
Abstract
Despite extensive prior research on prostate cancer (PCa) transcriptomics, the molecular mechanisms underlying the disease’s progression, particularly in the castration-resistant or metastatic stages, remain incompletely understood. The majority of recent research has concentrated on bulk RNA sequencing, which could mask the variation found in tumor microenvironments. This study aims to address this gap by integrating single-cell RNA sequencing (scRNA-seq) and bulk RNA sequencing with weighted gene co-expression network analysis (WGCNA) to investigate the molecular mechanisms of PCa at a higher resolution. In order to further individualized treatment plans for PCa, we aim to discover important genes and signaling pathways that could be used as therapeutic targets. We first preprocessed expression profile data from prostate cancer tissue samples, selecting 9,809 high-quality cells from a dataset. Following batch correction with Harmony and dimensionality reduction with principal component analysis (PCA), we used the Louvain clustering algorithm to divide the cells into discrete subtypes. The clusters were then visualized using t-SNE. This resulted in 16 cellular subtypes categorized into five major cell types: epithelial cells, monocytes, endothelial cells, CD8 + T-cells, and fibroblasts. Analysis of receptor-ligand pairs uncovered significant interactions between monocytes and both tumor cells and endothelial cells. Applying the high-dimensional WGCNA (hdWGCNA) method to construct a gene co-expression network, we detected seven gene modules, four of which were highly expressed in tumor cell subtypes and contained 380 key genes. Combining pathway analysis, we ultimately screened six key genes: CNPY2, CPE, DPP4, IDH1, NIPSNAP3A, and WNK4. We used Cox univariate regression and least absolute shrinkage and selection operator (lasso) regression techniques to build a prognostic prediction model that included these six important genes based on clinical data gathered from PCa patients. The prognostic prediction model constructed in this study demonstrated excellent predictive performance in both the training set and an external validation set, with the high-risk group showing significantly lower overall survival (OS) than the low-risk group. Furthermore, there was a substantial correlation found between risk scores and several immune-related gene sets, chemotherapeutic drug sensitivity, and tumor immune infiltration. High- and low-risk groups exhibited significant differences in immune cell content, immune factor levels, and immune dysfunction. Further analysis revealed significant correlations between the expression levels of model genes and multiple disease-related genes. Through Gene Set Variation Analysis (GSVA) and Gene Set Enrichment Analysis (GSEA), we uncovered perturbations in multiple signaling pathways in high- and low-risk groups, potentially impacting the prognosis of PCa patients. This study uncovers key genes and signaling pathways in the prostate cancer tumor microenvironment, particularly genes such as CNPY2, CPE, DPP4, IDH1, NIPSNAP3A and WNK4, which have potential as therapeutic targets. Our findings provide new insights into personalized treatment strategies for PCa and warrant further clinical validation in the future.
Similar content being viewed by others

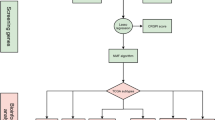

Introduction
Prostate cancer (PCa) remains a leading cause of cancer-related morbidity and mortality in men, with castration-resistant (CRPC) and metastatic variants (mPCa) posing critical therapeutic challenges due to intrinsic heterogeneity and adaptive resistance mechanisms1,2,3. While bulk RNA sequencing has provided foundational insights into PCa biology, recent advances in single-cell RNA sequencing (scRNA-seq) have revolutionized our understanding of tumor microenvironment (TME) dynamics. Seminal studies by Chen et al.4 and Karthaus et al.5 established foundational single-cell atlases of PCa, delineating epithelial cell plasticity and neuroendocrine differentiation in CRPC. Subsequent work by Zhang et al.6 further mapped TME heterogeneity, revealing myeloid cell-driven immunosuppression in mPCa, while Wang et al.7 implicated fibroblast-derived extracellular matrix remodeling in therapy resistance. Despite these advances, critical gaps persist in resolving how metabolic-immune crosstalk regulates therapeutic vulnerabilities and lineage plasticity.
Recent studies have begun addressing these gaps: Li et al.8 identified ASCL1-mediated lineage reprogramming in neuroendocrine PCa, while Kim et al.9 uncovered chemotherapy-induced epithelial-mesenchymal transition signatures via scRNA-seq. Furthermore, Zhao et al.10 characterized ligand-receptor networks driving immune evasion, and Nguyen et al.11 decoded T-cell exhaustion trajectories in CRPC. However, these studies lacked systematic integration of multi-scale transcriptome networks and instead concentrated on isolated pathways or distinct cell populations.
Building on this framework, our research makes three significant contributions to the field: Integration of Multi-Omic Networks: In contrast to previous single-cell atlases, we unveiled coordinated gene modules spanning the epithelial, immunological, and metabolic axes by combining scRNA-seq with high-dimensional weighted gene co-expression network analysis (hdWGCNA). This revealed CNPY2/IDH1-enriched networks regulating calcium-WNT signaling, complementing Wang et al.7 on stromal-tumor interactions but extending to metabolic reprogramming mechanisms. Lineage Plasticity Drivers: While Li et al. highlighted ASCL1 in neuroendocrine differentiation, our hdWGCNA identified DPP4 and WNK4 as novel regulators of epithelial plasticity, linking lineage transitions to immune evasion via CXCL12-CXCR4 signaling. Therapeutic Vulnerability Mapping: Expanding Kim et al.’s chemotherapy resistance insights, we integrated drug sensitivity data to demonstrate Axitinib susceptibility in high-risk patients with CNPY2/NIPSNAP3A overexpression, providing actionable biomarkers for combination therapies. By contextualizing our findings within this evolving landscape, we bridge critical gaps in understanding PCa’s metabolic-immune axis while delivering clinically translatable tools for risk stratification and targeted intervention.
Materials and methods
Data acquisition
The largest cancer genomics data source at the moment, The Cancer Genome Atlas (TCGA) database (https://portal.gdc.cancer.gov/), has a wealth of information about cancer, such as SNPs, copy number variants, gene expression profiles, miRNA expression, and DNA methylation. For this study, we accessed the processed mRNA expression data of PCa, encompassing both normal (n = 52) and tumor groups (n = 502).
The Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) is a public gene expression database maintained by the National Center for Biotechnology Information (NCBI). Seven tumor samples and eight control samples for single-cell analysis were included in the GSE176031 dataset, which we obtained from the GEO public database. Additionally, we downloaded the Series Matrix File data files for GSE70769, annotated with platform GPL10558, extracting data from 92 PCa patients with complete expression profiles and survival information. In a similar manner, we acquired the Series Matrix File data files for GSE54460, annotated with platform GPL11154, and harvested information from 55 PCa patients, including survival statistics and detailed expression profiles.
The disease-specific gene set for PCa utilized in this analysis was sourced from the GeneCards database (https://www.genecards.org), which provides a comprehensive resource for gene annotations and functional pathways related to various diseases, including cancer.
Single-cell analysis
We initiated our analysis by importing the expression profiles using the “Seurat” package12 and filtered out low-expressing genes (nFeature_RNA > 300 & percent.mt < 20)13. The data underwent normalization, scaling, principal component analysis (PCA), and Harmony analysis. ElbowPlot was utilized to determine the optimal number of principal components14 and t-SNE analysis was performed to visualize the inter-cluster relationships. Cell type annotation was conducted using the “Celldex” package15 assigning clusters to cell types significantly associated with disease pathology16. Finally, we extracted marker genes for each cell subtype from the single-cell expression profiles with FindAllMarkers, setting the logfc. Threshold to 1 and min. pct to 0.25. Genes with adjusted p_val_adj < 0.05 and |avg_log2FC| > 1 were considered as specific marker genes for each cell subtype.
Ligand-receptor interaction analysis
CellPhoneDB (database version: 4.0) is a publicly available repository of curated receptor-ligand interactions15. It includes subunit structures for both ligands and receptors, accurately representing heterodimeric complexes. The database integrates with UniProt, Ensembl, PDB, and IUPHAR and stores 1,885 protein-protein interactions, allowing for a comprehensive and systematic analysis of cell-cell communication molecules and studying the interactions and communication networks between different cell types. We performed a significance analysis of ligand-receptor relationships in the single-cell expression profiles using the statistical_analysis function of the “CellphoneDB” package17. We randomized the cluster labels of all cells 1000 times and determined the average expression levels of receptors in clusters and the average expression levels of ligands in interaction clusters. For each receptor-ligand pair in every comparison between two cell types, this generated a null distribution. We then selected several ligand-receptor pairs of interest for visualization.
HdWGCNA analysis
hdWGCNA provides built-in functions for network inference, gene module identification, functional gene enrichment analysis, network reproducibility statistical tests, and data visualization18. In addition to traditional single-cell RNAseq, hdWGCNA can also perform homotypic-level network analysis using long-read single-cell data19. We constructed a co-expression network of genes expressed in at least 5% of cells in the Seurat object using SetupForWGCNA, setting the soft threshold to 820. Modules with high median expression (ME) levels met the criteria of PercentExpressed > 75% and Average Expression > 1.521. PlotDendrogram was used to visualize the dendrogram of WGCNA, a common visualization to display the different co-expression modules produced by network analysis. Each leaf on the dendrogram represents a gene, and the color at the bottom indicates the assignment of co-expression modules. Finally, GetMEs were used to obtain the module eigengenes and visualize them. We set n_hubs = 100 to obtain hub genes in the modules22.
Gene functional enrichment analysis
We utilized the Metascape database (www.metascape.org) to functionally annotate significant gene sets, exploring their functional correlations comprehensively23. Certain genes were subjected to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis and Gene Ontology (GO) analysis24,25,26. A minimum overlap of 3 and a p-value ≤ 0.01 were considered statistically significant.
Model building and prognosis
Prognostically relevant genes were selected, and a lasso regression model was further constructed. A risk score formula, weighted by the estimated regression coefficient in the lasso regression analysis, was constructed for every patient after taking into account the expression values of each individual gene. The median risk score was used as the cutoff to separate patients into low-risk and high-risk groups based on the risk score algorithm. Kaplan-Meier was used to assess survival differences between the two groups, and the log-rank statistical method was employed for comparison. The prognostic value of the risk score was evaluated using Lasso regression analysis with 10-fold cross-validation, followed by stratified analysis. The receiver operating characteristic (ROC) curves were used to study the predictive accuracy of the model27.
Immune cell infiltration analysis
The CIBERSORT algorithm was applied to analyze RNA-seq data from different subgroups of PCa patients to infer the relative proportions of 22 immune-infiltrating cells28. Significance analysis of immune cell content between high- and low-risk groups was performed, with P < 0.05 considered statistically significant.
Drug sensitivity analysis
Based on the largest pharmacogenomics database (Genomics of Drug Sensitivity in Cancer (GDSC); Cancer Drug Sensitivity Genomics Database, https://www.cancerrxgene.org/), we used the R package “pRRophetic“29 to predict the chemosensitivity of each tumor sample. The estimated IC50 values for treatment with particular chemotherapeutic agents were obtained using a regression approach and confirmed using the GDSC training set with 10-fold cross-validation. Default parameters were used for all analyses, including the removal of batch effects with “combat” and averaging repeated gene expressions.
To standardize the expression levels of TCGA (RNA-seq) and GDSC (microarray) data using TPM normalization (RNA-seq) and RMA pretreatment (microarray). Use the removeBatchEffect function (method="combat”) built-in in pRRophetic to correct batch effects based on research sources (TCGA vs. GDSC) and sequencing platforms while preserving biological variations. Only genes that are coexpressed in the GDSC training set and target dataset (12508 intersecting genes) are retained to ensure model transferability. The prediction covers 253 chemotherapy/targeted drugs in the GDSC database, with priority given to FDA-approved drugs for prostate cancer or drugs in clinical trials (such as Axitinib, Mitomycin C, Bleomycin, etc.). Based on pathway enrichment analysis results (such as PI3K-AKT and WNT), drugs targeting relevant pathways were selected (n = 42). Perform 10-fold cross validation tests on all drugs, and only retain drugs with a cross-fold AUC consistency > 0.8.
Gene set variation analysis (GSVA) analysis
GSVA is a non-parametric, unsupervised method for assessing the enrichment of transcriptome gene sets. By combining changes at the gene level with changes at the pathway level, GSVA rates gene sets and measures the biological functions of the sample30. In this study, gene sets were downloaded from the Molecular Signatures Database (v7.0), and the GSVA algorithm was used to score each set, evaluating potential biological function changes in different samples.
Gene set enrichment analysis (GSEA) Enrichment analysis
GSEA was performed on the expression profiles of PCa patients to identify differentially expressed genes between high-risk and low-risk groups31. Gene sets with a maximum and minimum size of 500 and 15 genes, respectively, were filtered. After 100 permutations, enriched gene sets were obtained based on a P-value < 0.05 and a false discovery rate (FDR) value of 0.2532.
Statistical analysis
Survival curves were generated using the Kaplan-Meier method and compared using the log-rank test33. R language 4.2.0 was used for all statistical studies. P < 0.05 was regarded as statistically significant, and all statistical tests were two-tailed.
Results
Single-cell transcriptomic data preprocessing
We used transcriptome profiles from PCa-related tissue samples for our investigation. For additional analysis, we specifically kept 9,809 cells with a nFeature_RNA count greater than 300 and a percent.mt value less than 20%. The expression levels of features across these cells were examined, and the five genes with the highest normalized variance were highlighted (Supplementary Fig. 1A-C).
We performed PCA on 20 genes and observed varying scores across different dimensions (Supplementary Fig. 1D). PCA analysis among samples revealed significant overall differences (Supplementary Fig. 1E). To account for batch effects, we employed Harmony analysis for dimensionality reduction (Supplementary Fig. 1F). The ideal number of principle components, according to an ElbowPlot, was 16 (Supplementary Fig. 1G). Subsequently, t-distributed stochastic neighbor embedding (t-SNE) analysis was conducted, resulting in the identification of 16 distinct subtypes (Fig. 1A).

Single-cell dimension reduction clustering and cell annotation (A) t-SNE dimension reduction analysis of the sample, showcasing the clustering of cells. Each color represents a different cell subset, with a total of 16 cell populations identified. (B) Cell type annotation results, mark the five main cell types, including epithelial cells, monocytes, endothelial cells, CD8 + T cells, and fibroblasts. Each color represents a specific cell type. (C) Proportional analysis of the five cell types in various samples. Stacked bar charts display the distribution of proportions for the five cell types across different samples, with the x-axis representing different cell types and the y-axis representing proportions. N denotes normal tissue, and T denotes tumor tissue.
Cluster annotation
We employed the R package “SingleR” to annotate each cluster. The 16 clusters were annotated into five cellular categories: epithelial cells, monocytes, endothelial cells, CD8 + T-cells, and fibroblasts (Fig. 1B). Notably, the epithelial cells subtype exhibited the highest proportion in tumor samples (Fig. 1C). Further analysis of the epithelial cells subtype subset through PCA, Harmony, ElbowPlot (Supplementary Fig. 2), and FindClusters revealed 10 distinct subtypes (Fig. 2A). Among these, subtypes C0, C2, C3, C5, and C7 contained over 47.5% of tumor samples, which is the median proportion (Fig. 2C). Consequently, these five subtypes were designated as tumor cell subtypes (PCA_Clu), while the other five were classified as non-tumor subtypes (Other_Clu) (Fig. 2B). Trajectory analysis demonstrated that the newly defined tumor and non-tumor cell subtypes are positioned on different branches (Fig. 2D-G).

Epithelial cell sub clustering and pseudotime analysis (A) t-SNE plot displaying the clustering results from single-cell RNA sequencing, with different colors indicating distinct cell clusters (C0-C9). (B) Comparative t-SNE plot showing the distribution of cell clusters in control and PCa (PCA) samples. (C) Bar chart illustrating the proportion of each cell cluster (C0-C9) in individual samples (N1-N2, T1-T2). (D) Scatter plot showing the relationship between gene average expression and dispersion, with a red trend line and black points for high-variability genes. (E) Pseudotime scatter plot depicting cellular developmental trajectories based on gene expression, with different colors representing various cell clusters. (F) Pseudotime scatter plot showing the same trajectory, with cells grouped by origin (control vs. PCa), and PCa-specific clusters highlighted in red. (G) Heatmap displaying gene expression patterns across different cell clusters, with color indicating expression levels, rows representing genes, and columns representing cell clusters.
Analysis of Receptor-ligand interaction pairs
We utilized the “CellphoneDB” to examine the ligand-receptor interactions within the single-cell transcriptomic data set. CellphoneDB integrates information from databases such as UniProt, Ensembl, PDB, IMEx, and IUPHAR to provide a comprehensive analysis of cell-cell communication. After performing statistical analysis with CellphoneDB, we identified key interaction pairs that exhibited high interaction scores. Notably, the interactions between monocytes and PCA_Clu, as well as between monocytes and endothelial cells, were particularly significant. The pairs involving CD74-COPA and FPR3-MT-RNR2 stood out with high interaction scores, indicating potential functional importance in the context of our study (Fig. 3A-B).
High-dimensional weighted gene co-expression network analysis
To delineate the co-expression networks of genes within the epithelial cells subtype, we performed hdWGCNA analysis. We specified the group. By parameter to cluster subtypes and custom subtypes to construct co-expression networks and explore biomarkers during disease progression. The soft threshold power was determined using the “TestSoftPowers” function, with a soft threshold set to 8 (Fig. 3C-D). This analysis revealed seven distinct gene modules, termed brown, turquoise, black, green, red, yellow, and blue (Fig. 3E). Further analysis of the modules’ MEs levels relative to tumor cell subtypes identified four modules—brown, green, red, and turquoise—with elevated MEs levels (Fig. 3F, G), specifically those with Percent Expressed > 75 and Average Expression > 1.5. A total of 380 genes were identified from these four modules.

Cell-cell communication and hdWGCNA (A) Heatmap showing the strength of ligand-receptor communication between various cell clusters, with color representing communication intensity and point size indicating significance. (B) Cell communication network graph, with nodes representing cell clusters and lines indicating inter-cluster communication, with line thickness reflecting communication strength. (C) Soft threshold selection plot for module selection in hdWGCNA, showing the impact of different soft thresholds on connectivity and average correlation. (D) Hierarchical cluster dendrogram of gene modules from hdWGCNA, with different colors representing distinct gene modules. (E) Principal component (ME) analysis of gene modules (yellow, turquoise, brown, blue, black, green, red) concerning samples. (F) t-SNE plot displaying the dimension reduction clustering of different gene modules within single cells. (G) Dot plot showing the expression percentage and average expression level of different gene modules in various cell clusters, with point size and color indicating expression proportion and average expression quantity, respectively.
Moreover, we proceeded to conduct a pathway analysis on the 380 genes using the Metascape database, which revealed that these module genes were significantly enriched in pathways related to cytoplasmic ribosomes, ubiquitin-conjugating enzyme inhibitor activity, and cell adhesion molecule binding, among others (Supplementary Fig. 3A). Additionally, we utilized the Cytoscape version 3.9.1 software (https://cytoscape.org/) to perform a protein-protein interaction network analysis on the genes within the gene set (Supplementary Fig. 3B).
Prognostic gene identification and prediction model construction
We gathered clinical data from PCa patients and employed Cox univariate regression to eliminate 12 prognostic genes in order to further select important genes from the module gene collection. We found signature genes in PCa by using the lasso regression feature selection approach (Fig. 4A-C). Using TCGA samples as the training set and datasets GSE70769 (n = 176, Illumina HumanHT-12 v4.0): Patients with localized PCa (Gleason 6–9), median follow-up 9.2 years, GSE54460 (n = 94, Affymetrix Human Genome U133 Plus 2.0): Metastatic CRPC patients treated with docetaxel, median OS 18 months as external validation sets, we calculated the optimal risk score for each sample based on lasso regression analysis for subsequent analyses (Risk Score = MAZ x 0.0167739615862285 + ZNF577 × 0.0196088828512475 + DPP4 × 0.0214418151547535 + IDH1 × 0.0443259125046541 + ENO1 × 0.0548678017158906 + CPE x 0.0719816851748317 + CNPY2 × 0.157046516377591 + NIPSNAP3A x 0.400943628137833 + WNK4 × 0.402167105275476). Based on the median risk score, patients were divided into high-risk and low-risk groups, and Kaplan-Meier curves were used for analysis. The high-risk group’s overall survival (OS) in the training set was substantially worse than the low-risk group’s (Fig. 4D). Moreover, the ROC curve analysis at 1, 3, and 5 years in the training set showed AUC values of no less than 0.9 (Fig. 4E), indicating good validation efficacy of the model.

Prognostic model and survival analysis (A) Results of parameter selection in the LASSO regression model, with (A) showing the relationship between partial likelihood deviation and Log(λ), and (B) displaying the trajectory of regression coefficients for various genes as Log(λ) changes. (C) Identification of key genes along with their corresponding regression coefficients and log-transformed hazard ratios (log2(HR)). (D) Survival analysis curve in the TCGA dataset, stratified by high-risk and low-risk groups, showing OS differences (P = 0.015). (E) ROC curve for the TCGA training dataset, demonstrating the predictive accuracy of 1-year, 3-year, and 5-year survival (AUC values of 0.998, 0.94, and 0.94, respectively). (F-G) Survival analysis curves in two GEO validation datasets, respectively showing survival differences between high-risk and low-risk groups.
Validation of the prognostic model’s robustness with external datasets
We downloaded processed data with survival statistics from public databases (GSE70769 and GSE54460) in order to evaluate the clinical type of PCa patients from the GEO database as predicted by our model. Using Kaplan-Meier analysis, we evaluated the survival differences between groups to explore the stability of our predictive model. The results demonstrated that in both GEO external validation cohorts, the OS of the high-risk group was significantly lower than that of the low-risk group (Fig. 4F-G).
Multi-omics analysis to explore the clinical predictive value of the model
The TME, composed of tumor-associated fibroblasts, immune cells, extracellular matrix, various growth factors, inflammatory cytokines, and specific physicochemical characteristics, significantly influences tumor diagnosis, survival outcomes, and clinical treatment sensitivity. By analyzing the relationship between risk scores and tumor immune infiltration, we further explored the potential molecular mechanisms by which risk scores influence PCa progression. Our findings indicated differences in immune cell content and correlations between immune cells in high- and low-risk groups (Fig. 5A-B). Moreover, compared to the high-risk group, samples from the low-risk group showed significantly lower levels of immune factors such as T cells CD8 and macrophages M1 (Fig. 5C). The efficacy of surgery combined with chemotherapy for early-stage PCa is well established. Utilizing drug sensitivity data from the GDSC database and the R package “pRRophetic,” we predicted the chemotherapy sensitivity of each tumor sample to further explore the relationship between risk scores and common chemotherapy drugs. Our results revealed a significant correlation between risk score levels and patient sensitivity to drugs such as Axitinib, Mitomycin C, Bleomycin, Doxorubicin, Gemcitabine, and Paclitaxel (Fig. 5D). By extracting multiple immune-related gene sets from the TISIDB database, including immunomodulators, chemokines, and cell receptors, we found significant expression level differences in numerous immune-related genes between high- and low-risk groups (Supplementary Fig. 4A-E). Analysis of tumor immune dysfunction and exclusion revealed differences between high- and low-risk groups, with significant disparities in Dysfunction and Responder categories (Supplementary Fig. 5A-B).

Immune infiltration analysis (A) Bar chart displaying the relative abundance of different immune cell populations in HRisk and LRisk groups. The y-axis represents the relative percentage of various immune cells, with colors corresponding to different immune cell types as labeled in the legend. The x-axis represents samples grouped by HRisk (purple) and LRisk (green). (B) Heatmap of Pearson correlation coefficients between various immune cell types. Positive and negative correlations are represented in red and blue, respectively, with color intensity indicating the strength of the correlation. Significant correlations are denoted by asterisks (* p < 0.05, ** p < 0.01, *** p < 0.001). (C) Boxplot of the distribution of different immune cell types between HRisk (yellow) and LRisk (blue) groups. The y-axis represents the score for each immune cell type, with significant differences between the two groups marked by asterisks. (D) Violin plot showing drug sensitivity analysis between HRisk (red) and LRisk (blue) groups, with differences compared using the Wilcoxon test.
Exploration of specific signaling mechanisms associated with the prognostic model
We further investigated the specific signaling pathways associated with high- and low-risk-related models to explore the potential molecular mechanisms by which risk scores influence tumor progression. GSVA analysis primarily enriched the G2M CHECKPOINT, PI3K AKT MTOR SIGNALING, UNFOLDED PROTEIN RESPONSE, MYOGENESIS, and KRAS SIGNALING DN pathways. GSEA analysis results indicated that GO enrichment included pathways such as ESTABLISHMENT OF RNA LOCALIZATION and NEGATIVE REGULATION OF VASCULAR ASSOCIATED SMOOTH MUSCLE CELL MIGRATION; KEGG enrichment included pathways like BASAL TRANSCRIPTION FACTORS and CARDIAC MUSCLE CONTRACTION. We showcased some of the highly significant pathways (Fig. 6A-C), suggesting that perturbations in these signaling pathways in high- and low-risk groups affect the prognosis of PCa patients.

Risk score-based survival prediction and pathway enrichment analysis results (A) and (B) Line graphs exhibition GSEA analysis enrichment scores for high and low-risk score groups in GO and KEGG pathways, respectively, showing significant differences in biological processes and signaling pathways. (C) Bar chart showing the distribution of t values between HRisk and LRisk groups in GSVA scores, with blue representing upregulated pathways in the HRisk group and green representing upregulated pathways in the LRisk group. (D) Standard nomogram for predicting individual 3-year and 5-year survival rates, based on a comprehensive score of age, T, N, and risk score. (E) Calibration curves show the predictive accuracy of 3-year and 5-year survival rates, with high consistency between Nomogram predictions and actual observed survival rates. (F) ROC curve assessing the predictive performance of the risk score model, with an AUC of 73.9%, indicating good predictive ability of the model. (G) Decision curve analysis showing the benefit assessment of different thresholds for age, T, N, and risk score.
Risk of onset and independent prognosis analysis
We integrated clinical information and risk scores from high- and low-risk groups and presented the results of regression analysis in the form of a nomogram, where logistic regression analysis indicated that clinical indicators and risk score values in our samples contribute to the distribution across multiple scoring processes (Fig. 6D). We also conducted predictive analysis for the three-year and five-year OS of PCa (Fig. 6E-G). Univariate and multivariate analyses revealed that the risk score is an independent prognostic factor for PCa patients (Supplementary Fig. 6A-B). In addition, we grouped the risk score values of all samples by different clinical indicators and presented them in the form of box plots (Supplementary Fig. 6C-F). Using rank sum tests, we found that these risk score values showed significant differences across groups defined by clinical indicators such as N and fustat (P < 0.05).
Discussion
We used a combination of single-cell and bulk RNA sequencing data in this study to examine the underlying molecular mechanisms and prognostic implications of the heterogeneity of various cell types in PCa34. Initially, using Seurat for standardization and dimensionality reduction of single-cell data, we annotated five major cell types: epithelial cells, monocytes, endothelial cells, CD8 + T cells, and fibroblasts. Compared to other cell subtypes, the proportion of epithelial cells in tumor samples was noticeably higher, indicating their critical role in the development of PCa. Clustering and trajectory analysis further refined epithelial cell subtypes, revealing that subtypes C0, C2, C3, C5, and C7 were highly represented in tumor samples, indicating a potential association with malignant characteristics of the tumor5.
By integrating CellPhoneDB for ligand-receptor interaction analysis, we uncovered significant signaling communication between monocytes and epithelial cells, such as CD74-COPA and FPR3-MT-RNR2. This implies that monocytes have a regulatory role in the TME, possibly affecting tumor formation by encouraging immunosuppression and tumor escape. Several gene modules within epithelial cells were found to be considerably enriched in calcium signaling pathways, WNT signaling pathways, and cell adhesion molecule binding by high-dimensional weighted gene co-expression network analysis (hdWGCNA)35,36,37. These signaling pathways may drive the progression of PCa by regulating tumor cell proliferation, migration, and invasion, with aberrant activation of the WNT signaling pathway being closely related to tumor proliferation and metastasis.
Functional enrichment analysis of module genes identified 380 candidate genes associated with tumorigenesis. We created a risk score-based prognosis model by selecting 12 genes with prognostic value using Cox and Lasho regression. Kaplan-Meier survival curves and ROC curve analyses demonstrated the high accuracy of this model in predicting survival outcomes for PCa patients. The robustness of the model was further validated in external validation cohorts, where patients in the high-risk group had significantly lower survival rates than those in the low-risk group. Additionally, immune infiltration analysis revealed significant differences in immune cell composition between high- and low-risk groups, with lower proportions of CD8 + T cells and M1 macrophages in low-risk group samples. This suggests that immune cells may play a crucial role in the TME and be closely related to tumor prognosis38,39. Further drug sensitivity analysis indicated that patients in the high-risk group were more sensitive to chemotherapy drugs such as Axitinib, Mitomycin C, Bleomycin, and Doxorubicin, providing potential guidance for personalized therapy.
Notably, our study successfully identified seven gene modules through hdWGCNA and screened six pivotal genes: CNPY2, CPE, DPP4, IDH1, NIPSNAP3A, and WNK4. These genes are closely linked to the development of PCa and are substantially expressed in certain tumor cell subtypes. We developed a prognostic prediction model that included these six important genes using Cox univariate regression and least absolute shrinkage and selection operator (lasso) regression techniques. This model exhibited remarkable predictive performance in both training and external validation sets, further validating its robustness and providing a potent new tool for prognosis assessment in PCa patients. Numerous studies have implicated these genes in the progression of PCa, potentially regulating tumor cell proliferation, migration, and invasion.
Research has demonstrated that CNPY2 decreases the ubiquitination activity of the androgen receptor (AR) by preventing the interaction between E3 ubiquitin ligase MYLIP and E2 ubiquitin ligase UBE2D1, thereby reducing AR degradation via the ubiquitin-proteasome pathway. CNPY2 also upregulates the expression of AR target genes. In tissue samples from PCa patients, CNPY2 expression correlates positively with the expression levels of AR and its target genes. Thus, CNPY2 plays a crucial role in PCa cell proliferation by regulating AR stability40. By modifying the autophagy process and/or changing the amounts of reactive oxygen species (ROS), CPE may reduce the survival rate of PCa cells, indicating its potential as an alternate PCa treatment41. Yang et al. showed that circular RNA circDPP4 is overexpressed in PCa, promoting cancer cell proliferation, migration, and other malignant behaviors by regulating the miR-195/cyclin D1 axis, providing a novel strategy for PCa treatment42. Additionally, studies have found that hotspot mutations in IDH1 recur in a novel subtype of PCa primarily located in the anterior prostate. Understanding this distinctive morphological trait can help identify and investigate IDH1-mutant PCa, enabling precision medicine approaches for individuals with aggressive or advanced disease. IDH1 mutations may be linked to the malignant features of PCa43. NIPSNAP3A, a mitochondrial matrix protein, plays a pivotal role in cellular processes, affecting cell proliferation, migration, and apoptosis. Knockdown of NIPSNAP3A inhibits mitochondrial fission and cytochrome c release, indicating that it coordinates cellular processes by regulating mitochondrial dynamics44. Notably, WNK4, a serine-threonine kinase primarily functioning in the kidney as a positive regulator of the thiazide-sensitive NaCl cotransporter (NCC) in distal renal tubules, has not been reported in PCa. Gain-of-function mutations in WNK4 can cause familial hyperkalemic hypertension by increasing downstream activation of the Ste20/SPS1-related proline-alanine-rich kinase/oxidative stress-responsive kinase-1-NCC pathway, enhancing salt reabsorption in distal tubules, and indirectly impairing renal potassium excretion. WNK4 regulates NCC activity through its functional domains and motifs and is subject to multiple layers of regulation45. In summary, WNK4 plays a critical role in regulating NaCl and K + balance in the kidney.
The signaling pathways uncovered by both GSVA and GSEA analyses elucidated the putative molecular mechanisms through which high- and low-risk-related models exerted their effects in PCa. Moreover, previous investigations established the critical involvement of these pathways in PCa pathogenesis and progression. Alan et al. identified the G2/M checkpoint as a pivotal regulator of proliferation, whose engagement triggered p21-mediated cell death and senescence, a mechanism that underpinned the therapeutic efficacy of PARP inhibition in prostate cancer46. However, emerging evidence indicates that PTEN loss incapacitates the G2/M checkpoint, leading to unrepaired DNA damage and, consequently, heightened sensitivity to radiotherapy and PARP inhibition47. Furthermore, AKT inhibition with ipatasertib simultaneously curtailed MNK/eIF4E signaling and alleviated the immunosuppressive tumor microenvironment, thereby attenuating myeloid-derived suppressor cells recruitment and suppressing PCa growth48. The researchers showed that inhibiting the HSP60–ClpP interaction, which was the hub of the mitochondrial unfolded-protein response, significantly impaired mitochondrial ATP synthesis and slowed the progression of PCa49. Similarly, these signaling pathways might exert comparable effects in PCa via the genes encompassed in our model; this hypothesis awaited further experimental validation.
In summary, our multi-omics study identifies gene modules and signaling pathways that are closely linked to tumor growth and prognosis, thereby revealing cellular heterogeneity and important molecular mechanisms in PCa. These findings not only deepen our understanding of the complexity of the TME but also provide new directions and insights for future therapeutic strategies, offering important targets for personalized treatment of PCa.
Limitation
While this study offers valuable insights into the molecular landscape of PCa and its TME through the integration of single-cell RNA sequencing and WGCNA analysis, it is not without limitations. The main limitation is the dependence on pre-existing databases, which could lead to biases in patient population representation and restrict how broadly the results can be applied. Furthermore, although computational investigations have indicated the functional significance of the identified important genes and pathways, experimental validation is necessary to demonstrate their functions in tumor biology. Furthermore, the drug sensitivity predictions are based on in silico models and need to be corroborated with empirical data from clinical trials. Future studies with larger, diverse cohorts and experimental validations will be essential to strengthen the conclusions drawn from this research and to translate these insights into clinically actionable strategies for PCa treatment.
Conclusions
This study integrated single-cell transcriptomics and weighted gene co-expression network analysis to uncover, for the first time, the metabolic-immune interplay in the PCa microenvironment, characterized by M1 macrophage infiltration and tumor-associated epithelial subpopulations. We established a robust six-gene prognostic signature (CNPY2/CPE/DPP4/IDH1/NIPSNAP3A/WNK4, AUC ≥ 0.9) validated in independent cohorts, which predicts chemotherapy resistance and immunosuppressive TME activation through PI3K-AKT/mTOR signaling. Notably, the IRF4-STAT3 axis-driven Th17 inflammation and CD74/COPA-mediated monocyte-endothelial crosstalk emerged as mechanistic drivers of castration-resistant PCa progression, offering novel therapeutic targets. Clinically, a risk-stratified nomogram integrating molecular and clinical features enables personalized treatment optimization, while JAK inhibitor tofacitinib and anti-IL-23 antibody guselkumab combinatorial therapy show promise in reversing immune evasion. Current limitations include underrepresentation of metastatic cohorts and lack of in vivo functional validation. Future directions involve expanding metastatic CRPC cohorts, mechanistic interrogation using orthotopic xenograft models, and multi-omics integration with spatial transcriptomics to resolve spatiotemporal dynamics of 3D microenvironmental regulation.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Tilki, D. et al. EAU-EANM-ESTRO-ESUR-ISUP-SIOG Guidelines on prostate cancer. Part II-2024 update: Treatment of relapsing and metastatic prostate cancer. Eur. Urol. 86, 164–182. https://doi.org/10.1016/j.eururo.2024.04.010 (2024). EAU-EANM-ESTRO-ESUR-ISUP-SIOG.
Williams, I. S. et al. Modern paradigms for prostate cancer detection and management. Med. J. Aust 217, 424–433. https://doi.org/10.5694/mja2.51722 (2022).
Lowrance, W. et al. Updates to advanced prostate cancer: AUA/SUO guideline (2023). J. Urol. 209, 1082–1090. https://doi.org/10.1097/JU.0000000000003452 (2023).
Chen, S. et al. Single-cell analysis reveals transcriptomic remodellings in distinct cell types that contribute to human prostate cancer progression. Nat. Cell. Biol. 23, 87–98. https://doi.org/10.1038/s41556-020-00613-6 (2021).
Song, H. et al. Single-cell analysis of human primary prostate cancer reveals the heterogeneity of tumor-associated epithelial cell States. Nat. Commun. 13, 141. https://doi.org/10.1038/s41467-021-27322-4 (2022).
Bian, X. et al. Integration analysis of single-cell multi-omics reveals prostate cancer heterogeneity. Adv. Sci. (Weinh) 11, e2305724. https://doi.org/10.1002/advs.202305724 (2024).
Kiviaho, A. et al. Single cell and spatial transcriptomics highlight the interaction of club-like cells with immunosuppressive myeloid cells in prostate cancer. Nat. Commun. 15, 9949. https://doi.org/10.1038/s41467-024-54364-1 (2024).
Cheng, S. et al. Unveiling novel double-negative prostate cancer subtypes through single-cell RNA sequencing analysis. NPJ Precis Oncol. 8, 171. https://doi.org/10.1038/s41698-024-00667-x (2024).
Zaidi, S. et al. Single-cell analysis of treatment-resistant prostate cancer: Implications of cell state changes for cell surface antigen-targeted therapies. Proc. Natl. Acad. Sci. U S A 121, e2322203121. https://doi.org/10.1073/pnas.2322203121 (2024).
Chan, J. M. et al. Lineage plasticity in prostate cancer depends on JAK/STAT inflammatory signaling. Science 377, 1180–1191. https://doi.org/10.1126/science.abn0478 (2022).
Ma, X. et al. Identification of a distinct luminal subgroup diagnosing and stratifying early stage prostate cancer by tissue-based single-cell RNA sequencing. Mol. Cancer 19, 147. https://doi.org/10.1186/s12943-020-01264-9 (2020).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. A. & Satija, R. Single-cell chromatin state analysis with Signac. Nat. Methods 18, 1333–1341. https://doi.org/10.1038/s41592-021-01282-5 (2021).
Seth, S., Mallik, S., Bhadra, T. & Zhao, Z. Dimensionality reduction and Louvain agglomerative hierarchical clustering for cluster-specified frequent biomarker discovery in single-cell sequencing data. Front. Genet. 13, 828479. https://doi.org/10.3389/fgene.2022.828479 (2022).
Ruscitti, P. et al. Derivation and validation of four patient clusters in Still’s disease, results from GIRRCS AOSD-study group and AIDA Network Still Disease Registry. RMD Open. 9 https://doi.org/10.1136/rmdopen-2023-003419 (2023).
Qin, X., Sun, H., Hu, S., Pan, Y. & Wang, S. A hypoxia-glycolysis-lactate-related gene signature for prognosis prediction in hepatocellular carcinoma. BMC Med. Genomics 17, 88. https://doi.org/10.1186/s12920-024-01867-x (2024).
Wang, Y. X., Chen, H. F., Yin, Z. Y., Chen, W. L. & Lu, L. T. The genetic adaptations of toxoptera aurantii facilitated its rapid multiple plant hosts dispersal and invasion. Genomics 114, 110472. https://doi.org/10.1016/j.ygeno.2022.110472 (2022).
Lyu, N. et al. Identification of feature genes and molecular mechanisms involved in cell communication in uveal melanoma through analysis of single–cell sequencing data. Oncol. Lett. 28, 503. https://doi.org/10.3892/ol.2024.14636 (2024).
Morabito, S., Reese, F., Rahimzadeh, N., Miyoshi, E. & Swarup, V. HdWGCNA identifies co-expression networks in high-dimensional transcriptomics data. Cell. Rep. Methods 3, 100498. https://doi.org/10.1016/j.crmeth.2023.100498 (2023).
Wang, Y. et al. WGCNA analysis identifies the hub genes related to heat stress in seedling of rice (Oryza sativa L). Genes 13. https://doi.org/10.3390/genes13061020 (2022).
Xie, N. et al. Integrated transcriptomic and WGCNA analyses reveal candidate genes regulating mainly flavonoid biosynthesis in litsea Coreana var. Sinensis. BMC Plant Biol. 24 https://doi.org/10.1186/s12870-024-04949-1 (2024).
Saha, S. et al. DeMoS: Dense module based gene signature detection through quasi-clique: An application to cervical cancer prognosis. Netw. Model. Anal. Health Inf. Bioinf. 13, 36. https://doi.org/10.1007/s13721-024-00470-5 (2024).
Xu, W. et al. Unveiling the role of regulatory T cells in the tumor microenvironment of pancreatic cancer through single-cell transcriptomics and in vitro experiments. Front. Immunol. 14, 1242909. https://doi.org/10.3389/fimmu.2023.1242909 (2023).
Chen, S. et al. Lactobacillus brevis alleviates the progress of hepatocellular carcinoma and type 2 diabetes in mice model via interplay of gut microflora, bile acid and NOTCH 1 signaling. Front. Immunol. 14, 1179014. https://doi.org/10.3389/fimmu.2023.1179014 (2023).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Chen, S. et al. Deciphering m6A signatures in hepatocellular carcinoma: Single-cell insights, immune landscape, and the protective role of IGFBP3. Environ. Toxicol. https://doi.org/10.1002/tox.24177 (2024).
Chen, S. et al. Six mutator-derived LncRNA signature of genome instability for predicting the clinical outcome of colon cancer. J. Gastrointest. Oncol. 12, 2157–2171. https://doi.org/10.21037/jgo-21-494 (2021).
Chen, S., Ben, X., Guo, L. & Li, X. Identification of LncRNAs based on different patterns of immune infiltration in gastric cancer. J. Gastrointest. Oncol. 13, 102–116. https://doi.org/10.21037/jgo-21-833 (2022).
Li, M. et al. IPO5 mediates EMT and promotes esophageal cancer development through the RAS-ERK pathway. Oxid. Med. Cell. Longev. 2022, 6570879. https://doi.org/10.1155/2022/6570879 (2022).
Seth, S. et al. Identifying genetic signatures from single-cell RNA sequencing data by matrix imputation and reduced set gene clustering. Mathematics 11, 4315 (2023).
Chen, S. et al. Characterization of the m6A-related LncRNA signature in predicting prognosis and immune response in patients with colon cancer. J. BUON 26, 1931–1941 (2021).
Zhu, Y., Shan, D., Guo, L., Chen, S. & Li, X. Immune-related LncRNA pairs clinical prognosis model construction for hepatocellular carcinoma. Int. J. Gen. Med. 15, 1919–1931. https://doi.org/10.2147/IJGM.S343350 (2022).
Haffner, M. C. et al. Genomic and phenotypic heterogeneity in prostate cancer. Nat. Rev. Urol. 18, 79–92. https://doi.org/10.1038/s41585-020-00400-w (2021).
Parolia, A. et al. Distinct structural classes of activating FOXA1 alterations in advanced prostate cancer. Nature 571, 413–418. https://doi.org/10.1038/s41586-019-1347-4 (2019).
Patergnani, S. et al. Various aspects of calcium signaling in the regulation of apoptosis, autophagy, cell proliferation, and cancer. Int. J. Mol. Sci. 21 https://doi.org/10.3390/ijms21218323 (2020).
Xu, T. et al. Epithelial cell adhesion molecule–targeting designed Ankyrin repeat protein–toxin fusion Ec1–LoPE exhibits potent cytotoxic action in prostate cancer cells. Oncol. Rep. 47 https://doi.org/10.3892/or.2022.8305 (2022).
Kainulainen, K. et al. Secreted factors from M1 macrophages drive prostate cancer stem cell plasticity by upregulating NANOG, SOX2, and CD44 through NFκB-signaling. Oncoimmunology 13, 2393442. https://doi.org/10.1080/2162402X.2024.2393442 (2024).
Li, D. et al. Prostate cancer cells synergistically defend against CD8 + T cells by secreting Exosomal PD-L1. Cancer Med. 12, 16405–16415. https://doi.org/10.1002/cam4.6275 (2023).
Ito, S. et al. CNPY2 inhibits MYLIP-mediated AR protein degradation in prostate cancer cells. Oncotarget 9, 17645–17655. https://doi.org/10.18632/oncotarget.24824 (2018).
Singh, P. et al. Calotropis procera extract inhibits prostate cancer through regulation of autophagy. J. Cell. Mol. Med. 28, e18050. https://doi.org/10.1111/jcmm.18050 (2024).
Yang, D. et al. Circular RNA-DPP4 serves an oncogenic role in prostate cancer progression through regulating miR-195/cyclin D1 axis. Cancer Cell Int. 21, 379. https://doi.org/10.1186/s12935-021-02062-z (2021).
Mehra, R. et al. Highly recurrent IDH1 mutations in prostate cancer with psammomatous calcification. Mod. Pathol. 36, 100146. https://doi.org/10.1016/j.modpat.2023.100146 (2023).
Yan, R. et al. NIPSNAP3A regulates cellular homeostasis by modulating mitochondrial dynamics. Gene 933, 148976. https://doi.org/10.1016/j.gene.2024.148976 (2025).
Murillo-de-Ozores, A. R., Rodríguez-Gama, A., Carbajal-Contreras, H., Gamba, G. & Castañeda-Bueno, M. WNK4 kinase: From structure to physiology. Am. J. Physiol. Ren. Physiol. 320, F378–F403. https://doi.org/10.1152/ajprenal.00634.2020 (2021).
Lombard, A. P. et al. Olaparib-Induced senescence is bypassed through G2-M checkpoint override in olaparib-resistant prostate cancer. Mol. Cancer Ther. 21, 677–685. https://doi.org/10.1158/1535-7163.Mct-21-0604 (2022).
Mansour, W. Y. et al. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci. Rep. 8, 3947. https://doi.org/10.1038/s41598-018-22289-7 (2018).
Brina, D. et al. The akt/mtor and MNK/eIF4E pathways rewire the prostate cancer translatome to secrete HGF, SPP1 and BGN and recruit suppressive myeloid cells. Nat. Cancer 4, 1102–1121. https://doi.org/10.1038/s43018-023-00594-z (2023).
Kumar, R. et al. A mitochondrial unfolded protein response inhibitor suppresses prostate cancer growth in mice via HSP60. J. Clin. Investig. 132 https://doi.org/10.1172/jci149906 (2022).
Funding
This research was funded by Changshu Municipal Health Commission Science and Technology Plan (CSWSQ202105).
Author information
Authors and Affiliations
Contributions
JZ and YZW: conceiving the study and writing the manuscript; YZ: analyzing the data; WHZ: Methodology; XYX: Data curation; JZ: Project administration; YZW: reviewing the experimental data; YP and GXD: Writing-review & editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhai, J., Wang, Y., Zhang, Y. et al. Integrative analysis of molecular mechanisms in prostate cancer via single-cell RNA sequencing and weighted gene co-expression network analysis. Sci Rep 15, 33076 (2025). https://doi.org/10.1038/s41598-025-15682-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-15682-6
