaves continued to sustain photosynthesis while becoming increasingly involved in sugar metabolism and secondary metabolic processes. In ovaries, enrichment in pathways related to flower regulation, pollen development, and early fruit formation highlighted their central role in reproductive success and the initiation of fruit development.
As the plant entered the fruit development stage, regulatory complexity increased, with modules coordinating growth and metabolic processes across organs. Roots maintained essential physiological functions, including DNA replication and RNA transcription, while stems played a pivotal role in structural reinforcement, material transport, and defense mechanisms. Leaves actively contributed to secondary metabolism, synthesizing metabolites essential for fruit development. Within the developing fruits, the enriched pathways reflected their expanding size and complexity, with BIA biosynthesis becoming more pronounced during this stage.
In the fruit maturation stage, the regulatory networks displayed further specialization, supporting both metabolic and defensive processes. Roots became more focused on pathogen defense and signal transduction, while continuing to contribute to alkaloid biosynthesis. Stems facilitated material redistribution and structural maintenance, with a notable emphasis on alkaloid metabolism. Leaves balanced roles in pathogen defense and secondary metabolism, maintaining significant contributions to BIA biosynthesis. Fruits, serving as the central hub of metabolic activity, exhibited prominent enrichment in lipid synthesis and alkaloid biosynthesis pathways, marking the culmination of metabolic processes and the peak of alkaloid accumulation.
These enrichment analyses provided a comprehensive overview of the biological processes and metabolic pathways that drive P. somniferum development. The results revealed distinct stage-specific and organ-specific regulatory mechanisms, offering new insights into the molecular basis of growth, reproduction, and alkaloid biosynthesis. This understanding lays a foundation for strategies to optimize alkaloid yields through targeted genetic and metabolic interventions.
Dynamic Expression of BIA-Related Genes During Development
The biosynthesis of BIAs in P. somniferum is governed by a network of key genes whose expression varies across developmental stages and organs. Heatmap analyses revealed significant spatiotemporal differences in gene activity, particularly during flowering, fruit development, and maturation stages, reflecting the coordinated regulation of primary and secondary metabolism (Fig. 4A).

BIA Biosynthesis Gene Expression and Localization During Development. (A) Heatmap of BIA biosynthesis-related gene expression during P. somniferum development. The heatmap shows gene expression patterns across roots R, stems S, leaves L, ovaries C, and fruits F during vegetative stage VP, flowering stage FLP, fruit development stage FRDP, and fruit maturation stage FRMP. NA indicates genes with no annotation. (B) Localization of BIA-related genes during development. Red indicates genes significantly enriched in the BIA biosynthesis pathway, black represents genes enriched in other pathways, and blank indicates genes with no enrichment.
During the flowering stage, reproductive organs such as leaves and ovaries exhibited high expression of BIA-related genes, including TyrAT2, TYDC, and PPO. These genes, which catalyze the conversion of tyrosine into intermediates for morphine, codeine, and papaverine synthesis, were significantly enriched in BIA biosynthesis pathways 29,30. Leaves maintained their photosynthetic functions while also engaging in secondary metabolism, highlighting their dual role in energy production and alkaloid biosynthesis. Ovaries concentrated on reproductive development, with gene enrichment in pathways related to flower regulation and early fruit formation, demonstrating organ-specific metabolic allocation.
As the plant transitioned to the fruit development stage, gene expression patterns diversified to support both structural growth and alkaloid production. Stems displayed increased activity in pathways linked to substance transport and pathogen defense, emphasizing their role as conduits for nutrient and metabolite distribution. Fruits progressively activated genes involved in size expansion and alkaloid synthesis, supported by leaves, which remained active in secondary metabolism. Roots primarily focused on DNA replication and RNA transcription while providing foundational metabolic support.
In the fruit maturation stage, regulatory networks became more specialized, with fruits emerging as key sites for lipid metabolism, energy storage, and alkaloid biosynthesis. Genes such as CODM, SalR, and 7OMT, though not exclusive to primary BIA biosynthesis, were significantly involved in secondary metabolite pathways, reflecting their broader roles in metabolic regulation 31. Roots and stems intensified their contributions to pathogen defense and alkaloid metabolism, highlighting their adaptive responses to environmental and developmental cues. Meanwhile, leaves maintained their dual roles in photosynthesis and secondary metabolism, ensuring metabolic balance during this critical phase.
In contrast to the reproductive stages, the vegetative stage exhibited isolated activity of BIA-related genes such as TYDC in roots. These genes lacked co-expression with other metabolic pathways, suggesting limited involvement in secondary metabolism during early growth. This finding underscores the dynamic nature of BIA biosynthesis, where gene activity is tightly aligned with developmental and metabolic priorities.
Overall, the coordinated expression of BIA-related genes across organs and developmental stages illustrates a finely tuned regulatory network driving alkaloid biosynthesis in P. somniferum. During the reproductive stages, genes involved in the initial steps of BIA biosynthesis (TyrAT2, TYDC, and PPO) exhibited significant co-expression and pathway enrichment (Fig. 4B). Conversely, genes associated with downstream biosynthesis, such as CODM, SalR, and 7OMT, displayed expression patterns more aligned with secondary metabolic pathways. These results provide critical insights into the molecular mechanisms underlying BIA biosynthesis, offering a foundation for further exploration of its regulatory processes.
Differential Expression Analysis of BIA-Related Genes During Fruit Development and Maturation
The transition from the fruit development stage (FRDP) to the fruit maturation stage (FRMP) in P. somniferum involves substantial changes in the expression of genes associated with BIAs biosynthesis. Differential expression analysis revealed organ-specific and stage-specific patterns across roots, stems, leaves, and fruits, supported by GO and KEGG enrichment analyses (Fig. 5, Supplementary Figure S9-S16).

Differential Expression and Fold Change of BIA-Related Genes During Development. (A-D) Volcano plots showing differentially expressed genes between the fruit development stage FRDP and fruit maturation stage FRMP in P. somniferum across different tissues: A Roots, B Stems, C Leaves, and D Fruits. Upregulated genes are labeled as Up, and downregulated genes as Down. (E) Position of TYDC and PPO genes in the initial steps of the BIA biosynthesis pathway. (F) Fold change in BIA-related gene expression in roots R, stems S, and fruits F during FRDP and FRMP. Numbers -1, -2, -3, etc., indicate different gene copies.
In roots, 962 genes were differentially expressed, with 883 upregulated and 79 downregulated. Upregulated genes were enriched in transcription factor activity, ion binding, and transcriptional regulation, as well as KEGG pathways linked to plant-pathogen interactions, isoquinoline alkaloid biosynthesis, and signaling pathways. Conversely, downregulated genes were associated with stress-response pathways such as lysine degradation and monoterpene biosynthesis. These findings suggest that roots prioritize alkaloid precursor synthesis during maturation while scaling back stress-related mechanisms.
Stems exhibited 680 differentially expressed genes, with 371 upregulated and 309 downregulated. Upregulated genes were enriched in oxidoreductase activity, defense responses, and redox processes, reflecting the stem’s roles in structural support and intermediate transport. Downregulated genes were primarily associated with photosynthesis and protein oxidoreductase activity, indicating a metabolic shift towards maturation.
Leaves displayed more limited differential expression, with only 114 genes identified. Upregulated genes were linked to kinase activity and MAPK signaling, highlighting their involvement in signal transduction and secondary metabolism. Downregulated genes were enriched in photosynthesis and carbohydrate metabolism pathways, suggesting resource reallocation to reproductive tissues.
Fruits showed the most extensive changes, with 1,390 differentially expressed genes. Among the 668 upregulated genes, enrichment was observed in oxidoreductase activity, lipid storage, and defense responses, while KEGG analysis highlighted isoquinoline alkaloid biosynthesis and secondary metabolic pathways. The 722 downregulated genes were associated with hydrolase activity, chromosome assembly, and photosynthetic membrane processes, reflecting a shift from growth to energy storage and alkaloid accumulation.
Key BIA biosynthesis genes, including TYDC, PPO, and GsSRK, exhibited tissue-specific expression patterns. TYDC and PPO, crucial for tyrosine condensation reactions, were upregulated in roots and stems, indicating increased precursor synthesis during maturation. In contrast, PPO was downregulated in fruits, suggesting a shift in alkaloid biosynthesis activity towards vegetative tissues. GsSRK displayed dynamic changes across tissues, emphasizing its regulatory role in secondary metabolism.
Validation of RNA-seq results through qRT-PCR on 15 differentially expressed genes showed strong concordance (R2 = 0.9401, p < 0.0001) (Fig. 6). These results confirm the reliability of the transcriptomic analysis and underscore the biological significance of observed gene expression patterns.

Differential Gene Expression Levels from qRT-PCR and RNA-seq. (A) Log2 Fold Change of target genes as measured by qRT-PCR and RNA-seq. (B) Correlation and significance testing of log2 Fold Change values from qRT-PCR and RNA-seq. Gene names with suffixes such as -1, -2, -3 indicate different gene copies. Target genes include: PPO, GsSRK, 11S globulin subunit beta (11SB), Glutamine synthetase cytosolic (EPI10_032201), GDSL esterase/lipase (GLIP1), Putative EG45-like domain containing protein 1 (EGC1), leucoanthocyanidin reductase (LAR), Probable polygalacturonase At3g15720 (PGLR3), Aspartic proteinase CDR1 (CDR1), Transcription factor LHW (LHW), and Plastid division protein PDV2 (PDV2).
This analysis revealed distinct organ-specific and stage-specific changes in BIA-related gene expression during fruit development and maturation. Roots and stems were identified as primary sites for alkaloid precursor synthesis, while fruits focused on energy storage and downstream biosynthetic processes. Enrichment analyses further highlighted the dynamic regulation of key biosynthetic pathways, providing a molecular framework for optimizing alkaloid production in P. somniferum.
Discussion
This study utilized transcriptome sequencing combined with WGCNA to investigate the spatiotemporal regulation of benzylisoquinoline alkaloid (BIA) biosynthesis-related genes in P. somniferum. By integrating gene expression profiling with co-expression module analysis, 23 distinct modules were identified, each associated with specific developmental stages and organs. These findings offer valuable insights into the transcriptional control of alkaloid biosynthesis and establish a systematic framework for future research and targeted metabolic engineering strategies 32.
Previous studies, including Facchini and Park, have demonstrated that BIA biosynthesis is regulated developmentally, with differential expression patterns observed in distinct tissues during opium poppy growth23. Building on this foundational knowledge, our study employs transcriptome-wide profiling and network-based analysis to dissect organ- and stage-specific regulatory dynamics at higher resolution.
Key BIA biosynthesis genes, such as TYDC, PPO, and CODM, exhibited clear spatiotemporal specificity in their expression patterns. During fruit maturation, TYDC and PPO were significantly upregulated in roots and stems, reflecting their pivotal roles in precursor biosynthesis and metabolite transport. Interestingly, these genes showed reduced expression in fruits, supporting the hypothesis that fruits serve primarily as storage sites, while alkaloid biosynthesis is concentrated in vegetative tissues. Furthermore, previous investigations have shown that BIAs tend to accumulate predominantly in vegetative organs. For example, studies on sanguinarine localization report significant accumulation in root tissues33,34, while noscapine is primarily found in stems, and morphine, codeine and thebaine have been detected at higher levels in leaves compared to reproductive organs15,35,36. These observations align well with our findings and further support the notion of spatial separation between biosynthesis (in roots/stems/leaves) and storage (in fruits).
This division of labor aligns with findings in Nelumbo nucifera, where biosynthesis occurs predominantly in non-reproductive tissues, and reproductive organs focus on transport and storage 18,37,38,39. Such observations underscore the dynamic metabolic prioritization among different tissues during development and highlight the intricate coordination required for efficient alkaloid production.
The WGCNA-based approach enabled the identification of co-expression modules enriched in pathways associated with DNA repair, secondary metabolism, and stress responses. Notably, modules linked to root and stem tissues during vegetative and reproductive stages reflected dual roles in supporting growth and environmental adaptation 40,41. Similar co-expression networks have been reported in plants such as Solanum lycopersicum and Gossypium hirsutum, where secondary metabolism and stress adaptation are tightly interconnected 25,42,43. While WGCNA provides robust predictions about gene interactions, the correlation-based nature of this method limits its ability to establish causal relationships. Experimental studies, such as gene knockouts or overexpression analyses, are necessary to validate the regulatory roles of key hub genes like TYDC and PPO and their contributions to BIA biosynthesis.
Several BIA biosynthesis-related genes demonstrated multifunctionality, suggesting their broader biological significance beyond alkaloid production. For instance, TYDC participates in lysine degradation pathways associated with stress resistance, while PPO plays a role in oxidative stress responses and pathogen defense 11,44,45,46. These findings emphasize the integration of BIA-related genes into broader regulatory networks that optimize resource allocation during development and environmental adaptation. Additionally, organ-specific activation of modules, such as the enrichment of the blue module in fruit tissues during maturation, reveals a sophisticated balance between primary growth processes and secondary metabolism. This regulatory complexity is comparable to observations in medicinal plants like Dendrobium officinale and Catharanthus roseus, where secondary metabolite production is closely linked to developmental and environmental cues 47,48.
Moreover, the use of a CHM-type P. somniferum cultivar in this study provides an important contextual advantage. This locally preserved and genetically less-domesticated germplasm retains ancestral transcriptional features that may have been modified in intensively bred commercial varieties. As such, the regulatory patterns uncovered here likely reflect more native, evolutionarily conserved mechanisms of BIA biosynthesis. These findings not only broaden our understanding of transcriptional regulation in opium poppy but also highlight the potential of traditional germplasm resources for discovering untapped regulatory complexity relevant to secondary metabolism.
The methodology employed in this study demonstrates the power of combining transcriptome sequencing with WGCNA to uncover complex regulatory networks in plants 49. The rigorous quality control measures and high reproducibility of gene expression patterns across biological replicates enhance the reliability of these findings. However, certain methodological aspects present opportunities for refinement. For instance, while the exclusion of four outlier samples improved dataset consistency, these samples may have contained biologically meaningful variations driven by subtle environmental or developmental cues. Revisiting these samples with larger datasets or under varied conditions could uncover additional patterns in gene expression. Similarly, the geographic and environmental homogeneity of the sample source facilitated the identification of consistent regulatory modules but may limit the generalizability of these results. Future studies involving diverse environments and genotypes would provide a broader understanding of how external factors influence alkaloid biosynthesis 50,51,52. Furthermore, while this study focused on transcriptional regulation, comprehensive metabolite profiling across developmental stages and organs will be incorporated in future studies to validate gene expression findings and elucidate alkaloid accumulation patterns.
Future research could expand on these findings through several promising avenues. Functional validation of key genes, including TYDC, PPO, and CODM, using CRISPR/Cas9 or RNA interference, would confirm their roles in alkaloid biosynthesis and uncover regulatory interactions. Integrating transcriptomic data with metabolomic profiles would provide direct links between gene expression changes and metabolic outcomes, refining our understanding of transcriptional regulation. Targeted metabolic engineering strategies, such as enhancing precursor synthesis in roots and stems or optimizing transport mechanisms, could significantly improve alkaloid yields. Additionally, exploring the roles of transcription factors, such as WRKY and bZIP, in coordinating co-expression modules may reveal new layers of regulation and offer further opportunities for genetic manipulation. These transcription factors are known to play crucial roles in transcriptional regulation of alkaloid biosynthesis53. For instance, based on transcriptomic and functional analyses in Eschscholzia californica, WRKY transcription factors were shown to participate in the transcriptional regulation of BIA biosynthesis by responding to methyl jasmonate and exhibiting co-expression with biosynthetic enzyme genes; certain subgroup IIc WRKYs may also contribute to the transport and secretion of alkaloids54. bZIP transcription factors, which are evolutionarily conserved across plant species, play vital roles in regulating plant development, stress responses, and the biosynthesis of secondary metabolites including alkaloids. Recent studies have shown that bZIPs can either activate or repress the expression of genes involved in alkaloid biosynthetic pathways, thereby modulating the accumulation of medicinal compounds under varying environmental conditions55.
Future studies may also explore post-transcriptional regulation by small RNAs such as miRNAs, which have been shown to fine-tune the expression of alkaloid biosynthetic genes in various medicinal plants. In P. somniferum, miRNAs such as miR-13, miR-2161, and miR-408 have been identified to target key BIA biosynthetic genes56, while in Piper nigrum, miRNAs including miR-166, miR-396, and miR-397 were found to modulate piperine biosynthesis through interaction with structural enzyme transcripts57. Integrating small RNA sequencing with transcriptome and co-expression network analyses could help uncover novel regulatory layers contributing to the spatiotemporal control of BIA biosynthesis. Moreover, deciphering the miRNA-target interactions may reveal how environmental cues and developmental signals are transduced into fine-tuned metabolic outputs, offering additional targets for metabolic engineering strategies.
It should be noted that seeds were not included in the transcriptomic analysis in this study. This decision was based on the observation that BIA accumulation is primarily concentrated in vegetative and reproductive organs such as roots, stems, leaves, and capsules, whereas seeds, serving mainly a reproductive role, exhibit minimal secondary metabolic activity. Nevertheless, future investigations may explore seed tissues to assess whether any residual or regulatory BIA-related gene expression occurs during late developmental stages.
This study establishes a comprehensive molecular framework for understanding the transcriptional regulation of alkaloid biosynthesis in P. somniferum. By identifying stage- and organ-specific regulatory mechanisms and characterizing key biosynthesis genes, it highlights the complexity of regulatory networks underlying growth, development, and secondary metabolism. These findings not only advance our understanding of the spatiotemporal regulation of BIA biosynthesis but also provide a solid foundation for future research aimed at enhancing alkaloid yields through targeted genetic engineering and molecular breeding strategies. Ultimately, this work contributes to both fundamental plant science and applied pharmaceutical research, paving the way for innovative approaches in alkaloid production.
Materials and methods
Sample collection
Experimental plants used in this study were cultivated and maintained under controlled conditions at the Wuhan Botanical Garden, Chinese Academy of Sciences. The plant material represents a traditional Chinese Herbal Medicine (CHM)-type cultivar of P. somniferum, characterized by its primitive genetic background and lack of modern artificial selection.
Tissue samples were collected at four defined developmental stages, determined based on time after germination or anthesis in individually labeled plants:
Vegetative Period (VP): Day 140 after germination.
Flowering Period (FLP): Two days after the onset of flowering (anthesis) in marked individual plants.
Fruit Development Period (FRDP): Eighteen days after anthesis.
Fruit Maturity Period (FRMP): Thirty-six days after anthesis.
At each stage, specific organs were sampled as follows:
Vegetative Period: Root, stem, and leaf tissues.
Flowering, Fruit Development, and Maturity Periods: Root, stem, leaf, and ovary/fruit tissues.
Tissue sampling details:
Root: Collected from the apical region of the main root.
Stem: Composite sample from upper, middle, and lower segments.
Leaf: Young leaves harvested from the apex.
Ovary: Collected during the flowering period.
Fruit: Pericarp of the capsule sampled during fruit development and maturity.
Each organ sample was collected in triplicate from independent biological replicates, yielding a total of 45 samples (> 0.5 g per sample) for transcriptomic analysis. All fresh samples were immediately frozen in liquid nitrogen and stored at − 80 °C until RNA extraction.
RNA extraction and transcriptome library construction
Total RNA was extracted from samples using a commercial RNA extraction kit, followed by quality assessment for purity, concentration, and integrity. Poly(A) mRNA was enriched using Oligo dT magnetic beads and fragmented into smaller fragments. Single-stranded cDNA was synthesized from mRNA using random hexamer primers and reverse transcriptase, then converted to double-stranded cDNA. The cDNA underwent end repair, adapter ligation, and PCR amplification to construct sequencing libraries. Library quality was assessed, and qualified libraries were pooled and sequenced on the Illumina HiSeq 2000 platform.
Transcriptome sequencing, data quality control, and quality assessment
The sequencing data for this study have been deposited in the NCBI database under BioProject number PRJNA1148676. cDNA libraries were sequenced using an Illumina platform, employing bridge PCR amplification followed by fluorescently labeled dNTP incorporation. Sequencing was conducted with iterative cycles of fluorescence detection, azide group removal, and base addition, ensuring high accuracy for base identification.
Raw sequencing data were converted into FASTQ format and subjected to stringent quality control. This included adapter trimming, duplicate removal, and filtering of low-quality reads. Sequencing quality was assessed through statistical analysis of base quality scores (Q) and nucleotide composition stability across reads. The average single-base error rate was maintained below 1%, with consistent A-T and G-C base pair distributions across all samples. High-quality clean reads generated through this process were used for downstream analysis.
Sequence alignment and gene expression quantification
The genome published by Pei et al. (2021) (Bioproject ID: PRJNA503959) was used as the reference for alignment. Clean reads were aligned to the reference genome using Hisat2 v2.0.1, and alignment results were processed into BAM format with Samtools v1.9. Gene expression levels were quantified using featureCounts v2.0.1 by calculating the number of reads mapped to genomic regions.
To account for differences in gene lengths, experimental conditions, and sequencing depth, gene expression levels were normalized using FPKM (Fragments Per Kilobase of transcript per Million mapped fragments). FPKM values were computed with the R package CountToFPKM v1.0, ensuring accurate normalization and comparability across samples.
Principal component analysis and sample clustering
Principal Component Analysis (PCA) was conducted to reduce the dimensionality of gene expression data and compare variation among samples. PCA was performed using the fast.prcomp function from the gmodels package in R (v4.0.3). Scatter plots were generated to illustrate correlations between biological replicates and assess sample variability.
Pearson correlation coefficients were calculated using the COR function in R to evaluate expression similarities across growth stages and organs. Correlation coefficients close to 1 indicated high consistency among samples, confirming experimental reliability. Hierarchical clustering was performed using the hclust function, with results visualized in a heatmap and dendrogram. Outlier samples were identified and excluded based on clustering and correlation analysis.
Weighted gene co-expression network analysis
WGCNA was performed using the WGCNA package v1.70 in R (v4.0.3), following established protocols (Langfelder et al., 2012). Gene expression data with FPKM ≥ 1 and significant differential expression from 41 samples were used as input.
The analysis included the following steps: missing values and outliers were removed using the goodSamplesGenes and hclust functions. The pickSoftThreshold function was employed to identify the optimal soft-thresholding power (β), ensuring the network exhibited scale-free topology (R2 > 0.8). The powerEstimate function provided the final β value. Gene clustering was performed with the blockwiseModules function, grouping genes with similar expression patterns into modules. Module identification and visualization were completed using the labels2colors function to assign module names and colors.
Identification of modules associated with developmental stages
Gene modules identified through WGCNA were analyzed for their relevance to developmental stages. Module eigengenes were computed, and Pearson correlation coefficients with corresponding p-values were calculated to evaluate their associations with phenotypic traits. A module-trait correlation heatmap was generated to visualize these relationships.
Modules with the highest correlation coefficients (> 0.5) were identified as primary modules closely associated with poppy growth and development. Secondary modules, containing genes linked to supplementary processes in plant growth, were also identified based on their correlations. These results provide insights into the genetic regulation of developmental stages in P. somniferum.
GO and KEGG enrichment analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)58,59,60 enrichment analyses were conducted for genes within the identified modules using the clusterProfiler package v3.14.3 in R. Significantly enriched terms were identified using a threshold of P-adj < 0.05.
The analyses highlighted key biological processes, signaling pathways, and critical genes associated with developmental stages. Results provided insights into the expression patterns of BIAs synthesis-related genes across various organs and developmental stages, contributing to a comprehensive understanding of their roles in poppy growth and metabolism.
Differential gene expression analysis
Differential gene expression analysis was conducted for samples across various organs during fruit development and maturation stages using the DESeq2 package v1.32.0 in R. Gene expression levels during the fruit development stage served as the control. Differentially expressed genes were identified based on the criteria |log2(fold change)|> 2 and P-adj < 0.05.
Validation of differentially expressed genes by qRT-PCR
To validate RNA-seq results, qRT-PCR was conducted on fruit peel samples from poppy during fruit development and maturation. Fifteen genes, including PPO, GsSRK, 11SB, and GLIP1, were selected for validation.
RNA was extracted using the FastPure® Plant Total RNA Isolation Kit (RC411-C1). The RNA was reverse-transcribed into cDNA using the PrimeScript™ RT Reagent Kit with gDNA Eraser (Perfect Real Time) from TaKaRa. qRT-PCR was performed on a LightCycler® 480 system under the following conditions: initial denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 10 s, annealing at 60°C for 30 s, and extension at 72°C for 32 s. Relative gene expression levels were calculated using the 2−ΔΔCT method.
Data availability
The sequencing data for this study have been deposited in the NCBI database under BioProject number PRJNA1148676.
References
Vasek, J. et al. New EST-SSR Markers for Individual Genotyping of Opium Poppy Cultivars (Papaver somniferum L.). Plants (Basel) https://doi.org/10.3390/plants9010010 (2019).
Thangavel, T., Scott, J., Jones, S., Gugalothu, R. & Wilson, C. Effect of physio-chemical seed treatments on opium poppy downy mildews caused by Peronospora meconopsidis and P. somniferi. PLoS ONE 15, e0230801. https://doi.org/10.1371/journal.pone.0230801 (2020).
Chain, E. P. O. C. et al. Update of the Scientific Opinion on opium alkaloids in poppy seeds. EFSA J 16, e05243. https://doi.org/10.2903/j.efsa.2018.5243 (2018).
Diaz-Barcena, A., Fernandez-Pacios, L. & Giraldo, P. Structural Characterization and Molecular Dynamics Study of the REPI Fusion Protein from Papaver somniferum L.. Biomolecules https://doi.org/10.3390/biom14010002 (2023).
Ashrafi, S. et al. Papaverine: A miraculous alkaloid from opium and its multimedicinal application. Mol. Basel Switzerland https://doi.org/10.3390/molecules28073149 (2023).
Hong, U. V. T., Tamiru-Oli, M., Hurgobin, B. & Lewsey, M. G. Genomic and cell-specific regulation of benzylisoquinoline alkaloid biosynthesis in opium poppy. J Exp Bot https://doi.org/10.1093/jxb/erae317 (2024).
Beaudoin, G. A. & Facchini, P. J. Benzylisoquinoline alkaloid biosynthesis in opium poppy. Planta 240, 19–32. https://doi.org/10.1007/s00425-014-2056-8 (2014).
Dastmalchi, M. et al. Neopinone isomerase is involved in codeine and morphine biosynthesis in opium poppy. Nat. Chem. Biol. 15, 384–390. https://doi.org/10.1038/s41589-019-0247-0 (2019).
Gudin, J. & Fudin, J. A narrative pharmacological review of buprenorphine: a unique opioid for the treatment of chronic pain. Pain Ther. 9, 41–54. https://doi.org/10.1007/s40122-019-00143-6 (2020).
Pergolizzi, J. V. Jr. & Raffa, R. B. Safety and efficacy of the unique opioid buprenorphine for the treatment of chronic pain. J. Pain Res. 12, 3299–3317. https://doi.org/10.2147/JPR.S231948 (2019).
Desgagne-Penix, I. et al. Integration of deep transcriptome and proteome analyses reveals the components of alkaloid metabolism in opium poppy cell cultures. BMC Plant Biol. 10, 252. https://doi.org/10.1186/1471-2229-10-252 (2010).
Putranto, R., Mudjaddid, E., Shatri, H., Adli, M. & Martina, D. Development and challenges of palliative care in Indonesia: Role of psychosomatic medicine. Biopsychosoc. Med. 11, 29. https://doi.org/10.1186/s13030-017-0114-8 (2017).
Hagel, J. M. & Facchini, P. J. Dioxygenases catalyze the O-demethylation steps of morphine biosynthesis in opium poppy. Nat. Chem. Biol. 6, 273–275. https://doi.org/10.1038/nchembio.317 (2010).
Wijekoon, C. P. & Facchini, P. J. Systematic knockdown of morphine pathway enzymes in opium poppy using virus-induced gene silencing. Plant J. 69, 1052–1063. https://doi.org/10.1111/j.1365-313X.2011.04855.x (2012).
Gurkok, T. et al. Functional Characterization of 4’OMT and 7OMT Genes in BIA Biosynthesis. Front Plant Sci 7, 98. https://doi.org/10.3389/fpls.2016.00098 (2016).
AlagÖZ, Y. et al. Identification and sequence analysis of alkaloid biosynthesisgenes in Papaver section Oxytona. Turkish J. Biol. 40, 174–183. https://doi.org/10.3906/biy-1505-22 (2016).
Guo, L. et al. The opium poppy genome and morphinan production. Science 362, 343–347. https://doi.org/10.1126/science.aat4096 (2018).
Li, Q. et al. Gene clustering and copy number variation in alkaloid metabolic pathways of opium poppy. Nat. Commun. 11, 1190. https://doi.org/10.1038/s41467-020-15040-2 (2020).
Pei, L. et al. Genome and transcriptome of Papaver somniferum Chinese landrace CHM indicates that massive genome expansion contributes to high benzylisoquinoline alkaloid biosynthesis. Hortic. Res. 8, 5. https://doi.org/10.1038/s41438-020-00435-5 (2021).
Agarwal, P. et al. 3’O-Methyltransferase, Ps3’OMT, from opium poppy: involvement in papaverine biosynthesis. Plant Cell Rep. 38, 1235–1248. https://doi.org/10.1007/s00299-019-02439-5 (2019).
Desgagne-Penix, I., Farrow, S. C., Cram, D., Nowak, J. & Facchini, P. J. Integration of deep transcript and targeted metabolite profiles for eight cultivars of opium poppy. Plant Mol Biol 79, 295–313. https://doi.org/10.1007/s11103-012-9913-2 (2012).
Gurkok, T., Turktas, M. & Gulsen, G. In silico comparative transcriptome analysis of Papaver somniferum cultivars. ACTA Scientiarum Polonorum Hortorum Cultus 22, 10. https://doi.org/10.24326/asphc.2023.5165 (2023).
Facchini, P. J. & Park, S. U. Developmental and inducible accumulation of gene transcripts involved in alkaloid biosynthesis in opium poppy. Phytochemistry 64, 177–186. https://doi.org/10.1016/s0031-9422(03)00292-9 (2003).
Scialo, E., Sicilia, A., Continella, A., Gentile, A. & Lo Piero, A. R. Transcriptome profiling and weighted gene correlation network analysis reveal hub genes and pathways involved in the response to polyethylene-glycol-induced drought stress of two citrus rootstocks. Biology (Basel) https://doi.org/10.3390/biology13080595 (2024).
Shinozaki, Y. et al. High-resolution spatiotemporal transcriptome mapping of tomato fruit development and ripening. Nat.Commun. 9, 364. https://doi.org/10.1038/s41467-017-02782-9 (2018).
Li, X., Zheng, Y., Liu, M., Wang, K. & Chen, H. Weighted gene co-expression network analysis and identification of ginsenoside biosynthesis candidate genes for ginseng adventitious roots under MeJA treatment. Genes Genomics 46, 1473–1485. https://doi.org/10.1007/s13258-024-01577-9 (2024).
Qiao, L. et al. Identification of salt-stress-responding genes by weighted gene correlation network analysis and association analysis in wheat leaves. Plants (Basel) https://doi.org/10.3390/plants13182642 (2024).
Cheng, S. et al. High-resolution temporal dynamic transcriptome landscape reveals a GhCAL-mediated flowering regulatory pathway in cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 19, 153–166. https://doi.org/10.1111/pbi.13449 (2021).
Fang, Y. et al. Identification of differentially expressed genes involved in amino acid and lipid accumulation of winter turnip rape (Brassica rapa L.) in response to cold stress. PLoS ONE 16, e0245494. https://doi.org/10.1371/journal.pone.0245494 (2021).
Park, C. H. et al. Transcriptome analysis and metabolic profiling of lycoris radiata. Biology (Basel) https://doi.org/10.3390/biology8030063 (2019).
Ozber, N. & Facchini, P. J. Phloem-specific localization of benzylisoquinoline alkaloid metabolism in opium poppy. J. Plant Physiol. 271, 153641. https://doi.org/10.1016/j.jplph.2022.153641 (2022).
Hong, U. V. T., Tamiru-Oli, M., Hurgobin, B. & Lewsey, M. G. Genomic and cell-specific regulation of benzylisoquinoline alkaloid biosynthesis in opium poppy. J. Exp. Bot. 76, 35–51. https://doi.org/10.1093/jxb/erae317 (2024).
Weid, M., Ziegler, J. & Kutchan, T. M. The roles of latex and the vascular bundle in morphine biosynthesis in the opium poppy, Papaver somniferum. Proc. Natl. Acad. Sci. U S A 101, 13957–13962. https://doi.org/10.1073/pnas.0405704101 (2004).
Samanani, N., Liscombe, D. K. & Facchini, P. J. Molecular cloning and characterization of norcoclaurine synthase, an enzyme catalyzing the first committed step in benzylisoquinoline alkaloid biosynthesis. Plant J. 40, 302–313. https://doi.org/10.1111/j.1365-313X.2004.02210.x (2004).
Dang, T. T. & Facchini, P. J. Characterization of three O-methyltransferases involved in noscapine biosynthesis in opium poppy. Plant Physiol. 159, 618–631. https://doi.org/10.1104/pp.112.194886 (2012).
Frick, S., Kramell, R., Schmidt, J., Fist, A. J. & Kutchan, T. M. Comparative qualitative and quantitative determination of alkaloids in narcotic and condiment Papaver somniferum cultivars. J. Nat. Prod. 68, 666–673. https://doi.org/10.1021/np0496643 (2005).
Li, J. et al. Jasmonate-responsive transcription factors NnWRKY70a and NnWRKY70b positively regulate benzylisoquinoline alkaloid biosynthesis in lotus (Nelumbo nucifera). Front. Plant. Sci. 13, 862915. https://doi.org/10.3389/fpls.2022.862915 (2022).
Yang, M. et al. Digital gene expression analysis provides insight into the transcript profile of the genes involved in aporphine alkaloid biosynthesis in lotus (Nelumbo nucifera). Front. Plant. Sci. 8, 80. https://doi.org/10.3389/fpls.2017.00080 (2017).
Pathak, S. et al. Comparative transcriptome analysis using high papaverine mutant of Papaver somniferum reveals pathway and uncharacterized steps of papaverine biosynthesis. PLoS ONE 8, e65622. https://doi.org/10.1371/journal.pone.0065622 (2013).
Wang, F. et al. Weighted gene co-expression network analysis reveals hub genes regulating response to salt stress in peanut. BMC Plant. Biol. 24, 425. https://doi.org/10.1186/s12870-024-05145-x (2024).
Xu, Y. et al. Genetic regulatory networks for salt-alkali stress in Gossypium hirsutum with differing morphological characteristics. BMC Genomics 21, 15. https://doi.org/10.1186/s12864-019-6375-9 (2020).
Liu, C. et al. The bZIP73 transcription factor controls rice cold tolerance at the reproductive stage. Plant Biotechnol. J. 17, 1834–1849. https://doi.org/10.1111/pbi.13104 (2019).
Zhang, G. et al. Genome-wide and expression pattern analysis of the HIT4 gene family uncovers the involvement of GHHIT4_4 in response to verticillium wilt in gossypium hirsutum. Genes (Basel) https://doi.org/10.3390/genes15030348 (2024).
Liu, Y. et al. Consequences of gene flow between oilseed rape (Brassica napus) and its relatives. Plant Sci. 211, 42–51. https://doi.org/10.1016/j.plantsci.2013.07.002 (2013).
Jena, S. et al. Spatio-temporal expression of polyphenol oxidase unveils the dynamics of L-DOPA accumulation in faba bean (Vicia faba L.). Physiol. Mol. Biol. Plants 30, 839–850. https://doi.org/10.1007/s12298-024-01449-2 (2024).
Demir, D., Çağlayan, K. & Eken, C. Polyphenol oxidase activities in Japanese Pear (Pyrus pyrifolia (Burm) Nakai) fruit at different development stages. Biol. Bull. 50, 1115–1124. https://doi.org/10.1134/S1062359022602397 (2023).
Jiao, C. et al. Transcriptomic analysis of genes related to alkaloid biosynthesis and the regulation mechanism under precursor and methyl jasmonate treatment in Dendrobium officinale. Front. Plant. Sci. 13, 941231. https://doi.org/10.3389/fpls.2022.941231 (2022).
Zhang, S. Recent advances of polyphenol oxidases in plants. Mol. Basel Switzerland https://doi.org/10.3390/molecules28052158 (2023).
Liu, Y. CWGCNA: an R package to perform causal inference from the WGCNA framework. NAR Gen. Bioinf. https://doi.org/10.1093/nargab/lqae042 (2024).
Desquilles, L. & Musso, O. in Metabolic Reprogramming: Methods and Protocols (eds Salvatore Papa & Concetta Bubici) 317–325 (Springer US, 2023).
Kumar, A., Kanak, K. R., Arunachalam, A., Dass, R. S. & Lakshmi, P. T. V. Comparative transcriptome profiling and weighted gene co-expression network analysis to identify core genes in maize (Zea mays L.) silks infected by multiple fungi. Front Plant Sci. 13, 985396. https://doi.org/10.3389/fpls.2022.985396 (2022).
Wang, H. et al. The overall regulatory network and contributions of ABC(D)E model genes in yellowhorn flower development. BMC Plant Biol. 24, 1081. https://doi.org/10.1186/s12870-024-05796-w (2024).
Yamada, Y. & Sato, F. Transcription factors in alkaloid engineering. Biomolecules https://doi.org/10.3390/biom11111719 (2021).
Yamada, Y., Nishida, S., Shitan, N. & Sato, F. Genome-wide profiling of WRKY genes involved in benzylisoquinoline alkaloid biosynthesis in california poppy (Eschscholzia californica). Front. Plant Sci. 12, 699326. https://doi.org/10.3389/fpls.2021.699326 (2021).
Han, H. et al. Role of bZIP transcription factors in the regulation of plant secondary metabolism. Planta 258, 13. https://doi.org/10.1007/s00425-023-04174-4 (2023).
Boke, H. et al. Regulation of the alkaloid biosynthesis by miRNA in opium poppy. Plant Biotechnol J 13, 409–420. https://doi.org/10.1111/pbi.12346 (2015).
Ding, Y. et al. Small RNA sequencing reveals various microRNAs involved in piperine biosynthesis in black pepper (Piper nigrum L.). BMC Genomics 22, 838. https://doi.org/10.1186/s12864-021-08154-4 (2021).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Funding
This research was funded by the National Science Foundation of China, grant number 81671876; the Key Laboratory of Forensic Genetics, Ministry of Public Security Open Research Project, grant number 2022FGKFKT02; and the Science and Technology Plan Projects in Liaoning Province, grant number 2023-MS-329.
Author information
Authors and Affiliations
Contributions
Conceptualization, B.W., B.C., and Z.S.W.; Methodology, J.H. and B.W.; Software, Q.Y.; Validation, Z.W., B.W., and B.C.; Formal Analysis, Q.Y.; Investigation, Z.S.W., J.H., B.W., and Y.W.; Resources, S.L., F.L., D.F., H.X., and L.F.; Data Curation, Q.Y., Z.S.W., and N.L.; Writing – Original Draft Preparation, Z.S.W., Q.Y., and J.H.; Writing – Review & Editing, B.W., B.C., Z.W., and Z.L.W.; Visualization, Q.Y.; Supervision, B.W. and B.C.; Project Administration, B.W.; Funding Acquisition, B.W. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Z., Yun, Q., Hu, J. et al. Spatiotemporal dynamics of benzylisoquinoline alkaloid gene expression and co-expression networks during Papaver Somniferum developmental stages. Sci Rep 15, 27406 (2025). https://doi.org/10.1038/s41598-025-11942-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-11942-7
