
Abstract
The damage of ferroptosis is related to the pathogenesis of intervertebral disc degeneration (IDD). N6-methyladenosine (m6A) modification accounts for more than 80% of RNA modifications in eukaryotic cells. However, the key role of m6A related ferroptosis-related genes (FRGs) in IDD remain explorable. Firstly, batch correction between datasets was performed. Weighted gene co-expression network analysis (WGCNA) was then conducted to acquire the most relevant module genes. Differentially expressed genes (DEGs) between the IDD and Normal Group were obtained by differential analysis subsequently. Next, according to the Pearson correlation and the intersection of m6A and FRGs related genes, m6A-FRGs related genes were selected. Additionally, we intersected module genes, DEGs and m6A-FRGs related genes to obtain DEGs of m6A-FRGs related module genes (m6A-FRGs-DEGs). Least absolute shrinkage and selection operator (LASSO) was applied to get HUBgenes. After, we conducted gene set enrichment analysis (GSEA) to gain function items and related pathways of HUBgenes. Single sample gene set enrichment analysis (ssGSEA) and wilcox.test were proceeded to analyse differences in relative abundance of immune cells between the IDD and Normal Groups, and a nomogram was constructed based on significantly different HUBgenes. The Comparative Toxicogenomics Database (CTDbase) was then applied to predict potential drugs or molecular compounds that could modulate HUBgenes. Last but not least, we performed the quantitative real-time fluorescence PCR (qRT-PCR) to verify HUBgenes. 181 m6A-FRGs-DEGs were acquired by the intersection of key module genes (from MEtan, MEsalmon, MEbrown and MEgreen), 362 DEGs and 15,678 m6A-FRGs related genes. Subsequent analysis showed that HUBgenes (ZNF595, PLXDC1, FNBP1L, KLRB1, NRCAM, PPCDC, C9orf139, SIGLEC17P, RRAS2 and DPRXP4) significantly participated in positive regulation of cytokine production, mitochondrial inner membrane and organellar ribosome. Besides, the relative abundance of neutrophils was found significantly different between IDD and normal groups. A nomogram was constructed based on ZNF595 and RRAS2, and there were 11 drugs targeted on ZNF595, while 118 drugs predicted based on RRAS2 such as Tetrachlorodibenzodioxin, Bisphenol A and Benzo(a)pyrene. Lastly, ZNF595 and RRAS2 were both obviously up-regulated in IDD according to the qRT-PCR. Our research suggested 10 HUBgenes were significantly associated with IDD, providing more evidence about the vital role of HUBgenes in IDD.
Similar content being viewed by others


Introduction
Intervertebral disc degeneration (IDD) is one of the most common degenerative diseases of musculoskeletal disorders (MSKs)1. It can lead to low back pain (LBP), a primary and universal cause of disability2. The incidence of IDD is mostly concentrated in middle-aged and elderly people, which increases in prevalence with age3. However, research implicated that early IDD in the young was nonnegligible as well4,5. Therefore, IDD not only seriously affects the quality of patients’ life but also contributes to huge social and economic burden6.
IDD results from a lot of factors, which include genetic inheritance, ever-increasing aging, mechanical injury, malnutrition, etc7. Pathological changes of intervertebral disc (IVD) mainly consist of the narrowing of intervertebral space, dysfunction of nucleus pulposus cell (NPC) like senescence and apoptosis, progressive degradation of extracellular matrix (ECM), rupture and fibrosis of annulus fibrosus (AF), calcification of cartilaginous endplates (CEPs) and so on8. However, although scientists have already made a lot of effort, the exact etiology and molecular mechanism of IDD remain unclear, which are still worth exploring in the foreseeable future. Meanwhile, as tissue engineering9, growth factor therapy10, gene therapy11, small molecule-based treatment approach12, and cell-based treatment13 are proposed to regenerate IVD, it is still a fully noteworthy direction to explore new therapies aiming at diverse targets for IDD.
Nowadays, ferroptosis, one of the forms of programmed cell death (PCD), has been increasingly attractive as a conceivable therapeutic target for IDD14,15,16. It is driven by iron-dependent phospholipid peroxidation and regulated by multiple cellular metabolic pathways, including redox homeostasis, iron handling, mitochondrial activity and metabolism of amino acids, lipids and sugars, in addition to various signaling pathways relevant to degenerative diseases17,18. As such, pharmacological modulation of ferroptosis, via both its induction and its inhibition, holds great potential for the treatment of IDD linked to extensive lipid peroxidation19. Several studies have shown that the pathogenesis of IDD involved ferritin degradation mediated by ferritin phagocytosis and subsequent lipid peroxidation. In a rat model of IDD, the levels of GPX4, an inhibitor of ferroptosis, and ferritin heavy chain were found significantly lower than the control group20,21. In addition, while injury-induced IDD process could be attenuated by deferoxamine, another inhibitor of ferroptosis20, it can be promoted by iron overload through inducing oxidative stress and ferroptosis in endplate chondrocytes22. Thus, the inhibition of NPC ferroptosis has been already suggested as a probable effective strategy to alleviate IDD23. Wenkai et al. (2025) found that through marked ferroptosis inhibition in NPCs that not only prevents LDH recurrence but also reverses the IDD symptoms, leading to robust restoration of NP structure and functions24. Jitian et al. (2025) also suggested that ferroptosis was identified as critical molecular entities and cellular pathways implicated in reactive oxygen species (ROS)-mediated IDD25. Therefore, studying the role of ferroptosis in IDD will help us to reveal the molecular mechanism of IDD to a higher degree and provide further references for therapeutic strategies.
Epigenetic modification means that gene expression and function change without changing the gene sequence, including chemical modifications of DNA, RNA and proteins. For instance, Yongzhao et al. (2025) constructed a novel nanoparticle named Hi-Exos to reduce the senescence of NPC and repair IDD via epigenetic modification, involving the epigenetic factor miR-221-3p26. Shibin et al. (2025) also found that the activator protein-1 (AP-1) transcription factor critical in driving the chromatin accessibility changes during IDD27. Among many epigenetic modifications of inflammation in IDD28, N6-methyladenosine (m6A) methylation is not only the most abundant internal modification of mRNA in eukaryotes, but also is widely discovered in non-coding RNAs, such as rRNAs and snRNAs29,30,31. Briefly said, m6A methylation transfers methyl to the N-6 position of adenosine in RNA32. It has a conserved modified gene sequence, which is enriched in long exons, near stop codons and 3’-untranslated regions (3’-UTR)33,34. Moreover, the m6A methylation process is dynamic and reversible, regulated by “writers”, “erasers” and “readers”35. Some researchers have elucidated the close relationship between m6A and ferroptosis, indicating that targeting m6A to influence ferroptosis might be a promising therapy for IDD36,37,38. Nevertheless, the key role of m6A related ferroptosis-related genes (FRGs) in IDD still remains unreported.
Therefore, in this study, we applied bioinformatic methods to identify m6A related FRGs and suggested HUBgenes, which were significantly related to IDD. Through a series of analyses, the study offered more evidence about the vital role of HUBgenes in IDD. In this way, we hoped to elucidate the molecular mechanism that underlies IDD, leading us to identify receptors and bridging molecules of m6A and ferroptosis, which can be useful in incidence or severity prediction of IDD and guide better health care for patients.
Materials and methods
Date sources and reduction
There were 17 normal and 17 IDD blood samples in transcriptome dataset GSE150408 obtained from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/). The transcriptome dataset GSE124272 has 8 normal and 8 IDD blood samples, as GSE167199 has 3 normal and 3 IDD nucleus pulposus samples. 27 m6A methylation regulatory factors were acquired from the research conducted by Lu, et al39. There were 9 FRGs in the ferroptosis database (FerrDb V2) (http://www.zhounan.org/ferrdb). Principal component analysis (PCA) was used to batch effect between GSE150408 and GSE124272 datasets. Batch correction between IDD and normal groups was performed by the limma package (version 3.44.3), and the batch effect between datasets were tested by PCA again40.
Weighted gene co-expression network analysis (WGCNA)
The WGCNA algorithm was applied to construct and analyze the co-expression network of the whole gene expression profiles of normal and IDD samples in the merged dataset. The IDD and normal samples were used as WGCNA trait data to screen the highest correlation modules and genes related to IDD. Samples of the merged dataset (25 IDD and 25 normal samples) were used to construct a co-expression network by WGCNA package41. The overall correlation of all samples in the dataset was obtained by clustering, with Z.k < −2.5 as the connectivity screening threshold, and outlier samples were eliminated to ensure the accuracy of the analysis. The soft threshold on the data was determined to ensure that the intergenic interaction between genes conforms to the scale-free distribution to the maximum extent. Adjacency and similarity between genes were calculated to derive the dissimilarity coefficient, then the system clustering tree among genes was obtained. According to the standard of mixed dynamic shear tree, the minimum number of genes of per gene module were set to 50. MEDissThres were set to 0.3 to merge similar modules. The correlation analyses of gene modules with IDD and normal samples were performed, we transformed phenotypic traits into quantitative traits, with |Cor|> 3 and p < 0.01 were set as the threshold to screen modules.
Differential expression analysis and correlation analysis
Limma package was applied to obtain the differentially expressed genes (DEGs) between the normal and IDD groups in the merged dataset (|log2FC|> 0.5, p < 0.05). The ggplot2 package (version 3.3.2) and the heatmap package (version 0.7.7) were used to plot the volcano and heatmap, respectively42. Pearson correlations between 27 m6A methylation regulators and all genes in the merged dataset were calculated, and correlations between 9 FRGs and all genes in the merged dataset were calculated by pearson (|Cor|> 0.3 and p < 0.01). m6A-FRGs related genes were obtained by the intersection of m6A related and FRGs related genes.
Differential expression module gene
Module genes, DEGs and m6A-FRGs related genes were intersected to obtain m6A-FRGs related DEGs (m6A-FRGs-DEGs). Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of m6A-FRGs-DEGs were performed by clusterProfiler (version 3.2–3)43,44,45,46. The STRING online database (http://cn.string-db.org) was applied to construct a protein–protein interaction (PPI) network of m6A-FRGs-DEGs47. The results were visualized by Cytoscape v.3.9.1 and used for constructing the enrichment analysis pathway network of m6A-FRGs-DEGs by the clueGO plug-in (p < 0.05).
Screening of HUBgenes
HUBgenes of IDD were screened by least absolute shrinkage and selection operator (LASSO) to further evaluate the value of m6A-FRGs-DEGs48. We performed 5 types of machine learning methods on HUBgenes, including Logistic Regression (LR), Random Forest (RF), eXtreme Gradient Boosting (XGBoost), Adaptive Boosting (AdaBoost), and Support Vector Machine (SVM)49,50,51,52,53. The receiver operating characteristic (ROC) curves of 5 types of machine learning were drawn by the survival ROC to verify the credibility of HUBgenes54.
Enrichment analysis of HUBgenes
GOSemSim package was employed to perform functional similarity (friend) analysis on HUBgenes55. Functionally similar significantly correlated HUBgenes were visualized by boxplots. We calculated the association of HUBgenes with other genes in the merged dataset, and according to the correlation ranking, Gene Set Enrichment Analysis (GSEA) was performed on HUBgenes by clusterProfiler (version 4.0.2) and org.Hs.eg.db (version 3.13.0)43. GO and KEGG gene sets were applied to assess enrichment analysis of related pathways and molecular mechanisms of IDD, the minimum and maximum gene set sizes were set as 10 and 500, respectively (p < 0.05 and q < 0.25)44,45,46.
Immune infiltration and cytokine analysis
In the study, we used 29 immune-related gene sets (including 16 gene sets of immune cells and 13 immune-related pathways), including immune cell species, immune-related pathways and functions56. In order to further explore the infiltration of immune cells in normal and IDD samples, we analyzed the relative abundance of immune cells in GSE124272 and GSE150408. The single sample GSEA (ssGSEA) algorithm was applied to estimate the relative abundance of 29 immune cells. Differences in relative abundance of immune cells between normal and IDD samples were calculated by wilcox.test, and the immune cells with significant differences in both datasets would be selected as the key differential immune cells. ROC curves were plotted to determine the credibility of key differential immune cells. The correlation between HUBgenes and differential immune cells in the two datasets was calculated, respectively. And correlation network map was drawn (|Cor|> 0.3 and p < 0.05). Studies have already shown that NPC degeneration could increase the secretion of proinflammatory cytokines23. Therefore, the pearson correlation between HUBgenes and common cytokines in the merged dataset was analyzed. And significant correlation between HUBgenes and common cytokines were visualized by the heatmap.
Relationship between HUBgenes and clinical features, and the identification of potential drugs
In order to verify the effectiveness of the selected genes, the expression of HUBgenes were verified in GSE167199. And the correlation between HUBgenes obtained by verification (p < 0.05) and clinical features in GSE167199 was calculated. Next, SVM prediction model and ROC curve were constructed for HUBgenes verificated. A nomogram was constructed based on HUBgenes that were significantly different in the validation set, and the calibration curve was drawn based on the prediction model. Then, we applied the Comparative Toxicogenomics Database (CTDbase) (http://ctdbase.org/) to predict potential drugs or molecular compounds that could modulate HUBgenes that were significantly different in the validation set. And genes-drugs regulatory network was drawn based on all drugs and HUBgenes.
Experimental verification
All samples were obtained from IDD patients with knowledge and consent from Sun Yat-sen Memorial Hospital, Sun Yat-sen University (SYSU). The study has already been approved by the Sun Yat-sen Memorial Hospital Medical Ethics Committee. 20 frozen tissue samples of IVDs were divided into two groups, of which 10 normal and 10 IDD samples. Firstly, total RNA of samples was isolated and purified by TRIzol (Ambion) reagent following the instruction manual. Then, the extracted RNA was tested for concentration by NanoPhotometer N50. Next, reverse transcription via SureScript-First-strand-cDNA-synthesis-kit (Servicebio) by an ordinary PCR instrument. Reverse transcription product cDNA was diluted 5 to 20 times with ddH2O (RNase/ DNase free). Subsequently, polymerase chain reaction (PCR) amplification reaction was performed by CFX96 real-time quantitative PCR instrument. 1 min at 95 ℃ (pre-denaturation), followed by at 95 ℃ for 20 s (denaturation), 55 ℃ for 20 s (annealing) and 72 ℃ for 30 s (elongation). The above reactions were subjected to forty cycles. Primer sequences were shown in Table 1. The quantitative real-time fluorescence PCR (qRT-PCR) was performed to verify the expression levels of HUBgenes that were significantly different in the validation set in IDD.
Statement
All methods in the present article were performed in accordance with the relevant guidelines and regulations. Also, as for the involving human tissue samples used in the study, informed consent was obtained from all subjects and/or their legal guardian(s).
Results
Data consolidation and batch correction
There were significant systematic differences in gene expression between GSE150408 and GSE124272 before batch correction (Fig. 1a). PCA showed that samples were obviously divided into two clusters (Fig. 1b). After batch correction, the system differences between datasets GSE150408 and GSE124272 were eliminated (Fig. 1c). PCA showed that the samples clearly have been fused into one cluster (Fig. 1d). It was indicated that two datasets have been effectively merged, and there was no systematic error in the merged new dataset for subsequent analysis.

Dataset batch correction: (a) Dataset expression box plot before batch correction; (b) Dataset PCA main component scatter plot before batch correction; (c) Dataset expression box plot after batch correction; (d) PCA main component scatter plot after batch correction.
Screening module genes by WGCNA
Sample clustering result showed that GSM4548717 and GSM3526786 among all samples were outliers (Fig. 2a). The soft threshold was determined as 5, so that the network was closest to the distribution without network scale (Fig. 2b). 14 modules were obtained by mixed dynamic shear tree, and there were 11 modules left after merging (Fig. 2c). Among the 11 co-expression modules, MEtan has the highest correlation with normal and IDD phenotypic traits (normal: Cor = -0.48 and p = 6e-04, IDD: Cor = 0.48 and p = 6e-04), with a negative correlation with normal while a positive correlation with IDD. In addition, MEsalmon (|Cor|= 0.42, p = 0.003), MEbrown (|Cor|= 0.4, p = 0.006) and MEgreen (|Cor|= 0.36, p = 0.01) were also highly correlated with phenotypic traits. Therefore, MEtan, MEsalmon, MEbrown and MEgreen modules were selected as candidate key modules (2749 genes) for subsequent analyses (Fig. 2d).

(a) Sample clustering tree; (b) Scale-free soft threshold distribution. The higher the square of the correlation coefficient, the closer the network is to the distribution without network scale; (c) Module merging. Genes are divided into various modules through hierarchical clustering. Different colors represent different modules. Gray is the default gene that cannot be classified into any module; (d) Heat map of module and trait correlation. Red in the figure represents positive correlation, green represents negative correlation, and the color depth represents the degree of correlation.
Differential expression analysis and correlation analysis between normal and IDD group
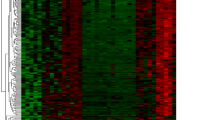
There were 362 DEGs between normal and IDD groups, of which 159 were up-regulation and 203 were down-regulation (Fig. 3a). Heat map showed the expression of DEGs between IDD and Normal group (Fig. 3b). Then, 15,678 m6A-FRGs related genes were obtained by intersection and correlation analysis (Fig. 3c).

(a, b) Volcano plot and heat map of differentially expressed genes between sample groups: (a) Volcano plot of differential genes. Each point in the figure represents a gene, green and red points represent significantly differentially expressed genes. The red dots indicate that the gene expression level is up-regulated, the green indicate down-regulated, and the black indicate that there is no significant difference between these genes; (b) Differential gene heat map. Each square in the heat map represents the relative expression of a gene in different samples. Red indicates high relative expression and blue indicates low; (c) The intersection and correlation analysis of m6A-FRGs related genes.
Enrichment analysis and construction of m6A-FRGs-DEGs
There were 181 m6A-FRGs-DEGs obtained by the intersection (Fig. 4a). In the GO functional enrichment analysis, m6A-FRGs-DEGs were mainly enriched in regulation of immune effector process, natural killer (NK) cell mediated immunity and immune receptor activity (Fig. 4b, Supplementary Data). Then in the KEGG functional enrichment analysis, m6A-FRGs-DEGs were involved in osteoclast differentiation, NK cell mediated cytotoxicity, antigen processing, and presentation (Fig. 4c). Notably, there were 12 m6A-FRGs-DEGs (KLRB1, IL2RB, KLRC1, COMMD6, GZMA, SH2D1B, NCR3, KLRD1, KLRK1, KLRF1, KLRC3 and KLRC4) in the PPI network (Fig. 4d). Moreover, enrichment analysis pathway network showed that m6A-FRGs-DEGs were mainly participated in ficolin-1-rich granule, osteoclast differentiation and immune receptor activity (Fig. 4e).

(a) DE-COR-ME Venn. The blue circle represents DEGs, the yellow represents MEgenes, and the green represents the COR gene set; (b) GO enrichment results of differentially expressed genes: (b1) GO enrichment bar chart (the top 10 were screened and drawn according to significance ranking). The color represents p value. The redder the color, the smaller the value; (b2) GO enrichment chord diagram (top 16 were filtered for visualization based on significance ranking); (c) KEGG functional enrichment analysis; (d) PPI of m6A-FRGs-DEGs. Red nodes are up-regulated proteins, blue nodes are down-regulated, color depth and node size are distributed according to logFC, and the thickness of the connecting line is sorted according to the confidence of protein interaction; (e) Enrichment network analysis of m6A-FRGs-DEGs: (e1) The node colors represent different enriched pathway categories, the color ratio represents the number of genes overlapped by each pathway, and the thickness of the connecting line represents the degree of association between pathways; (e2) The bar graph corresponding to the enriched network. The length of the bar graph represents the number of enriched genes; (e3) The pie chart corresponding to the network diagram, the area of the pie chart represents the proportion of enriched pathway categories.
Screening HUBgenes by machine learning methods
There were 10 HUBgenes (ZNF595, PLXDC1, FNBP1L, KLRB1, NRCAM, PPCDC, C9orf139, SIGLEC17P, RRAS2 and DPRXP4) screened out by LASSO (Fig. 5a). Through the ROC curves, it was clearly found that 10 HUBgenes had favorable reliability (Fig. 5b). Furthermore, it was found that 7 HUBgenes (ZNF595, PLXDC1, FNBP1L, KLRB1, NRCAM, PPCDC and RRAS2) have significant functional similarity (Fig. 5c).

(a) Cross-validated LASSO model plot: (a1, a2). In actual analysis, we hope to find the position with the smallest cross-validation error. In a1, the dotted line is the position with the smallest cross-validation error. According to this position (λ.min), the corresponding horizontal axis log (λ) is determined. The number of characteristic genes is shown above. After finding the optimal log (λ) value, the corresponding gene and its coefficient are found in a2, as well as the proportion of residuals explained by the model; (a3) ROC curve for LASSO screening genes; (b) Machine Learning Model ROC Curves: (b1) ROC curves of five machine learning models; (b2) ROC curve of HUBgenes in GSE124272; (b3) ROC curve of HUBgenes in GSE150408; (c) HUBgene functional similarity analysis.
GSEA of HUBgenes
In the GO enrichment analysis, ZNF595 was mainly enriched in telomere maintenance, RNA splicing, via transesterification reactions and nuclear chromosome. PLXDC1 was involved in cytoplasmic translation, mitochondrial matrix and olfactory receptor activity. In addition, FNBP1L was mainly participated in cytoplasmic translation, mitochondrial inner membrane and mitochondrial matrix. KLRB1 was associated with mRNA splicing, via spliceosome, organellar ribosome and positive regulation of cytokine production. NRCAM was mainly enriched in maturation of LSU-rRNA, organellar large ribosomal subunit and organellar ribosome. PPCDC and RRAS2 took part in cytoplasmic translation, mRNA splicing, via transesterification reactions and organellar ribosome. And in the KEGG enrichment analysis, ZNF595 was mainly enriched in neuroactive ligand-receptor interaction, ribosome and spliceosome. PLXDC1 mainly participated in NOD-like receptor signaling pathway and ribosome. FNBP1L was mainly involved in DNA replication, ribosome biogenesis in eukaryotes and ribosome. KLRB1 and RRAS2 were associated with osteoclast differentiation and ribosome. NRCAM and PPCDC took part in NOD-like receptor signaling pathway, osteoclast differentiation and ribosome (Fig. 6a–g, Supplementary Data).

GSEA enrichment analysis of HUBgenes: (a1) ZNF595.GSEA.GO; (a2) ZNF595.GSEA.KEGG; (b1) PLXDC1.GSEA.GO; (b2) PLXDC1.GSEA.KEGG; (c1) FNBP1L.GSEA.GO; (c2) FNBP1L.GSEA.KEGG; (d1) KLRB1.GSEA.GO; (d2) KLRB1.GSEA.KEGG; (e1) NRCAM.GSEA.GO; (e2) NRCAM.GSEA.KEGG; (f1) PPCDC.GSEA.GO; (f2) PPCDC.GSEA.KEGG; (g1) RRAS2.GSEA.GO; (g2) RRAS2.GSEA.KEGG.
Immune infiltration and cytokine analysis of HUBgenes
Subsequently, neutrophil was screened out as key differential immune cell (Fig. 7a). The key differential immune cell that screened out had certain accuracy (Fig. 7b). In the correlation network, it was found that neutrophil related to all HUBgenes in two datasets (Fig. 7c). PPCDC, DPRXP4 and C9orf139 were positively correlated with neutrophil in two datasets, while ZNF595, SIGLEC17P, RRAS2, NRCAM, PLXDC1, FNBP1L and KLRB1 had a negative correlation (Fig. 7d). The significant correlation between HUBgenes and common cytokines were visualized by the heatmap (Fig. 7e).

(a) Box plots of 29 immune gene sets in GSE124272 and GSE150408. a1 and a2 are box plots of 29 immune gene set contents in the IDD and normal groups of GSE124272 and GSE150408, respectively. (* p < 0.05, ** p < 0.01, *** p < 0.001); (b) ROC curves of key immune cells. b1 and b2 are the ROC curves of key immune cells in GSE124272 and GSE150408 of IDD and normal groups, respectively; (c) HUBgenes correlation network with key immune cells. The red nodes represent up-regulated genes, the blue represent down-regulated, the green represent differential immune cells, the yellow represent immune cells with significant differences in both datasets, and the thickness of the connection represents the magnitude of the correlation; (d) Lollipop charts of HUBgenes’ correlation with key immune cells. d1 and d2 are lollipop charts of the correlation between HUBgenes and key immune cells in GSE124272 and GSE150408 of IDD and normal groups, respectively; (e) Heat map of the correlation between HUBgenes and common cytokines.
The construction of nomogram
The correlation between HUBgenes (ZNF595, RRAS2, PPCDC, PLXDC1 and FNBP1L) and clinical features in the GSE167199 validation set were shown by heatmap (5 HUBgenes were not matched in GSE167199 validation set) (Fig. 8a). RRAS2 and ZNF595 were significantly different in the GSE167199 validation set, and they were also significantly related to clinical features (Fig. 8b). ROC curves also showed that two HUBgenes had favorable reliability (Fig. 8c). A nomogram was constructed based on RRAS2 and ZNF595 (Fig. 8d). The calibration curve was plotted based on the above nomogram, and it indicated that the prediction ability of the model was excellent (Fig. 8e). We obtained a total of 123 drugs According to the CTDbase. Among them, RRAS2 predicted 118 drugs such as Tetrachlorodibenzodioxin, bisphenol A and Benzo(a)pyrene. There were also 11 drugs (abrine, Aflatoxin M1, bisphenol A, etc.) based on ZNF595 (Fig. 8f, Supplementary Data).

(a) Heatmap of correlation between significantly different HUBgenes and clinical features obtained in GSE167199; (b1) Expression box plot of HUBgenes in GSE167199; (b2) Expression box plot of HUBgenes in different age groups in GSE167199; (c1) SVM validation ROC curve in GSE167199; (c2) Validation ROC curve of HUBgenes in GSE167199; (d) Patient HUBgenes Nomogram; (e) Patient HUBgenes calibration curve; (f) Gene-drug network of HUBgenes. The blue nodes are drugs or small molecule compounds targeting HUBgenes, and the yellow are verified HUBgenes.
QuantitativeReal-timePCR (qPCR) identification
It can be seen that both RRAS2 and ZNF595 were significantly upregulated in IDD, which is not absolutely consistent with our above analyses results (Fig. 9, Table 2).

Validation of the expression of HUBgenes: (a) qRT-PCR detection of RRAS2 expression levels; (b) qRT-PCR detection of ZNF595 expression levels.
Discussion
Degeneration of IVDs is a major contributor to back, neck and radicular pain, which has already caused large socio-economic impact in the whole world10. Despite its universality, the initiation and progression of IDD are still not well understood, and a generic disease prediction model is still lacking57. In recent years, many clinical studies have indicated an association of iron overload with IDD’s incidence and pathological progression, which can be considered as an independent risk factor. Through an iron overload mouse model, researchers have observed that it promoted IDD and cartilage endplate degeneration in a dose dependent manner22. Moreover, a high dose of excess iron promoted chondrocytes ferroptosis22. Thus, iron overload is strongly related with the onset and development of IDD via ferroptosis, inhibiting ferroptosis could therefore be a promising therapeutic strategy for IDD induced by iron overload. As the most abundant internal modification of mRNA in eukaryotes, the relationship between m6A and ferroptosis in a variety of diseases has been an extremely hot research direction. It has already been proved that m6A-ferroptosis genes play important roles in diseases such as osteoporosis, acute kidney injury, cancers and so on58,59,60. Therefore, we have brought the question that how m6A-ferroptosis genes act their roles in IDD into the present study, aiming to identify m6A-ferroptosis genes in IDD and further hope to reveal the immune characteristics.
The research suggested that 7 HUBgenes (ZNF595, PLXDC1, FNBP1L, KLRB1, NRCAM, PPCDC and RRAS2) have significant functional similarity. ZNF595 was found to be methylation status associated with ambient temperature, which influences human epigenetic modifications61. It indicates that whether temperature is an influential factor in the mechanism of IDD might be a research direction in the future. While ZNF595 has already been found crucial in human gastric cancer and Dengue, its role in IDD remains unclear62,63. Our results suggested that the expression of ZNF595 is related to age, one of the high-risk factors for IDD. Whether its post-transcriptional regulation via m6A might contribute to IDD in relation to age or not is a research direction worth exploring.
PLXDC1 was suggested that it may regulate the biological functions of lipid metabolism and transport-related proteins through a series of biological processes, which is also significant in progression of IDD64. Therefore, it has potential to be a prognostic marker of IDD as of gastric cancer and colon cancer65,66. Moreover, the expression of PLXDC1 is significantly up-regulated in IDD group and down-regulated in older group, suggesting that it can be applied as therapeutic target in IDD via regulation of aging-related pathways. FNBP1L was predicted to function as a scaffold protein for microtubule, Rho family proteins, Formin-homology proteins and WAPS family proteins67. Researchers should study its role in IDD along the direction. Recent research found that expressions of favorably prognostic genes, including KLRB1 (encoding CD161), largely reflect tumor-associated leukocytes, indicating that KLRB1 could be a great break point for the following studies to elucidate the effects of IDD on body immunity68. NRCAM has already been found functioning in multiple diseases, such as liver cancer, Alzheimer’s disease, thyroid carcinoma and so on69,70,71. PPCDC is highly related to breast cancer72. However, there is still a lack of research on the mechanism of NRCAM and PPCDC in IDD.
As we all know, the incidence of IDD is mostly concentrated in middle-aged and elderly people, which increases in prevalence with age3. Cellular senescence has been viewed as a contributor to individual aging. In our results, the expression of RRAS2 was higher in normal group and younger group in comparative to IDD group and older group. Therefore, RRAS2 might be considered as a protective gene in IDD. RRAS2 preferred longer 3’ UTR usage and exhibited decreased expression in senescent cells73. Therefore, it was suggested that depletion of RRAS2 promoted senescence, while rescue of RRAS2 reversed senescence-associated phenotypes. It suggested that although RRAS2 has not been reported in any degeneration diseases, it could have important functions in the IDD prediction and treatment. However, much other research also reported that RRAS2 was overexpressed in various cancers, indicating its carcinogenic potential74,75,76. Thus, promoting the anti-aging mechanism of RRAS2 without leading to cancers is a research direction worth exploring as well.
We have also noticed that in the KEGG functional enrichment analysis, m6A-FRGs-DEGs were also involved in leishmaniasis and malaria. Although it may be in relation to environmental exposure or characteristics of the GEO datasets used in the present paper, it has been proposed that the association of m6A-FRGs-DEGs with leishmaniasis and malaria pathways is essentially the intersection of host epitranscriptome regulation and pathogen immune escape strategies77,78,79.
As for the experimental validation results, there were several factors that may cause the situation as we consider. First of all, our selection of the test samples is one of the most significant influence factors. For ethical and practical reasons, it was extremely difficult to obtain normal IVDs of young people, which has led to insufficient sample heterogeneity. Moreover, our samples were all from Chinese patients and mostly female, which were not consistent with the data from the GEO. Different human races and genders were other important reasons that may bring opposite gene expression results in our experiment. Last but not least, although we have already included 20 samples in our validation test, there were still not enough samples to prove our HUBgenes practically. In order to explore the expression and function of the selected genes in our study, future experiments using statistically large numbers of IVDs, applying multiple technical methods for verification, and conducting in-depth exploration of the specific mechanism of action of RRAS2 and ZNF595 in IDD will be definitely necessary. Besides, short-read sequencing platforms applied in our study might not fully capture repetitive elements of genes, it would be very helpful to use long-read sequencing technologies in future work.
Conclusion
The focus of the study is to select a set of genes for further research on IDD. Through bioinformatic technologies, our research indicates that 10 HUBgenes (ZNF595, PLXDC1, FNBP1L, KLRB1, NRCAM, PPCDC, C9orf139, SIGLEC17P, RRAS2 and DPRXP4) are significantly associated with IDD, providing more evidence about the vital role of ZNF595 and RRAS2 in IDD. RRAS2 and ZNF595, which are genes highly differently expressed in IDD, on which our prediction model of IDD based. Further experimental mechanistic studies and clinical application studies are still necessary, and we will continuously focus on the role of HUBgenes in our research. We hope our work will provide new ideas for IDD diagnosis and management in the near future.
Data availability
Data is provided within the manuscript or supplementary information files.
Abbreviations
- SYSU:
-
Sun Yat-Sen University
- IDD:
-
Intervertebral disc degeneration
- m6A:
-
N6-methyladenosine
- FRGs:
-
Ferroptosis-related genes
- WGCNA:
-
Weighted gene co-expression network analysis
- DEGs:
-
Differentially expressed genes
- m6A-FRGs-DEGs:
-
DEGs of m6A-FRGs related module genes
- LASSO:
-
Least absolute shrinkage and selection operator
- GSEA:
-
Gene set enrichment analysis
- ssGSEA:
-
Single sample gene set enrichment analysis
- CTDbase:
-
The comparative toxicogenomics database
- qRT-PCR:
-
The quantitative real-time fluorescence PCR
- MSKs:
-
Musculoskeletal disorders
- IVD:
-
Intervertebral disc
- NPC:
-
Nucleus pulposus cell
- ECM:
-
Extracellular matrix
- AF:
-
Annulus fibrosus
- CEPs:
-
Cartilaginous endplates
- PCD:
-
Programmed cell death
- ROS:
-
Reactive oxygen species
- DNA:
-
Deoxyribonucleic acid
- RNA:
-
Ribonucleic acid
- AP-1:
-
Activator protein-1
- rRNA:
-
Ribosomal ribonucleic acid
- snRNA:
-
Small nuclear ribonucleic acid
- 3’-UTR:
-
3’-Untranslated regions
- GEO:
-
Gene Expression omnibus
- PCA:
-
Principal component analysis
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PPI:
-
Protein–protein interaction
- LR:
-
Logistic regression
- RF:
-
Random forest
- XGBoost:
-
EXtreme gradient boosting
- Adaboost:
-
Adaptive boosting
- SVM:
-
Support vector machine
- ROC:
-
Receiver operating characteristic
- PCR:
-
Polymerase chain reaction
- NK:
-
Natural killer
References
Zhang, Z. et al. Hub genes and key pathways of intervertebral disc degeneration: Bioinformatics analysis and validation. Biomed. Res. Int. 2021, 5340449. https://doi.org/10.1155/2021/5340449 (2021).
Zehra, U. et al. Mechanisms and clinical implications of intervertebral disc calcification. Nat. Rev. Rheumatol. 18, 352–362. https://doi.org/10.1038/s41584-022-00783-7 (2022).
Boden, S. D., Davis, D. O., Dina, T. S., Patronas, N. J. & Wiesel, S. W. Abnormal magnetic-resonance scans of the lumbar spine in asymptomatic subjects. A prospective investigation. J. Bone Joint Surg. Am. 72, 403–408 (1990).
Salminen, J. J., Erkintalo, M. O., Pentti, J., Oksanen, A. & Kormano, M. J. Recurrent low back pain and early disc degeneration in the young. Spine (Phila Pa 1976) 24, 1316–1321. https://doi.org/10.1097/00007632-199907010-00008 (1999).
Francisco, V. et al. A new immunometabolic perspective of intervertebral disc degeneration. Nat. Rev. Rheumatol. 18, 47–60. https://doi.org/10.1038/s41584-021-00713-z (2022).
Zhang, G. Z. et al. Sirtuins and intervertebral disc degeneration: Roles in inflammation, oxidative stress, and mitochondrial function. Clin. Chim. Acta 508, 33–42. https://doi.org/10.1016/j.cca.2020.04.016 (2020).
Xin, J. et al. Treatment of intervertebral disc degeneration. Orthop. Surg. 14, 1271–1280. https://doi.org/10.1111/os.13254 (2022).
Dai, S. et al. Sodium tanshinone IIA sulfonate ameliorates injury-induced oxidative stress and intervertebral disc degeneration in rats by inhibiting p38 MAPK signaling pathway. Oxid. Med. Cell Longev. 2021, 5556122. https://doi.org/10.1155/2021/5556122 (2021).
Mohd Isa, I. L. et al. Intervertebral disc degeneration: biomaterials and tissue engineering strategies toward precision medicine. Adv. Healthc. Mater. 11, e2102530. https://doi.org/10.1002/adhm.202102530 (2022).
Risbud, M. V. & Shapiro, I. M. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat. Rev. Rheumatol. 10, 44–56. https://doi.org/10.1038/nrrheum.2013.160 (2014).
Roh, E. J. et al. Genetic therapy for intervertebral disc degeneration. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22041579 (2021).
Kamali, A. et al. Small molecule-based treatment approaches for intervertebral disc degeneration: Current options and future directions. Theranostics 11, 27–47. https://doi.org/10.7150/thno.48987 (2021).
Krut, Z., Pelled, G., Gazit, D. & Gazit, Z. Stem cells and exosomes: New therapies for intervertebral disc degeneration. Cells https://doi.org/10.3390/cells10092241 (2021).
Zhang, C., Liu, X., Jin, S., Chen, Y. & Guo, R. Ferroptosis in cancer therapy: a novel approach to reversing drug resistance. Mol. Cancer 21, 47. https://doi.org/10.1186/s12943-022-01530-y (2022).
Jiang, X., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282. https://doi.org/10.1038/s41580-020-00324-8 (2021).
Yan, H. F. et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 6, 49. https://doi.org/10.1038/s41392-020-00428-9 (2021).
Jiang, M. et al. A risk-scoring model based on evaluation of ferroptosis-related genes in osteosarcoma. J. Oncol. 2022, 4221756. https://doi.org/10.1155/2022/4221756 (2022).
Tang, D., Chen, X., Kang, R. & Kroemer, G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125. https://doi.org/10.1038/s41422-020-00441-1 (2021).
Zhang, Y. et al. Ferroptosis and musculoskeletal diseases: “Iron Maiden” cell death may be a promising therapeutic target. Front. Immunol. 13, 972753. https://doi.org/10.3389/fimmu.2022.972753 (2022).
Yang, R. Z. et al. Involvement of oxidative stress-induced annulus fibrosus cell and nucleus pulposus cell ferroptosis in intervertebral disc degeneration pathogenesis. J. Cell Physiol. 236, 2725–2739. https://doi.org/10.1002/jcp.30039 (2021).
Stockwell, B. R., Jiang, X. & Gu, W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 30, 478–490. https://doi.org/10.1016/j.tcb.2020.02.009 (2020).
Wang, W. et al. Iron overload promotes intervertebral disc degeneration via inducing oxidative stress and ferroptosis in endplate chondrocytes. Free Radic. Biol. Med. 190, 234–246. https://doi.org/10.1016/j.freeradbiomed.2022.08.018 (2022).
Li, Y. et al. Silencing ATF3 might delay TBHP-induced intervertebral disc degeneration by repressing NPC ferroptosis, apoptosis, and ECM degradation. Oxid. Med. Cell Longev. 2022, 4235126. https://doi.org/10.1155/2022/4235126 (2022).
Wang, W. et al. Injectable ECM-mimetic dynamic hydrogels abolish ferroptosis-induced post-discectomy herniation through delivering nucleus pulposus progenitor cell-derived exosomes. Nat. Commun. 16, 3131. https://doi.org/10.1038/s41467-025-58447-5 (2025).
Li, J. et al. Quercetin nanoformulation-embedded hydrogel inhibits osteopontin mediated ferroptosis for intervertebral disc degeneration alleviation. J. Nanobiotechnol. 23, 492. https://doi.org/10.1186/s12951-025-03574-w (2025).
Zhao, Y. et al. Exosomes derived from MSCs exposed to hypoxic and inflammatory environments slow intervertebral disc degeneration by alleviating the senescence of nucleus pulposus cells through epigenetic modifications. Bioact. Mater. 49, 515–530. https://doi.org/10.1016/j.bioactmat.2025.02.046 (2025).
Shu, S. et al. Upregulated CEMIP promotes intervertebral disc degeneration via AP-1-mediated change in chromatin accessibility. Clin. Transl. Med. 15, e70322. https://doi.org/10.1002/ctm2.70322 (2025).
Kang, L., Zhang, H., Jia, C., Zhang, R. & Shen, C. Epigenetic modifications of inflammation in intervertebral disc degeneration. Ageing Res. Rev. 87, 101902. https://doi.org/10.1016/j.arr.2023.101902 (2023).
Sendinc, E. & Shi, Y. RNA m6A methylation across the transcriptome. Mol. Cell 83, 428–441. https://doi.org/10.1016/j.molcel.2023.01.006 (2023).
Yang, B. et al. RNA methylation and cancer treatment. Pharmacol. Res. 174, 105937. https://doi.org/10.1016/j.phrs.2021.105937 (2021).
He, L. et al. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 18, 176. https://doi.org/10.1186/s12943-019-1109-9 (2019).
Sun, T., Wu, R. & Ming, L. The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 112, 108613. https://doi.org/10.1016/j.biopha.2019.108613 (2019).
Linder, B. et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772. https://doi.org/10.1038/nmeth.3453 (2015).
Ke, S. et al. A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev. 29, 2037–2053. https://doi.org/10.1101/gad.269415.115 (2015).
Wu, Y. et al. The role of m6A methylation in osteosarcoma biological processes and its potential clinical value. Hum. Genomics 16, 12. https://doi.org/10.1186/s40246-022-00384-1 (2022).
Song, Z., Jia, G., Ma, P. & Cang, S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 276, 119399. https://doi.org/10.1016/j.lfs.2021.119399 (2021).
Ma, L. et al. Targeting SLC3A2 subunit of system X(C)(-) is essential for m(6)A reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic. Biol. Med. 168, 25–43. https://doi.org/10.1016/j.freeradbiomed.2021.03.023 (2021).
Liu, L. et al. Insights into N6-methyladenosine and programmed cell death in cancer. Mol. Cancer 21, 32. https://doi.org/10.1186/s12943-022-01508-w (2022).
Lu, T. et al. Systematic profiling of ferroptosis gene signatures predicts prognostic factors in esophageal squamous cell carcinoma. Mol. Ther. Oncolytics 21, 134–143. https://doi.org/10.1016/j.omto.2021.02.011 (2021).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Ito, K. & Murphy, D. Application of ggplot2 to pharmacometric graphics. CPT Pharmacometrics Syst. Pharmacol. 2, e79. https://doi.org/10.1038/psp.2013.56 (2013).
Wu, T. et al. clusterProfiler 40: A universal enrichment tool for interpreting omics data. Innovation (Cambridge) 2, 100141. https://doi.org/10.1016/j.xinn.2021.100141 (2021).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672-d677. https://doi.org/10.1093/nar/gkae909 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Szklarczyk, D. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362-d368. https://doi.org/10.1093/nar/gkw937 (2017).
Lu, J., Wang, X., Sun, K. & Lan, X. Chrom-Lasso: a lasso regression-based model to detect functional interactions using Hi-C data. Brief Bioinform. https://doi.org/10.1093/bib/bbab181 (2021).
Meurer, W. J. & Tolles, J. Logistic regression diagnostics: Understanding how well a model predicts outcomes. JAMA 317, 1068–1069. https://doi.org/10.1001/jama.2016.20441 (2017).
Asadi, S., Roshan, S. & Kattan, M. W. Random forest swarm optimization-based for heart diseases diagnosis. J. Biomed. Inform. 115, 103690. https://doi.org/10.1016/j.jbi.2021.103690 (2021).
Shin, H. XGBoost regression of the most significant photoplethysmogram features for assessing vascular aging. IEEE J. Biomed. Health Inform. 26, 3354–3361. https://doi.org/10.1109/jbhi.2022.3151091 (2022).
Dou, L., Li, X., Zhang, L., Xiang, H. & Xu, L. iGlu_AdaBoost: Identification of lysine glutarylation using the AdaBoost classifier. J. Proteome Res. 20, 191–201. https://doi.org/10.1021/acs.jproteome.0c00314 (2021).
Ding, C., Bao, T. Y. & Huang, H. L. Quantum-inspired support vector machine. IEEE Trans. Neural Netw. Learn. Syst. 33, 7210–7222. https://doi.org/10.1109/tnnls.2021.3084467 (2022).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Yu, G. et al. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics 26, 976–978. https://doi.org/10.1093/bioinformatics/btq064 (2010).
Zhao, S. et al. Machine learning-based characterization of cuprotosis-related biomarkers and immune infiltration in Parkinson’s disease. Front. Genet. 13, 1010361. https://doi.org/10.3389/fgene.2022.1010361 (2022).
Vergroesen, P. P. et al. Mechanics and biology in intervertebral disc degeneration: a vicious circle. Osteoarthritis Cartilage 23, 1057–1070. https://doi.org/10.1016/j.joca.2015.03.028 (2015).
Lin, Y. et al. Activation of osteoblast ferroptosis via the METTL3/ASK1-p38 signaling pathway in high glucose and high fat (HGHF)-induced diabetic bone loss. Faseb J. 36, e22147. https://doi.org/10.1096/fj.202101610R (2022).
Ni, L. et al. The correlation between ferroptosis and m6A methylation in patients with acute kidney injury. Kidney Blood Press. Res. 47, 523–533. https://doi.org/10.1159/000524900 (2022).
Xie, H. et al. Identification of m6A- and ferroptosis-related lncRNA signature for predicting immune efficacy in hepatocellular carcinoma. Front. Immunol. 13, 914977. https://doi.org/10.3389/fimmu.2022.914977 (2022).
Xu, R. et al. Environmental temperature and human epigenetic modifications: A systematic review. Environ. Pollut. 259, 113840. https://doi.org/10.1016/j.envpol.2019.113840 (2020).
Cui, J. et al. Comprehensive characterization of the genomic alterations in human gastric cancer. Int. J. Cancer 137, 86–95. https://doi.org/10.1002/ijc.29352 (2015).
Xiong, N. & Sun, Q. Identification of stage-related and severity-related biomarkers and exploration of immune landscape for Dengue by comprehensive analyses. Virol. J. 19, 130. https://doi.org/10.1186/s12985-022-01853-8 (2022).
Guo, J. et al. Association between greenness and dyslipidemia in patients with coronary heart disease: A proteomic approach. Ecotoxicol. Environ. Saf. 231, 113199. https://doi.org/10.1016/j.ecoenv.2022.113199 (2022).
Li, X. et al. PLXDC1 can be a biomarker for poor prognosis and immune evasion in gastric cancer. J. Inflamm. Res. 15, 5439–5455. https://doi.org/10.2147/jir.S383191 (2022).
Fu, Y., Zhang, X. & Qiao, Q. PLXDC1 serves as a potential prognostic marker and involves in malignant progression and macrophage polarization in colon cancer. J. Biochem. Mol. Toxicol. 38, e23832. https://doi.org/10.1002/jbt.23832 (2024).
Katoh, M. & Katoh, M. Identification and characterization of human FNBP1L gene in silico. Int. J. Mol. Med. 13, 157–162 (2004).
Gentles, A. J. et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 21, 938–945. https://doi.org/10.1038/nm.3909 (2015).
Zhou, L. et al. Liver cancer stem cell dissemination and metastasis: uncovering the role of NRCAM in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 42, 311. https://doi.org/10.1186/s13046-023-02893-w (2023).
Brummer, T. et al. NrCAM is a marker for substrate-selective activation of ADAM10 in Alzheimer’s disease. EMBO Mol. Med. https://doi.org/10.15252/emmm.201809695 (2019).
Wang, C. et al. CircNRCAM up-regulates NRCAM to promote papillary thyroid carcinoma progression. J. Endocrinol. Invest. 47, 1215–1226. https://doi.org/10.1007/s40618-023-02241-x (2024).
Ruddy, K. J. et al. Genetic predictors of chemotherapy-related amenorrhea in women with breast cancer. Fertil. Steril. 112, 731-739.e731. https://doi.org/10.1016/j.fertnstert.2019.05.018 (2019).
Chen, M. et al. 3’ UTR lengthening as a novel mechanism in regulating cellular senescence. Genome Res. 28, 285–294. https://doi.org/10.1101/gr.224451.117 (2018).
Gutierrez-Erlandsson, S. et al. R-RAS2 overexpression in tumors of the human central nervous system. Mol. Cancer 12, 127. https://doi.org/10.1186/1476-4598-12-127 (2013).
Cifuentes, C. et al. Unmutated RRAS2 emerges as a key oncogene in post-partum-associated triple negative breast cancer. Mol. Cancer 23, 142. https://doi.org/10.1186/s12943-024-02054-3 (2024).
Hortal, A. M. et al. Overexpression of wild type RRAS2, without oncogenic mutations, drives chronic lymphocytic leukemia. Mol. Cancer 21, 35. https://doi.org/10.1186/s12943-022-01496-x (2022).
Yue, J. et al. AlkB RNA demethylase homologues and N(6) -methyladenosine are involved in Potyvirus infection. Mol. Plant Pathol. 23, 1555–1564. https://doi.org/10.1111/mpp.13239 (2022).
Hua, X. et al. ALKBH5 promotes non-small cell lung cancer progression and susceptibility to anti-PD-L1 therapy by modulating interactions between tumor and macrophages. J. Exp. Clin. Cancer Res. 43, 164. https://doi.org/10.1186/s13046-024-03073-0 (2024).
Yang, X. et al. m(6)A promotes R-loop formation to facilitate transcription termination. Cell Res. 29, 1035–1038. https://doi.org/10.1038/s41422-019-0235-7 (2019).
Acknowledgements
This research was partially supported by the China Scholarship Council (CSC) under Grant 202406380074. Open access funding provided by the Open Access Publishing Fund of RWTH Aachen University. The author Z. C. would like to express his gratitude to RWTH Aachen University for their invaluable support and resources that significantly contributed to the success of this research.
Funding
Open Access funding enabled and organized by Projekt DEAL. The author, Z.C., is financially funded and supervised by the China Scholarship Council (CSC) under Grant 202406380074.
Author information
Authors and Affiliations
Contributions
Conceptualization, Z.C.; Data curation, Z.C., R.C. and Q.Z.; Formal analysis, D.Y., Y.L. (Yuwei Liang), G.W. and Q.Z.; Investigation, Y.L. (Yuwei Liang), Y.L. (Yuxi Li), K.S. and C.L.; Methodology, Z.C. and R.C.; Project administration, L.H.; Resources, Y.L. (Yuxi Li), G.W., T.X., K.S. and C.L.; Software, Z.C. and R.C.; Supervision, L.H. and Q.Z.; Validation, M.L., Z.L., K.W. and D.Y.; Writing—original draft, Z.C.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Che, Z., Chen, R., Li, M. et al. Identification and immune characteristics of N6-methyladenosine related ferroptosis-related genes in intervertebral disc degeneration. Sci Rep 15, 33659 (2025). https://doi.org/10.1038/s41598-025-18011-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18011-z
