
Abstract
Immune checkpoint inhibitors targeting programmed cell death ligand 1 (PD-L1) have transformed cancer therapy but have been linked to increased cardiovascular risk, particularly atherosclerosis (AS). This study hypothesized that anti-PD-L1 therapy promotes atherosclerosis progression by modulating macrophage phenotypes and enhancing foam cell formation via gene-level changes. Single-cell RNA sequencing (scRNA-seq) analysis of macrophages post-anti-PD-L1 immunotherapy was conducted using the GSE169246 dataset. Differential expression, GO/KEGG enrichment, and transcription factor analyses were performed. Cellular communication patterns were examined, and in vitro validation included foam cell assays and protein-level assessments. Anti-PD-L1 treatment promoted a shift toward pro-inflammatory M1 macrophages, increased foam cell formation, and upregulated EGR1 and HSP90AB1. These gene changes correlated with altered cellular interaction patterns, particularly between macrophages and endothelial cells. PD-L1 inhibition reprograms macrophage behavior through EGR1 and HSP90AB1-mediated pathways, driving M1 polarization and foam cell development. These findings reveal a mechanistic link between immunotherapy and AS progression and underscore the need for cardiovascular monitoring in patients undergoing PD-L1 blockade.
Similar content being viewed by others

Introduction
Immune checkpoint inhibitors targeting programmed cell death ligand 1 (PD-L1) have markedly improved survival across various cancers1,2,3. However, their benefits come with an increasing incidence of immune-related adverse events, including cardiovascular complications4,5. Notably, PD-L1 inhibitors have been implicated in promoting inflammation and accelerating atherosclerosis (AS), potentially through heightened macrophage activity6,7.
Emerging evidence highlights that PD-L1 blockade intensifies immune responses within atherosclerotic plaques, exacerbates necrotic core formation, and elevates the risk of acute coronary events8,9,10,11,12. Despite these observations, the precise mechanisms linking anti-PD-L1 therapy to AS progression remain incompletely understood.
Macrophages are central to AS development, mediating inflammation, lipid uptake, and foam cell formation13,14,15,16. The balance between pro-inflammatory M1 and anti-inflammatory M2 macrophages is particularly critical17,18,19. Given the role of macrophages in both immune regulation and plaque evolution, we hypothesized that PD-L1 inhibition disrupts this balance and promotes foam cell formation by modulating key macrophage-associated genes.
To test this, we analyzed single-cell transcriptomic data from anti-PD-L1-treated models, identified key regulatory genes including EGR1 and HSP90AB1, and validated their roles experimentally. Furthermore, we explored intercellular signaling between macrophages and endothelial cells, offering a broader view of PD-L1 inhibitor impact on the atherosclerotic microenvironment. This study aims to elucidate how PD-L1 immunotherapy may influence AS progression through macrophage reprogramming.
Methods
Single-Cell sequencing data and analysis
For this study, we utilized scRNA-seq data of chemotherapy treated (CHEMO) and ANTI-PD-L1 + CHEMO treated samples from the publicly available GSE169246 dataset20. Researchers of the dataset selected 489,490 high-quality cells and 22 blood samples from individuals with Triple-negative breast cancer (TNBC), with 11 receiving single-agent chemotherapy paclitaxel and 11 receiving a first-line treatment of paclitaxel in combination with the PD-L1 inhibitor atezolizumab.
For 10X data, Cell Ranger 3.0.0 was used to quantify gene expression levels, identify T cell receptor (TCR sequences), and quantify ATAC-seq peaks. The R package Seurat (v.4.3.0.1) was used to integrate all sample data21. Inadequate cells, defined as either those expressing genes detected in five or less cells, or expressing less than 500 genes, were excluded from subsequent analysis. The integrated dataset resulted in a sparse matrix containing 20,619 genes across 308,992 cells. A sparse matrix containing 308,992 cells was obtained after filtering. The gene expression of all cells was normalized using a linear regression model, and the top 2,000 genes with highly variable features were selected. Principal component analysis was conducted on the single-cell samples, and the top 20 principal components were chosen for subsequent analysis. The UMAP algorithm was employed for global dimensionality reduction analysis22.
The FindAllMarkers function was utilized for detecting changes in the gene expression of specific markers. Markers for each cluster were computed based on the following criteria: logfc.threshold = 0.25, min.pct = 0.25, only.pos = TRUE. To accurately identify cell types within GSE169246, cells were manually annotated as follows: natural killer (NK) cells (GNLY, KLRB1), CD4+ T cells (CD3D, CD3E, IL7R, CCR7), CD8+ T cells (CD3D, CD3E, CD8A, CD8B), B cells (CD79A, CD79B), monocytes (LYZ, VCAN, FCN1), M1 macrophages (NLRP3), M2 macrophages (S100A8, TGFB1), cDC2 (CD1C, FCER1A), pDC (GZMB, SCT), mast cells (CPA3), endothelial cells (PECAM1), fibroblasts (ACTA2), plasmablasts (MZB1), platelets (PF4, NRGN). The FindMarkers function in Seurat was employed for the differential gene expression analysis of macrophages, with parameters set as min.pct = 0.1, logfc.threshold = 0.25, and the detection method as Wilcoxon. CellChat analysis was carried out by R package ‘cellchat’ (version 1.6.1)23.
The likelihood ratio test was used to identify genes with differential expression in macrophages between the CHEMO and ANTI-PD-L1 + CHEMO groups, with a p-value of 0.05 considered statistically significant. The ClusterProfiler package (v4.8.1) was utilized to analyze significantly enriched Gene Ontology (GO) pathways and Kyoto Encyclopedia of Genes and Genomes (KEGG) between the CHEMO and ANTI-PD-L1 + CHEMO treatment groups, with gene identifiers plotted using the org.Hs.eg.db package24,25,26. The KEGG pathway analysis related to this project was conducted under an authorized license (see Supplementary Materials for details), in accordance with the guidelines described by Kanehisa et al.27.
A gene expression matrix averaging gene expression changes between the CHEMO and ANTI-PD-L1 + CHEMO treatment groups was constructed, and 50 hallmark gene sets were retrieved using the gene set variation analysis (GSVA) package (v1.48.1). Variations in enriched gene sets were assessed using standard settings; gene sets with a p-value < 0.05 were considered significantly enriched. To conduct transcription factor analysis on macrophage subtypes, we employed the Dorothea package (v1.12.0) to analyze the enrichment of transcription factors in each cell and calculate their activity scores28.
Cell culture and cell model establishment
The human THP-1 monocyte cell line (SCSP-567) was purchased from the Chinese Academy of Sciences cell bank (Shanghai, China) on February 23rd, 2023. (Ethics Statement is not applicable). Cells were cultured in 10% (v/v) fetal bovine serum (FBS; Sijiqing Bioengineering Material Co., Ltd, Zhejiang, China) and RPMI 1640 medium (Gibco, Shanghai, China) in a humidified incubator with 5% CO2 at 37 °C.
THP-1 cells were seeded at a density of 2 × 105 cells/well in a 24-well plate. Cells were incubated with 5 ng/mL PMA for 24 h (Sigma-Aldrich, St. Louis, MO, USA), followed by a rest period in fresh medium for 72 h to differentiate THP-1 cells into adherent THP-1- macrophages (Mφ). Subsequent incubation with 20 ng/mL IL-4 for 48 h was performed to induce the differentiation of THP-1-Mφ into THP-1-M229. Foam cells were generated by growing THP-1-M2 cells with 50 µg/mL ox-LDL (Yiyuan Biotech. Co. Ltd, Guangdong, China) for 24 h. Next, 1 µM BMS-1 (MedChemExpress, Monmouth Junction, NJ, USA), an inhibitor of PD-1/PD-L1 protein-protein interaction, was added to cells cultured with ox-LDL for 24 h prior to subsequent downstream analyses.
Oil red O staining
Oil Red O staining was performed to assess lipid accumulation in macrophages, following the protocol established in our previous work30.
Western blotting
Western blotting was performed as previously described by our laboratory30 using the antibodies listed below: anti-β-Actin (13E5) rabbit mAb (CST, MA, USA, #4970,1:1000 dilution), anti-human CD86 (Huabio, Hangzhou, Zhejiang, China, ET1606-50, 1:1000 dilution), anti-human CD80 (Huabio, Hangzhou, Zhejiang, China, M1007-10, 1:1000 dilution), anti-human CD206 (Huabio, Hangzhou, Zhejiang, China, ET1702-04, 1:1000 dilution), Rabbit anti-human CD163 (abcam, MA, USA, ab182422, 1:1000 dilution), early growth response-1 (EGR‐1) Rabbit pAb (ABclone, MA, USA, A2722, 1:1000 dilution), and heat shock protein 90 kDa alpha family class B member 1 (HSP90AB1) Polyclonal antibody (Proteintech, Wuhan, Hubei, China, 11405-1-AP, 1:1000 dilution).
Immunofluorescence analysis
After the treatments described above, macrophages were fixed with 4% paraformaldehyde for 15 min and washed with 0.05% Tween-20 1 × PBS three times. Nonspecific binding was blocked with 10% normal goat serum at room temperature for 30 min. Overnight incubation with mouse anti-human CD86 (Huabio, Hangzhou, Zhejiang, China, ET1606-50, 1:500 dilution), and mouse anti-human CD206 (Huabio, Hangzhou, Zhejiang, China, ET1702-04, 1:200 dilution) at 4 °C was performed and washed with 0.05% Tween-20 PBS three times. Prior to anti-iNOS antibody incubation, 0.25% Triton X-100 was used for 20 min at room temperature. Samples were incubated with appropriate secondary antibodies (FITC-conjugated goat anti-rabbit-IgG secondary antibody; Zhongshanjinqiao, 1:200) for 2 h at room temperature protected from light. Plates were washed and incubated with DAPI as a nuclear stain. Finally, the plates were observed under a fluorescence microscope (Olympus, Tokyo, Japan) and analyzed using Image J software (NIH, Bethesda, Maryland) as previously described by our laboratory30.
Statistical analysis
All data are expressed as mean ± SEM unless otherwise noted. In total, four groups of samples were prepared, and two-way analysis of variance was used to establish statistical significance. GraphPad Prism 9 software was used to conduct all statistical analyses (GraphPad, USA). All experiments were performed independently in triplicate.
Results
Quality control of single-cell RNA sequencing and cell- specific classification based on characteristic gene expression
By utilizing scRNA-seq, we successfully identified fourteen distinct cell types using the markers described in the methods (Fig. 1A). First, our quality control analysis unveiled 308,992 cells expressing a total of 20,619 genes. Subsequently, the Uniform Manifold Approximation and Projection (UMAP) algorithm was applied to classify these cell types based on their unique gene expression profiles (Fig. 1B). Following this, the expression levels of marker genes specific to each cell cluster were examined, culminating in the creation of a heatmap illustrating the expression patterns of these genes across the clusters (Fig. 1C, D). Quality control and classification of scRNA-seq data led to the identification of 14 distinct cell types using characteristic gene markers, followed by UMAP-based classification and heatmap visualization of cell-specific marker gene expression patterns across clusters.

Identification of 14 cell types was achieved through single-cell RNA sequencing (scRNA-seq). (A) Quality control revealed a total of 308,992 cells expressing 20,619 genes. (B) Fourteen cell types were classified using the Uniform Manifold Approximation and Projection (UMAP) algorithm, based on their characteristic gene expression profiles. The expression levels of marker genes for each cell cluster were analyzed (C), leading to the generation of a heatmap (D) reflecting the expression levels of the marker genes within each cluster.
Circulating macrophage responses following anti-PD-L1 immunotherapy treatment are centered on pathways associated with immune responses, signaling cascades, and metabolic regulation
Utilizing scRNA-seq, we conducted GO, KEGG, and GSVA analyses to investigate the impact and directional influence of anti-PD-L1 immunotherapy treatment on macrophages. The top 10 GO-biological process items were as follows: cytoplasmic translation, response to virus, cell activation involved in immune response, leukocyte activation involved in immune response, positive regulation of cytokine production, positive regulation of defense response, viral process, antigen processing and presentation, and positive regulation of leukocyte activation (Fig. 2A). The top 10 KEGG pathways were as follows: coronavirus disease- COVID-19, Fluid shear stress and AS, Leishmaniasis, Leukocyte trans-endothelial migration, pertussis, osteoclast differentiation, oxytocin signaling pathway, cellular senescence, Epstein-Barr virus infection, and the Rap1 signaling pathway (Fig. 2B). Thus, following anti-PD-L1 immunotherapy, circulating macrophages engage in various pathways and critical processes associated with the immune response, viral clearance, vascular health, infectious diseases, and cellular activation.

Analysis of circulating macrophage enrichment and transcription factor activity in the chemotherapy (CHEMO) group and the group receiving chemotherapy in combination with a PD-L1 inhibitor (ANTI-PD-L1 + CHEMO) therapy. (A) GO enrichment analysis of differentially expressed genes in macrophages between the CHEMO group and ANTI-PD-L1 + CHEMO group. (B) KEGG enrichment analysis of differentially expressed genes in macrophages between the CHEMO group and ANTI-PD-L1 + CHEMO group. (C) GSVA enrichment analysis of differentially expressed genes in macrophages between the CHEMO group and ANTI-PD-L1 + CHEMO group. (D) Heatmap depicting transcription factor enrichment in macrophage subsets in both groups.
The top 10 GSVA pathways were as follows: interferon alpha response, phosphatidylinositol 3-kinase (PI3K) -protein kinase B (Akt) - mammalian target of Rapamycin (MTOR) signaling, apical surface, interferon gamma response, pancreatic beta cells, gap 2/mitosis phase (G2/M) DNA damage checkpoint, cholesterol homeostasis, notch signaling, and bile acid metabolism (Fig. 2C). The highest activity scores of transcription factor activity were regulatory factor X5 (RFX5), activating transcription factor 6 (ATF6), and homeobox B13 (HOXB13) (Fig. 2D). These results suggest the involvement of circulating macrophages in pathways related to immune responses, cell signaling, cellular processes, and metabolism following anti-PD-L1 immunotherapy treatment. In summary, circulating macrophage responses post anti-PD-L1 immunotherapy treatment primarily center on immune responses, signaling cascades, and metabolic regulation.
Cell chat responses to anti-PD-L1 immunotherapy: focusing on ECs and macrophages
CellChat analysis was performed using the R package ‘CellChat’ (version 1.6.1)25 to investigate the impact of anti-PD-L1 immunotherapy on the crosstalk between macrophages and ECs during AS.
Compared to the CHEMO therapy group, patients receiving ANTI-PD-L1 + CHEMO treatment showed an increase in both the total number and intensity of cell interactions (Fig. 3A). Specifically, interactions between myeloid cells and fibroblasts, were more pronounced in the ANTI-PD-L1 + CHEMO therapy group, and involved monocytes, cDC2, pDC, M1, fibroblasts, and platelets (Fig. 3B). In contrast, communication with CD4+ T cells was reduced (Fig. 3B). Subsequently, a total of 17 activated signaling pathways were analyzed and identified in the ANTI-PD-L1 + CHEMO therapy group, with 16 pathways shared between both groups and the TNF signaling pathway uniquely activated in the combination therapy group (Fig. 3C). Comparison of the strength of all incoming signaling patterns in each cell type between the two patient groups revealed that the TNF signaling pathway was predominantly received by ECs, M1 macrophages, and M2 macrophages in the ANTI-PD-L1 + CHEMO therapy group (Fig. 3D). These findings were consistent with the pathological processes of AS, where the phenotypic changes of ECs and macrophages are directly linked19. Based on these data, an analysis was conducted on all cell communication signaling pathway-related receptor-ligand pairs from these three cell sources, revealing predominant signaling through the TNF superfamily (TNFS) 13B-TNF receptor superfamily (TNFSF) 17 (TNFSF13B-TNFRSF17), TNFSF13B-TNFRSF13B, TNFSF13-TNFRSF17, and TNFSF13-TNFRSF13B networks (Fig. 3E). These data confirmed the involvement of the TNF pathway in mediating interactions between ECs and macrophages.

Comparison of cellular communication patterns between chemotherapy (CHEMO) patients and those receiving combination therapy with PD-L1 inhibitors (ANTI-PD-L1 + CHEMO). (A) Histograms depicting the quantity and strength of cell-to-cell communication in the CHEMO and ANTI-PD-L1 + CHEMO groups. (B) Variances in the quantity and strength of cell communication across different cell types between CHEMO and ANTI-PD-L1 + CHEMO treated patients. (C) Disparities in the number and strength of signaling pathways between the CHEMO and ANTI-PD-L1 + CHEMO groups. (D) Heatmap comparing cell-incoming signaling patterns between CHEMO and ANTI-PD-L1 + CHEMO treated patients. (E) Scatter plot analysis of receptor-ligand pairs from endothelial cells, M1 macrophages, and M2 macrophages between both groups.
In summary, these findings indicate that cellular communication-based combination therapy promoted cell-cell interactions between circulating macrophages and ECs.
EGR1 and HSP90AB1 are upregulated in Circulating macrophages post anti-PD-L1 immunotherapy
We investigated the impact of anti-PD-L1 + CHEMO treatment on the proportion of M1 and M2 macrophage subsets and associated changes in gene expression. The proportion of M1 and M2 macrophage clusters relative to the total clustered cells was analyzed. Following anti-PD-L1 + CHEMO treatment, an increase in M1 macrophages and a decrease in M2 macrophages was observed (Fig. 4A). This suggested an increase in inflammatory macrophages. However, the distinction between M1 and M2 was not absolute. Therefore, we proceeded to further study gene expression changes. In response to the changes in M1 and M2 phenotypes, EGR1 and HSP90AB1 emerged as the most significantly upregulated genes (Fig. 4B, C).

Anti-PD-L1 immunotherapy drives foam cell formation through EGR1 and HSP90AB1. EGR1 and HSP90AB1 were key genes responsible for circulating macrophage phenotypes in in the CHEMO and ANTI-PD-L1 + CHEMO groups. (A) The percentage of M1 and M2 macrophage clustering relative to all clustered cells was compared. (B) A violin plot was utilized to display the standardized expression levels of EGR1 and HSP90AB1 in macrophage clusters between both groups. (C) A volcano plot was used to visualize the differential gene expression of macrophage clusters in the CHEMO group and ANTI-PD-L1 + CHEMO group, where red indicates upregulated genes and blue indicates downregulated genes. (D) Western blots indicating total EGR1, HSP90AB1, and β-Actin levels in whole cell lysates. (E) Quantification of EGR1 protein levels from panel (D). *** denotes p-value < 0.001, ** denotes p-value < 0.01 by Two-way ANOVA (n = 4, > 1000 cells per trial). (F) Quantification of HSP90AB1 protein levels from panel (D). ***denotes p-value < 0.01, * denotes p-value < 0.05 by Two-way ANOVA (n = 4, > 1000 cells per trial).
Additionally, it is possible that EGR1 specifically interacts with PD-L1. Activation of EGR1 has been shown to increase the expression of PD-L131, while nuclear translocation of PD-L1 can activate EGR1 and promote angiogenesis32. The expression of EGR1 following anti-PD-L1 immunotherapy remains unclear. Furthermore, there is also a potential interaction between HSP90 and PD-L1. For example, an upregulation in PD-L1 expression was observed in tumor tissues after use of inhibitors targeting HSP90 and Caspase-933. Previous research has demonstrated that inhibiting HSP90 leads to alterations in the surface expression of PD-L134. However, the expression of HSP90 following anti-PD-L1 immunotherapy remains uncertain. To confirm these findings in our study, THP-1 cells were differentiated into M2 macrophages (referred to as THP-1-M2 cells), exposed to 50 µg/mL ox-LDL and/or 1 µM BMS-1, and then analyzed by western blotting. Our study revealed elevated levels of EGR1 and HSP90 protein after treatment with anti-PD-L1 immunotherapy (Fig. 4D-F). In conclusion, these data indicate that anti-PD-L1 + CHEMO treatment leads to an increase in inflammatory M1 macrophages, a decrease in M2 macrophages, and increased expression of EGR1 and HSP90AB1.
Anti-PD-L1 immunotherapy drives foam cell formation and alters macrophage phenotypes
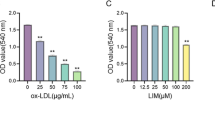
Our data to this point indicated that anti-PD-L1 + CHEMO treatment resulted in an increase in M1 macrophages and a decrease in M2 macrophages, with BMS-1 and ox-LDL influencing macrophage polarization and lipid accumulation, suggesting a dynamic interplay between these factors in modulating macrophage phenotypes (Fig. 4). To confirm these findings, 50 µg/mL ox-LDL and/or 1 µM BMS-1 were used to treat THP-1-M2 cells and simulate AS. Interestingly, the lipid deposition area of the ox-LDL group, the BMS-1 group, and the ox-LDL + BMS-1 group were all increased compared to the control group, indicating an accumulation of lipid droplets within treated cells (Fig. 5A and C). The protein levels (Fig. 5B, F, and G) and immunocytochemistry staining areas (Fig. 6A-C) of CD86 and CD206 decreased under ox-LDL and/or BMS-1 intervention, while there was no significant difference in the total levels of CD163 and CD80 (Fig. 5B, D, and E). These data suggest that ox-LDL and BMS-1 both play a role in promoting the formation of M1 macrophages and increasing intracellular lipid droplets in macrophages. These trends were accompanied by a decrease in CD86-positive and CD206-positive cells, providing additional evidence that the distinction between M1 and M2 macrophages was not absolute.

BMS-1 plays a crucial role in driving foam cell formation and inducing changes in macrophage phenotypes, particularly affecting CD206 and CD86 expression. (A) The area of Oil Red O staining was quantified in THP-1 cells after 24 h of treatment with or without 50 µg/mL ox-LDL or 1 µM BMS-1. Scale bar: 50 μm. (B) Western blots indicating total CD206, CD86, CD80, CD163, and β-Actin levels in whole cell lysates. (C) Quantification of lipid staining area from panel (A). **** denotes p-value < 0.0001 by Two-way ANOVA (n = 7, > 1000 cells per trial). (D-G) Quantification of CD163, CD80, CD86, and CD206 protein levels from panel (B). **** denotes p-value < 0.0001, ** denotes p-value < 0.01 by Two-way ANOVA (n = 7, > 1000 cells per trial).

BMS-1 induces changes in immunofluorescence intensity of CD206 and CD86. (A) The intensity of immunofluorescence staining for CD86 and CD206 was quantified in THP-1 cells post- treatment with or without 50 µg/mL ox-LDL or 1 µM BMS-1. Scale bar: 50 μm. (B-C) Quantification of CD86 and CD206 staining area from panel (A). **** denotes p-value < 0.0001, and * denotes p-value < 0.05 by Two-way ANOVA (n = 4, > 1000 cells per trial).
Discussion
In this study, we examined alterations in macrophages following anti-PD-L1 immunotherapy utilizing comprehensive scRNA-seq data. These alterations were likely a result of the involvement of two upregulated genes, EGR1 and HSP90AB1, which exhibited a synergistic effect on the expression patterns and polarization of M1 macrophages35,36. We recapitulated the effects of anti-PD-L1 immunotherapy on macrophages in in vitro studies and demonstrated their potential to induce macrophage foaming and phenotypic changes, albeit not entirely aligning with the conventional M1 and M2 classification criteria. This study supports our previous hypothesis that PD-L1 inhibitors modulate macrophage phenotypes and foam cell formation by targeting specific genes. The aim of this study was to explore the association between anti-PD-L1 immunotherapy and AS, providing further insights for the clinical application of anti-PD-L1 immunotherapy.
Through rigorous quality control screening of the scRNA-seq data, we investigated the expression levels of specific marker genes within each cell cluster. The subsequent top 10 GO-BP analyses predominantly focused on the immune response against viruses, encompassing processes such as antigen processing and presentation, as well as leukocyte activation involved in immune response. In KEGG pathway analysis, a similar trend emerged, highlighting immune pathways associated with various viruses including COVID-19, pertussis, and Epstein-Barr virus. We speculate that this pattern may be attributed to sample collection during the peak of the COVID-19 pandemic, potentially influencing potential viral effects on our study outcomes. However, a unique pathway, “Fluid shear stress and atherosclerosis,” was identified. This finding was corroborated by GSVA analysis, where pathways such as PI3K-AKT-MTOR signaling, interferon gamma response, and cholesterol homeostasis showed relevance to AS37,38,39. In subsequent studies, the transcription factors RFX5, ATF6, and HOXB13 were found to be associated with macrophage phenotype alterations40,41,42. Thus, we hypothesized that anti-PD-L1 immunotherapy may exert a beneficial effect on AS by modulating macrophage phenotypes through engagement in immune processes.
Cellular communication-based combination therapy plays a pivotal role in enhancing cell-cell interactions between circulating macrophages (M1 and M2) and ECs, potentially influencing the stability of AS plaques in human arteries through the modulation of macrophage phenotypes. The polarization of macrophage phenotypes can influence the stability of atherosclerotic plaques in human arteries13. Macrophages are commonly classified as M1, M2, Mox (oxidized phospholipid induced macrophages), or Mhem (heme induced macrophages), and are characterized based on a limited set of markers19. In murine AS, scRNA-Seq has been utilized to categorize aortic macrophages from inflammatory macrophages, resident-like macrophages (Res-like), and triggered receptor expressed on myeloid cells (TREM2hi) macrophages43. Foam cells, which are critical to the development and extension of necrotic cores in atherosclerotic plaques, are formed when ox-LDL is recognized and taken up by scavenger receptor- expressing macrophages38. Through the phagocytosis of ox-LDL in AS, monocytes accumulate under vascular ECs and differentiate into macrophages (Qin 2012).
To determine changes in the proportion of macrophage populations following anti-PD-L1 immunotherapy, we assessed changes in macrophage phenotypes and observed a significant increase in M1 macrophages and a relative decrease in M2 macrophages. Atherogenesis has been linked to the generation of unstable plaques, primarily influenced by M1 macrophages, whereas M2 macrophages are associated with the formation of stable plaques44. Therefore, we utilized a foam cell model in conjunction with the PD-L1 inhibitor BMS-1 to evaluate the lipid phagocytosis area of macrophages and macrophage markers in vitro. The area of lipid phagocytosis by macrophages increased significantly with BMS-1 and/or ox-LDL treatment. These data suggests that PD-L1 inhibitor treatment may stimulate foam cell formation. While CD86 and CD206 were notably suppressed, there was no significant change in the levels of CD163 and CD80, highlighting the boundaries between M1 and M2 are not absolute. Validation of CD86 and CD206 expression profiles was obtained through immunofluorescence-based assays.
Although CD86 and CD206 are classical markers of M1 and M2 macrophages respectively, recent studies have shown that during foam cell formation, especially under prolonged ox-LDL exposure or metabolic reprogramming, canonical markers can be significantly downregulated. Nogieć et al. demonstrated that human macrophages undergoing foam transformation after chronic ox-LDL stimulation exhibit reduced expression of typical surface markers and adopt a distinct, metabolically altered phenotype45. Similarly, macrophage phenotypes in atherosclerotic plaques span a broad spectrum, and foam cells may no longer express either M1- or M2-defining markers, consistent with our observation of concurrent CD86 and CD206 suppression46,47. This pattern may reflect a non-classical polarization state triggered by BMS-1-induced immunometabolic changes.
Ox-LDL can have stimulatory effects on macrophages, thus enhancing their ability to phagocytose lipids48. However, the degree of foam formation when ox-LDL was used in combination with the PD-L1 inhibitor was not higher than when cells were treated with BMS-1 alone, making it challenging to speculate on the role of ox-LDL in macrophage function after immunotherapy treatment. PD-L1 inhibitor treatment alone does not inherently contain lipid components. Therefore, we speculated that BMS-1 may promote lipid synthesis in macrophages via use of glucose in the culture medium.
In subsequent studies, we found that EGR1 and HSP90AB1 emerged as the most significantly upregulated genes. EGR1 has been confirmed to play a critical role in macrophage polarization and AS and is involved in regulating biological processes such as inflammation, cell proliferation, and apoptosis49,50. Upregulation of the proatherogenic transcription factor EGR1 may closely link to the activation of vascular ECs, inflammatory responses, and upregulation of cholesterol metabolism50. Research has been conducted to demonstrate that inhibiting HSP90 leads to alterations in the surface expression of PD-L134. Inhibition of HSP90 has been shown to effectively reduce inflammation levels and alleviate the severity of AS progression51. Previous studies have also demonstrated that inhibiting the activity of HSP90AB1 can lead to reduced M1 activation, while increased activity of HSP90AB1 can result in enhanced M1 activation36. These findings highlight the potential implications of targeting EGR1 and HSP90AB1 in modulating inflammatory responses and AS progression.
Our findings raise important clinical considerations regarding the cardiovascular safety of anti-PD-L1 immunotherapy. As immune checkpoint inhibitors are increasingly used in long-term cancer management, understanding their effects on non-malignant immune processes becomes critical. This study reveals that PD-L1 blockade may reprogram macrophages toward a pro-inflammatory M1-like phenotype and enhance foam cell formation, thereby potentially contributing to atherosclerotic plaque instability. Given the widespread use of anti-PD-L1 agents like atezolizumab in solid tumors, including breast, lung, and urothelial cancers, these immunomodulatory effects on macrophages may have unanticipated consequences for patients with pre-existing cardiovascular risk factors. Our results therefore underscore the need for cardiovascular monitoring in cancer patients undergoing PD-L1-targeted therapies, particularly in those with known or suspected subclinical atherosclerosis. In addition, EGR1 and HSP90AB1 may serve as molecular markers or therapeutic targets for mitigating cardiovascular risk associated with checkpoint inhibitor use.
Currently used checkpoint inhibitors for patients (anti-CTLA-4, anti-LAG3, anti-PD-1 antibodies) have also been reported to promote the progression of AS. For example, mice treated with a combination of anti-CTLA-4 and anti-PD-1 showed higher endothelial activation compared to control mice, with the aortic arch containing more pathological intimal thickening and AS plaques8. Plasma levels of LAG3 serve as a potential novel independent predictive factor for coronary heart disease risk52. Further, deficiency or blockade of LAG3 promotes activation and recruitment of T cells to atherosclerotic plaques, and the combined blockade of LAG3 and PD-1 has an additive effect on T cell activation53. Although LAG3 deficiency or blockade does not affect the size of atherosclerotic lesions, increased T cell activation and infiltration suggest plaque inflammation and AS risk54.
Despite these insights, several limitations should be acknowledged. First, the scRNA-seq dataset was derived from patients with TNBC, which may bias the sample toward female patients and limit the generalizability of our findings across genders. Second, we sourced 22 peripheral blood samples from TNBC patients treated with paclitaxel, either alone or in combination with atezolizumab (anti–PD-L1), from the publicly available dataset GSE169246. Metadata analysis revealed variation in treatment timing (pre- versus post-therapy) and individual immune profiles, such as baseline B-cell levels and CXCL13⁺ T-cell enrichment, which may influence macrophage gene expression patterns and cell proportions. Although demographic annotations (e.g., age, sex, and prior treatments) were limited in the dataset, these inter-sample differences represent potential confounders that could impact the interpretation of macrophage dynamics and pathway enrichment results. Third, although we observed altered expression of key genes and changes in macrophage polarization, the functional validation of EGR1 and HSP90AB1 remains incomplete and warrants further investigation in in vivo AS models. Fourth, the classification of macrophage subtypes in this study was limited to established markers and may not fully capture the spectrum of macrophage heterogeneity observed in AS. In addition, although foam cell formation was assessed in vitro, the study could not definitively establish whether these changes translate to plaque formation or progression in vivo. Finally, the presence of underlying AS in the original patient cohort was not confirmed, limiting our ability to directly associate the observed molecular alterations with clinical cardiovascular outcomes. Future studies should address these limitations through longitudinal clinical cohort analyses and mechanistic investigations in AS-prone animal models.
Conclusions
In this study, we conducted single-cell analysis on samples from patients receiving anti-PD-L1 immunotherapy. The cells were annotated and grouped by cell type, and key differentially expressed genes were identified via subsequent GO, KEGG, GSVA, and transcription factor analyses. We then performed intercellular communication analysis, elucidating a strong interaction between macrophages and ECs. Subsequently, we examined and validated a role for the two genes most relevant to AS in the single-cell analysis, EGR1 and HSP90AB1, through in vitro cell experiments, further confirming their consistent high expression trends. Finally, exploration of macrophage phenotypes post Anti-PD-L1 immunotherapy revealed high expression of CD86 and CD206. In conclusion, Anti-PD-L1 immunotherapy potentially promotes AS progression by altering macrophage phenotypes, targeting EGR1 and HSP90AB1, and participating in macrophage foam formation.
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author.
References
Andrews, L. P., Yano, H. & Vignali, D. A. A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat. Immunol. 20 (11), 1425–1434 (2019).
Sharma, N., Vacher, J. & Allison, J. P. TLR1/2 ligand enhances antitumor efficacy of CTLA-4 Blockade by increasing intratumoral Treg depletion. Proc. Natl. Acad. Sci. U S A. 116 (21), 10453–10462 (2019).
Larkin, J. et al. Five-Year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 381 (16), 1535–1546 (2019).
Liu, S. et al. Cardiovascular toxicity with PD-1/PD-L1 inhibitors in cancer patients: A systematic review and Meta-Analysis. Front. Immunol. 13, 908173 (2022).
Johnson, D. B., Nebhan, C. A., Moslehi, J. J. & Balko, J. M. Immune-checkpoint inhibitors: long-term implications of toxicity. Nat. Rev. Clin. Oncol. 19 (4), 254–267 (2022).
Chen, Y. et al. Prevotellaceae produces butyrate to alleviate PD-1/PD-L1 inhibitor-related cardiotoxicity via PPARα-CYP4X1 axis in colonic macrophages. J. Exp. Clin. Cancer Res. 41 (1), 1 (2022).
Lyon, A. R. et al. 2022 ESC guidelines on cardio-oncology developed in collaboration with the European hematology association (EHA), the European society for therapeutic radiology and oncology (ESTRO) and the international Cardio-Oncology society (IC-OS). Eur. Heart J. 43 (41), 4229–4361 (2022).
Poels, K. et al. Immune checkpoint inhibitor therapy aggravates T Cell-Driven plaque inflammation in atherosclerosis. JACC CardioOncol. 2 (4), 599–610 (2020).
Stein-Merlob, A. F., Rothberg, M. V., Ribas, A. & Yang, E. H. Cardiotoxicities of novel cancer immunotherapies. Heart 107 (21), 1694–1703 (2021).
Drobni, Z. D. et al. Association between immune checkpoint inhibitors with cardiovascular events and atherosclerotic plaque. Circulation 142 (24), 2299–2311 (2020).
Hu, Y. B. et al. Evaluation of rare but severe immune related adverse effects in PD-1 and PD-L1 inhibitors in non-small cell lung cancer: a meta-analysis. Transl Lung Cancer Res. 6 (Suppl 1), S8–s20 (2017).
Oren, O. et al. Cardiovascular health and outcomes in cancer patients receiving immune checkpoint inhibitors. Am. J. Cardiol. 125 (12), 1920–1926 (2020).
Filipek, A., Mikołajczyk, T. P., Guzik, T. J. & Naruszewicz, M. Oleacein and foam cell formation in human Monocyte-Derived macrophages: A potential strategy against early and advanced atherosclerotic lesions. Pharmaceuticals (Basel) 13(4), 64 (2020).
Wolf, D. & Ley, K. Immunity and inflammation in atherosclerosis. Circ. Res. 124 (2), 315–327 (2019).
Song, P. et al. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: a systematic review, meta-analysis, and modelling study. Lancet Glob Health. 8 (5), e721–e9 (2020).
Zhou, M. et al. Mortality, morbidity, and risk factors in China and its provinces, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet (London, England). 394(10204), 1145–1158 (2019).
Xiao, M., Zhang, J., Chen, W. & Chen, W. M1-like tumor-associated macrophages activated by exosome-transferred THBS1 promote malignant migration in oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 37 (1), 143 (2018).
Porcheray, F. et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin. Exp. Immunol. 142 (3), 481–489 (2005).
Colin, S., Chinetti-Gbaguidi, G. & Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 262 (1), 153–166 (2014).
Schmid, P. et al. Atezolizumab and Nab-Paclitaxel in advanced Triple-Negative breast cancer. N. Engl. J. Med. 379 (22), 2108–2121 (2018).
Gribov, A. et al. SEURAT: visual analytics for the integrated analysis of microarray data. BMC Med. Genomics. 3, 21 (2010).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol.. https://doi.org/10.1038/nbt.4314 (2018).
Jin, S. et al. Inference and analysis of cell-cell communication using cellchat. Nat. Commun. 12 (1), 1088 (2021).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45 (D1), D353–d61 (2017).
Gene Ontology Consortium. Going forward. Nucleic Acids Res. 43 (Database issue), D1049–D1056 (2015).
Wu, T. et al. ClusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (Camb). 2 (3), 100141 (2021).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44 (D1), D457–D462 (2016).
Garcia-Alonso, L., Holland, C. H., Ibrahim, M. M., Turei, D. & Saez-Rodriguez, J. Benchmark and integration of resources for the Estimation of human transcription factor activities. Genome Res. 29 (8), 1363–1375 (2019).
Baxter, E. W. et al. Standardized protocols for differentiation of THP-1 cells to macrophages with distinct M(IFNγ + LPS), M(IL-4) and M(IL-10) phenotypes. J. Immunol. Methods. 478, 112721 (2020).
Shou, X., Wang, Y., Jiang, Q., Chen, J. & Liu, Q. miR-126 promotes M1 to M2 macrophage phenotype switching via VEGFA and KLF4. PeerJ 11, e15180 (2023).
Yu, J. et al. Nuclear PD-L1 promotes EGR1-mediated angiogenesis and accelerates tumorigenesis. Cell. Discov. 9 (1), 33 (2023).
Yan, C. et al. PD-L1 Expression Is Increased in LPS-Induced Acute Respiratory Distress Syndrome by PI3K-AKT-Egr-1/C/EBPδ Signaling Pathway. Inflammation. 47(4), 1459–1478. https://doi.org/10.1007/s10753-024-01988-6 (2024).
Li, J. et al. Caspase-9 Inhibition triggers Hsp90-based chemotherapy-mediated tumor intrinsic innate sensing and enhances antitumor immunity. J. Immunother Cancer 11(12), e007625 (2023).
Wickenberg, M. et al. Hsp90 Inhibition leads to an increase in surface expression of multiple immunological receptors in cancer cells. Front. Mol. Biosci. 11, 1334876 (2024).
Zhi, Y. K. et al. Sinomenine inhibits macrophage M1 polarization by downregulating α7nAChR via a feedback pathway of α7nAChR/ERK/Egr-1. Phytomedicine 100, 154050 (2022).
Ma, S. et al. 1,8-cineole ameliorates colon injury by downregulating macrophage M1 polarization via inhibiting the HSP90-NLRP3-SGT1 complex. J. Pharm. Anal. 13 (9), 984–998 (2023).
Karar, J. & Maity, A. PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 4, 51 (2011).
Ball, R. Y. et al. Evidence that the death of macrophage foam cells contributes to the lipid core of atheroma. Atherosclerosis 114 (1), 45–54 (1995).
Moore, K. J., Sheedy, F. J. & Fisher, E. A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 13 (10), 709–721 (2013).
Agrafioti, P. et al. Decoding the role of macrophages in periodontitis and type 2 diabetes using single-cell RNA-sequencing. Faseb j. 36 (2), e22136 (2022).
Luo, L. et al. Precisely regulating M2 subtype macrophages for renal fibrosis resolution. ACS Nano. 17 (22), 22508–22526 (2023).
Tong, H. et al. Tumor-associated macrophage-derived CXCL8 could induce ERα suppression via HOXB13 in endometrial cancer. Cancer Lett. 376 (1), 127–136 (2016).
Cochain, C. et al. Single-Cell RNA-Seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 122 (12), 1661–1674 (2018).
Wu, L. et al. Angiotensin II induces RAW264.7 macrophage polarization to the M1–type through the connexin 43/NF–κB pathway. Mol. Med. Rep. 21 (5), 2103–2112 (2020).
Nogieć, A., Bzowska, M., Demczuk, A., Varol, C. & Guzik, K. Phenotype and response to pamps of human Monocyte-Derived foam cells obtained by Long-Term culture in the presence of OxLDLs. Front. Immunol. 11, 1592 (2020).
Medbury, H. J., Williams, H. & Fletcher, J. P. Clinical significance of macrophage phenotypes in cardiovascular disease. Clin. Transl Med. 3 (1), 63 (2014).
Hou, P. et al. Macrophage polarization and metabolism in atherosclerosis. Cell. Death Dis. 14 (10), 691 (2023).
Liao, Y., Zhu, E. & Zhou, W. Ox-LDL aggravates the oxidative stress and inflammatory responses of THP-1 macrophages by reducing the Inhibition effect of miR-491-5p on MMP-9. Front. Cardiovasc. Med. 8, 697236 (2021).
Akuzawa, N., Kurabayashi, M., Ohyama, Y., Arai, M. & Nagai, R. Zinc finger transcription factor Egr-1 activates Flt-1 gene expression in THP-1 cells on induction for macrophage differentiation. Arterioscler. Thromb. Vasc Biol. 20 (2), 377–384 (2000).
Manta, C. P. et al. Targeting of scavenger receptors Stabilin-1 and Stabilin-2 ameliorates atherosclerosis by a plasma proteome switch mediating Monocyte/Macrophage suppression. Circulation 146 (23), 1783–1799 (2022).
Zhao, S. et al. Hsp90 S-nitrosylation at Cys521, as a conformational switch, modulates cycling of Hsp90-AHA1-CDC37 chaperone machine to aggravate atherosclerosis. Redox Biol. 52, 102290 (2022).
Golden, D. et al. Lymphocyte activation gene 3 and coronary artery disease. JCI Insight. 1 (17), e88628 (2016).
Jimenez, J. & Prabhu, S. D. LAG3 Blockade expands T cells within atherosclerotic plaque: an ongoing need for cardiovascular disease risk assessment. JACC CardioOncol. 4 (5), 646–648 (2022).
Mulholland, M. et al. LAG3 regulates T cell activation and plaque infiltration in atherosclerotic mice. JACC CardioOncol. 4 (5), 635–645 (2022).
Acknowledgements
We thank LetPub (www.letpub.com.cn) for linguistic assistance.
Funding
The study was funded by Medical Science and Technology Project of Zhejiang Province (Grant Number: 2024KY1233) and the Zhejiang Provincial Department of Education’s Special Project for Reforming the Training Model of Professional Graduate Programs in Universities (Grant Number: Y202351281).
Author information
Authors and Affiliations
Contributions
X.S., Y.W., and J.L. contributed to conceptualization and data curation. X.S., Y.W., and Z.Z. performed formal analysis and methodology. Q.L. acquired funding and supervised the project. J.L. led the investigation. Resources were provided by Z.Y. and Q.L. Software development was done by X.S., Y.W., and Z.Z. Validation was conducted by X.S. and Z.Y., and Y.W. was responsible for visualization. X.S., Y.W., and D.Z. wrote the original draft, and Z.Y. and Q.L. reviewed and edited the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors read and approved the final manuscript before submission.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shou, X., Wang, Y., Zhou, Z. et al. Anti PD-L1 immunotherapy alters macrophage phenotypes via EGR1 and HSP90AB1 supported by integrated methodologies. Sci Rep 15, 36873 (2025). https://doi.org/10.1038/s41598-025-20826-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-20826-9
