
Abstract
Sarcopenia and obesity, two prevalent metabolic disorders in aging populations, often coexist and share overlapping pathophysiological mechanisms, yet the molecular mechanisms underlying their comorbidity remain elusive. This study aimed to identify key gene expression signatures and pathways underlying their comorbidity through integrative transcriptomic and bioinformatics analyses. Gene expression datasets from sarcopenia (GSE111016, skeletal muscle) and obesity (GSE152991, adipose tissue) were downloaded from the GEO database. Differentially expressed genes (DEGs) were identified using the limma package, and 208 common differentially expressed genes (CDEGs) were selected via Venn diagram intersection. Functional enrichment analyses (GO and KEGG) were performed to explore shared biological processes and pathways. A protein-protein interaction (PPI) network was constructed using STRING and Cytoscape, and key CDEGs were identified via ten topological algorithms (e.g., MCC, Degree) in the CycloHubba plugin. Pearson correlation analysis and qPCR were used to validate gene co-expression patterns and expression levels in tissue samples. GO and KEGG analyses revealed that CDEGs were significantly enriched in mitochondrial oxidative phosphorylation, electron transport chain, and thermogenesis pathways, with overlap in neurodegenerative disease pathways. The PPI network and multi-algorithm integration identified four key CDEGs: SDHB, SDHD, ATP5F1A, and ATP5F1B, all of which are components of mitochondrial respiratory chain complexes. These genes exhibited strong positive correlations (r > 0.86, p < 10⁻¹²) in both datasets and were significantly downregulated in sarcopenia and obesity tissues, as validated by qPCR. This study confirms mitochondrial dysfunction, particularly impaired oxidative phosphorylation, as a common pathological mechanism linking sarcopenia and obesity. The key genes SDHB, SDHD, ATP5F1A, and ATP5F1B represent potential therapeutic targets for managing these comorbid metabolic disorders. Future research should explore their functional roles in energy metabolism and cross-tissue crosstalk to develop targeted interventions.
Similar content being viewed by others


Introduction
Sarcopenia, characterized by age-related loss of skeletal muscle mass and function, and obesity, defined by excessive adipose tissue accumulation, are two major public health challenges increasingly prevalent in aging populations1,2. Both conditions share overlapping pathophysiological mechanisms, including chronic inflammation, oxidative stress, and mitochondrial dysfunction, which contribute to metabolic derangements and functional decline3. Notably, sarcopenia often coexists with obesity (termed sarcopenic obesity), exacerbating the risk of frailty, mobility impairment, and cardiovascular complications4. Despite their clinical relevance, the molecular interplay between sarcopenia and obesity remains incompletely understood, hindering the development of targeted therapeutic strategies.
Recent advancements in transcriptomic profiling have enabled the identification of disease-specific gene signatures and pathways5. In this study, we integrated gene expression datasets from sarcopenia (GSE111016) and obesity (GSE152991) to uncover shared molecular mechanisms. By leveraging bioinformatics analyses, including differential expression analysis, functional enrichment, protein-protein interaction (PPI) network construction, we aimed to identify key genes and pathways underlying the comorbidity of sarcopenia and obesity.
Sarcopenia and obesity are multifactorial conditions influenced by genetic, environmental, and lifestyle factors6,7. Emerging evidence suggests that mitochondrial dysfunction plays a central role in both disorders8,9. For instance, impaired mitochondrial respiration and reduced ATP production contribute to muscle atrophy in sarcopenia, while adipose tissue mitochondrial dysfunction is linked to insulin resistance and metabolic inflexibility in obesity10,11. However, the specific genes and pathways mediating this crosstalk remain unclear.
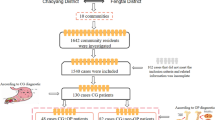
Transcriptomic studies have identified numerous differentially expressed genes (DEGs) in sarcopenia and obesity. For example, GSE111016, a sarcopenia dataset, revealed DEGs associated with muscle remodeling and energy metabolism, while GSE152991, an obesity dataset, highlighted genes involved in adipogenesis and inflammation. By intersecting these DEGs, we sought to identify conserved molecular signatures that may drive the co-occurrence of sarcopenia and obesity (Fig. 1).

Study workflow diagram. DEGs, differentially expressed genes; CDEGs, common differentially expressed genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, protein-protein interaction; RT-PCR, reverse transcription-polymerase chain reaction.
Methods
Data collection
The datasets GSE152991 and GSE111016 used in this study were both obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). For GSE152991, to minimize confounding by metabolic abnormalities, only adipose tissue samples from 11 metabolically healthy lean (non-obese) individuals and 14 metabolically healthy obese individuals (with normal insulin sensitivity) were included, while excluding 20 metabolically unhealthy obese samples with abnormal insulin sensitivity. GSE111016 included skeletal muscle samples from 20 sarcopenia patients and 20 controls, which were used for the identification of DEGs.
Identification of common DEGs between sarcopenia and obesity
The R language “limma” package was used to analyze the data from GSE152991 and GSE111016 to identify DEGs. The filtering criteria were set as an absolute value of log2 Fold Change (log2 FC) > 0.25 and an –log₁₀(p‑value) > 1.25. A Venn diagram was employed to intersect the DEGs based on GSE152991 with those based on GSE111016, thereby obtaining Common Differentially Expressed Genes (CDEGs).
Enrichment analysis of gene ontology and pathways
GO and KEGG enrichment analyses were performed on 208 CDEGs and four identified key CDEGs using the R package “clusterProfiler”. GO analysis categorized gene functions into three categories: biological processes (BP), cellular components (CC), and molecular functions (MF). KEGG analysis mapped genes to molecular interaction networks encompassing seven major categories, with significant pathways defined by a threshold of p < 0.05. Visualization using the R package “ggplot2” showed that enrichment results for both the full CDEG set and key CDEGs were significantly enriched in energy metabolism-related processes (e.g., oxidative phosphorylation, mitochondrial electron transport chain) and showed overlap with neurodegenerative disease pathways.
Construction of protein-protein interaction network
PPI network analysis of the CDEGs was conducted based on the STRING database (https://cn.string-db.org/), a commonly used tool for evaluating protein interactions. The results were further imported into the software Cytoscape v 3.10.3 (https://cytoscape.org/) to construct the PPI network. In the PPI network, nodes represent the CDEGs from the STRING database, while edges (connections between nodes) represent the interactions among different CDEGs.
Screening and correlation analysis of key CDEGs
Based on the constructed PPI network map, ten algorithms (namely MCC, DMNC, MNC, Degree, EPC, BottleNeck, Closeness, Radiality, Betweenness, and Stress) were used to calculate the top 30 ranked CDEGs, respectively. The intersection of these results was taken as the key CDEGs. Pearson correlation analysis was employed to evaluate the correlations among the key CDEGs. In Pearson correlation analysis, the r value (correlation coefficient) was used to assess the magnitude of the effect. The R package “ggplot2” was utilized to plot the correlation scatter plots.
Tissue sample collection
Patients meeting the diagnostic criteria for sarcopenia and obesity, along with age- and sex-matched healthy control individuals, were recruited clinically. Specifically, sarcopenia patients (n = 5, age 65 ± 5 years, BMI 22 ± 3, diagnosed per EWGSOP2 criteria) and obesity patients (n = 6, age 65 ± 5 years, BMI 32 ± 4, metabolically healthy per IDF criteria) were recruited, with matched controls. Full clinical characteristics are in Supplementary material. Following the acquisition of informed consent from all participants, skeletal muscle tissue and subcutaneous adipose tissue samples were collected via surgical resection. Collected samples were immediately frozen in liquid nitrogen and subsequently transferred to an -80 °C freezer for storage until subsequent experiments.
RNA extraction and RT-PCR protocols
Total RNA was extracted from cryopreserved tissue samples ground in liquid nitrogen using the SteadyPure RNA Extraction Kit (AG, AG21024). RNA concentration and purity were assessed via spectrophotometry, ensuring A260/A280 ratios between 1.8 and 2.0. Reverse transcription of RNA to cDNA was performed using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, K1622), following the manufacturer’s specifications for reaction setup and conditions. Specific primers for SDHB, SDHD, ATP5F1A, and ATP5F1B genes were designed (sequences provided in Table 1). Quantitative PCR (qPCR) was conducted using LightCycler 480 SYBR Green I Master (Roche, 04707516001) on a LightCycler 480 II Real-Time PCR System (Roche). GAPDH served as the internal reference gene. Relative expression levels of each selected gene were calculated using the 2−△ΔCt method, comparing sarcopenia patients vs. healthy controls and obesity patients vs. healthy controls. Statistical analysis was performed using Student’s t-test, with p < 0.05 considered statistically significant.
Statistical methods
The R software (version 4.4.2) and Cytoscape software (v 3.10.3) were utilized for data processing and graphical representation. Pearson correlation analysis was employed to evaluate the correlations among key CDEGs. In all data processing, a p-value of less than 0.05 was considered statistically significant.
Results
Identification of DEGs and shared genes between sarcopenia and obesity
Using the limma R package, we analyzed the sarcopenia dataset GSE111016 and identified a total of 582 DEGs, including 271 significantly upregulated genes and 311 significantly downregulated genes (Fig. 2a). In the obesity dataset GSE152991, 7,332 DEGs were detected, comprising 4,190 upregulated genes and 3,142 downregulated genes. The wider distribution of fold changes indicated more significant perturbation of gene expression associated with obesity (Fig. 2b).
Further analysis revealed that the DEG expression patterns in both sarcopenia and obesity groups were significantly separated from those in the control group, exhibiting disease-specific modular distribution characteristics (Fig. 2d and e). Additionally, intersection analysis of DEGs from the two datasets showed that 208 overlapping DEGs were shared between the 582 DEGs in sarcopenia and the 7,332 DEGs in obesity (Fig. 2c). This result suggests that the two metabolic diseases may share partial core molecular mechanisms, providing important clues for exploring their underlying shared pathophysiological basis.

Analysis and intergroup comparison of DEGs in sarcopenia and obesity datasets. (a) Volcano plot of DEGs in the sarcopenia dataset (GSE111016). (b) Volcano plot of DEGs in the obesity dataset (GSE152991). (c) Venn diagram of DEG intersection analysis between the two datasets. (d) Hierarchical clustering heatmap of DEG expression profiles in the sarcopenia group (Sarcopenia) and non-sarcopenia group (Normal). (e) Hierarchical clustering heatmap of DEG expression profiles in the obesity group (Obesity) and non-obesity group (Normal).
GO and KEGG analyses of CDEGs
Further GO and KEGG enrichment analyses were performed on the 208 CDEGs (Fig. 2c), yielding the following results: KEGG pathway analysis showed that the CDEGs were significantly enriched in 10 key pathways (Fig. 3d)12,13,14. Among these, the thermogenesis and oxidative phosphorylation pathways exhibited the highest enrichment significance, involving 28 and 32 CDEGs, respectively. Additionally, the analysis revealed significant enrichment in neurodegenerative disease-related pathways, including Parkinson’s disease and Alzheimer’s disease, which overlapped with oxidative stress-related pathways.
Combined with the GO analysis results (Figs. 3a–c), energy metabolism processes such as oxidative phosphorylation and mitochondrial electron transport chain were significantly enriched in biological processes (p < 0.001). In cellular components, gene expression associated with respiratory chain complexes (e.g., cytochrome complex, NADH dehydrogenase complex) was significantly altered. Molecular function analysis also showed significant enrichment of energy metabolism-related functions. Collectively, these results suggest that energy metabolism disorders and activation of neurodegenerative pathways may serve as common molecular foundations for the two metabolic diseases.

Construction of PPI network and acquisition of key CDEGs
A PPI network of the 208 intersecting CDEGs was constructed based on the STRING database and visualized using Cytoscape, revealing complex synergistic regulation among genes (Fig. 4a). Ten topological algorithms (MCC, DMNC, MNC, Degree, EPC, BottleNeck, Closeness, Radiality, Betweenness, Stress) from the CycloHubba plugin were used to rank gene importance (Fig. 4b). By taking the intersection of the top 30 ranked CDEGs from each algorithm, four Key CDEGs were identified: SDHB, SDHD, ATP5F1A, and ATP5F1B (Fig. 4c). All four genes were significantly downregulated in both sarcopenia and obesity groups.

Construction of the PPI network and identification of Key CDEGs. (a) The PPI network of 208 CDEGs was constructed based on the STRING database and visualized using Cytoscape. (b) Ten topological algorithms from the CycloHubba plugin in Cytoscape were used to rank the importance of genes. (c) Four Key CDEGs were identified.
Pearson correlation analysis of the four Key CDEGs revealed strong positive correlations in the GSE111016 (sarcopenia) dataset: ATP5F1B showed a robust positive correlation with SDHB (r = 0.97, p = 8.5 × 10⁻²⁴), while SDHD and ATP5F1A exhibited lower but significant correlation (r = 0.86, p = 2.2 × 10⁻¹²), indicating tight co-expression among mitochondrial respiratory chain-related genes (Figs. 5a–c). In the GSE152991 (obesity) dataset, ATP5F1A and ATP5F1B showed the highest correlation (r = 0.94, p = 6.8 × 10⁻¹³), with SDHB also strongly correlated with ATP5F1B (r = 0.91, p = 6.8 × 10⁻¹³) (Figs. 5d–e). Both datasets validated the conserved co-regulatory patterns of core genes across different tissues. The functions of these genes were enriched in mitochondrial oxidative phosphorylation, electron transport chain, and other energy metabolism pathways, suggesting that mitochondrial dysfunction may serve as a common pathological basis for sarcopenia and obesity.

Pearson correlation analysis of key CDEGs in sarcopenia and obesity datasets. (a–c) Correlation matrices and scatter plots of four Key CDEGs in the GSE111016 sarcopenia dataset. (d–f) Correlation matrices and scatter plots of four Key CDEGs in the GSE152991 obesity dataset.
Functional enrichment analysis of key CDEGs
GO and KEGG enrichment analyses showed that the four Key CDEGs (SDHB, SDHD, ATP5F1A, ATP5F1B) were significantly enriched in energy metabolism processes such as oxidative phosphorylation, proton motive force-driven mitochondrial ATP synthesis, and electron transport chain in biological processes (Fig. 6a). In cellular components, they were primarily enriched in mitochondrial structural components such as respiratory chain complexes and proton-transporting ATP synthase complexes (Fig. 6b). In molecular functions, they were closely associated with proton-transporting ATPase activity, electron transfer activity, and quinone binding (Fig. 6c).
KEGG pathway analysis indicated that these genes were significantly enriched in oxidative phosphorylation, thermogenesis pathways, and neurodegenerative disease pathways such as Parkinson’s disease and Alzheimer’s disease, with overlaps in oxidative stress pathways (Fig. 6d)12,13,14. Collectively, the functional and pathway enrichment results of the core genes all point to mitochondrial oxidative phosphorylation and energy metabolism regulation, suggesting that mitochondrial dysfunction may be the common key pathological basis for sarcopenia and obesity.

Verification of key CDEGs expression by qPCR
To validate the expression patterns of core genes identified through bioinformatics analysis in sarcopenia and obesity, qPCR was used to quantitatively analyze the expression of four Key CDEGs (SDHB, SDHD, ATP5F1A, ATP5F1B) in tissue samples. Results showed that the expression levels of these four genes were significantly lower in skeletal muscle samples from sarcopenia patients and adipose tissue samples from obesity patients compared to healthy controls, with the downregulation trend consistent with transcriptome data analysis (GSE111016 and GSE152991). The magnitude of downregulation was slightly greater in sarcopenia (approximately 40–50%, p < 0.001) than in obesity (approximately 50–60%, p < 0.001) (Figs. 7a–h). These results provide direct experimental evidence for the hypothesis that mitochondrial dysfunction serves as a shared pathological basis for the two diseases.

qPCR validation of Key CDEGs in sarcopenia and obesity. (a–d) Relative expression levels of SDHB, SDHD, ATP5F1A, and ATP5F1B in skeletal muscle samples from sarcopenia patients and healthy controls; (e–h) Relative expression levels of the above genes in adipose tissue samples from obesity patients and healthy controls. *Data are presented as mean ± standard deviation (Mean ± SD). *p < 0.05, **p < 0.01, ***p < 0.001, **** p < 0.0001 (independent samples t-test).
Discussion
This study integrated transcriptomic data from sarcopenia (GSE111016) and obesity (GSE152991) to identify 208 CDEGs, of which four core genes (SDHB, SDHD, ATP5F1A, ATP5F1B) were significantly enriched in the mitochondrial oxidative phosphorylation pathway. SDHB and SDHD, as key subunits of mitochondrial respiratory chain complex II, participate in electron transport, while ATP5F1A and ATP5F1B, components of the ATP synthase, directly influence energy production15,16,17,18. The significant downregulation of these genes in skeletal muscle from sarcopenia patients and adipose tissue from obesity patients suggests that mitochondrial energy metabolism defects may serve as a common molecular mechanism for the two diseases.
This finding is consistent with previous studies: in sarcopenia, impaired mitochondrial respiration and reduced ATP production lead to muscle atrophy19, with studies showing that decreased ATP5F1A expression increases muscle fat infiltration and inhibits myogenic differentiation17; in obesity, mitochondrial dysfunction in adipose tissue is associated with insulin resistance and metabolic inflexibility20,21,22. SDHB gene mutations are associated with various diseases, including hereditary paraganglioma, pheochromocytoma, gastrointestinal stromal tumor (GIST), and renal cell carcinoma23,24,25. Furthermore, SDHB mutations can induce mitochondrial dysfunction, accumulation of reactive oxygen species (ROS), and tumorigenesis26. SDHD, like SDHB, is also a component of the SDH complex, and its mutations are also associated with the aforementioned diseases. It is worth noting that SDHB mutations often lead to metastatic tumors outside the adrenal glands, while tumors associated with SDHD mutations are mostly benign and frequently occur in the head and neck region. The mechanism underlying this is related to the accumulation of succinate and the stabilization of HIF proteins15,27,28. ATP5F1B abnormalities are associated with an increased risk of metabolic syndrome in obese patients29. This study further validated the expression trends of core genes through qPCR, providing direct experimental evidence for the hypothesis that mitochondrial dysfunction drives the comorbidity of metabolic diseases.
PPI network analysis showed that the four core genes were consistently identified as key nodes by algorithms such as MCC and Degree, and exhibited strong positive correlations, indicating functional synergy. GO/KEGG enrichment analyses revealed their primary involvement in oxidative phosphorylation, electron transport chain, and mitochondrial ATP synthesis, with cross-talk to neurodegenerative disease pathways (e.g., Parkinson’s disease, Alzheimer’s disease)30,31. This may explain the common comorbidity of neuromuscular decline and metabolic disorders in sarcopenia and obesity, suggesting that mitochondrial dysfunction could affect multisystem pathology through an energy metabolism-neuroinflammation axis. Notably, the core genes all belong to mitochondrial complexes II and V, implying that targeting mitochondrial complex assembly or function may represent a therapeutic strategy for metabolic diseases. For example, small-molecule compounds targeting ATP synthase have shown promise in improving energy metabolism in metabolic disease models32,33,34.
qPCR validation revealed a greater downregulation amplitude (40–50%) in sarcopenia skeletal muscle than in obesity adipose tissue (50–60%), possibly reflecting the higher dependency of skeletal muscle on mitochondrial function and thus greater sensitivity to energy metabolism abnormalities35,36. This difference provides a clue for disease stratification: sarcopenia patients may rely more on mitochondrial function restoration to improve muscle quality, while obesity patients require simultaneous regulation of mitochondrial metabolism and inflammatory responses in adipose tissue. Additionally, the PPI network revealed indirect interactions between core genes and inflammation-related genes (e.g., IL-6, TNF-α), supporting the “metabolism-inflammation axis” theory, where the release of mitochondrial damage-associated molecular patterns (DAMPs) triggers immune activation and exacerbates metabolic disorders37,38,39.
This study has several limitations. The sample size in the qPCR validation was small, which inevitably reduces statistical power and increases the possibility of false-negative results; consequently, the results require verification in larger cohorts to ensure generalizability. To address this issue, we plan to validate these genes in an independent cohort of 100 sarcopenia and 100 obesity patients to collect more skeletal muscle and adipose tissue samples, which will enable us to confirm the robustness of the observed gene expression trends and enhance the translational potential of our discoveries.
Focusing solely on skeletal muscle (GSE111016) and subcutaneous adipose tissue (GSE152991) may bias pathway interpretation, as mitochondrial dysfunction in liver or visceral fat could exhibit distinct molecular signatures. For instance, liver-specific oxidative phosphorylation defects are linked to insulin resistance in obesity, but were not captured here, potentially oversimplifying the shared mechanism40,41,42. This limitation highlights that our identified CDEGs may represent tissue-specific markers, and their role in cross-organ crosstalk requires validation in multi-tissue models. Additionally, the specific mechanisms underlying how core genes regulate mitochondrial function—including epigenetic regulation, post-translational modification, and potential transcriptional control—remain unclear and require functional validation in cell models like C2C12 myocytes and 3T3-L1 adipocytes through gene knockout or overexpression experiments43,44,45,46. Looking ahead, integrating single-cell sequencing could dissect cell-type-specific expression heterogeneity of mitochondrial genes, while animal models such as high-fat diet-induced obese mice or muscle-specific mitochondrial deficiency models may help explore the therapeutic potential of intervention targets47,48,49.
In conclusion, this study reveals a shared mitochondrial dysfunction mechanism between sarcopenia and obesity through multi-omics integration and experimental validation, identifying a key gene network centered on SDHB, SDHD, ATP5F1A, and ATP5F1B. Clinically, the downregulation of SDHB and ATP5F1A in both tissues suggests their utility as minimally invasive biomarkers. For example, blood-based assays of these genes could stratify sarcopenic obesity risk or monitor intervention efficacy, given their high correlation (r > 0.86) across datasets. Beyond biomarker potential, targeting mitochondrial oxidative phosphorylation, particularly through small-molecule modulators such as ATP synthase activators, could be prioritized for dual-disease therapy.
Conclusion
Our study reveals mitochondrial oxidative phosphorylation dysfunction as a shared pathological mechanism in sarcopenia and obesity through integrative transcriptomic analysis. Four key genes (SDHB, SDHD, ATP5F1A, ATP5F1B) in mitochondrial respiratory chain complexes are identified and validated by qPCR, showing significant downregulation in both diseases. These findings may provide new biomarkers or potential therapeutic targets for sarcopenia and obesity, highlighting mitochondrial energy metabolism as a cross-disease intervention axis.
Data availability
The gene expression datasets (GSE111016 and GSE152991) used in this study are publicly available in the Gene Expression Omnibus (GEO) database. All other raw data generated in this study are provided as supplementary materials uploaded with the manuscript.
References
Cruz-Jentoft, A. J. et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 48 (1), 16–31 (2019).
Perdomo, C. M. et al. Contemporary medical, device, and surgical therapies for obesity in adults. Lancet (London England) 401 (10382), 1116–1130 (2023).
Bilski, J. et al. Multifactorial mechanism of sarcopenia and sarcopenic obesity. Role of physical exercise, microbiota and myokines. Cells 11 (1) (2022).
Zou, Y. et al. Obesity, sarcopenia, sarcopenic obesity, and hypertension: Mediating role of inflammation and insulin resistance. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 80 (3) (2025).
Hrdlickova, R., Toloue, M. & Tian, B. RNA-Seq Methods for Transcriptome Analysis 8 (Wiley interdisciplinary reviews RNA, 2017). 1.
Kalyani, R. R., Corriere, M. & Ferrucci, L. Age-related and disease-related muscle loss: The effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol. 2 (10), 819–829 (2014).
Carbone, S. et al. Lean mass abnormalities in heart failure: The role of sarcopenia, sarcopenic obesity, and cachexia. Curr. Probl. Cardiol. 45 (11), 100417 (2020).
Hood, D. A. et al. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu. Rev. Physiol. 81, 19–41 (2019).
Liao, T. et al. Mitochondrial Disorders as a Mechanism for the Development of Obese Sarcopenia15224 (Diabetology & metabolic syndrome, 2023). 1.
Lee, H. et al. Mitochondrial dysfunction in skeletal muscle contributes to the development of acute insulin resistance in mice. J. Cachexia Sarcopenia Muscle 12 (6), 1925–1939 (2021).
Wen, X. et al. Signaling pathways in obesity: Mechanisms and therapeutic interventions. Signal. Transduct. Target. Therapy. 7 (1), 298 (2022).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. Public. Protein Soc. 28 (11), 1947–1951 (2019).
Kanehisa, M. et al. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–d7 (2025).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Garrett, A. et al. Quantifying evidence toward pathogenicity for rare phenotypes: The case of succinate dehydrogenase genes, SDHB and SDHD. Genet. Med. Off. J. Am. Coll. Med. Genet. 24 (1), 41–50 (2022).
Xu, M. et al. Overexpression of ATP5F1A in cardiomyocytes promotes cardiac reverse remodeling. Circ. Heart Fail. 17 (7), e011504 (2024).
You, W. et al. Growth arrest and DNA damage-inducible alpha regulates muscle repair and fat infiltration through ATP synthase F1 subunit alpha. J. Cachexia Sarcopenia Muscle 14 (1), 326–341 (2023).
Nasca, A. et al. Variants in ATP5F1B are associated with dominantly inherited dystonia. Brain: J. Neurol. 146 (7), 2730–2738 (2023).
Mills, K. F. et al. Long-Term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metabol. 24 (6), 795–806 (2016).
Xia, W. et al. Obesity causes mitochondrial fragmentation and dysfunction in white adipocytes due to RalA activation. Nat. Metab. 6 (2), 273–289 (2024).
Boutari, C., DeMarsilis, A. & Mantzoros, C. S. Obesity and diabetes. Diabetes Res. Clin. Pract. 202, 110773 (2023).
Nakamura, M. & Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 598 (14), 2977–2993 (2020).
Ibrahim, A. & Chopra, S. Succinate Dehydrogenase-Deficient Gastrointestinal stromal tumors. Arch. Pathol. Lab. Med. 144 (5), 655–660 (2020).
Usatii, M. et al. Complex immunophenotypes: Renal cell carcinoma with concurrent loss of SDHB/INI1 and FH/INI1. Hum. Pathol. 139, 135–137 (2023).
Choi, H. R. et al. Efficacy of immunohistochemistry for SDHB in the screening of hereditary pheochromocytoma-paraganglioma. Biology 10 (7). (2021).
Yang, W. et al. 4-Octyl Itaconate Inhibits Aerobic Glycolysis by Targeting GAPDH To Promote Cuproptosis in Colorectal Cancer 159114301 (Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 2023).
Liu, C. et al. Research Progress on the Pathogenesis of the SDHB Mutation and Related Diseases167115500 (Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 2023).
Pawlu, C., Bausch, B. & Neumann, H. P. Mutations of the SDHB and SDHD genes. Fam. Cancer 4 (1), 49–54 (2005).
Souza, A. F. P. et al. Maternal Isocaloric High-Fat Diet Induces Liver Mitochondria Maladaptations and Homeostatic Disturbances Intensifying Mitochondria Damage in Response To Fructose Intake in Adult Male Rat Offspring 66e2100514 (Molecular Nutrition & Food Research, 2022). 8.
Hossain, M. A. et al. Systems biology and in silico-based analysis of PCOS revealed the risk of metabolic disorders. Heliyon 8 (12), e12480 (2022).
Zhong, J. et al. Bioinformatics and system biology approach to identify potential common pathogenesis for COVID-19 infection and sarcopenia. Front. Med. 11, 1378846 (2024).
Arosio, B. et al. Sarcopenia and cognitive decline in older adults: Targeting the muscle-brain axis. Nutrients 15 (8) (2023).
Almeida-Oliveira, F. et al. Reference Genes for Quantitative PCR in the Adipose Tissue of Mice with Metabolic Disease 88948–955 (Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 2017).
Foutz, M. A. et al. Design, synthesis, and biological evaluation of [1,2,5]Oxadiazolo[3,4-b]pyridin-7-ol as mitochondrial uncouplers for the treatment of obesity and metabolic Dysfunction-Associated steatohepatitis. J. Med. Chem. 67 (23), 21486–21504 (2024).
Pesta, D. Mitochondrial density in skeletal and cardiac muscle. Mitochondrion 75, 101838 (2024).
Halling, J. F. & Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutrition Metab. 45 (9), 927–936 (2020).
Glass, C. K. & Olefsky, J. M. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metabol. 15 (5), 635–645 (2012).
McGarry, T. et al. Hypoxia, Oxidative Stress and Inflammation 12515–24 (Free radical biology & medicine, 2018).
Marchi, S. et al. Mitochondrial control of inflammation. Nat. Rev. Immunol. 23 (3), 159–173 (2023).
Ajnakina, O. et al. The long-term effects of a polygenetic predisposition to general cognition on healthy cognitive ageing: Evidence from the english longitudinal study of ageing. Psychol. Med. 53 (7), 2852–2860 (2023).
Loh, K. P. et al. Associations of interleukin-6 with functional trajectories in older adults with cancer: Findings from the health, aging, and body composition study. Exp. Gerontol. 177, 112185 (2023).
Moon, J. H. et al. Serum 25-hydroxyvitamin D level and the risk of mild cognitive impairment and dementia: The Korean longitudinal study on health and aging (KLoSHA). Clin. Endocrinol. 83 (1), 36–42 (2015).
Gu, X. et al. Pb inhibited C2C12 myoblast differentiation by regulating HDAC2. Toxicology 499, 153639 (2023).
Wong, C. Y., Al-Salami, H. & Dass, C. R. C2C12 cell model: Its role in Understanding of insulin resistance at the molecular level and pharmaceutical development at the preclinical stage. J. Pharm. Pharmacol. 72 (12), 1667–1693 (2020).
Zebisch, K. et al. Protocol for effective differentiation of 3T3-L1 cells to adipocytes. Anal. Biochem. 425 (1), 88–90 (2012).
Yudhani, R. D. et al. In vitro insulin resistance model: A recent update. J. Obes. 2023, 1964732 (2023).
de Wouters d’Oplinter, A. et al. Gut Microbes participate in food preference alterations during obesity. Gut Microbes 13 (1), 1959242 (2021).
Murakami, T., Inagaki, N. & Kondoh, H. Cellular senescence in diabetes mellitus: Distinct senotherapeutic strategies for adipose tissue and pancreatic β cells. Front. Endocrinol. 13, 869414 (2022).
Zhang, S. et al. Skeletal muscle-specific DJ-1 ablation-induced atrogenes expression and mitochondrial dysfunction contributing to muscular atrophy. J. Cachexia Sarcopenia Muscle 14 (5), 2126–2142 (2023).
Acknowledgements
The authors thank the providers of GEO datasets (GSE111016 and GSE152991) for enabling this analysis, and the clinical staff involved in sample collection for their assistance.
Funding
This study was funded by the Beijing Municipal Health Commission (BJRITO-RDP) (Code: QN-2025-09); the Beijing Municipal Public Welfare Development and Reform Pilot Project for Medical Research Institutes (Code: JYY2023-8, JYY2023-11); the Beijing Physician Scientist Training Project (Code: BJPSTP-2024-08); and the Capital Medical University Outstanding Youth Talent Program (Category A) (Code: A2413).
Author information
Authors and Affiliations
Contributions
R.Y. and J.W. co-led tissue collection; R.Y. performed qPCR and co-drafted the manuscript. M. conducted bioinformatics analyses (including DEG, GO/KEGG, and PPI), with support from D.W. and Y.L. in data processing. R.W. and L.W. (corresponding authors) oversaw study design and manuscript revision. All authors contributed to data interpretation and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
This study was approved by the Ethics Committee of Beijing Jishuitan Hospital, Capital Medical University (Ethics Approval No. [K2025] NO. [018]-00). All participants provided written informed consent prior to sample collection, in accordance with the Declaration of Helsinki and institutional guidelines. All methods were performed in accordance with the relevant guidelines and regulations.
Consent for publication
All participants were informed of the study’s purpose and agreed to the publication of clinical data and research findings. No identifiable patient information is included in the manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, R., Miao, L., Wang, J. et al. Integrated transcriptomics unveils mitochondrial oxidative phosphorylation dysfunction as a shared mechanism in sarcopenia and obesity. Sci Rep 15, 34810 (2025). https://doi.org/10.1038/s41598-025-18824-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-18824-y
