
Abstract
Glioblastoma (GBM) is an aggressive brain tumour with limited treatment options and poor patient survival, largely due to the blood-brain barrier (BBB) restricting effective drug delivery. In this study, we focused on two small molecular weight polyfluoroalkyl phosphonates, ZOT5-1-Me and ZOT5-1-Et, designed to permeate the BBB. Comprehensive in vitro analyses using U-87 MG cells and a panel of glioma cell lines revealed that both compounds exhibit potent cytostatic and cytotoxic activities. Mechanistically, they induce reactive oxygen species (ROS) production, triggering both intrinsic and extrinsic apoptotic pathways via caspase-dependent and caspase-independent mechanisms. Additionally, ZOT5-1-Me and ZOT5-1-Et induced DNA damage, including single-strand breaks and alkali-labile sites. Notably, ZOT5-1-Me also caused significant DNA double-strand breaks and impaired DNA repair. Furthermore, both compounds exhibited antiproliferative effects by inducing cell cycle arrest in the S phase and activating p53-p21 signalling pathway. Pre-treatment with the ROS scavenger N-acetyl-L-cysteine (NAC) effectively abrogated these cytotoxic effects, underscoring the central role of oxidative stress in mediating the compounds’ antitumour activity. Collectively, our findings suggest that these polyfluoroalkyl phosphonates represent promising ROS-modulating chemotherapeutic candidates with unique mechanisms of action that may complement existing GBM treatment strategies.
Similar content being viewed by others

Introduction
Glioblastoma (GBM, IDH-wildtype diffuse astrocytoma, WHO grade IV) is the most prevalent primary tumour of the central nervous system (CNS), comprising 14.5% of all and 48.6% of malignant CNS tumours1. GBM remains the most aggressive primary malignant brain tumour in adults with a median survival time in registry databases of 6–10 months and 14.6–21.1 months when anticancer treatment is included2. Only 3% to 5% of patients survive more than three years, and reports of survival exceeding five years are sporadic3. Despite the efforts made by clinicians and researchers, no major clinical advances have been achieved in increasing overall patient survival since establishing Stupp protocol 20 years ago (12.1 vs. 14.6 months)4,5. This standard of care includes maximal safe resection and radiotherapy with concurrent and adjuvant chemotherapy with temozolomide (TMZ). Despite multimodal treatments, recurrence is almost inevitable, partially due to the difficulty of complete surgical removal, with a median interval of less than 10 months6,7,8. The treatment options are limited at recurrence, with the unavailability of a universal standard of care for recurrent GBM. Therefore, re-resection, re-irradiation and systemic chemotherapy with TMZ rechallenge, nitrosoureas, bevacizumab, and tumour treating fields (TTF) or clinical trial enrolment to test experimental drugs are considered for all recurrent patients.
TMZ remains the only available first-line chemotherapeutic agent for GBM. As one of the few chemotherapeutic drugs capable of crossing the blood–brain barrier (BBB), with approximately 20% of the injected dose reaching the brain, TMZ offers a modest survival benefit by increasing patient lifespan by only about 2.5 months4. TMZ chemotherapy and radiotherapy share common pathways to GBM cell death, inducing DNA damage either directly or indirectly by generating reactive oxygen species (ROS). Although the cytotoxic effects of TMZ is mainly attributed to the induction of DNA damage, O6-methylguanine, oxidative stress induced by TMZ was demonstrated to play a decisive role in its cytotoxic effects9,10,11. Inhibition of reactive oxygen species (ROS) prevented cytotoxic effects of TMZ9,11. Moreover, TMZ-sensitive and resistant GBM cells display differences in antioxidant system with the resistant cells displaying upregulation of the antioxidant system indicating key role of ROS detoxification in the TMZ resistance mechanism9,10,11.
ROS play a dual role in GBM, exerting both tumour-promoting and tumour-suppressing effects. This paradox has spurred the development of ROS-inducing agents designed to elevate ROS levels beyond the threshold of cellular tolerance, thereby selectively targeting GBM cells10. Small molecule compounds that modulate redox homeostasis have been investigated in both preclinical and early-phase clinical settings10,12,13,14. In preclinical models these compounds have individually induced GBM cell death or enhanced GBM cell sensitivity to the conventional therapies10. Early-phase clinical trials have begun to assess their efficacy, though no ROS-modulating small molecule has yet significantly improved the standard of care15. Nonetheless, small molecules remain the predominant drug class under clinical evaluation, and the rational design of novel compounds, particularly in combination therapies, holds promise for improving GBM treatment outcomes.
The focus on small molecule compounds in the design of GBM chemotherapeutic candidates are highly desired due to the presence of the BBB, which allows only certain drugs (i.a. with molecular mass below 400–500 Da) to enter the brain16. This restricts the clinical application of most anticancer drugs for treating brain tumours. Therefore, the development of new small molecule compounds that can modulate the redox status of GBM cells and induce cell death through oxidative stress is urgently needed.
Simple dialkyl 2,2,3,3,3-pentafluoropropylphosphonates (referred to as ZOTs, Fig. 1) were designed in our laboratory as small molecular weight anticancer drug candidates with the potential to permeate the BBB. We hypothesized that an optimal scaffold for GBM therapy should combine: (a) low molecular weight; (b) lipophilic fragments to facilitate BBB permeation; (c) a brain-targeting element; and (d) an innovative lead structure distinct from existing drugs. Polyfluoroalkylated motifs, known to increase lipophilicity and BBB passage, were therefore incorporated17. Because poly/perfluoroalkyl substances are broadly membrane-permeant rather than BBB-specific, these fragments were coupled to a phosphonate group to exploit the anionic chemical delivery system for brain targeting and retention18. Initial cytotoxicity screening revealed that two ZOTs, specifically ZOT5-1-Me and ZOT5-1-Et, exhibit high potency against GBM cells19. ZOTs represent a new class of small molecular weight compounds that combine polyfluoroalkyl and phosphonic moieties. Due to their unique chemical structure and simplicity, predicting the molecular mechanism behind their cytotoxic properties is challenging, as no studies have been conducted on similarly structured compounds. The promising cytotoxicity results of ZOT5-1-Me and ZOT5-1-Et against GBM cells, along with computational study, encouraged us to further explore the molecular mechanism underlying their cytotoxic effects in GBM19.

Chemical structure of ZOT5-1-Me and ZOT5-1-Et.
Materials and methods
Cell line
Human GBM cell line, U-87 MG was purchased from ECACC (Sigma-Aldrich Chemie GmbH, Munich, Germany) and was maintained in Eagle’s Minimum Essential Medium (EMEM) medium supplemented with 10% inactivated fetal bovine serum (FBS) (Biowest, Nuaillé, France), 2 mM L-glutamine (Merck Life Science, Darmstadt, Germany), 1% non-essential amino acids (NEAA, Merck Life Science), 1 mM sodium pyruvate (NaP, Merck Life Science), and 100 units/mL penicillin (Merck Life Science) and 100 μg/mL streptomycin (Merck Life Science). Cells were incubated in a humidified atmosphere of 5% CO2 at 37 °C and sub-cultured once to twice per week to maintain exponential growth. The cultured U-87 MG cells from passages 3–20 were used for experiments.
Compounds
ZOT5-1-Me and ZOT5-1-Et were synthesized according to the general procedures published elsewhere19 with minor modifications. The intermediate hydroxyphosphonates were additionally purified by distillation. The final distilled ZOT5-1-Me and ZOT5-1-Et were purified by column chromatography using sequential elution with n-heptane, dichloromethane and acetonitrile. The NMR spectra were consistent with the literature. For biological assays ZOT5-1-Me and ZOT5-1-Et were freshly dissolved in DMSO to a stock concentration of 100 mM. Subsequent dilutions were conducted in cell culture medium.
Compound screening
Synthesized compounds ZOT5-1-Me and ZOT5-1-Et were submitted to National Cancer Institute (NCI, Bethesda, Maryland, U.S.A.) under the Developmental Therapeutic Program (DTP). Compounds were screened in a panel of 59 cancer cell lines, including glioma cell lines (SF-268, SF-295, SF-539, SNB-19, SNB-75, and U251) at a concentration of 10 µM in the one-dose screen and concentrations 0.01, 0.1, 1, 10 and 100 µM in the five-dose screen according to the procedure (https://dtp.cancer.gov/discovery_development/nci-60/methodology_HTS384.htm). The sulforhodamine B assay was used for the determination of cell density relative to the no-drug control and relative to the number of cells at time zero. The results were utilized to generate dose–response curves, plotting the logarithm (base 10) of the compound concentration against the percentage of growth inhibition. For each cell line, three parameters were determined: GI50, TGI, and LC50. The GI50 (growth inhibitory activity) indicates the concentration at which cell growth is reduced by 50%. The TGI (total growth inhibition) represents the concentration required to completely halt cell proliferation. The LC50 (cytotoxic activity) reflects the compound concentration that leads to a 50% reduction in the initial cell population after 48 h of exposure.
Cell treatment
U-87 MG cells were seeded onto tissue culture plates or dishes at a density of 5 × 104/mL in EMEM medium and allowed to attach for 24 h. After overnight growth, cells were treated with 0.03125% DMSO (as a vehicle) and ZOTs in a concentration range 0.98–250 µM for 4–48 h as indicated beneath. Negative control cells were incubated in medium only.
Cell viability assay
Logarithmically growing cells (5 × 103) were seeded into 96-well plates and treated with ZOTs in the concentration range 0.98–250 µM for 4–48 h. After the desired time elapsed, 10 μL of Cell Counting Kit-8 (CCK-8, Merck Life Science) was added to each well to obtain a final concentration of 0.5 mg/mL CCK-8 and plates again were incubated in 5% CO2 at 37 °C for 2 h. Next, absorbance was measured at 450 nm with a microplate reader Synergy HT (Bio-Tek Instruments, Winooski, VT, USA).
Annexin V and propidium iodide apoptosis assay
Double staining of cells with Annexin V-FITC and Propidium Iodide (PI) was used to assay apoptosis. This method is a useful tool for distinguishing viable cells (Annexin V-/PI-), early apoptotic cells (Annexin V+/PI-), late apoptotic cells (Annexin V+/PI+), and necrotic cells (Annexin V-/PI+). Visualization of cells stained with Annexin V-FITC and PI was applied according to the manufacturer’s protocol (FITC Annexin V Apoptosis Detection Kit I, Becton Dickinson, Franklin Lakes, NJ, USA). Briefly, 1.5 × 105 cells were seeded onto 6-well plates and allowed to attach. Next, the cells were treated with the indicated concentrations of ZOTs for 4, 24 and 48 h. After treatment, the cells were washed twice with cold PBS and resuspended in 300 μL binding buffer containing 3 μL of Annexin V-Alexa Fluor™ 488 at 27 µg/mL and 3 μL of PI at 50 μg/mL and stained for 15 min at room temperature in the dark. Finally, at least 104 cells were analysed using a BD FACSymphony™ A1 Cell Analyzer (Becton Dickinson, Franklin Lakes, NJ, USA) and BD FACSDiva™ Software v9.0.2. FITC conjugated antibody was excited with the 488 nm laser and emission detected with a 530/30 bandpass filter. PI was excited with the 561 nm laser and its emission detected with a 610/20 bandpass filter. A dual parameter dot plot of forward (FSC-A) and side scatter peak areas (SSC-A) were used to identify intact cells from debris. Forward scatter peak area and height (FSC-A and FSC-H) and side scatter peak area and height (SSC-A and SSC-H) were used to identify single cells and exclude doublets. Data were analysed in FlowJo v7.6.4 software (FlowJo LLC, Ashland, OR, USA). The positive control cells were incubated with 20 μM camptothecin (CPT) for 48 h at 37 °C.
Caspase activity assay
U-87 MG cells were seeded on a 96-well black plate in a count of 3.750 per well in 75 µL medium. Then the cells were incubated with ZOTs for 4, 24 and 48 h at 37 °C in 5% CO2. Following the treatments the cells were subjected to caspase 3/7, 8, and 9 activities measurement with Caspase-Glo assay kits (Promega, Madison, WI, USA) according to the manufacturer’s protocol. Briefly, the plates containing cells were removed from the incubator and allowed to equilibrate to room temperature for 30 min. One volume of Caspase-Glo reagent was added to each well, the content of well was gently mixed with a plate shaker at 400 rpm for 30 s. The plate was then incubated at room temperature for 1 h. The luminescence of each sample was measured with a microplate reader Synergy HT (Bio-Tek Instruments, USA). The positive control cells were incubated with 20 μM camptothecin (CPT) at 37 °C. Caspase activities were expressed as a percentage compared to that of control cells (100%).
Caspase inhibition
The inhibition of caspases was conducted by a cell-permeant pan-caspase inhibitor Z-VAD-FMK (Promega, Madison, WI, USA) that irreversibly binds to the catalytic site of caspase proteases. Z-VAD-FMK was added to the culture medium at a concentration of 10 μM, according to the manufacturer’s recommendations for 48 h. Controls were generated using DMSO as an inhibitor vehicle at the same concentration. The effectiveness of the caspase inhibitor was verified with Caspase-Glo assays.
Intracellular ROS assay
The analysis of total intracellular ROS was conducted using 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Invitrogen, Waltham, MA, USA) as described previously20. Briefly, the U-87 MG cells were detached with trypsin on the day of the analysis. Cells in suspension (5 × 105 cells per 1 mL medium) were washed twice with HBSS (Biowest, Nuaillé, France) and stained with 9 µM CM-H2DCFDA in HBSS for 30 min at 37 °C in the dark. Then, the cells were washed twice with HBSS and treated with an increasing concentration of ZOTs for 2 h at 37 °C in the dark. Hydrogen peroxide (H2O2) at a concentration of 500 µM was used as positive control. Pre-incubation with 1 mM N-acetyl-L-cysteine (NAC) for 1 h was used to scavenge ROS. Then, the fluorescence intensity of 2’,7'-dichlorofluorescein (DCF) was measured with the excitation and emission set at 485/20 nm and 528/20 nm, respectively, using a Synergy HT spectrophotometer (BioTek Instruments, Winooski, VT, USA).
Mitochondrial membrane potential (ΔΨm)
The change in the ΔΨm was detected by staining with 5,5ʹ,6,6ʹ-tetrachloro-1,1ʹ,3,3ʹ-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) (Sigma-Aldrich Chemie GmbH, Munich, Germany) as described elsewhere21. Briefly, 0.86 × 106 cells were seeded onto 100 mm cell culture dishes and allowed to attach. Next, the cells were treated with the indicated concentrations of ZOTs for 48 h at 37 °C. The positive control cells were incubated with 25 μM carbonyl cyanide 3-chlorophenylhydrazone (CCCP), a mitochondrial uncoupling agent, for 48 h at 37 °C. After the treatment, the cells were washed with PBS and detached with trypsin on the day of the analysis. Cells in suspension (1 × 106 cells per 1 mL) were washed twice with HBSS and stained with 5 µM JC-1 in HBSS (Biowest, Nuaillé, France) for 30 min at 37 °C in the dark. After this incubation period, cells were washed twice with HBSS and 105 cells were transferred to a 96-well black plate for fluorescence measurement. The fluorescence intensity was measured with Synergy HT spectrophotometer (BioTek Instruments, Winooski, VT, USA) with the following settings: excitation at 530/25 nm and emission at 590/35 nm for JC-1 dimers and excitation at 485/20 nm and emission at 528/20 nm for JC-1 monomers. The value of ΔΨm was expressed as the ratio of JC-1 dimer fluorescence to monomer fluorescence (measurements at 590 nm/528 nm). The JC-1 stained cells were imaged immediately after the dye was washed with HBSS with an inverted Olympus IX70 fluorescence microscope (Olympus, Tokyo, Japan).
Comet assay
On the day of the experiment, U-87 MG cells were harvested using trypsin and subsequently resuspended at a concentration of 5 × 104 cells per mL of culture medium. The cells were then exposed to a gradient of ZOT compound concentrations for 2 h. DNA damage was assessed using the comet assay under three different pH conditions: neutral (pH 9), pH 12.1, and alkaline (pH > 13), following previously published protocols20. The neutral version of the assay is selective for detecting DNA double-strand breaks (DSBs), while the pH 12.1 condition allows visualization of both single- and double-strand breaks. In contrast, the alkaline version identifies a broader spectrum of lesions, including alkali-labile sites and DNA strand breaks.
Following treatment, cells were mixed with 0.75% low melting point (LMP) agarose at 37 °C and rapidly layered onto microscope slides precoated with 0.5% normal melting point (NMP) agarose. A cover slip was applied to evenly distribute the cell-agarose suspension. The slides were then placed on ice to allow the agarose to solidify. Once solidified, the coverslips were carefully removed, and the slides were immersed for 1 h in lysis buffer containing 2.5 M NaCl, 100 mM EDTA, 1% Triton X-100, and 10 mM Tris at pH 10 to remove membranes and proteins.
For the alkaline version of the assay, slides were transferred into ice-cold buffer consisting of 300 mM NaOH and 1 mM EDTA (pH > 13) for 20 min to unwind the DNA. Electrophoresis was then conducted in the ice-cold buffer of 30 mM NaOH, 1 mM EDTA for 20 min under an electric field of 0.73 V/cm (32 mA).
In the pH 12.1 variant, the denaturation step was similarly performed for 20 min in a buffer made of 300 mM NaOH and 1 mM EDTA, with the pH adjusted to 12.1 using glacial acetic acid. Electrophoresis followed under the same conditions: 20 min at 4 °C and 0.73 V/cm.
For the neutral conditions, the slides were equilibrated for 20 min in an ice-cold buffer containing 100 mM Tris and 300 mM sodium acetate, with pH adjusted to 9.0 using glacial acetic acid. This was followed by electrophoresis for 60 min at 4 °C, using a field of 0.41 V/cm (100 mA).
Following electrophoresis, the slides were rinsed with distilled water, air-dried, stained with DAPI at a concentration of 4 µg/mL, and covered with cover slips. Fluorescent imaging was conducted at 200 × magnification using an Eclipse fluorescence microscope (Nikon, Tokyo, Japan) equipped with a ProgRes MF cool monochrome camera (JENOPTIK, Jena, Germany) and connected to a Lucia Comet Assay 7.30 image analysis system (Laboratory Imaging, Prague, Czech Republic). For each sample, 50 comets were randomly selected for analysis, and the percentage of DNA in the tail region was quantified as an indicator of DNA damage, including strand breaks and alkali-labile sites22.
DNA repair
To examine DNA repair, the cells were prepared as for comet assay and after the treatment with ZOTs, cells were washed and resuspended in fresh medium containing 10% FBS. Cells were allowed to repair for 5, 10, 15, 30, 60, 90, and 120 min and the alkaline version of comet assay was performed. The kinetics of DNA repair was quantified by determination of the extent of residual DNA damage at each time-point.
Plasmid relaxation assay
The plasmid relaxation assay was performed according to established methodology23. Plasmid pUC19 was extracted from E.coli DH5α using the Isolate II Plasmid Mini Kit (Meridian Bioscience, Cincinnati, OH, USA) in accordance with the manufacturer’s protocol. The concentration and purity of the obtained plasmid DNA were assessed using the spectrophotometer and verified by agarose gel electrophoresis. In its native state, pUC19 is predominantly in a supercoiled (covalently closed circular, CCC) conformation, which exhibits elevated electrophoretic mobility. To generate the linear (L) form, the plasmid was digested with the PstI restriction enzyme (New England Biolabs, Ipswich, MA, USA) according to the manufacturer’s protocol. Changes in topological structure between the CCC and L forms results in distinct migration patterns during electrophoresis. To evaluate the capacity of ZOT compounds to cause DNA strand breaks, 250 ng of plasmid DNA was incubated with increasing concentrations of the compounds at 37 °C for 24 h. Post-incubation, the samples were immediately subjected to electrophoresis on 0.8% agarose gels followed by ethidium bromide staining. DNA bands were visualized under UV illumination at 302 nm, captured using a CCD imaging system, and analysed using GeneTools software (Syngene, Cambridge, UK). A 1 kb DNA ladder (GeneRuler 1 kb DNA Ladder, Thermo Scientific, Waltham, MA, USA) served as a molecular weight marker.
γH2AX analysis using flow cytometry
The analysis of γH2AX with flow cytometry was conducted as described previously24. On the day of the treatment, U-87 MG cells were detached with trypsin. Cells in suspension (3 × 105 cells in 5 mL medium) were treated with increasing concentrations of ZOTs for 2 h. After treatment, cells were washed with PBS, fixed and permeabilized with freshly prepared 1× Cytofix/Cytoperm Buffer (BD Biosciences, San Diego, CA, USA) for 40 min at 4 °C. Then, cells were washed twice with 1× Perm/Wash Buffer (BD Biosciences) and incubated with rabbit monoclonal anti-phospho-H2AX (Ser139) antibody (#9718, Cell Signaling Technology, Danvers, MA, USA) diluted 1:250 in 1× Perm/Wash Buffer (BD Biosciences) for 30 min at 37 °C. Next, cells were washed twice with 1× Perm/Wash Buffer (BD Biosciences) and incubated with secondary goat anti-rabbit IgG (H + L) Alexa Fluor 488-conjugated antibody (#A-11070, Invitrogen, Waltham, MA, USA) diluted 1:400 in 1× Perm/Wash Buffer (BD Biosciences) for 30 min at 37 °C in the dark. Cells were washed again twice with 1× Perm/Wash Buffer (BD Biosciences), re-suspended in PBS and subjected to flow cytometric analysis at an excitation wavelength of (488 nm) employing BD FACSymphony™ A1 Cell Analyzer (Becton Dickinson, San Jose, CA, USA) equipped with BD FACSDiva™ Software v9.0.2. FITC conjugated antibody was excited with the 488 nm laser and emission was detected with a 530/30 bandpass filter. A dual parameter dot plot of forward (FSC-A) and side scatter peak areas (SSC-A) were used to identify intact cells from debris. Forward scatter peak area and height (FSC-A and FSC-H) followed by side scatter peak area and height (SSC-A and SSC-H) were used to identify single cells and exclude doublets. Data were analysed in FlowJo v7.6.4 software (FlowJo LLC, Ashland, OR, USA). The positive control cells were incubated with 50 μM etoposide (Etop) for 4 h at 37 °C.
Western blot
Western blot was conducted as described previously25. On the day of the treatment, U-87 MG cells were detached with trypsin. Cells in suspension (2 × 105 cells per 1 mL medium) were treated with increasing concentrations of ZOTs for 2 h and the Western blot for the assessment of γH2AX level was performed.
For the analysis of cell cycle proteins, 0.86 × 106 cells were seeded onto 100 mm cell culture dishes and allowed to attach. Next, the cells were treated with the 7.8 µM ZOT5-1-Me and 15.6 µM ZOT5-1-Et for 24 or 48 h at 37 °C. After the treatment, the cells were washed with PBS and detached with trypsin.
Following the treatments, cells were washed with ice cold PBS and lysed in ice cold RIPA buffer (50 mM Tris HCl pH 8, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS, 1 mM EDTA) containing 1 mM phenylmethanesulfonyl fluoride (PMSF) and 1× protease and phosphatase inhibitor cocktail (Sigma-Aldrich Chemie GmbH, Munich, Germany). The protein concentration in cell lysates was determined using the Bradford method26. The 80 µg (for γH2AX) or 40 µg (for p53 and p21) of proteins was separated on 10–12% SDS-PAGE and transferred to PVDF transfer membrane (ThermoFisher Scientific, Waltham, MA, USA). The membranes were blocked with 5% milk in 0.1% Tween-20/TBS (Tris-buffered saline) for 1 h at room temperature and they were incubated with rabbit monoclonal anti-phospho-H2AX (Ser139) antibody (#9718, Cell Signaling Technology, Danvers, MA, USA) diluted 1:1000 in 5% milk and 5% BSA in 0.1% TBST or anti-phospho-p53 (Ser15) (#9284, CST) diluted 1:1000 in 5% milk in 0.1% TBST, or anti-p21 (#2947, CST) or anti-p53 (#2527, CST) diluted 1:1000 in 5% BSA in 0.1% TBST overnight at 4 °C. Goat anti-rabbit antibody conjugated with horseradish peroxidase (#7074, CST) were used as secondary antibodies. For loading, the control detection of β-actin was performed with anti-β-actin antibody (sc-477778, SantaCruz Biotechnology, Santa Cruz, CA, USA) diluted 1:4000 in 5% milk in 0.1% TBST. The bands were visualized with Clarity Western ECL Substrate (Bio-Rad, Hercules, CA, USA) and Gel-Pro Analyzer (Microchem Laboratory, Round Rock, TX, USA). The band quantification was performed using ImageJ software v1.54 g (National Institutes of Health, Bethesda, MD, USA) and the band intensities were normalized against β-actin.
Cell proliferation assay
The analysis of proliferation rate was conducted as described elsewhere27. Briefly, 5 × 104 cells were seeded onto 6-well plates and allowed to attach. The number of cells on the day of seeding was considered as the number of cells at the starting time point (time 0). Next, cells were treated with the indicated concentrations of ZOTs for 24, 48, 72, and 96 h. Following the treatment, cells were detached with trypsin and the number of viable and non-viable cells was manually counted in a hemocytometer using trypan blue exclusion method. Proliferation curves were generated by plotting viable cell counts against the time of culture. Population doubling times (PDTs) were determined based on the cell proliferation assay results and calculated using the doubling time online calculator28.
Cell cycle analysis
The analysis of cell cycle was conducted as described previously20. Briefly, 1.5 × 105 cells were seeded onto 6-well plates and allowed to attach. Next, the cells were treated with the indicated concentrations of ZOTs for 24 and 48 h. After treatment, the cells were collected, washed twice with PBS, resuspended in PBS to a final concentration of 106 cells/mL and allowed to cool on ice for 15 min. One volume of − 20 °C absolute ethanol was added to each sample and the samples were stored at 4 °C until analysis. At that time, the cells were pelleted (400 × g, 20 min) and resuspended in a staining solution containing 50 µg/mL PI (81845, Sigma-Aldrich Chemie GmbH, Munich, Germany) and 50 U/mL RNase A (EURx, Gdansk, Poland) in PBS. Samples were incubated at 37 °C for at least 30 min in the dark prior to analysis by fluorescence-activated cell sorting (FACS) performed on the BD FACSymphony™ A1 Cell Analyzer (Becton Dickinson, San Jose, CA, USA) and BD FACSDiva™ Software v9.0.2. PI was excited with the 561 nm laser and its emission detected with a 610/20 bandpass filter. A dual parameter dot plot of forward (FSC-A) and side scatter peak areas (SSC-A) were used to identify intact cells from debris. Forward scatter peak area and height (FSC-A and FSC-H), side scatter peak area and height (SSC-A and SSC-H), and PI peak area and half wide (PE-A and PE-W) were used to identify single cells and exclude doublets. Data were analysed in FlowJo v7.6.4 software (FlowJo LLC, Ashland, OR, USA). The positive control cells were incubated with 200 ng/mL nocodazole (NOC) for 18 h at 37 °C.
Data analysis
Statistical analyses were conducted using GraphPad Prism 8.0.1 Software (GraphPad Software, San Diego, CA, USA). For datasets with sample sizes n < 30, comparisons were conducted using the Mann–Whitney U test. For sample sizes n > 30, an unpaired Student’s t-test was applied, provided that data distribution satisfied the Shapiro–Wilk normality criterion. Results are reported as mean ± standard error of the mean (SEM). Unless otherwise specified, tests were two-tailed, and a statistical significance was assigned with *p-value ≤ 0.05, **p-value ≤ 0.01, ***p-value ≤ 0.001, ns, not significant.
Data availability
The data generated in this study are available at Zenodo (https://doi.org/10.5281/zenodo.16272119). Drug response profiles obtained in this study are available at NCI/DTP repository (https://dtp.cancer.gov/dtpstandard/dwindex/index.jsp); at NSC 835966 (ZOT5-1-Me; https://dtp.cancer.gov/dtpstandard/servlet/dwindex?searchtype=NSC&searchlist=835966) and NSC 835967 (ZOT5-1-Et, https://dtp.cancer.gov/dtpstandard/servlet/dwindex?searchtype=NSC&searchlist=835967).
Results
ZOT5-1-Me and ZOT5-1-Et are cytostatic and cytotoxic for glioblastoma and astrocytoma cells
The tumour growth inhibition properties of ZOT5-1-Me and ZOT5-1-Et were evaluated on human cancer cell lines at the NCI as part of the Developmental Therapeutics Program (DTP). Both compounds met the NCI-60 one-dose (10 μM) screening criteria allowing them to advance to the five-dose testing stage. Table 1 and Fig. 2A present the five-dose assay results for the anticancer activity of these compounds against glioma cell lines. Results presented are expressed as the 50% growth inhibition concentration (GI50), 50% lethal concentration (LC50) and total growth inhibition (TGI), compared to the values obtained for untreated control cells (Table 1). The GI50 results indicated that both ZOT5-1-Me and ZOT5-1-Et exhibited antiproliferative effects against all tested GBM (SF-295, SF-539, and SNB-75) and astrocytoma (SF-268, SNB-19, and U251) cell lines. ZOT5-1-Me achieved total growth inhibition across all tested cell lines, while ZOT5-1-Et inhibited 100% of growth in all the cell lines except astrocytoma SNB-19 cell line. The LC50 data showed that ZOT5-1-Me was cytotoxic for the SF-295, SF-539, and U251 cell lines, and ZOT5-1-Et demonstrated a significant lethal effect on the GBM SF-295 cell line.

ZOT5-1-Me and ZOT5-1-Et induced growth inhibition and lethality in glioblastoma and astrocytoma cell lines and COMPARE analysis of their mechanisms of action relative to reference anticancer drugs. (A) Cells were treated with an increasing concentration (10–8 to 10–4 M) of ZOT5-1-Me and ZOT5-1-Et for 48 h, and the analysis was performed thanks to the NCI-60 Cell Screen. Five dose–response curves in 3 glioblastoma (SF-295, SF-539, and SNB-75) and 3 astrocytoma (SF-268, SNB-19, and U251) cell lines are shown. NCI-60 analysis allows for the detection of both growth inhibition (values between 0 and 100) and lethality (values less than 0). A value of 100% represents no growth inhibition. The value of 0% indicates no cell growth throughout the experiment, corresponding to the number of cells at the beginning. The value of −50% means 50% lethality, and the value of 100% means that all cells are dead by treatment. GI50, the concentration required for 50% cell growth inhibition; TGI, the concentration required for total inhibition (0%); LC50, the concentration required for 50% cell death. (B) Comparison of the mechanism of ZOT5-1-Me and ZOT5-1-Et to FDA-approved and non-FDA-approved GBM anticancer drugs by COMPARE plots. ZOT5-1-Me and ZOT5-1-Et have a unique mechanism compared to known chemotherapeutics. High PCC (Pearson correlation coefficient) (> 0.8) shown in red in the matrix COMPARE figure, indicates these two drugs have similar mechanisms of anticancer action.
To compare the efficacy of these compounds with existing therapies, GI50, TGI, and LC50 values for three FDA-approved GBM drugs (TMZ, lomustine [CCNU], and carmustine [BCNU]) were retrieved from the DTP database and compared to the values for ZOT5-1-Me and ZOT5-1-Et (Table 1). The TGI values revealed that both ZOT5-1-Me and ZOT5-1-Et showed higher efficacy than the FDA-approved GBM drugs across all cell lines, except for the ZOT5-1-Et in SNB-19 cell line (data not available). Overall, both ZOT5-1-Me and ZOT5-1-Et demonstrated greater efficacy than most FDA-approved drugs against GBM.
Additionally, the COMPARE algorithm was used to evaluate the similarity of the biological response pattern of ZOT5-1-Me and ZOT5-1-Et with both FDA-approved drugs and chemotherapeutics undergoing combination clinical trials for GBM (Fig. 2B). The algorithm was used to assess similarities in anticancer mechanisms, quantified as the Pearson correlation coefficient (PCC). For both compounds, nearly all PCC values were below or equal to 0.6 for GI50, TGI, and LC50 values, indicating minimal correlation with the mechanisms of known chemotherapeutics. These findings suggest that the mechanisms of action of ZOT5-1-Me and ZOT5-1-Et are unique and distinct from those of established chemotherapeutic agents. Consequently, further investigations were undertaken to explore the novel anticancer mechanisms of ZOT5-1-Me and ZOT5-1-Et.
ZOT5-1-Me and ZOT5-1-Et induced caspase-dependent and -independent cell death in U-87 MG cells
Based on our previous findings19, U-87 MG was identified as the most sensitive cell line to both ZOT5-1-Me and ZOT5-1-Et, prompting its selection for further investigation. To assess the cytotoxic effects of these compounds, U-87 MG cells were exposed to increasing concentrations of ZOT5-1-Me and ZOT5-1-Et (0–250 μM) for 4 h, 24 h, and 48 h, followed by cell viability assessment using CCK-8 assay. Significant cytotoxicity was detected as early as 4 h post-treatment with both compounds (Fig. 3A). After 24 h of treatment, dose-dependent cytotoxic effects were evident, with IC50 values of 9.57 μM for ZOT5-1-Me and 14.12 μM for ZOT5-1-Et, indicating higher potency of ZOT5-1-Me. Extending the treatment to 48 h led to IC50 values of 8.70 μM for ZOT5-1-Me and 12.00 μM for ZOT5-1-Et, reflecting a marginal increase in cytotoxicity compared to the 24-h exposure. These findings suggest that ZOTs effectively exert their cytotoxic action within relatively short incubation periods, and extending treatment beyond 48 h may not provide significant additional benefits.

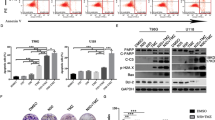
ZOT5-1-Me and ZOT5-1-Et induced cell death and apoptosis in U-87 MG cells. (A) The decreased viability of U-87 MG cells after treatment with an increasing concentration (0.95–250 µM) of ZOT5-1-Me and ZOT5-1-Et for 4–48 h was evaluated by CCK-8 assay. Cell viability curves and corresponding IC50 values are visible. The dashed line represents a 50% reduction in CCK-8 signal. The data were normalized to control cells (CTRL, 100%, n = 9). (B,C) ZOT5-1-Me and ZOT5-1-Et induced externalisation of phosphatidylserine in U-87 MG cells as evaluated by Annexin V/PI assay followed by flow cytometry (FACS) quantification (n = 6). Camptothecin (CPT, 20 µM) was used as positive control. (B) Representative FACS dot plots after 48 h treatment with ZOTs are presented with the indicated percentages of necrotic (Q1), late-apoptotic (Q2), early-apoptotic (Q3), and viable cells (Q4). Apoptosis was presented as a percentage of Annexin V-positive cells (Q2 + Q3). FACS dot plots after 4 h and 24 h treatments are presented in Supplementary material (D) ZOT5-1-Me and ZOT5-1-Et activated caspase 3/7 in U-87 MG cells as estimated by Caspase-Glo 3/7 assay (n = 6). The data were normalized to control cells (CTRL, 100%, n = 6). CPT (20 µM) was used as positive control. (E–F) Pre-incubation with pan-caspase inhibitor Z-VAD-FMK (10 µM, 48 h) decreased ZOT5-1-Me and ZOT5-1-Et-evoked apoptosis in U-87 MG cells as determined by Caspase-Glo 3/7 assay (E) and Annexin V/PI assay (F) after 48 h treatment with ZOTs (n = 6). (G) Representative FACS dot plots are presented. DMSO was used as a solvent control. Results presented as bar plots with mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001, ns – not statistically significant.
To investigate whether the observed cytotoxicity was associated with apoptosis induction, an Annexin V-FITC/PI assay was performed following the exposure of U-87 MG cells to ZOT5-1-Me and ZOT5-1-Et for 4 h, 24 h, and 48 h. Flow cytometry analysis revealed a significant increase in the proportion of cells undergoing early- and late-phase apoptosis in a dose- and time-dependent manner after treatment with both compounds (Fig. 3B-C). Consistent with the cytotoxic assay, a significant increase in the apoptosis rate was detectable after 4 h of treatment and became more pronounced after 24 h, with a slight increase at 48 h. The lowest concentrations inducing apoptosis were 7.8 μM for ZOT5-1-Me and 15.6 μM for ZOT5-1-Et.
To further explore the molecular mechanism underlying ZOT-induced apoptosis, caspase-3 and -7 activation was analysed following 4 h, 24 h, and 48 h of treatment. Caspase3/7 activity was undetectable after 4 h treatment but became evident after 24 h and 48 h in a dose- and time-dependent manner for both compounds (Fig. 3D). The minimum concentrations required to activate caspase-3/7 were 7.8 μM for ZOT5-1-Me and 15.6 μM for ZOT5-1-Et.
To confirm that ZOTs induce apoptosis in U-87 MG cells through caspase activation, a pan-caspase inhibitor, Z-VAD-FMK, was employed. As expected, pre-treatment with Z-VAD-FMK (10 μM) completely blocked caspase 3/7 activation (Fig. 3E). Moreover, Z-VAD-FMK reduced the apoptotic rate induced by ZOT5-1-Me from 32.80 ± 1.15 to 13.19 ± 0.50% and by ZOT5-1-Et from 65.08 ± 0.25 to 42.35 ± 0.25%, as evaluated by the Annexin V-FITC/PI assay (Fig. 3F-G). The partial attenuation of cytotoxic effects in the presence of Z-VAD-FMK suggests that ZOTs can trigger both caspase-dependent and caspase-independent cell death pathways.
Our findings demonstrate that ZOT5-1-Me and ZOT5-1-Et exhibit cytotoxic effects against U-87 MG cells, with ZOT5-1-Me showing a more pronounced impact. The induction of cell death by these compounds involves both caspase-dependent and caspase-independent mechanisms.
ZOT5-1-Me and ZOT5-1-Et induced intrinsic and extrinsic apoptotic pathways in U-87 MG cells
Activation of caspases, which drive apoptotic changes, can occur through two distinct pathways: the intrinsic pathway, involving the mitochondria, and the extrinsic pathway, mediated by death receptors. To identify which apoptotic pathways are induced by ZOTs, we analysed the activation of caspase-8 and caspase-9, which are key markers of the extrinsic and intrinsic pathways, respectively. Our results showed that both ZOT5-1-Me and ZOT5-1-Et activated caspase-8 and caspase-9, suggesting that these compounds trigger apoptosis through a dual mechanism involving both the intrinsic and extrinsic pathways (Fig. 4A-B). To confirm the involvement of the intrinsic pathway, we assessed mitochondrial function by measuring changes in mitochondrial membrane potential (ΔΨm) using the JC-1 assay. The observed loss of ΔΨm further supported the involvement of the intrinsic pathway, as dissipation of ΔΨm is a hallmark of mitochondrial-mediated apoptosis (Fig. 4D-E). Given that mitochondrial dysfunction is often associated with oxidative stress, we also measured ROS levels using the CM-H2DCFDA assay. A brief 2-h treatment with ZOT5-1-Me and ZOT5-1-Et significantly increased ROS levels in a dose-dependent manner, with a more pronounced effect observed for ZOT5-1-Me (Fig. 4C). These findings support the notion that both ZOT5-1-Me and ZOT5-1-Et exert cytotoxic effects via induction of extrinsic and intrinsic apoptotic pathways. Additionally, these findings suggest that oxidative stress may contribute to the induction of apoptosis through mitochondrial dysfunction. The increased ROS could further promote the intrinsic pathway by exacerbating mitochondrial damage and facilitating the release of apoptogenic factors.

ZOT5-1-Me and ZOT5-1-Et activated both intrinsic and extrinsic apoptotic pathways in U-87 MG cells. (A, B) ZOT5-1-Me and ZOT5-1-Et activated caspase-8 and caspase-9 in U-87 MG after 48 h treatment as evaluated by Caspase-Glo 8 and Caspase-Glo 9 assays, respectively. The data were normalized to control cells (CTRL, 100%, n = 6). (C) ZOT5-1-Me and ZOT5-1-Et increased intracellular reactive oxygen species (ROS) in U-87 MG after 2 h treatment as estimated by CM-H2DCFDA assay. Hydrogen peroxide (H2O2) was used as a positive control. (D) ZOT5-1-Me and ZOT5-1-Et reduced mitochondrial membrane potential in U-87 MG after 48 h treatment as assayed by JC-1 staining (n = 6). Carbonyl cyanide 3-chlorophenylhydrazone (CCCP, 25 µM, 48 h) was used as a positive control. The value of ΔΨm was expressed as the ratio of JC-1 dimer fluorescence to monomer fluorescence. The data were normalized to control cells (CTRL, 100%, n = 6). (E) Representative images of JC-1 assay are presented. DMSO was used as a solvent control. Results presented as bar plots with mean ± SEM; **p < 0.01; ns – not statistically significant.
ZOT5-1-Me and ZOT5-1-Et induced DNA damage, including DNA double-strand breaks in U-87 MG cells
Given that apoptosis can often result from DNA damage, particularly double-strand breaks (DSBs), we further assessed the DNA-damaging potential of these compounds using the comet assay. A brief 2-h treatment with ZOTs was conducted to evaluate whether DNA damage could serve as a primary mechanism underlying their cytotoxicity. The neutral comet assay revealed that ZOT5-1-Me induced DSBs, whereas ZOT5-1-Et did not exhibit this effect (Fig. 5A-B). Additionally, the pH 12.1 version of the assay demonstrated the presence of single-strand breaks (SSBs), and the alkaline version identified alkali-labile sites (ALS) as the predominant type of DNA damage induced by both compounds. The induction of DSBs, recognized as the most lethal form of DNA lesion, was further validated by γH2AX analysis. Western blotting (Fig. 5C-D) and flow cytometry (Fig. 5E-F) findings confirmed that ZOT5-1-Me, but not ZOT5-1-Et, significantly induced DSBs in U-87 MG cells.

ZOT5-1-Me and ZOT5-1-Et induced DNA damage, including DNA double-strand breaks in U-87 MG cells. (A) ZOT5-1-Me and ZOT5-1-Et induced DNA damage (percentage of DNA in the comet tail) in U-87 MG cells after 2 h treatment as determined by neutral, pH 12.1, and alkaline versions of the comet assay (n = 100). (B) Representative images of comets from the alkaline version of the comet assay are presented. (C) ZOT5-1-Me, but not ZOT5-1-E,t induced phosphorylation of histone H2AX (Ser139) in U-87 MG cells after 2 h treatment as assayed by Western blot (n = 4). The intensity of bands corresponding to proteins was analysed by densitometry. The results are shown as the fold change of γH2AX levels of treated cells vs. control cells (CTRL). β-actin served as a loading control. (D) Representative Western blot images are presented. (E) ZOT5-1-Me, but not ZOT5-1-Et, induced phosphorylation of histone H2AX (Ser139) in U-87 MG cells after 2 h treatment as evaluated by immunofluorescence staining followed by flow cytometry (FACS) quantification (n = 6). Etoposide (Etop, 50 µM, 4 h) was used as a positive control. (F) Representative FACS histograms are presented. (G) ZOT5-1-Me and ZOT5-1-Et do not induce DNA breaks in isolated supercoiled plasmid as determined by plasmid relaxation assay following 24 h treatment. Lane M – DNA ladder; CTRL – negative control, supercoiled plasmid, PstI – positive linear control (plasmid incubated with PstI restriction enzyme for the induction of DNA double-strand break. (H) Time course of repair of DNA damage induced by ZOT5-1-Me and ZOT5-1-Et in U-87 MG cells after 2 h treatment. After the exposure, the cells were washed and incubated in medium at 37 °C for the indicated time points. The extent of DNA damage at each time point was estimated by the alkaline version of the comet assay (n = 100). DMSO was used as a solvent control. Results are presented as bar plots with mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001, ns – not statistically significant.
To further investigate the ability of ZOT5-1-Me and ZOT5-1-Et to induce DNA damage, we performed an in vitro cell-free plasmid relaxation assay. This assay evaluates the induction of relaxed and linear forms of plasmid DNA, indicative of SSBs and DSBs, respectively, using gel electrophoresis. Neither ZOT5-1-Me nor ZOT5-1-Et altered the relative proportions of relaxed and linear plasmid DNA, as determined by their electrophoretic migration profiles. This indicates that both compounds require metabolic activation to exert their genotoxic effects and are incapable of directly inducing DNA breaks in isolated plasmid DNA (Fig. 5G).
Given the extensive and heterogeneous nature of DNA damage induced by ZOT5-1-Me and ZOT5-1-Et, we further evaluated the capacity of cells to repair these lesions. Using the alkaline version of the comet assay, we monitored the progression of DNA repair over a 2-h recovery period following ZOT treatment, a timeframe sufficient for DSB resolution. DNA damage induced by ZOT5-1-Et was fully repaired within this period, with damage levels returning to those observed in untreated controls. In contrast, cells treated with ZOT5-1-Me retained significant levels of DNA damage after 2 h of recovery (14.79 ± 2.09), suggesting that ZOT5-1-Me may interfere with DNA repair pathways (Fig. 5H).
ZOT5-1-Me and ZOT5-1-Et arrested U-87 MG cells in S phase by activating p53-p21 pathway
Unrepaired DNA damage, particularly DSBs, activates the DNA damage response (DDR), which arrests cell cycle at G1/S and G2/M checkpoints to allow time for DNA repair, preventing progression into the S and M phases with damaged DNA and reducing the risk of mutagenesis. Given that i) ZOT5-1-Me and ZOT5-1-Et induce significant and heterogeneous DNA lesions, ii) ZOT5-1-Me generates DSBs and iii) DNA repair pathways failed to resolve ZOT5-1-Me-induced DNA lesions within 2 h, we investigated the potential of ZOTs to induce cytostatic effects in greater detail.
Initially, we evaluated the ability of ZOT5-1-Me and ZOT5-1-Et to reduce cell proliferation rate and extend PDTs. The treatment with an increasing concentrations of ZOT5-1-Me and ZOT5-1-Et up to 4 days led to a significant reduction in the cell proliferation rate of U-87 MG cells (Fig. 6A). Notably, proliferation rates dropped below zero at concentrations above 7.8 μM, indicating cytotoxic effects. Calculations at the highest measurable concentrations (3.9 μM) yielded PDTs of 39.4 h for ZOT5-1-Me and 39.8 h for ZOT5-1-Et, which were significantly longer compared to control cells, which exhibited a PDT of 26.3 h. These findings indicate that both ZOT5-1-Me and ZOT5-1-Et exert antiproliferative effects in U-87 MG cell line.

ZOT5-1-Me and ZOT5-1-Et induced cell cycle arrest in U-87 MG cells. (A) Decreased proliferation rates of U-87 MG cells after treatment with an increasing concentration (0.95–15.6 µM) of ZOT5-1-Me and ZOT5-1-Et for 1–4 days were evaluated by cell proliferation assay. Proliferation curves allowed for the calculation of population doubling times. (B) ZOT5-1-Me and ZOT5-1-Et induced cell cycle arrest in U-87 MG cells after 48 h treatment as determined by PI staining followed by FACS analysis. Nocodazole (NOC, 200 ng/mL, 18 h) was used as a positive control of G2/M arrest. (C) Representative FACS dot plots after 48 h treatment with ZOTs are presented. FACS dot plots after 24 h treatments are presented in the Supplementary material. (D) ZOT5-1-Me and ZOT5-1-Et evoked changes to proteins regulating cell cycle, including p21 and phosphorylation of p53 (Ser15), in U-87 MG cells after 48 h treatment as evaluated by Western blot (n = 4). The intensity of bands corresponding to proteins was analysed by densitometry. The results are shown as the fold change of protein levels of treated cells vs. control cells (CTRL). β-actin served as a loading control. (E) Representative Western blot images are presented. DMSO was used as a solvent control. Results are presented as bar plots with mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001, ns – not statistically significant.
To elucidate the underlying mechanisms of ZOT-induced cytostatic effects, we analysed cell cycle distribution via flow cytometry. The treatment with ZOT5-1-Me and ZOT5-1-Et caused a significant accumulation of cells in the S phase, accompanied by a reduction in the G0/G1 population (Fig. 6B-C). Additionally, the G2/M phase population remained relatively unaffected, suggesting that the primary arrest occurs during DNA synthesis and may be associated with the persistence of DNA damage.
To further investigate the molecular mechanisms underlying the antiproliferative activity of ZOTs, we analysed the expression of cell cycle regulatory proteins with Western blot. We focused on the phosphorylation of p53 at Ser15 and the expression of p21, a potent inhibitor of cell cycle progression. The data revealed that ZOT5-1-Me and ZOT5-1-Et induced phosphorylation of p53 at Ser15, a marker of DDR activation. As expected, the expression levels of total p53 remained unchanged across treatments. Notably, only ZOT5-1-Me significantly upregulated p21 expression, indicating that this compound may have a stronger impact on the p53-p21 axis of cell cycle regulation (Fig. 6D-E).
Taken together, these findings suggest that ZOT5-1-Me and ZOT5-1-Et exert cytostatic effects by arresting U-87 MG cells in the S phase of the cell cycle. This effect appears to be mediated through the activation of the p53-p21 signalling pathway, primarily through activation of the p53 pathway, with ZOT5-1-Me showing a more pronounced effect via p21 upregulation.
The cytotoxic and genotoxic effects of ZOT5-1-Me and ZOT5-1-Et are mediated by ROS
ZOT5-1-Me and ZOT5-1-Et were demonstrated to induce ROS accumulation and mitochondrial damage (Fig. 4C-E), suggesting that their cytotoxic and genotoxic effects might be mediated through ROS upregulation. To explore the role of ROS in the mechanism of action of these compounds, we utilized N-acetyl-l-cysteine (NAC), a potent ROS scavenger. Cells were pre-incubated with NAC (1 mM, 1 h) prior to the treatment with ZOTs. NAC pre-treatment effectively suppressed ROS production induced by both ZOT5-1-Me and ZOT5-1-Et (Fig. 7A). Furthermore, NAC completely abrogated the cytotoxic effects of these compounds, as evidenced by the restoration of cell viability levels comparable to untreated controls (Fig. 7B). This protective effect was corroborated by the suppression of apoptotic markers, including Annexin V externalisation (Fig. 7C-D) and caspase 3/7 activation (Fig. 7E). Additionally, NAC mitigated the genotoxic effects of ZOT5-1-Me and ZOT5-1-Et, as demonstrated by the absence of detectable DNA damage (Fig. 7F-G). Overall, these results support a model in which ZOT5-1-Me and ZOT5-1-Et induce apoptotic cell death with oxidative stress potentially serving as a key trigger in these processes.

Pre-treatment with ROS scavenger completely blocked the cytotoxic and genotoxic effect of ZOT5-1-Me and ZOT5-1-Et in U-87 MG cells. (A) Pre-incubation with N-acetyl-l-cysteine (NAC, 1 mM, 1 h) decreased intracellular reactive oxygen species (ROS) induced by ZOT5-1-Me and ZOT5-1-Et in U-87 MG as estimated by CM-H2DCFDA assay. The data were normalized to control cells (CTRL, 100%, n = 6). (B) Pre-incubation with NAC restored cell viability of U-87 MG cells after 48 h treatment with ZOT5-1-Me and ZOT5-1-Et as evaluated by CCK-8 assay. The data were normalized to control cells (CTRL, 100%, n = 6). (C) Pre-incubation with NAC reduced the externalisation of phosphatidylserine in U-87 MG cells as evaluated by Annexin V/PI assay followed by flow cytometry (FACS) quantification (n = 6). (D) Representative FACS dot plots after 48 h treatment with ZOTs are presented with the indicated percentages of necrotic (Q1), late-apoptotic (Q2), early-apoptotic (Q3), and viable cells (Q4). Apoptosis was presented as a percentage of Annexin V-positive cells (Q2 + Q3). (E) Pre-incubation with NAC diminished ZOT5-1-Me and ZOT5-1-Et-evoked apoptosis in U-87 MG cells as determined by Caspase-Glo 3/7 assay (n = 6). The data were normalized to control cells (CTRL, 100%, n = 6). (F) Pre-incubation with NAC decreased DNA damage (percentage of DNA in the comet tail) induced by ZOT5-1-Me and ZOT5-1-Et in U-87 MG cells as determined by the alkaline versions of comet assay (n = 100). (G) Representative images of comets from the alkaline version of the comet assay are presented. Results are presented as bar plots with mean ± SEM; *p < 0.05; **p < 0.01; ***p < 0.001, ns – not statistically significant.
Discussion
GBM is among the most aggressive and lethal primary brain tumours1. Despite multimodal treatment strategies, long-term survival remains limited, with median survival ranging from 14.6 to 21.1 months4. The management of GBM is hampered by several challenges, including tumour heterogeneity, the presence of therapy-resistant glioma stem cells, the highly infiltrative growth pattern of tumour, primary and acquired drug resistance, and BBB, which restricts drug delivery7,8,27,28,29. These factors significantly complicate the development of effective therapies.
The majority of approved anticancer drugs are unable to cross the BBB, reducing therapeutic options. U.S. Food and Drug Administration (FDA) has approved only a few treatments for GBM, including the chemotherapeutics lomustine (CCNU, 1976), carmustine (BCNU, 1977), and TMZ (2005), as well as anti-vascular endothelial growth factor (VEGF) antibody bevacizumab (2009), and tumour treating fields (TTF, 2011)30,31. Among these, TMZ can cross the BBB and shows bioavailability of 98% when orally administered32,33,34. Although TMZ is the first-line chemotherapy for GBM, its effectiveness is limited, particularly against recurrent tumours35,36. Lomustine and carmustine, despite their ability to penetrate the BBB, offer limited benefits for GBM patients37,38. On the other hand, bevacizumab – a monoclonal antibody that inhibits angiogenesis by targeting VEGF-A – cannot penetrate the BBB, and thus its efficacy is limited, resulting in no improvement in the overall survival of GBM patients39,40.
Given that no current treatment regimen for GBM is curative, the National Comprehensive Cancer Network (NCCN) recommends clinical trials as the preferred option for eligible patients, reflecting the need for innovative approaches41. Currently, there are 286 ongoing clinical trials focused on GBM, underscoring the scientific community’s recognition of the urgent need for new therapies42.
Although numerous compounds have demonstrated cytotoxic effects against GBM cells in vitro, translating these preclinical findings into clinical success has proven exceptionally rare. Nevertheless, novel agents are currently being developed, either as monotherapies or in combination regimens28,36,43. Most therapeutic advances in GBM therapy have been hampered due to the restricted permeability across the BBB, which permits only small (< 500 Da and < 400 nm) and lipophilic molecules to pass diffusely across, while larger or hydrophilic compounds are typically excluded8,44,45. Consequently, GBM therapy has seen little advancement in the past two decades, with the Stupp protocol remaining the standard of care4,5. The simplest polyfluoroalkyl phosphonates were designed as small molecular weight (< 250 Da) anticancer candidates with the potential to cross the BBB19. These compounds were initially screened against GBM cells, and the most cytotoxic compounds, ZOT5-1-Me and ZOT5-1-Et, were selected for further analysis. To expand our knowledge of the biological activity of the simplest polyfluoroalkyl phosphonates, this study focuses on elucidating the molecular mechanism of ZOT5-1-Me and ZOT5-1-Et, as promising candidates for chemotherapeutics against GBM.
Mechanisms of action of ZOT compounds
In this study, we first examined the anticancer potential of ZOT5-1-Me and ZOT5-1-Et in the U-87 MG cell line. We found that ZOT5-1-Me and ZOT5-1-Et induced cell death through both intrinsic and extrinsic apoptotic pathways. This dual activation is a critical finding, as it suggests the compounds can trigger apoptosis from multiple cellular signals. Apoptosis is often dysregulated in cancer cells, allowing them to evade normal cell death mechanisms. These compounds can effectively decrease tumour cell survival by activating initiator caspase 3/7, and executive caspase-8 and caspase-9. Additionally, pan-caspase inhibitor Z-VAD-FMK has not completely prevented cell death, although caspase activity was completely inhibited (Fig. 3E-G), indicating the activation of caspase-independent cell death pathways.
DNA is among the principal targets of many anticancer drugs. Multiple DDR inhibitors are now being tested in clinical trials for GBM46. All clinically used FDA-approved chemotherapeutics against GBM belong to DNA alkylating agents. The cytotoxic effect of TMZ is primarily exerted by methylation of O6 position of guanine (O6-meG) which escapes O6-meG-DNA-methyl transferase (MGMT) repair resulting in futile cycles of mismatch repair leading to DSBs and triggering cell cycle arrest and apoptosis32. MGMT down-regulation is observed in 40% of GBM patients due to MGMT gene promoter methylation. Not all GBM patients respond to TMZ, and drug resistance has been correlated to MGMT overexpression and/or malfunctioning mismatch repair (MMR)47. Lomustine and carmustine are bifunctional DNA alkylating agents and give rise to the formation of interstrand crosslinks (ICLs), which are critical cytotoxic DNA lesions that block DNA replication and transcription48. Besides ICLs, DSBs are considered the most lethal form of DNA damage and a primary cause of cell death. Recently, SSBs are also becoming recognized as important lesions for lethality46. We have demonstrated that both ZOT5-1-Me and ZOT5-1-Et induced DNA damage, including SSBs and ALS, in the U-87 MG cell line (Fig. 5A-B). Interestingly, ZOT5-1-Me was effective at inducing DSBs compared to ZOT5-1-Et, indicating potential differences in their mechanisms of action (Fig. 5). Both compounds appear to act as prodrugs, requiring metabolic activation to induce DNA damage, as they were chemically inert in their initial state during plasmid relaxation assays (Fig. 5G).
Targeting the cell cycle remains a mainstay in the treatment of cancer. All FDA-approved chemotherapeutics against GBM are cytostatic drugs inhibiting the proliferation of GBM cells, which are characterized by rapid division and growth. Both ZOT5-1-Me and ZOT5-1-Et had an antiproliferative effect on U-87 MG cells, which was associated with cell cycle arrest at the S phase (Fig. 6A-C). The ability of ZOT5-1-Me and ZOT5-1-Et to arrest the cell cycle correlated with their capacity to induce DNA damage. Additionally, both compounds induced phosphorylation of p53, a tumour suppressor protein, and ZOT5-1-Me increased the expression of p21, a cyclin-dependent kinase inhibitor, indicating that ZOTs at least partially arrest cell cycle via p53-p21 signalling pathway (Fig. 6D-E).
ZOT5-1-Me and ZOT5-1-Et significantly increased intracellular ROS levels in U-87 MG cells, with a concentration-dependent relationship, suggesting oxidative stress as a potential mechanism of cytotoxicity (Fig. 4C). Indeed, the cytotoxic and genotoxic effects of ZOT5-1-Me and ZOT5-1-Et were strongly dependent on their ability to elevate ROS (Fig. 7). Redox imbalance plays a complex and dualistic role in GBM, contributing to both tumour progression and resistance to therapy7,49,50. In this context, both antioxidant and pro-oxidant therapeutic strategies have been explored in GBM. Antioxidants, particularly at high or pharmacological doses, can paradoxically exert pro-oxidant effects by inducing oxidative stress sufficient to trigger cancer cell death, while potentially reducing non-specific toxicity associated with chemotherapy50. A prominent example is ascorbate (pharmacological vitamin C), which exhibited pro-oxidant activity at high concentrations. A phase II clinical trial is currently ongoing to assess the efficacy of high-dose ascorbate in combination with radiotherapy and adjuvant chemotherapy with TMZ51,52,53,54. Nevertheless, reducing ROS levels within tumours can be a double-edged sword. Antioxidants may inadvertently protect cancer cells from ROS-mediated cell death, thereby attenuating the efficacy of chemo-radiation, as demonstrated in some preclinical studies10,55. Conversely, pro-oxidant therapies aim to increase ROS levels beyond a critical threshold, selectively inducing cancer cell death. Standard therapies such as radiotherapy and chemotherapy rely in part on ROS-mediated mechanisms to achieve their cytotoxic effects. Although preclinical findings are promising, pro-oxidants are yet to progress through the clinical trial phase10,12,13,14. Given the importance of ROS in mediating the cytotoxic effects of ZOTs, combining these compounds with conventional therapies that further elevate oxidative stress may enhance their therapeutic efficacy as noted for other pro-oxidants56,57,58. Nonetheless, this approach requires careful consideration of potential off-target effects on surrounding normal brain tissue50. Collectively, the available evidence suggests that a therapeutic window exists for ROS-modulating strategies in GBM, either as monotherapy or in combination with conventional treatments. While ZOTs exhibit strong anticancer potential through ROS generation, successful translation into clinical practice will depend on achieving a delicate balance – maximizing cytotoxic effects within GBM cells while preserving the integrity and function of healthy brain tissue.
Molecular determinants of ZOT sensitivity in glioma
To elucidate the relationship between genetic alterations and the sensitivity of glioma cell lines to the novel compounds ZOT5-1-Me and ZOT5-1-Et, we analysed the mutation status of key biomarkers, including TP53, PTEN, CDKN2A, and IDH, across seven glioma cell lines: SF-268, SNB-19, SNB-75, U251, SF-539, SF-295, and U-87 MG (Table 2). Differential sensitivity was observed, with higher sensitivity towards GBM cell lines SF-539, SF-295, and U-87 MG, as well as the U251 astrocytoma cell line, and lower sensitivity towards astrocytoma SF-268 and SNB-19, and GBM SNB-75 cell lines (Fig. 2A, Fig. 3A, Table 1).
Both PTEN and TP53 are frequently mutated in GBM, contributing to aggressive behaviour, treatment resistance, and poor prognosis64,65,66,67. Among the analysed cell lines, only U-87 MG retained wild-type TP53, which corresponded with the highest cytotoxic response to ZOT5-1-Me and ZOT5-1-Et (Fig. 3A, Table 2), indicating that TP53 status may influence drug sensitivity. ZOT efficacy was primarily influenced by PTEN mutation status, with PTEN-deficient cell lines—particularly SF-539—showing heightened sensitivity, while cell lines with mutated TP53 and intact PTEN—SF-268, SNB-75—were less sensitive (Fig. 2A, Table 1–2). In U-87 MG, the synergy between functional p53 and PTEN loss could lead to the improved cytotoxic outcomes (Fig. 3A, Table 2). Indeed, PTEN is known to cooperate with p53 in the repair of DSBs, a mechanism potentially leveraged by ZOT activity (Fig. 5H)59. These results suggest that ZOTs could be most effective in GBM cells with wild-type TP53 and mutated PTEN, a genotype common in IDH-wildtype GBM (~ 90% of adult gliomas)68. Conversely, TP53-mutant/PTEN-intact cells were less sensitive, indicating limited efficacy in other adult-type diffuse gliomas (Fig. 2A, Table 1–2)69. Sensitivity was further modulated by mutations in genes like CDKN2A and IDH, suggesting a multifactorial basis for ZOT responsiveness (Fig. 2A, 3A; Table 1–2). These insights highlight the need for molecular profiling and potential combination therapies in less responsive cases.
Implications for clinical application
The ability of ZOT5-1-Me and ZOT5-1-Et to induce cell death through multiple pathways, generate ROS, cause DNA damage, and cell cycle arrest in U-87 MG cells suggests that they hold promise as therapeutic agents for GBM. Their ability to activate a few cell death pathways is particularly significant, as they could overcome some of the primary resistance mechanisms that have hindered the efficacy of other treatments like TMZ. Moreover, the study highlights the potential for ZOTs to be used in combination with other therapies. Given the promising role of ROS in mediating the cytotoxic effects of ZOTs, their combination with agents that further promote oxidative stress in GBM cells may enhance their cytotoxic potential. Additionally, their use alongside traditional DNA-damaging agents like TMZ could further disrupt GBM cell survival by overwhelming their DNA repair mechanisms.
While the results are promising, several limitations warrant further investigation. Firstly, the experiments were conducted in vitro using established GBM cell lines, which may not fully capture the heterogeneity of patient tumours. Moreover, detailed analyses were restricted to the U-87 MG cell line, selected as the most sensitive model for in-depth mechanistic studies, and future work will therefore extend to resistant lines and primary GBM cultures to more accurately capture tumour heterogeneity and mechanisms of therapy resistance. A further limitation is the absence of a non-tumoural neural control cell line, which would help determine whether the observed effects are preferentially directed against transformed cells. Although our previous work included cytotoxicity screening in peripheral blood mononuclear cells (PBMCs) as a representative non-tumoural model, additional data on non-neoplastic neural cells are needed to strengthen the safety assessment19. Additionally, employing transwell assays with brain endothelial cells could provide a more physiologically relevant model to confirm the ability of ZOTs to penetrate the BBB. In vivo studies using animal models of GBM are also essential to evaluate the compounds’ capacity to cross the BBB, validate their therapeutic potential within the complex tumour microenvironment, and refine their clinical applications. Finally, comprehensive assessments of the long-term toxicity and pharmacokinetics of ZOT5-1-Me and ZOT5-1-Et are necessary to establish their safety profile and identify potential side effects in a clinical setting.
Future research should also focus on understanding the precise molecular mechanisms that differentiate ZOT5-1-Me and ZOT5-1-Et, particularly their differential effects on DSBs. Further exploration of these mechanisms could lead to the optimization of these compounds for enhanced efficacy against GBM. Moreover, the potential for these compounds to synergize with existing therapies, including TMZ and radiotherapy, should be explored in preclinical models.
Conclusion
In conclusion, ZOT5-1-Me and ZOT5-1-Et represent promising new candidates for GBM therapy. Their ability to induce cell death through multiple pathways, generate ROS, and cause DNA damage and cell cycle arrest in GBM cells highlights their potential to overcome some of the most significant challenges in GBM treatment (Fig. 8). However, further studies are necessary to validate these findings in vivo and to fully explore their therapeutic potential in combination with existing treatments. If successful, these compounds could represent a significant advancement in the treatment of GBM, offering new hope for patients with this devastating disease.

Proposed molecular mechanism underlying anticancer activity of ZOT5-1-Me and ZOT5-1-Et in glioblastoma (GBM) cells. ZOT5-1-Me and ZOT5-1-Et elevate the level of reactive oxygen species (ROS) in GBM cells, causing DNA damage and subsequent activation of TP53 pathway. Phosphorylated TP53 upregulates the expression of p21, resulting in cell cycle arrest at G1/S and G2/M checkpoints, and initiates apoptosis through intrinsic (caspase-9) pathway. Additionally, ZOT5-1-Me and ZOT5-1-Et can trigger extrinsic (caspase-8) apoptotic pathway and caspase-independent cell death pathways (created in BioRender. Wozniak, K. (2025) https://BioRender.com/35yqjr4).
Data availability
The data generated in this study are available at Zenodo (https://doi.org/10.5281/zenodo.16272119). Drug response profiles obtained in this study are available at NCI/DTP repository (https://dtp.cancer.gov/dtpstandard/dwindex/index.jsp); at NSC 835966 (ZOT5-1-Me; https://dtp.cancer.gov/dtpstandard/servlet/dwindex?searchtype=NSC&searchlist=835966) and NSC 835967 (ZOT5-1-Et, https://dtp.cancer.gov/dtpstandard/servlet/dwindex?searchtype=NSC&searchlist=835967).
References
Grochans S, Cybulska AM, Simińska D, Korbecki J, Kojder K, Chlubek D et al. Epidemiology of Glioblastoma Multiforme-Literature Review. Cancers (Basel) 2022; 14(10).
Brown NF, Ottaviani D, Tazare J, Gregson J, Kitchen N, Brandner S et al. Survival Outcomes and Prognostic Factors in Glioblastoma. Cancers (Basel) 2022; 14(13).
Krex, D. et al. Long-term survival with glioblastoma multiforme. Brain 130(Pt 10), 2596–2606 (2007).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl. J. Med. 352(10), 987–996 (2005).
Obrador E, Moreno-Murciano P, Oriol-Caballo M, López-Blanch R, Pineda B, Gutiérrez-Arroyo JL et al. Glioblastoma Therapy: Past, Present and Future. Int J Mol Sci 2024; 25(5).
Perry, J. R. et al. Short-course radiation plus temozolomide in elderly patients with glioblastoma. N Engl. J. Med. 376(11), 1027–1037 (2017).
Olivier, C., Oliver, L., Lalier, L. & Vallette, F. M. Drug resistance in glioblastoma: The two faces of oxidative stress. Front. Mol. Biosci. 7, 620677 (2020).
Ou A, Yung WKA, Majd N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int J Mol Sci 2020; 22(1).
Lo Dico A, Salvatore D, Martelli C, Ronchi D, Diceglie C, Lucignani G et al. Intracellular Redox-Balance Involvement in Temozolomide Resistance-Related Molecular Mechanisms in Glioblastoma. Cells 2019; 8(11).
Campos-Sandoval, J. A. et al. Antioxidant responses related to temozolomide resistance in glioblastoma. Neurochem. Int. 149, 105136 (2021).
Rocha, C. R. R., Kajitani, G. S., Quinet, A., Fortunato, R. S. & Menck, C. F. M. NRF2 and glutathione are key resistance mediators to temozolomide in glioma and melanoma cells. Oncotarget 7(30), 48081–48092 (2016).
Ling, Z. et al. Small-molecule Molephantin induces apoptosis and mitophagy flux blockage through ROS production in glioblastoma. Cancer Lett. 592, 216927 (2024).
Orlicka-Płocka M, Fedoruk-Wyszomirska A, Gurda-Woźna D, Pawelczak P, Krawczyk P, Giel-Pietraszuk M et al. Implications of Oxidative Stress in Glioblastoma Multiforme Following Treatment with Purine Derivatives. Antioxidants (Basel) 2021; 10(6).
Buccarelli, M. et al. Elesclomol-induced increase of mitochondrial reactive oxygen species impairs glioblastoma stem-like cell survival and tumor growth. J. Exp. Clin. Cancer Res. 40(1), 228 (2021).
Zanders, E. D., Svensson, F. & Bailey, D. S. Therapy for glioblastoma: Is it working?. Drug Discov Today 24(5), 1193–1201 (2019).
Pitz, M. W., Desai, A., Grossman, S. A. & Blakeley, J. O. Tissue concentration of systemically administered antineoplastic agents in human brain tumors. J. Neurooncol. 104(3), 629–638 (2011).
Starnes, H. M., Rock, K. D., Jackson, T. W. & Belcher, S. M. A critical review and meta-analysis of impacts of per- and polyfluorinated substances on the brain and behavior. Front. Toxicol. 4, 881584 (2022).
Somogyi, G. et al. Targeted drug delivery to the brain via phosphonate derivatives. Int. J. Pharm. 166(1), 15–26 (1998).
Zagórski, P. M., Tokarz, P., Gostyński, B. & Tokarz, P. Experimental and computational studies on the formation and biological properties of the simplest polyfluoroalkyl phosphonates. New J. Chem. 44(26), 10933–10945 (2020).
Kluska, M., Piastowska-Ciesielska, A. W. & Tokarz, P. Cell cycle status influences resistance to apoptosis induced by oxidative stress in human breast cancer cells, which is accompanied by modulation of autophagy. Curr. Issues Mol. Biol. 45(8), 6325–6338 (2023).
Reers, M., Smith, T. W. & Chen, L. B. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 30(18), 4480–4486 (1991).
Ashby, J., Tinwell, H., Lefevre, P. A. & Browne, M. A. The single cell gel electrophoresis assay for induced DNA damage (comet assay): Measurement of tail length and moment. Mutagenesis 10(2), 85–90 (1995).
Juszczak, M. et al. Piano-stool ruthenium(II) complexes with maleimide and phosphine or phosphite ligands: Synthesis and activity against normal and cancer cells. Dalton Trans. 52(13), 4237–4250 (2023).
Firsanov, D. et al. Rapid detection of γ-H2AX by flow cytometry in cultured mammalian cells. Methods Mol. Biol. 1644, 129–138 (2017).
Hytti, M. et al. Inhibition of BET bromodomains alleviates inflammation in human RPE cells. Biochem. Pharmacol. 110–111, 71–79 (2016).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–254 (1976).
Korzyńska, A. & Zychowicz, M. A method of estimation of the cell doubling time on basis of the cell culture monitoring data. Biocybern. Biomed. Eng. 28(4), 75–82 (2008).
Roth V. Doubling Time Computing; 2006 [cited 2025 Apr 13]. Available from: URL: http://www.doubling-time.com/compute.php.
ATCC. Cell Lines by Gene Mutation; 2022 [cited 2025 Apr 7]. Available from: URL: https://www.atcc.org/-/media/resources/brochures/cell-lines-by-gene-mutation.pdf.
Seker-Polat F, Pinarbasi Degirmenci N, Solaroglu I, Bagci-Onder T. Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives. Cancers (Basel) 2022; 14(2).
Ichimura, K. et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro. Oncol. 11(4), 341–347 (2009).
Rana, M., Liou, K.-C., Thakur, A., Nepali, K. & Liou, J.-P. Advancing glioblastoma therapy: Learning from the past and innovations for the future. Cancer Lett 617, 217601 (2025).
Fisher JP, Adamson DC. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021; 9(3).
Newlands, E. S., Stevens, M. F., Wedge, S. R., Wheelhouse, R. T. & Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat Rev. 23(1), 35–61 (1997).
Ostermann, S. et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 10(11), 3728–3736 (2004).
Patel, M., McCully, C., Godwin, K. & Balis, F. M. Plasma and cerebrospinal fluid pharmacokinetics of intravenous temozolomide in non-human primates. J. Neurooncol. 61(3), 203–207 (2003).
Chen, C., Xu, T., Lu, Y., Chen, J. & Wu, S. The efficacy of temozolomide for recurrent glioblastoma multiforme. Eur. J. Neurol. 20(2), 223–230 (2013).
Gaiaschi, L., Bottone, M. G. & de Luca, F. Towards effective treatment of glioblastoma: The role of combination therapies and the potential of phytotherapy and micotherapy. Curr. Issues Mol. Biol. 46(12), 14324–14350 (2024).
Wick, W. et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl. J. Med. 377(20), 1954–1963 (2017).
Reithmeier, T. et al. BCNU for recurrent glioblastoma multiforme: Efficacy, toxicity and prognostic factors. BMC Cancer 10, 30 (2010).
Garcia, J. et al. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat Rev. 86, 102017 (2020).
Cha, G. D. et al. Advances in drug delivery technology for the treatment of glioblastoma multiforme. J Control Release 328, 350–367 (2020).
Nabors, L. B. et al. Central nervous system cancers, version 3.2020, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc. Netw. 18(11), 1537–1570 (2020).
Rajaratnam V, Islam MM, Yang M, Slaby R, Ramirez HM, Mirza SP. Glioblastoma: Pathogenesis and Current Status of Chemotherapy and Other Novel Treatments. Cancers (Basel) 2020; 12(4).
Thakur, A. et al. Glioblastoma: Current status, emerging targets, and recent advances. J. Med. Chem. 65(13), 8596–8685 (2022).
Pajouhesh, H. & Lenz, G. R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2(4), 541–553 (2005).
Banks WA. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol 2009; 9 Suppl 1(Suppl 1):S3.
Wen, P. Y. et al. Glioblastoma in adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol 22(8), 1073–1113 (2020).
Lee, S. Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 3(3), 198–210 (2016).
Nikolova, T., Roos, W. P., Krämer, O. H., Strik, H. M. & Kaina, B. Chloroethylating nitrosoureas in cancer therapy: DNA damage, repair and cell death signaling. Biochim. Biophys. Acta Rev. Cancer 1868(1), 29–39 (2017).
Salazar-Ramiro, A. et al. Role of redox status in development of glioblastoma. Front. Immunol. 7, 156 (2016).
Qi, X. et al. Antioxidants in brain tumors: Current therapeutic significance and future prospects. Mol. Cancer 21(1), 204 (2022).
Allen, B. G. et al. First-in-human phase I clinical trial of pharmacologic ascorbate combined with radiation and temozolomide for newly diagnosed glioblastoma. Clin. Cancer Res. 25(22), 6590–6597 (2019).
Allen B. A Phase 2 Trial of High-Dose Ascorbate in Glioblastoma Multiforme: University of Iowa; 2024 [cited 2025 Apr 7]. Available from: URL: https://clinicaltrials.gov/study/NCT02344355.
Schoenfeld, J. D. et al. O2⋅- and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 31(4), 487-500.e8 (2017).
Chen, Q. et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl. Acad. Sci. U S A 105(32), 11105–11109 (2008).
Arabzadeh, A. et al. Therapeutic potentials of resveratrol in combination with radiotherapy and chemotherapy during glioblastoma treatment: A mechanistic review. Cancer Cell Int. 21(1), 391 (2021).
Yuan, Y., Xue, X., Guo, R.-B., Sun, X.-L. & Hu, G. Resveratrol enhances the antitumor effects of temozolomide in glioblastoma via ROS-dependent AMPK-TSC-mTOR signaling pathway. CNS Neurosci. Ther. 18(7), 536–546 (2012).
Wang, X. et al. Gain of function of mutant TP53 in glioblastoma: Prognosis and response to temozolomide. Ann. Surg. Oncol. 21(4), 1337–1344 (2014).
Ohgaki, H. & Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 19(4), 764–772 (2013).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro. Oncol. 23(8), 1231–1251 (2021).
Abaan, O. D. et al. The exomes of the NCI-60 panel: A genomic resource for cancer biology and systems pharmacology. Cancer Res. 73(14), 4372–4382 (2013).
Ikediobi, O. N. et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol. Cancer Ther. 5(11), 2606–2612 (2006).
Grogan, P. T., Sarkaria, J. N., Timmermann, B. N. & Cohen, M. S. Oxidative cytotoxic agent withaferin A resensitizes temozolomide-resistant glioblastomas via MGMT depletion and induces apoptosis through Akt/mTOR pathway inhibitory modulation. Invest. New Drugs 32(4), 604–617 (2014).
Yin, H. et al. Curcumin sensitizes glioblastoma to temozolomide by simultaneously generating ROS and disrupting AKT/mTOR signaling. Oncol. Rep. 32(4), 1610–1616 (2014).
Zheng, H. et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455(7216), 1129–1133 (2008).
Lee, Y.-J. et al. Gene expression profiling of glioblastoma cell lines depending on TP53 status after tumor-treating fields (TTFields) treatment. Sci. Rep. 10(1), 12272 (2020).
Kato, H. et al. Functional evaluation of p53 and PTEN gene mutations in gliomas. Clin. Cancer Res. 6(10), 3937–3943 (2000).
Li, Y. et al. PTEN has tumor-promoting properties in the setting of gain-of-function p53 mutations. Cancer Res. 68(6), 1723–1731 (2008).
Acknowledgements
We would like to thank National Cancer Institute Developmental Therapeutics Program (NCI/DTP) https://dtp.cancer.gov for providing screening data of compounds present in this manuscript. Specifially, NSC #: 835966 (ZOT5-1-Me) and NSC 835967 (ZOT5-1-Et). We would also like to thank Klaudia Kwaśniewska, MSc, and Marcelina Strachanowska, MSc, for their technical assistance.
Funding
This research was funded by National Science Centre in Poland grant number 2020/39/D/NZ7/03417.
Author information
Authors and Affiliations
Contributions
Conceptualization, PT*; data curation, PT*; formal analysis, PT* and PW; methodology, PT*, PW, MJ and PT; investigation, PT*, PW, MJ, and PT; writing—original draft preparation, PT*; writing—review and editing, PT*, PW, MJ, PT, and KW; visualization, KW and PT*; supervision, PT* and KW; project administration, PT*; funding acquisition, PT*. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Generative AI and AI-assisted technologies in the writing process
During the preparation of this work the author(s) used chat.gpt in order to improve language and readability. After using this tool/service, the author(s) reviewed and edited the content as needed and take(s) full responsibility for the content of the publication.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wołodkiewicz, P., Juszczak, M., Tokarz, P. et al. Small molecular weight polyfluoroalkyl phosphonates induce ROS-mediated cytotoxicity in glioblastoma cells: a molecular mechanism study. Sci Rep 15, 38896 (2025). https://doi.org/10.1038/s41598-025-22754-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-22754-0
