
Abstract
Esophageal cancer (EC) is a highly aggressive malignancy. ID1, a transcription factor overexpressed in multiple cancers, promotes tumor progression, but its molecular mechanisms in EC remain unclear. We hypothesized that ID1 drives EC progression by regulating the PTEN/YAP/Galectin-3 axis to induce proliferation and immune suppression. Bioinformatics analysis was employed to predict and validate the interactions between genes in esophageal cancer; protein expression levels, including ID1 and PTEN, were assessed using western blotting and immunofluorescence; tumor cell proliferation was evaluated with the CCK-8 assay; and macrophage phagocytosis function was tested through engulfment assays. Additionally, the effects of ID1 on esophageal cancer were investigated using subcutaneous tumor transplantation experiments in nude mice and immunohistochemical staining experiments of human cancerous tissues and peritumoral tissues. ID1 was found to inhibit the action of NFIC. When ID1 was overexpressed, there was a decrease in the expression levels of NFIC, PTEN, and p-YAP, and an increase in the levels of Galectin-3, Myc, CD47, and cyclin-A1. Overexpression of ID1 significantly enhanced the proliferation of esophageal cancer cells and improved the phagocytic ability of macrophages, whereas after treatment with PTEN agonists and YAP inhibitors, the proliferation of esophageal cancer cells decreased markedly, and the phagocytic ability of macrophages was reduced. Furthermore, overexpression of ID1 promoted the progression of esophageal cancer, and the expression of ID1 in human cancerous tissues was significantly higher than that in peritumoral tissues. ID1 promotes the progression of esophageal cancer by inducing proliferation and immune suppression through regulation of the PTEN/YAP/Galectin-3 signaling pathway.
Similar content being viewed by others


Introduction
Esophageal cancer is one of the most common and deadly malignant tumors worldwide, ranking sixth among all cancer-related deaths globally1. Due to the absence of specific symptoms or signs in the early stages, its overall prognosis is extremely poor, with a five-year overall survival rate of approximately 15%. Histologically, esophageal cancer is primarily classified into squamous cell carcinoma (ESCC) and adenocarcinoma (EAC). While ESCC predominates globally, the incidence of esophageal adenocarcinoma (EAC) has risen dramatically in Western populations over recent decades and represents a distinct clinical entity2. The etiology and pathogenesis of esophageal adenocarcinoma may be associated with various factors, including chronic gastroesophageal reflux disease (GERD), Barrett’s esophagus, obesity, diet, and genetic predisposition3,4. In Western countries, EAC development is closely linked to these specific risk factors and exhibits unique molecular characteristics5,6, which remain incompletely understood. Therefore, it is essential to conduct in-depth studies on the molecular mechanisms associated with esophageal cancer to explore potential novel biomarkers and provide new strategies for its diagnosis and treatment.
ID1 is a transcription factor that plays a key role in cell proliferation, differentiation, and survival. Studies have shown that ID1 is highly expressed in various cancers, and its overexpression is associated with tumor invasiveness and poor prognosis.In esophageal cancer, high levels of ID1 are believed to promote tumor cell proliferation and metastasis, making ID1 inhibition a potential therapeutic strategy7.
Galectin-3 is a β-galactoside-binding protein that plays important roles in cell proliferation, apoptosis, adhesion, and metastasis. It exhibits abnormal expression in various tumors, including esophageal cancer.Studies have shown that Galectin-3 promotes the malignant phenotype of esophageal cancer cells by regulating multiple signaling pathways, including the Wnt/β-catenin pathway8. Additionally, Galectin-3 is associated with tumor chemotherapy resistance, making it an important therapeutic target. The HIPPO/YAP signaling pathway is a key regulator of cell proliferation and apoptosis. YAP (Yes-associated protein) is the primary effector of this pathway, and its abnormal activation has been observed in various cancers, including esophageal cancer. The HIPPO signaling pathway maintains the balance between cell proliferation and apoptosis by regulating YAP activity9. High expression of YAP in esophageal cancer is associated with tumor invasiveness and poor prognosis, and inhibiting YAP activity may be an effective strategy to control esophageal cancer progression.
NFIC is a transcription factor that plays an important role in gene expression regulation. Recent studies have shown that NFIC expression is significantly reduced in esophageal cancer, and its low expression is associated with tumor invasiveness and poor prognosis10. NFIC may influence tumor growth and metastasis by regulating the expression of downstream genes, and further investigation of its mechanisms will aid in the development of new therapeutic approaches. PTEN is a key tumor suppressor gene, regulates cell proliferation and apoptosis by inhibiting the PI3K/AKT signaling pathway through its phosphatase activity. PTEN is frequently inactivated or lost in various cancers, including esophageal cancer. Studies have found that the loss or mutation of PTEN is closely associated with the occurrence and development of esophageal cancer11,12,13. Restoring PTEN function or targeting its downstream signaling pathways may become an effective strategy for treating esophageal cancer.
In summary, ID1, Galectin-3, the HIPPO/YAP pathway, NFIC, and PTEN all play important roles in the progression of esophageal cancer. Based on existing evidence, we hypothesize that in esophageal adenocarcinoma, ID1 inhibits NFIC, leading to downregulation of PTEN expression, which in turn weakens the negative regulation of YAP, leading to YAP activation; activated YAP promotes Galectin-3 expression; elevated Galectin-3 not only directly promotes tumor cell proliferation but may also enhance tumor cells’ “don’t eat me” signal through regulating the Myc/CD47 signaling axis, thereby inducing immune escape and jointly driving esophageal cancer progression.This study aims to utilize bioinformatics and molecular biology methods to establish a cellular model and systematically validate the interactive relationships among ID1, NFIC, PTEN, YAP, and Galectin-3 in esophageal cancer, as well as their effects on tumor cell proliferation and immune suppression, thereby providing new insights and strategies for the diagnosis, prognosis assessment, and treatment of esophageal cancer.
Materials and methods
Bioinformatics analysis
Selected esophageal cancer-related datasets GSE20347 (gene expression data from 17 patients’ tumors and matched control tissues), GSE17351 (5 primary ESCC tumors and normal tissues), and GSE45168 (5 pairs of cancerous and adjacent tissues) from the GEO database, and performed differential expression analysis using the DESeq2 package. The screening criteria were set as follows: genes with |log2 Fold Change| > 2 and p-value < 0.05 were identified as differentially expressed genes (DEGs), and the Benjamini-Hochberg method was used for p-value multiple testing correction. The DEGs screened based on these criteria were further analyzed using the ClusterProfiler package for Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis. GO analysis covered three categories: Cellular Component (CC), Molecular Function (MF), and Biological Process (BP); KEGG pathway analysis was used to identify key biological pathway regulatory mechanisms.
Experimental cells and materials
Human esophageal adenocarcinoma cells OE19 and human monocytic THP-1 cells were purchased from Shangon Biotech. Fetal bovine serum and RPMI-1640 medium were obtained from Gibco. The PTEN agonist OroxinB was purchased from Beijing Solarbio Technology Co., Ltd. The YAP inhibitor verteporfin was sourced from MCE. Plasmids were purchased from Genescript (Nanjing, China). The INTERFERin® reagent kit was obtained from Selleck Chemicals. The CCK-8 assay kit was acquired from Beijing Bioatlas Technology Co., Ltd. A Western blot electrophoresis apparatus was also used.
Cell model construction
OE19 cells were divided into the NC group, ID1-KD group, ID1-OE group, OroxinB-NC group, OroxinB + ID1-OE group, verteporfin-NC group, verteporfin + ID1-OE group, ID1-OE + NFIC-OE group, ID1-OE + NFIC-OE + Galectin-3-OE group, and ID1-OE + NFIC-OE + Galectin-3-OE + SHP2-KD + SHP1-KD group.
We constructed a lentiviral vector pCDH-CMV-MCS-EF1-Puro expressing full-length human ID1-OE, NFIC-OE, and Galectin-3-OE. ID1-KD (5’-CCG GGC CAA CTC AGT GAC CAT CAA TCT CGA GAT TGA TGG TCA CTG AGT TGG CTT TTT G-3’), SHP2-KD (5’-CCG GGC CTG CAA CTT CAA GAT CAA ACT CGA GTT TGA TCT TGA AGT TGC AGG CTT TTT G-3’), and SHP1-KD (5’-CCG GCG CAA CAT CTA CAT CCA GAT AAC TCG AGT TAT CTG GAT GTA GAT GTT GCT TTT TG-3’) lentivirus were obtained from Shanghai Hanheng Biotechnology Co., Ltd. HEK293 cells were transfected with Lipofectamine 3000. After 72 h of infection, the culture supernatant was collected, centrifuged to remove cell debris, and then concentrated using a virus concentration kit from Jiangsu Feitian Biotechnology Co., Ltd. OE19 and THP-1 cells were seeded in a 6-well plate and cultured to 70% confluence. They were infected with 40 µL of virus stock solution and 400 µL of medium containing 6 µg/mL Polybrene for 24 h, with an MOI of 10–20, and incubated for 24 h. After 24 h, the medium was replaced with fresh medium, and the cells were continued to be cultured. After 48 h of transfection, the cells were screened with 2 µg/mL Puromycin for 7 days. OE19 cells were infected with ID1-OE, NFIC-OE, Galectin-3-OE, and ID1-KD lentiviruses, and THP-1 cells were infected with SHP2-KD and SHP1-KD lentiviruses.
The OroxinB-NC group and OroxinB-ID1-OE group were treated with the PTEN agonist OroxinB (2 µM) on the basis of ID1 overexpression; the verteporfin-NC group and verteporfin-ID1-OE group were treated with the YAP inhibitor verteporfin (3 µM).
THP-1 cells were stimulated with M-CSF (100 ng). After mixing THP-1 cells with OE19 cells, THP-1 cells were extracted using magnetic beads specific for THP-1 cell surface markers according to the product instructions. The beads were added to the mixed cell culture, and appropriate incubation time was followed. Elution removed unlabelled OE19 cells, while labelled THP-1 cells were captured by the magnetic beads in the column. The THP-1 cells were eluted using the specified buffer and collected for further experiments.
RT-qPCR
Total RNA was extracted from cells using Trizol reagent, and the procedure was carried out according to the provided instructions. Subsequently, the isolated total RNA was reverse-transcribed into cDNA using a reverse transcription kit. PCR amplification was performed with an RT-qPCR instrument in conjunction with Trans Start Top Green qPCR Super Mix. 2 µl of cDNA was used for qPCR, and the reaction conditions were as follows: 95 ℃ for 2 min, 95 ℃ for 15 s, 60 ℃ for 30 s, for a total of 40 cycles. Data were quantified using the 2−△△Ct method, with GAPDH as an internal reference, to analyze the relative mRNA expression levels. ID1: Forward primer: 5’-TAT ACC GAC GGG GAA ACG GA-3’; Reverse primer: 5’-CTT CAG CGA CAC AAG ATG CG-3’. GAPDH: Forward primer: 5’-AAA TGG TGA AGG TCG GTG TGA AC-3’; Reverse primer: 5’-CAA CAA TCT CCA CTT TGC CAC TG-3’.
CO-IP
Total cell lysates were mixed with IgG antibody and incubated overnight. Then 5 µL of agarose (A + G) was added and incubated for an additional 2 h at room temperature. Composites were rinsed 3 times with PBS. The purified protein samples were analyzed by Western blot to detect specific protein interactions.
Western blotting
Cell lysates were prepared using RIPA buffer to extract proteins. The RIPA buffer typically contains protease inhibitors and phosphatase inhibitors to prevent protein degradation. Protein concentration in the samples was measured using the BCA method to ensure equal loading of protein across samples. Polyacrylamide gels were prepared, consisting of separating and stacking gels, with common separating gel concentrations being 10% or 12%. The protein samples and molecular weight markers were loaded onto the gel. Electrophoresis was performed using a Tris-Glycine buffer, with the duration adjusted based on gel thickness and voltage, typically ranging from 60 to 90 min. Appropriate transfer membranes, such as PVDF (Polyvinylidene Fluoride) membranes, were selected. The gel and membrane were placed in a transfer apparatus, usually employing either wet or semi-dry transfer methods for 1 to 2 h. The transfer buffer was typically Tris-Glycine with added methanol to enhance transfer efficiency. The membrane was blocked at room temperature with 5% non-fat dry milk for 1 h to prevent non-specific binding. The membrane was then incubated with a primary antibody against the target protein, diluted to 1:2000, at 4 °C overnight. After incubation, the membrane was washed with TBST (Tris-buffered saline with Tween 20) three times for 10 min each to remove unbound antibodies. The membrane was subsequently incubated with a secondary antibody conjugated to an enzyme (typically horseradish peroxidase, HRP) for 1 h. Following this, the membrane was washed again with TBST to remove any unbound secondary antibody. Protein detection was performed using chemiluminescent reagents (ECL), which were applied to the membrane and exposed in a darkroom to capture the chemiluminescent signal. The signal was recorded using a chemiluminescence imaging system or X-ray film. Image analysis software, such as ImageJ, was used to quantify the Western blot images, comparing the band intensity of the target protein to that of a control protein (such as GAPDH) to determine protein expression levels.
Immunofluorescence staining
Cells were fixed on slides using 4% paraformaldehyde to preserve their morphology and internal structure. The cell membranes were permeabilized with 0.1–0.5% Triton X-100 to allow antibody access to the interior of the cells. Non-specific binding sites were blocked using a buffer containing 5–10% normal animal serum. The cells were then incubated with a primary antibody against ID1, labeled with a red fluorescent dye (Alexa Fluor 594). The incubation time and temperature were adjusted according to the antibody manufacturer’s instructions, typically overnight at 4 °C. Next, the cells were incubated with a primary antibody against PTEN, labeled with a green fluorescent dye (Alexa Fluor 488). Cells were washed multiple times with PBS (phosphate-buffered saline) to remove unbound antibodies. The samples were then mounted with a mounting medium containing anti-fade reagent to prevent fluorescence quenching. Fluorescence microscopy was used to observe the samples, with appropriate filter sets selected to separate the red and green fluorescence signals. The fluorescence intensity of ID1 and PTEN was recorded. Quantitative analysis can be performed using the software provided with the microscope or using image processing software such as ImageJ to measure fluorescence intensity.
CCK-8 assay
Cell suspensions were seeded in 96-well plates with approximately 10,000 cells per well. At least three replicates were used for each treatment group. After 24 h of incubation at 37 °C and 5% CO2, different concentrations of drugs or treatments were added according to the experimental design. Control groups received equal volumes of medium or solvent. After 48 h of incubation, 10 µl of CCK-8 reagent was added to each well, and the plate was gently shaken. Absorbance at 450 nm was measured using a microplate reader.
Phagocytosis assay
Macrophages were seeded on 96-well plates or cover slips and grown to 70–80% confluence. After 24 h, fluorescent beads were diluted in medium according to the manufacturer’s instructions and added to each well or cover slip. The cells were incubated at 37 °C and 5% CO2 for 4 h to allow phagocytosis. Cells were washed with PBS, fixed with 4% PFA, and mounted with anti-fade mounting medium containing DAPI. Fluorescence microscopy was used to observe phagocytosed fluorescent beads.
Subcutaneous tumor transplantation experiment in nude mice
Thirty male BALB/c mice aged 6 weeks were selected and purchased from Henan Skebes Biotechnology Co., Ltd. The mice weighed 18 ± 1 g and were acclimatized for 7 days before being randomly divided into 5 groups. The cells were divided into the following groups: NC group, ID1-OE group, ID1-OE + NFIC-OE group, ID1-OE + NFIC-OE + Galectin-3-OE group, and ID1-OE + NFIC-OE + Galectin-3-OE + SHP2-KD + SHP1-KD group. OE19 cells grown to the logarithmic phase were adjusted to a concentration of 1 × 104 cells/µL. Two hundred microliters of cell suspension were injected into the right dorsal subcutaneous tissue of nude mice. The tumor length and width were measured every 3 days, and the tumor volume was calculated. Tumor volume = (length × width)2/2.
Nude mice were euthanized 4 weeks post-injection using inhaled CO₂, with a CO₂ flow rate of 30% of the container volume per minute. Prior to euthanasia, anesthesia was induced with 3% isoflurane and maintained with 1.5% isoflurane to alleviate anxiety in the nude mice. Euthanasia was confirmed when the nude mice had no heartbeat for 15 min. Following euthanasia, tumor tissues were removed, and tumor weight and volume were measured. All animal experiments used in this experiment comply with the 3Rs principle and ARRIVE guidelines. And all methods in this experiment were conducted in accordance with relevant guidelines and regulations.
Clinical sample collection
A systematic retrospective study was conducted on 100 patients with esophageal adenocarcinoma who underwent radical esophagectomy and systemic lymph node dissection at our hospital from January 2022 to December 2023. Among them, 74 were male and 26 were female; aged 40–78 years, with an average age of (59.3 ± 9.15) years; postoperative TNM staging: Stage I, 25 cases; Stage II, 40 cases; Stage III, 30 cases; Stage IV, 5 cases; tumor location: upper segment, 10 cases; middle segment, 55 cases; lower segment, 35 cases; history of smoking, 48 cases; history of alcohol consumption, 42 cases;18 patients had a history of hypertension; 15 patients had a history of coronary heart disease; 10 patients had a history of diabetes; 72 patients had a tumor diameter ≥ 3 cm, and 28 patients had a tumor diameter < 3 cm; 34 patients had lymph node metastasis, and 66 patients did not have lymph node metastasis; differentiation grade: 14 cases were poorly differentiated, 71 cases were moderately differentiated, and 15 cases were well differentiated. Inclusion criteria: (1) Pathologically diagnosed as esophageal adenocarcinoma; (2) Meeting surgical criteria and undergoing radical esophageal cancer resection and systemic lymph node dissection for the first time; (3) Complete clinical records and informed consent and cooperation from the patient and their family; (4) No history of radiotherapy or chemotherapy before surgery; (5) No history of infection, hematological diseases, rheumatic autoimmune diseases, splenectomy, or liver disease before surgery; (6) No recent history of blood product transfusion or treatment. Exclusion criteria: (1) History of other malignant tumors; (2) Death caused by non-esophageal tumors or other diseases; (3) Preoperative anticoagulant or antiplatelet therapy; (4) Preoperative radiotherapy or chemotherapy; (5) Incomplete clinical records; (6) Incomplete or missing follow-up data. All subjects and their legal guardians consented to provide informed consent to the experiment. The human experiments in this study followed the Declaration of Helsinki.
Immunohistochemical staining
An appropriate amount was taken and placed in a tissue embedding tray. The tissue was fixed with 10% neutral buffered formalin solution, and then the fixed tissue was embedded in paraffin. A microtome was used to section the paraffin-embedded tissue into 4 μm thick sections. The tissue sections were dewaxed with xylene and ethanol, and antigen retrieval was performed using sodium citrate antigen retrieval solution. Next, an endogenous peroxidase inhibitor was added to the tissue surface, and the sections were blocked with 5% goat serum. The sections were incubated with the primary antibody at 4 °C overnight, followed by incubation with the secondary antibody at room temperature for 20 min. The prepared DAB staining solution was added to the tissue, ensuring complete coverage, and the staining was terminated when the reaction turned brownish-yellow. The cells were counterstained with hematoxylin. Differentiation was performed with hydrochloric acid and alcohol, and the sections were counterstained with blue. Dehydration was carried out with graded alcohol, and the sections were cleared with xylene. The tissue sections were mounted with neutral resin. Finally, the tissue sections were imaged using a Leica microscopic imaging system.
Statistical analysis
Statistical analysis was performed using SPSS 26.0 software, and GraphPad Prism 7 was used for plotting. Data were expressed as mean ± standard deviation. Comparisons between two groups were conducted using t-tests, and comparisons among multiple groups were performed using one-way ANOVA. A p-value of < 0.05 was considered statistically significant.
Results
Significant elevation of ID1 expression in esophageal cancer
Through DESeq2 analysis, a total of 12,548 differentially expressed genes (DEGs) were identified from the GSE20347 dataset, including 528 upregulated and 640 downregulated genes. Similarly, the GSE17351 dataset revealed 14,080 DEGs with 326 upregulated and 170 downregulated genes, while the GSE45168 dataset yielded 21,754 DEGs, including 509 upregulated and 743 downregulated genes. Analysis of these three datasets using a Venn diagram tool identified 26 upregulated and 44 downregulated DEGs. Among these, ID1 was found to be significantly upregulated in esophageal cancer tissues. GO and KEGG analyses conducted with ClusterProfiler showed that the DEGs were enriched in regulation of DNA-directed DNA polymerase activity, positive regulation of DNA-directed DNA polymerase activity, and mitotic cell cycle phase transition (BP); collagen-containing extracellular matrix, basement membrane, and Golgi lumen (CC); and extracellular matrix structural constituent, single-stranded DNA helicase activity, and serine-type endopeptidase activity (MF). The KEGG pathway analysis indicated that the DEGs were significantly enriched in Protein digestion and absorption, ECM-receptor interaction, and Cytokine-cytokine receptor interaction. According to the GEO database, ID1 expression was significantly upregulated in esophageal cancer samples compared to normal samples (Fig. 1).

Bioinformatics analysis28,29. A: Volcano plot of differentially expressed genes in GSE20347; B: Volcano plot of differentially expressed genes in GSE17351; C: Volcano plot of differentially expressed genes in GSE45168; D: Venn diagram of upregulated genes in GSE20347 and GSE17351; E: Venn diagram of downregulated genes in GSE20347 and GSE17351; F: Bar graph of GO enrichment analysis; G: Bubble plot of KEGG enrichment analysis; H: Expression of ID1 in normal tissues and esophageal cancer tissues based on GEO database analysis.
ID1 suppresses NFIC, PTEN, p-YAP, and promotes galectin-3 expression
To investigate the relationship between ID1 and NFIC, PTEN, p-YAP, and Galectin-3, we used western blot, RT-qPCR, and CO-IP experiments for verification. Western blot results showed that, compared with the NC group, ID1 expression was significantly decreased in the ID1-KD group, while NFIC, PTEN, p-YAP, and Galectin-3 expression was significantly increased. Conversely, ID1 and Galectin-3 expression was significantly increased in the ID1-OE group, while NFIC, PTEN, and p-YAP expression was significantly decreased (Fig. 2A).Additionally, compared with the NC group, PTEN levels were significantly elevated in the ID1-KD group, while PTEN levels were significantly reduced in the ID1-OE group (Fig. 2B). RT-qPCR experimental results showed that there was no significant difference in the relative mRNA expression levels of ID1 in the control group and NC group OE19 cells. Compared with the NC group, the relative mRNA expression levels of ID1 in the ID1-KD group OE19 cells were significantly reduced. Compared with the group, the relative mRNA expression levels of ID1 in the ID1-OE group OE19 cells were significantly increased (Fig. 2C). CO-IP experiments showed that ID1 interacts with Galectin-3, NFIC, and PTEN in OE19 cells (Fig. 2D). The above results showed that ID1 inhibited NFIC, PTEN, and p-YAP and promoted Galectin-3 expression.

ID1 inhibits NFIC and PTEN, while promoting Galectin-3 expression. A: Western blot analysis of ID1, NFIC, PTEN, p-YAP, and Galectin-3 proteins in four groups: NC group, ID1-KD group, NC group, and ID1-OE group; GAPDH as control protein; B: Immunofluorescence detection of ID1 (red fluorescence) and PTEN (green fluorescence) in the NC group, ID1-KD group, NC group, and ID1-OE group; C: RT-qPCR analysis of relative mRNA expression levels of ID1 in the control group, NC group, ID1-KD group, and ID1-OE group; D: CO-IP experimental results diagram. Data are expressed as mean ± standard deviation. N = 3; nsP>0.05; *P<0.05, **P<0.01.
PTEN facilitates YAP degradation and suppresses the “don’t eat me” signal
To explore the further functions of PTEN, we used western blot and CCK-8 experiments for verification. Western blot results showed that, compared with the NC group, the ID1-OE group exhibited increased expression of ID1, YAP, Galectin-3, Myc, CD47, and Cyclin-A1, while NFIC and PTEN expression decreased. In the OroxinB-NC group, PTEN expression was elevated, while YAP, Galectin-3, Myc, CD47, and Cyclin-A1 expression decreased without significant changes in ID1 and NFIC. The verteporfin-NC group exhibited reduced expression of YAP, Galectin-3, Myc, CD47, and Cyclin-A1 without affecting ID1, NFIC, or PTEN. Compared to the ID1-OE group, both the OroxinB-ID1-OE group and the verteporfin-ID1-OE group showed significantly increased PTEN expression and decreased expression of YAP, Galectin-3, Myc, CD47, and Cyclin-A1, with no significant changes in ID1 and NFIC (Fig. 3A, B). CCK-8 assay results showed that the IC50 values for OroxinB and verteporfin were 3.724 µM and 4.816 µM, respectively. Therefore, we used 2 µM OroxinB and 3 µM verteporfin to treat the cells in our experiments (Fig. 3C). These results showed that PTEN promotes YAP degradation and inhibits the “don’t eat me” signal.

PTEN promotes YAP degradation and inhibits the “Don’t eat me” signal. A: Western blot analysis of ID1, NFIC, PTEN, YAP, and Galectin-3 proteins in six groups: NC group, ID1-OE group, OroxinB-NC group, OroxinB-ID1-OE group, verteporfin-NC group, and verteporfin-ID1-OE group; B: Western blot analysis of Myc, CD47, and Cyclin-A1 proteins in the six groups; GAPDH as control protein; C: CCK-8 assay results for the IC50 values of OroxinB and verteporfin. Data are expressed as mean ± standard deviation. N = 3; nsP>0.05; *P<0.05, **P<0.01.
ID1 enhances esophageal cancer cell proliferation and macrophage phagocytic capacity, which are inhibited by PTEN activators and YAP inhibitors
To investigate the effects of ID1 on the proliferation capacity of OE19 cells and the phagocytic capacity of THP-1 cells, we validated the results using CCK-8 and fluorescent microsphere phagocytosis assays. CCK-8 experimental results showed that, compared with the NC group, the OD values of OE19 cells in the ID1-OE group were significantly higher. There were no significant differences between the OroxinB-NC group and the OroxinB-ID1-OE group, nor between the verteporfin-NC group and the verteporfin-ID1-OE group. However, the OD values in the NC group and the ID1-OE group were significantly higher than those in the OroxinB-NC group and the OroxinB-ID1-OE group, while the OD values in the OroxinB-NC group and the OroxinB-ID1-OE group were higher than those in the verteporfin-NC group and the verteporfin-ID1-OE group (Fig. 4A). Immunofluorescence phagocytosis experiment results showed that the number of phagocytosed fluorescent microspheres in the ID1-OE group was significantly lower than that in the NC group.The number of phagocytosed fluorescent microspheres in the NC group and ID1-OE group was significantly lower than that in the OroxinB-NC group and OroxinB-ID1-OE group, and the number of phagocytosed fluorescent microspheres in the NC group and ID1-OE group was also significantly lower than that in the OroxinB-NC group and OroxinB-ID1-OE group.The number of phagocytosed fluorescent microspheres in the OroxinB-ID1-OE group was slightly lower than that in the OroxinB-NC group.The number of phagocytosed fluorescent microspheres in the verteporfin-ID1-OE group was slightly lower than that in the verteporfin-NC group (Fig. 4B). These results showed that ID1 mediates the enhancement of PTEN/YAP-induced proliferation of esophageal cancer cells and inhibits the phagocytic capacity of macrophages.

ID1 promotes esophageal cancer cell proliferation and enhances macrophage phagocytic capacity. A: CCK-8 assay of OD values in OE19 cells from six groups: NC group, ID1-OE group, OroxinB-NC group, OroxinB-ID1-OE group, verteporfin-NC group, and verteporfin-ID1-OE group; B: Phagocytosis experiment observing fluorescent microspheres in macrophages from the six groups. Data are expressed as mean ± standard deviation. N = 3; **P<0.01.
ID1-Mediated NFIC/Galectin-3/SHP2/SHP1 signaling pathway promotes the progression of esophageal cancer
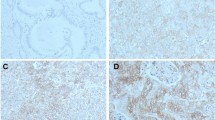
Finally, to investigate the role of the ID1-mediated NFIC/Galectin-3/SHP2/SHP1 signaling pathway in the progression of esophageal cancer.We validated this using subcutaneous tumor transplantation experiments in nude mice and human immunohistochemical staining experiments. The results of the subcutaneous tumor transplantation experiments in nude mice showed that, compared with the NC group, the tumor volume and weight were significantly increased in the ID1-OE group. Compared with the ID1-OE group, the tumor volume and weight were significantly reduced in the ID1-OE + NFIC-OE group.Compared with the ID1-OE + NFIC-OE group, the tumor volume and weight were significantly increased in the ID1-OE + NFIC-OE + Galectin-3-OE group. Compared with the ID1-OE + NFIC-OE + Galectin-3-OE group, the tumor volume and weight were significantly reduced in the ID1-OE + NFIC-OE + Galectin-3-OE + SHP2-KD + SHP1-KD group (Fig. 5A). Immunohistochemical staining results showed that ID1 expression was significantly reduced in peritumoral tissue compared to cancerous tissue (Fig. 5B). In conclusion, ID1 promotes esophageal cancer progression by regulating the PTEN/YAP/Galectin-3 signaling pathway to induce proliferation and immune suppression (Fig. 6).

ID1-mediated NFIC/Galectin-3/SHP2/SHP1 signaling pathway promotes esophageal cancer progression. A: Analysis of tumor volume and weight in the NC group, ID1-OE group, ID1-OE + NFIC-OE group, ID1-OE + NFIC-OE + Galectin-3-OE group, and ID1-OE + NFIC-OE + Galectin-3-OE + SHP2-KD + SHP1-KD group in the subcutaneous tumor transplantation experiment in nude mice; N = 6; B: Immunohistochemical staining analysis of ID1 expression in cancerous tissue and peritumoral tissue. N = 100; Data are expressed as mean ± standard deviation. **P<0.01.

ID1 promotes the progression of esophageal cancer by regulating PTEN/YAP/Galectin-3 signaling pathways to induce proliferation and immune suppression (Created with BioRender.com).
Discussion
Esophageal cancer (EC) is a highly aggressive malignancy with significant global incidence and mortality. Accumulating evidence indicates that multiple molecules and signaling pathways play critical roles in its pathogenesis and progression. This study employed bioinformatics and molecular biology approaches to elucidate the interactions and underlying molecular mechanisms involving ID1, NFIC, PTEN, Galectin-3, and the HIPPO/YAP pathway in EC.
ID1 (Inhibitor of Differentiation 1), a transcriptional regulator frequently overexpressed in various malignancies, is strongly associated with tumor invasiveness and poor prognosis. Studies have demonstrated that ID1 promotes tumor growth, invasion, and metastasis in diverse cancers, including non-small cell lung cancer, glioblastoma, small cell lung cancer, and colorectal cancer14,15,16. In EC, elevated ID1 expression correlates with adverse outcomes. Mechanistically, ID1 enhances cancer cell proliferation, migration, and anti-apoptotic capabilities by modulating key signaling pathways such as PI3K/AKT, MAPK, and NF-κB17,18. These findings collectively underscore the significant contribution of ID1 to EC development and progression, highlighting its potential as a therapeutic target.
Galectin-3, a β-galactoside-binding lectin, participates in numerous biological processes, including cell growth, apoptosis, adhesion, and migration. Within the tumor context, Galectin-3 is recognized as a pivotal factor promoting cancer cell survival, invasion, and metastasis19,20,21. Our findings suggest that ID1 may enhance Galectin-3 expression, potentially through transcriptional regulation and modulation of signaling cascades like NF-κB21. Beyond facilitating cancer cell dissemination, Galectin-3 interacts with the Myc oncoprotein, thereby supporting cancer cell proliferation and survival. Myc, a classical oncogene, is itself associated with poor prognosis across multiple cancers22. Crucially, Myc can transcriptionally activate the CD47 promoter, leading to elevated CD47 expression. CD47, functioning as a transmembrane “don’t eat me” signal, inhibits macrophage phagocytosis of cancer cells by binding to SIRPα on macrophages. Activation of the CD47-SIRPα axis triggers phosphorylation of SHP1 and SHP2 phosphatases, consequently suppressing macrophage phagocytic activity23,24. This mechanism enables EC cells to evade immune surveillance and achieve immune escape.
NFIC (Nuclear Factor I C) is a transcription factor influencing cell differentiation and development. Research indicates that NFIC regulates PTEN (phosphatase and tensin homolog) expression by acting on its promoter25. Consistent with bioinformatics analyses, our results demonstrate that ID1 suppresses NFIC, consequently downregulating PTEN expression. PTEN, a critical tumor suppressor gene, negatively regulates the PI3K/AKT signaling pathway via its phosphatase activity. Notably, PTEN loss or inactivation can indirectly impact HIPPO pathway activity. The HIPPO signaling pathway serves as a major regulator of cell growth and apoptosis. Its core components include MST1/2 and LATS1/2 kinases. NF2 (Merlin) is another key HIPPO regulator, directly binding to LATS1/2 to promote their MST1/2-mediated phosphorylation and activation, thereby stabilizing the MST1/2-LATS1/2 complex and enhancing LATS1/2 kinase activity. Activation of the HIPPO pathway culminates in the phosphorylation of YAP (Yes-associated protein), leading to its ubiquitination and proteasomal degradation. This prevents YAP nuclear accumulation and its interaction with the TEAD1 transcription factor, ultimately inhibiting cell proliferation and promoting apoptosis26,27. Conversely, when HIPPO signaling is inactive, unphosphorylated YAP accumulates in the nucleus, binds TEAD1, and drives the expression of target genes, including Galectin-3, thereby facilitating EC progression.
In summary, this study systematically delineates the interactions and molecular mechanisms linking ID1, NFIC, PTEN, Galectin-3, and the HIPPO/YAP pathway in EC. We demonstrate that ID1 promotes cancer cell proliferation and migration, in part by inducing immune escape through the Galectin-3/Myc/CD47/SHP1/SHP2 axis. Concurrently, NFIC exerts tumor-suppressive effects by upregulating PTEN expression, while the HIPPO pathway regulates cell growth dynamics through YAP phosphorylation. Collectively, our findings establish that ID1 drives EC progression by modulating the PTEN/YAP/Galectin-3 signaling axis, thereby inducing both tumor cell proliferation and immune suppression. This work identifies novel potential therapeutic targets and provides a theoretical foundation for developing improved EC treatment strategies. While we demonstrated the functional role of the ID1/PTEN/YAP/Galectin-3 axis in vitro and in vivo, the direct clinical translatability of targeting this pathway requires further validation. The therapeutic efficacy and potential side effects of modulating key nodes in clinically relevant models, including patient-derived organoids or immunocompetent models, remain to be thoroughly evaluated.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries [J]. Cancer J. Clin. 71 (3), 209–249 (2021).
Joseph, A. et al. Esophageal adenocarcinoma: A dire need for early detection and treatment [J]. Cleve Clin. J. Med. 89 (5), 269–279 (2022).
Croft, W. et al. The single cell transcriptional landscape of esophageal adenocarcinoma and its modulation by neoadjuvant chemotherapy [J]. Mol. Cancer. 21 (1), 200 (2022).
Ergun, P., Kipcak, S. & Bor, S. Epigenetic alterations from Barrett’s esophagus to esophageal adenocarcinoma. Int. J. Mol. Sci. 24(9), 7817 (2023).
Jiang, W. et al. Current status and perspectives of esophageal cancer: a comprehensive review [J]. Cancer Commun. (London England). 45 (3), 281–331 (2025).
Bos, V., Chan, M. W. & Pouw, R. E. Towards personalized management of early esophageal adenocarcinoma [J]. Curr. Opin. Gastroenterol. 40 (4), 299–304 (2024).
Li, Z. X. et al. Transcription factor 3 promotes migration and invasion potential and maintains cancer stemness by activating ID1 expression in esophageal squamous cell carcinoma [J]. Cancer Biol. Ther. 24 (1), 2246206 (2023).
Li, Y. S. et al. Roles of galectin-3 in metabolic disorders and tumor cell metabolism [J]. Int. J. Biol. Macromol. 142, 463–473 (2020).
Koo, J. H. & Guan, K. L. Interplay between YAP/TAZ and metabolism [J]. Cell Metabol. 28 (2), 196–206 (2018).
Yang, C. et al. Dihydroartemisinin suppresses the tumorigenesis of esophageal carcinoma by elevating DAB2IP expression in a NFIC-dependent manner [J]. Naunyn Schmiedebergs Arch. Pharmacol. 397 (10), 8117–8128 (2024).
Xing, Y. et al. Circular RNA circ-Foxo3 inhibits esophageal squamous cell cancer progression via the miR-23a/PTEN axis [J]. J. Cell. Biochem. 121 (3), 2595–2605 (2020).
Papa, A. et al. The PTEN⁻PI3K axis in cancer. Biomolecules 9(4), 153 (2019).
Wu, Y. R. et al. MicroRNA-21 promotes cell proliferation, migration, and resistance to apoptosis through PTEN/PI3K/AKT signaling pathway in esophageal cancer [J]. Tumour Biol. 37 (9), 12061–12070 (2016).
Zhao, Z. et al. Inhibitor of differentiation 1 (Id1) in cancer and cancer therapy [J]. Int. J. Med. Sci. 17 (8), 995–1005 (2020).
Huang, Y. H. et al. ID1 mediates escape from TGFβ tumor suppression in pancreatic cancer [J]. Cancer Discov. 10 (1), 142–157 (2020).
Meng, J. et al. ID1 Confers Cancer Cell Chemoresistance Through STAT3/ATF6-mediated Induction of Autophagy [J]11137 (Cell death & disease, 2020). 2.
Zhao, G. S. et al. High expression of ID1 facilitates metastasis in human osteosarcoma by regulating the sensitivity of Anoikis via PI3K/AKT depended suppression of the intrinsic apoptotic signaling pathway [J]. Am. J. Translational Res. 11 (4), 2117–2139 (2019).
Tsai, C. H. et al. Identification of Id1 as a downstream effector for arsenic-promoted angiogenesis via PI3K/Akt, NF-κB and NOS signaling [J]. Toxicol. Res. (Camb). 5 (1), 151–159 (2016).
Nangia-makker, P., Hogan, V. & Raz, A. Galectin-3 and cancer stemness [J]. Glycobiology 28 (4), 172–181 (2018).
Wang, Q. et al. Galectin-3 induces pathogenic immunosuppressive macrophages through interaction with TREM2 in lung cancer [J]. J. Experimental Clin. Cancer Research: CR. 43 (1), 224 (2024).
Yin, F. et al. Hippo-YAP signaling in digestive system tumors [J]. Am. J. Cancer Res. 11 (6), 2495–2507 (2021).
Kuo, H. Y. et al. Galectin-3 modulates the EGFR signalling-mediated regulation of Sox2 expression via c-Myc in lung cancer [J]. Glycobiology 26 (2), 155–165 (2016).
Logtenberg, M. E. W., Scheeren, F. A. & Schumacher, T. N. The CD47-SIRPα immune checkpoint [J]. Immunity 52 (5), 742–752 (2020).
Van Duijn, A., Van Der Burg, S. H. & Scheeren, F. A. CD47/SIRPα axis: bridging innate and adaptive immunity. J. Immunother. Cancer 10(7), e004589 (2022).
Jia, P., Zhang, W. & Shi, Y. NFIC Attenuates Rheumatoid arthritis-induced Inflammatory Response in Mice by Regulating PTEN/SENP8 Transcription [J]81102013 (Tissue & cell, 2023).
Vlashi, R. et al. The molecular biology of NF2/Merlin on tumorigenesis and development [J]. FASEB Journal: Official Publication Federation Am. Soc. Experimental Biology. 38 (13), e23809 (2024).
Wu, J. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling [J]. Nature 572 (7769), 402–406 (2019).
Kanehisa, M. & Sato, Y. Kegg mapper for inferring cellular functions from protein sequences [J]. Protein Science: Publication Protein Soc. 29 (1), 28–35 (2020).
Kanehisa, M., Sato, Y. & Kawashima, M. KEGG mapping tools for Uncovering hidden features in biological data [J]. Protein Science: Publication Protein Soc. 31 (1), 47–53 (2022).
Funding
Hebei Province Medical Science Research Project Plan, Project Number: 20201101.
Author information
Authors and Affiliations
Contributions
All the authors conceived and designed the experiments. Y.J: Design idea, experimental verification, writing manuscript. Z.Y.Y and Z.J.G. : Proof of concept, experimental methods, writing manuscripts. W.W.L and X.J.X: Advice and guidance on cell models, analysis of experimental data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical statement
The animal experiments in this study have been approved by the Animal Ethics Committee of the Fourth Hospital of Hebei Medical University (Approval No. IACUC-4th Hos Hebmu-2024010). All animal experiments used in this experiment comply with the 3Rs principle and ARRIVE guidelines. And all methods in this experiment were conducted in accordance with relevant guidelines and regulations.
The human experiments in this study have been approved by the Ethics Committee of the Fourth Hospital of Hebei Medical University (2022KY053). All subjects and their legal guardians consented to provide informed consent to the experiment. The human experiments in this study followed the Declaration of Helsinki.
Patient consent for publication
All subjects and their legal guardians have given their consent.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ji, Y., Yan, Z., Gao, Z. et al. ID1 promotes the progression of esophageal cancer via modulating PTEN/YAP/galectin-3 signaling pathway induced proliferation and immune suppression. Sci Rep 15, 34796 (2025). https://doi.org/10.1038/s41598-025-18546-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18546-1
