
Abstract
Sargassum horneri is rich in bioactive compounds, including phytosterols, exhibits antioxidant, anti-obesity and anti-inflammation properties; however, its underlying mechanisms remain unclear. In this study, we evaluated that the antioxidant and anti-inflammatory effect of S. horneri ethanolic extract and its subfractions using lipopolysaccharide (LPS)-treated RAW264.7 cells. Furthermore, we examined the in vivo anti-obesity efficacy of S. horneri using obese mice fed a high-fat diet. Results revealed that the in vitro treatment increases the inflammatory cytokines such as inducible nitric oxide synthase (iNOS), nitric oxide (NO), cyclooxygenase-2 (COX-2), prostaglandin E2 (PGE2), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) including nuclear factor kappa B (NF-κB) subunit p65 protein expressions in LPS treated RAW264.7 cells. However, these pro-inflammatory cytokines were significantly reduced by inhibiting the NF-κB- p65 translocation pathway in S. horneri treatment. In addition, the S. horneri extracts increased the nuclear factor erythroid 2-related factor 2 (Nrf2) translocation into the nucleus as well as their heme oxygenase (HO-1) target gene expression. Whereas, in vivo treatment with S. horneri reduces body and organ weight, including pathological damage in liver and adipose tissue. Moreover, S. horneri decreases serum triglycerides (TG), LDL-cholesterol, total cholesterol (TC), arteriosclerosis index (AI), and cardiovascular risk factor (CVRF), but increases HDL-cholesterol concentration-dependently. The liver antioxidant enzyme activities and AMP-activated protein kinase (AMPK) protein expressions were raised in S. horneri treated groups; while sterol regulatory element-binding protein-1 (SREBP-1), Fatty acid synthase (FAS), and Acetyl-CoA carboxylase (ACC) expressions was reduced with dose-dependent manner. These findings provide an innovative pharmacological basis for the antioxidant, anti-inflammatory and anti-obesity effect of S. horneri. It specifies the potential of S. horneri as a candidate for preventing inflammation, obesity and other related disorders.
Similar content being viewed by others


Introduction
Inflammation is a complex biological response linked to various diseases including obesity, and excessive inflammation can threaten a patient’s life1. The overproduction of pro-inflammatory cytokines, including prostaglandin E2 (PGE2), nitric oxide (NO), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6) has been linked to several inflammatory illnesses2. Normal physiological conditions result in either no expression of inducible nitric oxide synthase (iNOS) or very low expression of cyclooxygenase-2 (COX-2). However, massive numbers of inflammatory mediators (iNOS and COX-2) and their derivatives (NO and PGE2) were produced throughout the inflammatory response3. Pro-inflammatory cytokines can activate macrophages’ iNOS and COX-2, which can lead to vasodilation linked to inflammation4, whereas too much NO and PGE2 can cause inflammation and related illnesses5. Therefore, the identification of iNOS and COX-2 inhibitors is a positive approach toward inhibiting inflammatory diseases. The inflammatory cytokine TNF-α is generated from mononuclear macrophages, and it initiates cytokines production including IL-6 by inducing macrophages3. However, high TNF-α and IL-6 concentrations can have adverse effects, which potentially lead to pathological situations like inflammatory and autoimmune illnesses6.
Nuclear factor kappa B (NF-κB) contains p50 and p65 subunits, which are well-known transcription factors that respond to inflammation. The NF-κB complex is inactive in the resting cell cytoplasm. Lipopolysaccharide (LPS) treatment activates the NF-κB pathway by initiating intracellular signal transduction, which in turn initiates the transcription of various inflammatory genes, leading to the secretion of vast quantities of inflammatory factors, including TNF-α, thereby increasing the inflammatory response7. Therefore, the LPS-activated NF-κB pathway elicits an important inflammatory effect, leading to the development of several critical illnesses. On the other hand, the nuclear factor-erythroid factor 2-related factor 2 (Nrf2)/heme oxygenase (HO-1) signaling pathway is essential for controlling the antioxidant and anti-inflammatory qualities of natural products that contain bioactive substances. By modulating the NF-κB pathway, HO-1 expression can significantly lower the production of pro-inflammatory cytokines in response to LPS induced macrophages, and maintain intracellular redox homeostasis8,9. Therefore, Nrf2/HO-1 pathway initiation may provide a promising approach for controlling inflammation and related disorders. Reducing inflammation is crucial in the treatment of obesity, as evidenced by recent research that linked obesity to metabolic inflammation because of increased levels of pro-inflammatory cytokines in adipose tissue10,11.
One of the most prevalent metabolic illnesses in both industrialized and developing countries is obesity, which can be attributed to the prevalence of sedentary lifestyles and high-fat, high-glucose fast food12. Overweight and obesity are terms used to describe the excessive build-up of adiposity brought on by abnormalities in energy intake and expenditure13. Research has indicated that the production of some bioactive chemicals known as adipokines is influenced by the liver and adipose tissue14. Human obesity is associated with a number of chronic illnesses, including metabolic syndrome, hyperlipidemia, hyperglycemia, diabetes, inflammation, and hypertension12. Cardiovascular diseases (CVD) and metabolic syndrome can be brought on by a diet heavy in fat and carbohydrates15. Total cholesterol (TC), triacylglycerol (TG), and serum cholesterol profile (TC / high density lipoprotein cholesterol-HDL-C) are known as CVD biomarkers16. Lowering triglyceride (TG), TC, LDL-cholesterol (LDL-C), and atherosclerosis index (AI) can reduce CVD risk17. However, preventing inflammation, obesity and related diseases are difficult; thus, screening the anti-inflammatory and anti-obesity drugs for a practical approach using in vitro and in vivo studies is essential.
Currently, synthetic drugs are widely used to treat obesity, inflammation, and related diseases, although natural products and their derived extracts containing various bioactive compounds serve an important role in preventing obesity, and inflammation, and may offer alternative therapeutic approaches18. Sargassum horneri is an edible brown seaweed grown on the coasts of Japan, China, and Korea19. It is rich in polyphenols, flavonoids, polysaccharides, pigments, and chromanols, which are known for their antioxidant, anti-inflammatory, and anti-obesity effects20,39. Currently, we extracted the bioactive components from S. horneri and evaluated their antioxidant and anti-inflammatory effect of S. horneri extraction and its subfractions, in LPS treated RAW264.7 macrophages, and we found that S. horneri may inhibit inflammation via triggering the Nrf2/HO-1 pathway. We postulated that impeding the NF-κB pathway through the use of S. horneri as a medicine could be a helpful therapeutic strategy against conditions associated with inflammation. Furthermore, we investigated the biological mechanism of S. horneri anti-obesity capabilities by controlling the lipid profile in order to prevent liver damage caused by a high-fat diet in ICR mice.
Materials and methods
Materials
Gibco BRL Co. (Grand Island, NY, USA) provided the tissue culture reagents, fetal bovine serum (FBS) (#16000044), and RPMI-1640 medium (Roswell Park Memorial Institute, #11875093). The primary antibodies obtained from Santa Cruz biotechnology (Santa Cruz, CA, USA), which comprised anti-p65 (#sc-8008), anti-COX-2 (#sc-19999), anti-iNOS (#sc-7271), anti-HO-1 (#sc-10789), anti-β-actin (#sc-47778), anti-Nrf2 (#sc-518036), and anti-PCNA (anti-proliferating cell nuclear antigen; #sc-25280). Rabbit (#AP510), and mouse (#AP308P) secondary antibodies were available from Millipore (Billerica, MA, USA). To measure the levels of PGE2 (#KGE004B), TNF-α (#MTA00B), and IL-6 (#D6050B), we used enzyme-linked immunosorbent assay (ELISA) kits, which were produced from R&D Systems, Inc. (Minneapolis, MN, USA). Kits for transcription factor NF-κB (#10007889) and nuclear extraction (#10009277) were acquired from Cayman (Cayman, Ann Arbor, MI, USA).
Preparation of the S. horneri powder and their extractions
We prepared dried S. horneri powder, its 70% ethanol (EtOH) extract, and its subfractions following a previously described method, with minor modifications21. Briefly, the washed and dried S. horneri samples were provided by Prof. Sang Rul Park (collected in April 2022 from the Jeju coast, Jeju National University, South Korea). Thereafter, 570 g of dried S. horneri powder was extracted three times using 70% EtOH under reflux, and residue (112.517 g) collected. We suspended 70 g of the obtained residue in water, and the soluble extract was sequentially partitioned with equal volumes of ethyl acetate (EtOAc; #270989), n-hexane (#HX0293), n-butanol (n-BuOH; #71363), and dichloromethane (CH2Cl2; #75092) were acquired from Sigma-Aldrich (St. Louis, MO, USA). Each of the subfractions was evaporated using vacuo to yield EtOAc, n-BuOH, n-Hexane, CH2Cl2, and water residues.
High-performance liquid chromatography (HPLC) analysis
During this process, 9.86 mg of S. horneri 70% EtOH extract was dissolved in 1 mL of methanol, and the resulting mixture was subjected to HPLC analysis. An HPLC system containing a quaternary pump (#50400031, LPG-3400SD, Thermo Fisher Scientific) and diode array detector (#50820010, DAD-3000, Thermo Fisher Scientific) was connected to an HPLC column (#501218097, YMC-Triart C18, 5 µm, 4.6 × 250 mm; Thermo Fisher, Waltham, MA, USA) for analysis. In the isocratic solvents, 3:97% of channels (A) and (B) containing 0.1% acetic acid (#64197) with distilled water and methanol (#34860) respectively; which were acquired from Sigma-Aldrich (St. Louis, MO, USA) were used, and maintained at a 1-mL/min flow rate during the analysis. Thereafter, 50 µL of the S. horneri extract was injected, and 50 µL of fucosterol (PubChem CID: 5281328) was subsequently used as a standard (2.5 mg/mL fucosterol (≥ 98%) in methanol; Aktin Chemicals Inc., Chengdu, China). Absorbance was measured at a wavelength of 210 nm. Sample quantity was analyzed using a calibration graph at various fucosterol concentrations (10–500 µg mL−1)21.
Analysis of S. horneri component profiles
The components of the dried S. horneri sample were analyzed using ultra-high-performance liquid chromatography-mass spectrometry (UPLC-MS). A chromatographic column (2.1 mm × 150 mm; 1.6 μm, #176003170, Waters Technologies, Milford, MA, USA) was attached to an Dionex™ Ultimate 3000 column (#57300010, Thermo Dionex, Sunnyvale, CA, USA). The following mobile phases were used: 0.1% formic acid aqueous (A) (#64186) and 0.1% formic acid acetonitrile (B) (#75058), which were produced from Sigma-Aldrich (St. Louis, MO, USA). Briefly, 80% methanol was used to dissolve the S. horneri extract. Then, for component analysis, 3 μL of a 20 mg mL−1 S. horneri extract solution was added to the device. This was the gradient program, with the mobile phase flow rate set at 0.3 mL min−1. The solvent B concentrations were 5% (0–10 min), 30% (10–30 min), 100% (30–55 min), and 1% (55–60 min). To analyze the data, Elements Viewer (version 2.1) was utilized22,68.
Antioxidant activities of S. horneri 70% EtOH extract and its subtractions
S. horneri ethanolic extract and its subfractions antioxidant activity were evaluated by scavenging DPPH (2,2-diphenyl-1-picrylhydrazyl; #1898664, Sigma-Aldrich, St. Louis, MO, USA) and ABTS (2,2'-Azino-bis (3-ethylbenzothiazoline-6-sulfonic acid; #30931670, Sigma-Aldrich, St. Louis, MO, USA) radicals in vitro as described earlier23. In brief, methanol was used to dissolve 0.20 mM of DPPH solution. Samples were generated at different concentrations (0.0625, 0.125, 0.25, 0.5, 1, 2, and 4 mg mL−1). Next, 50 µL of each sample and 150 µL of DPPH solution were added. Following a 15 min dark incubation period, the combination as a whole was tested for absorbance at 517 nm for each sample. The formula for calculating the DPPH radical scavenging activity (%) is (Acontrol−Asample)/Acontrol × 100, where Asample denotes the test sample’s absorbance and Acontrol denotes the control group’s absorbance. Half inhibitory concentration (IC50) values were also calculated using the fraction concentration relationship curve and the DPPH radical scavenging radical activity of the relevant samples.
The ABTS reagent was prepared by adding 7 mM ABTS and 140 mM potassium persulfate solutions. After a 16-h dark period to encourage the production of free radicals, the mixture was diluted with water. A 96-well plate was filled with the sample (100 µL) and the ABTS reagent (100 µL). Following an incubation period of six minutes at room temperature, the absorbance at 734 nm was determined. The equation for the analysis of ABTS scavenging activity (%) is (Blank O.D.−Sample O.D.)/Blank O.D. × 100. The corresponding sample concentration curve against the radical scavenging activity percentage (%) was used to compute the IC50.
The viability assay for RAW264.7 macrophage cell culture
RPMI-1640 medium supplemented with heat-inactivated FBS (10%), streptomycin (100 mg mL−1) (#3810740), penicillin G (100 units mL−1) (#69578), and L-glutamine (2 mM) (#56859) were available from Sigma-Aldrich (St. Louis, MO, USA), and used to culture the RAW264.7 cells at a density of 5 × 105 cells mL−1. Subsequently, the cells were then incubated at 37 °C in a humidified atmosphere containing 5% CO221. To determine S. horneri 70% EtOH extract and its subfractions on macrophage cell viability by the assessment of mitochondrial reductase function. The basic principle of this method follows that the tetrazolium salt 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) (#298931) was reduced into crystal-type formazan24. To evaluate the viability of the cells, 1 mL of RAW264.7 cells (1 × 105 cell mL−1) from each group were placed in a 96-well microplate. 50 mg mL−1 of MTT was added, and the cells containing formazan crystals were allowed to dissolve in dimethyl sulfoxide (DMSO) (#67685) for 4 h. The optical density value of the samples was then measured at 540 nm wavelength.
Nitrite analysis as an indicator of NO production
To analyze NO production in macrophages, we measured the levels of stable nitrite, which is the end-product of NO oxidation, using the reagents were acquired from Sigma-Aldrich (St. Louis, MO, USA). We determined nitrite concentrations using the Griess reaction method25. In summary, the process involved adding 100 mL of the supernatant portion of the sample to 100 mL of Griess reagent (1% [w/v] sulfanilamide (#63741) and 0.1 percent (w/v) N-(1-naphthyl)-ethylene diamine (#1465254) in 5% (v/v) phosphoric acid (#7664382) and letting it sit at room temperature for 10 min. Next, determine the absorbance at 540 nm spectrophotometrically using an ELISA plate reader.
Determination of PGE2 levels
R&D Systems, Inc. (Minneapolis, MN, USA) used a commercially available kit approach to test the PGE2 concentration levels26. In short, 48-well microplates were used to cultivate the subfractions of RAW264.7 cells after they were subjected to varying doses of S. horneri extract for three hours. The macrophage cells were then exposed to LPS (1 μg mL−1) for 24 h. Following this, the cells were centrifuged for 2 min at 13,000 × g, after which the part of the cell culture supernatants was collected and the particulate debris was eliminated. A 96-well microplate that had been pre-coated with PGE2-specific polyclonal antibodies was filled with the collected supernatant portion. After that, fill the 96-microwell plates with enzyme-linked polyclonal antibodies and allow them to react for 20 h. Finally, wash the plates to get rid of any leftover unbound enzyme-antibody solution. Add a substrate solution after that, then evaluate the color intensity at 450 nm to determine how much PGE2 is present in the sample.
Determination of TNF-α and IL-6 levels
To assess each sample’s TNF-α and IL-6 levels using an ELISA kit in accordance with R-D Systems, Inc. (Minneapolis, MN, USA)21. In summary, 48-well microplates containing 5 × 105 cells/well were seeded with RAW264.7 macrophages, and the cells were pre-incubated for three hours using different doses of S. horneri extracts and its sub-fractions. Thus, LPS treatment (1 μg mL−1) was used to excite macrophages for a duration of 24 h. After that, the supernatant portion was separated and the cytokine ELISA kit method was used to determine the amounts of TNF-α and IL-6.
Preparation of cytosolic and nuclear fractions
The cytosolic and nuclear fractions of RAW264.7 macrophage cells were obtained by lysing each fraction in compliance with the manufacturer’s instructions using Cayman’s Nuclear Extraction Kits (Cayman, Ann Arbor, MI, USA)27.
NF-κB DNA-binding activity
As directed by the manufacturer, we used the NF-κB Transcription Factor Assay kit (Cayman, Ann Arbor, MI, USA) to examine nuclear extracts in order to determine the NF-κB DNA-binding activity21.
NF-κB p65 localization and immunofluorescence analysis
For the NF-κB p65 localization, 100 μg mL−1 S. horneri extracts and their fractions were added to cultured RAW264.7 macrophages in Lab-Tek II slides chambers (#C7057, Sigma-Aldrich) for three hours. The macrophages were then exposed to 1 μg mL−1 LPS (#EC2974730, Sigma-Aldrich) for an hour. The cells were permeabilized using cold acetone after they had been fixed in formalin. After that, macrophages were probed with p65 antibodies and treated with fluorescein isothiocyanate (FITC)-labeled secondary antibody (#174, Alexa Fluor 488, Invitrogen, Carlsbad, CA, USA). Using 1 µg mL−1 of DAPI (4ʹ,6-diamidino-2-phenylindole; #28718903, Sigma-Aldrich) for 30 min, a 5 min phosphate-buffered saline wash, and the addition of 50 μL of Vectashield® mounting medium (#H1800, Vector Laboratories, Burlingame, CA, USA) were the steps involved in the staining method for macrophages. Using a Zeiss fluorescent microscope (#Provis AX70, Olympus Optical Co., Tokyo, Japan), the stained macrophages were observed and photographed28.
Experimental animals and diet
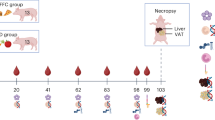
We obtained male ICR mice with an average weight of 32 g at five weeks of age from Orient Bio (Orient Bio Inc., Seongnam, Korea). The animals were housed in accordance with the Chonnam National University Guidelines for the Care and Use of Laboratory Animals. Before beginning the experiment, they took a week to acclimate to a room including a 12 h light/dark cycle and a 55.5% humidity control. After a week of acclimatization, the four groups of six mice each were randomly assigned to the mice in this study: (1) Standard diet-fed normal group (NOR), (2) high-fat/high-cholesterol diet-fed obesity control group (CON), (3) high-fat/high-cholesterol diet group fed with 1.5% S. horneri powder (SH1), and (4) high-fat/high-cholesterol diet group fed with 3% S. horneri powder (SH2). Furthermore, NOR mice were fed the following substances per kilogram of food: 200 g of casein lactic, 100 g of sucrose, 50 g of cellulose, 100 g of lard, 35 g of mineral mix, 10 g of vitamin mix, and 2 g of choline chloride (#AIN76, G-bio Co., Ltd., Gwangju, Korea) were all included. With the exception of maize starch (390 g), lard (200 g), and cholesterol (10 g), the diets (#D12451, RaonBio Co., Ltd., Gyeonggi-do, Korea) of the CON, SH1, and SH2 groups were also identical. In addition, the experimental groups’ diets included S. horneri powder (15 g kg−1 for SH1 and 30 g kg−1 for SH2).
After six weeks, the mice were euthanized by inhalation anesthesia using isoflurane (#1042, Hana Pharm Co., Ltd., Seongnam, Korea) following American Veterinary Medical Association (AVMA) guidelines29. Heparin tubes were used to collect blood samples, which were then centrifuged at 2,000 × g for 10 min at 4 °C. After centrifuging, the serum was then separated in order to evaluate its biochemical properties. After being gathered and cleansed with physiological saline, the organs were dried and weighed. They were stored at –80 °C for analysis after being quickly frozen with liquid nitrogen. The weights of fat came from different sources. For histological analysis, liver and epididymal tissues were preserved in 10% formalin (#50000, Sigma-Aldrich, St. Louis, MO, USA). Using a homogenizer, liver tissues were homogenized 1:9 in phosphate buffer (450 mM) at pH 7. Next, the mixture was centrifuged at 25,000 rpm for 20 min at 4 °C. The supernatant was gathered in order to assess the lipid and antioxidant activity. The Chonnam National University Animal Ethics Committee approved the experimental procedures employed in this study (CNU IACUC-YS-2020-9). This study is reported in compliance with the ARRIVE guidelines.
Analysis of serum biochemical indicators
Serum biochemical indicators were analyzed using an automatic blood analyzer (#06473245001, Cobas C702, Roche Diagnostics, Holliston, MA, USA). Using kit method by following the manufacturer guidelines of Sigma-Aldrich (St. Louis, MO, USA), aspartate aminotransferase (AST, #MAK055), alanine aminotransferase (ALT, #MAK052), blood urea nitrogen (BUN, #MAS008), and creatinine (#C4255) levels were analyzed. Moreover, protein was measured by the biuret reaction (#B3934), albumin was measured with a colorimetric method (#MAK124). These levels were used to assessment toxicities to liver and kidney23.
Serum lipids analysis
To determine the levels of TC (#EBCK109M), HDL-C (#EBCK222S), and TG (#EBCK261M) in serum, using the kit method of Elabscience Inc., (Houston, TX, USA). LDL-C content was computed using the formula TC – (HDL-C)—(TG/5). The formula for AI was (TC-HDL-C)/HDL-C. The formula for cardiac risk factor (CRF) was TC/HDL-C30. Elabscience Inc, Houston, TX, USA.
Antioxidant enzyme activities in liver
Using the kit of Abcam Inc., (Cambridge, MA, USA), the activities of glutathione peroxidase (GSH-Px, #ab102530), catalase (CAT, #ab83464), glutathione S-transferase (GST, #ab53942), superoxide dismutase (SOD, #ab65354), glutathione Reductase (GR, #ab83461), and glutathione (GSH, #ab239727) levels were measured31.
Western blot analysis
The levels of protein expression for iNOS, COX-2, NF-κB-p65, including Nrf2, and HO-1 were determined using Western blot analysis. To lyse the cells intact, a mixture of protease inhibitors (5 mg mL−1 pepstatin A, 5 mg mL−1 aprotinin, 1 mg mL−1 chymostatin, and 0.1 mM phenylmethylsulfonyl fluoride-PMSF) (#11206893001) was added to 20 mM of Tris–HCl buffer (#93313) at pH 7.4; the reagents were acquired from Sigma-Aldrich (St. Louis, MO, USA). For use in vivo studies, the liver tissues were homogenized in a buffer at 4 °C using a homogenizer (#Polytron CH-6010, Kinematica GmbH, Luzern, Switzerland). Once the homogenate was obtained, it was centrifuged (#MX-160, Tomy Seiko Co., Ltd., Tokyo, Japan) for 20 min at 4 °C at 12,000 rpm. Cell Signaling Technology (Danvers, MA, USA) provided the primary and secondary antibodies for Western blotting analysis. The primary antibodies SREBP-1 (#ab28481), FAS (#3180), ACC (# 3662), AMPK (#2532) and p-AMPK (#2535) protein expressions was performed on 100 μg of liver supernatant protein. The membranes were then incubated with secondary antibodies (IgG-HRP) of mouse (#7076S), and rabbit (#7074S)32. By adding protein test dye reagent (#5000006; Bio-Rad Laboratories, Hercules, CA, USA), we ascertained the protein content. After that, the protein expressions were examined using the Western blotting technique in accordance with a previously published protocol26. The quantitative analysis of Western blot band intensities was conducted using ImageJ (Image Processing and Analysis in Java) version 1.52 software (National Institutes of Health, Bethesda, MD, USA; https://imagej.net).
Real-time PCR (RT-PCR) for RNA isolation and quantification
Total RNA was extracted from liver tissue using the TRIzol RNA isolation reagent (#15596018, Invitrogen, Carlsbad, CA, USA). RNA reverse transcription was performed using the Prime Script™ 1st Strand cDNA Synthesis Kit (#2708891, Bio-Rad, CA, USA) in accordance with the manufacturer’s instructions. For quantitative RT-PCR, twenty microliters of Takara Bio Inc.’s SYBR® Premix Ex Taq were utilized. Adjusting for mRNA signals against glyceraldehyde-3-phosphate dehydrogenase (GAPDH), the findings were obtained. In this work, the primer sequences listed in Table 1 were employed33.
Histopathological analysis
For histopathological analysis, liver tissues were fixed with 10% formalin solution, washed with water, dehydrated, rendered transparent, and infiltrated. They were subsequently embedded with paraffin. After embedding, they were sectioned and stained with hematoxylin (#517282, Sigma-Aldrich, St. Louis, MO, USA) & eosin (#548243, Sigma-Aldrich, St. Louis, MO, USA) (H&E). The stained tissues were observed using a microscope (Olympus DP70, Olympus Optical Co., Tokyo, Japan) to determine liver tissue steatosis and inflammation levels in the experimental groups34.
Statistical analysis
GraphPad Prism (version 5.01; GraphPad Software, Inc., San Diego, CA, USA) was used for all statistical analyses. The findings were displayed as the average ± standard deviation (SD) of a minimum of three separate investigations. One-way analysis of variance (ANOVA) was used to analyze the data, and Tukey’s multiple test (p < 0.05) was used for comparison.
Results
Phytochemical profile of S. horneri extract and its cell viability assay
The approximate composition of the dried S. horneri samples was as follows: 14.19% moisture, 45.99% carbohydrates, 20.93% ash, 4.37% crude fat, and 14.52% crude protein. In this study, a 70% EtOH extract fraction was obtained from S. horneri dried powder, and samples of this extract were sequentially subdivided with equal volumes of n-BuOH, n-Hexane, EtOAc, CH2Cl2, and water. After vacuum evaporator of these solvents, the sequentially partitioned subfractions were collected, 6.9 g of n-Hexane (9.8%), 8.6 g of CH2Cl2 (12.1%), 0.25 g of EtOAc (0.3%), 5.1 g of n-BuOH (7.2%), and 47.15 g of water (67.3%). Thereafter, we evaluated the cytotoxicity of S. horneri 70% EtOH extract and its subfractions using RAW264.7 cells via the MTT assay as shown in Fig. 1a, b. After being seeded in microplates, the macrophages were treated with 25–200 μg mL−1 concentrations of S. horneri 70% EtOH extract and its subfractions. The results indicate that treatment with concentrations of up to 100 μg mL−1 of the 70% EtOH extract as well as its Hexane-, CH2Cl2-, and EtOAc-soluble subfractions exerted no toxic effect on cell viability. Concentrations of the BuOH- and water-soluble subfractions up to 200 μg mL−1 had no toxic effect on cell viability (Fig. 1b). Therefore, non-toxic ranges were selected for the subsequent evaluation of in vitro anti-inflammatory effect.

The S. horneri dried powder was extracted using, (a) 70% EtOH and then partitioned sequentially with equal volumes of n-hexane, CH2Cl2 (dichloromethane), EtOAc (ethyl acetate), and n-BuOH (n-butanol). (b) Cell viability assay of RAW264.7 cells. (c) The chromatography (HPLC) analysis results of S. horneri 70% EtOH extract and fucosterol as a standard compound. (d) Positive and (e) Negative mode of UPLC-QTOF-HRMS results of S. horneri extract. Data are expressed as means ± SD (n = 3); *p < 0.05 compared with the control group.
Among all the prepared fractions, the 70% EtOH extract was used to identify potential bioactive compounds using HPLC and UPLC-QTOF-HRMS analysis as shown in Fig. 1c–e. In S. horneri using an HPLC analysis UV-chromatogram at a 210 nm absorption wavelength, fucosterol was detected after a retention time of approximately 20.6 min. The fucosterol peak area for the S. horneri extract in the HPLC chromatogram was substituted with a fucosterol calibration curve to analyze the contents. The S. horneri 70% EtOH extract contained approximately 34.4 ± 0.75 µg mg−1 of fucosterol contents (C29H48O, tR 20.5, MW 412.7) are shown in Figs. 1c, and 2a.

The HPLC and UPLC-MS analysis of S. horneri dried powder, each peak represents a specific compound obtained at a specific retention time (tR). The chemical structures of the bioactive compounds are presented.
Moreover, ultra-performance liquid chromatography–tandem quadrupole time-of-flight–high-resolution mass spectrometry (UPLC-QTOF-HRMS) analysis of the positive and negative modes of the UPLC-MS analysis demonstrated significant bioactive compound diversity, as shown in Fig. 1d, e. The ion peaks intensity chromatograms in positive mode and MS information of the compounds (Figs. 1d, and 2b–h) were analyzed using Elements Viewer (version 2.1). The base peak (b) in Fig. 1d, benzophenone (C6H5COC6H5, tR 1.67, m/z 205.06 [M + Na]+) (PubChem CID:3102), peak (c) (2S,3S)-4-benzyloxy-1-(tert-butyldimethylsi1yloxy)-3-methylbutan-2-ol (C18H32O3Si, tR 25.34, m/z 325.22 [M + H]+), peak (d) denatonium (C21H29N2O+, tR 25.38, m/z 325.23 [M + H]+) (PubChem CID:15488), peak (e) D-(+)-Cellobiose octaacetate (C28H38O19, tR 30.68, m/z 701.18 [M + Na]+) (PubChem CID:107429), peak (f) 1-hydroxy Vitamin D3 (C27H44O2, tR 46.07, m/z 383.33 [M + H-H2O]+) (PubChem CID:5283731), peak (g) deoxykhivorin (C32H42O9, tR 46.10, m/z 593.27 [M + Na]+) (PubChem CID:6708722), and peak (h) pheophytin A (C55H74N4O5, tR 51.92, m/z 871.57 [M + H]+) (PubChem CID:135398712). The peak intensity chromatograms of S. horneri dried sample in negative UPLC-MS ionization mode (Figs. 1e, and 2i–l), peak (i) 5-methoxysalicylic acid sulfate (C8H8O7S, tR 3.19, m/z 246.99 [M-H]–), peak (j) palmitoleoyl 3-carbacyclic phosphatidic acid (C20H39O5P, tR 25.26, m/z 369.22 [M-H-H2O]–) (PubChem CID:10178491), peak (k) 3-Hydroxybenzoic acid sulfate (C7H6O6S, tR 5.14, m/z 216.98 [M-H-H2O]–) and peak (l) Asn-Thr-Lys (C14H27N5O6, tR 43.81, m/z 721.39 [2M-H]–) (PubChem CID:145454267) was identified.
S. horneri in vitro antioxidant activity
The antioxidant activity of the S. horneri 70% EtOH extract and its subfractions was evaluated by scavenging DPPH and ABTS radicals. This method is simple and widely used to ascertain S. horneri’s antioxidant properties. The results indicate that DPPH and ABTS radical-scavenging activities gradually increased with increasing concentrations of the S. horneri EtOH extract and its subfractions, as shown in Table 2. In particular, the CH2Cl2 subfraction yielded a relatively low IC50 value (0.28–0.44), exhibiting comparatively high scavenging ability for both DPPH and ABTS radicals. The EtOAc-soluble fraction also displayed superior potency for scavenging DPPH and ABTS radicals, yielding a relatively low IC50 value of 0.3–0.47. Overall, the CH2Cl2-subfraction exerted the most remarkable effect compared with the other fractions, suggesting that the presence of moderately polar bioactive compounds efficiently extracted by CH2Cl2 solvent.
Impact of the S. horneri extract and its subfractions on inflammatory cytokines production in LPS-treated RAW264.7 cells
NO is a short-lived free radical, and excessive NO can induce pro-inflammatory responses and inflammatory disorders35. Therefore, NO production inhibition may be of therapeutic benefit for diseases related to NO overproduction. In this study, the macrophages were pretreated with different concentrations (25–200 µg mL−1) of the S. horneri 70% EtOH extract and its subfractions and subsequently stimulated for 24 h with LPS (1 µg mL−1). The results reveal that NO levels increased in LPS-induced RAW264.7 macrophages, and they were subsequently attenuated by increasing the concentrations of the S. horneri 70% EtOH extract and its subfractions (Fig. 3a). These observations indicate that S. horneri’s bioactive compounds, fucosterol, may significantly reduce NO production in macrophages by inhibiting iNOS expression in a dose-related manner.

Effects of the S. horneri 70% EtOH extract and its subfractions on the levels of (a) NO, (b) PGE2, (c) TNF-α, (d) IL-6 in LPS-stimulated RAW264.7 cells, (e) iNOS and COX-2 protein expressions. β-actin served as the loading control. Red color dotted squares indicate the regions where β-actin bands were merged for iNOS and COX-2 panels for each group, to enhance clarity of presentation in the main figure; and (f) quantified iNOS and COX-2 protein expressions levels using ImageJ software and specific band intensity was normalized to β-actin. Data are expressed as means ± SD (n = 4). ***p < 0.001, ###p < 0.001, **p < 0.01, and *p < 0.05 compared with the LPS-treated group.
To determine whether a reduction in NO can regulate the inflammatory response, subsequent experiments were performed to analyze TNF-α, IL-6, and PGE2 levels in LPS-stimulated macrophages using the ELISA method. During this process, the cells were pretreated with S. horneri extract and its CH2Cl2- (12.1%) and water-soluble (67.3%) subfractions owing to their high yield percentage; thereafter, they were treated with LPS for 24 h. The results clarify that LPS treatment significantly induced IL-6, TNF-α, and PGE2 production, while pre-treatment with the S. horneri 70% EtOH extract and its CH2Cl2- and water-soluble subfractions significantly (p < 0.001) reduced PGE2 levels (Fig. 3b). In addition, LPS-induced pro-inflammatory cytokine (TNF-α and IL-6) expression was significantly suppressed (p < 0.001) by the 70% EtOH extract and its CH2Cl2- and water-soluble subfractions dose-dependently (Fig. 3c, d).
To determine whether the S. horneri 70% EtOH extract and its CH2Cl2- and water-soluble subfractions reduced NO and PGE2 production via protein expressions regulation, we evaluated the iNOS and COX-2 expression levels in RAW264.7 macrophage cells via Western blot analysis. Upon inflammation, vast qualities of these two inflammatory mediators were produced. These results reveal that LPS treatment stimulated iNOS and COX-2 expression in macrophages compared with the control, while pretreatment with the S. horneri 70% EtOH extract and its CH2Cl2- and water-soluble subfractions inhibited NO and PGE2 production via iNOS and COX-2 expression downregulation in a dose-dependent manner (Fig. 3e, f).
Effects of the S. horneri EtOH extract and its subfractions on NF-κB activation in LPS-treated RAW264.7 macrophages
To examine the anti-inflammatory of the S. horneri 70% EtOH extract and its CH2Cl2- and water-soluble subfractions, we analyzed NF-κB-p65 subunit protein expression and its DNA binding activity. In this study, we prepared RAW264.7 macrophages with or without the S. horneri 70% EtOH extract and its subfractions for 3 h and subsequently treated them with 1 µg mL−1 of LPS treatment for 24 h. The results indicate that pretreatment with the 70% EtOH extract (100 µg mL−1) and its CH2Cl2- (100 µg mL−1) and water-soluble (200 µg mL−1) subfractions, inhibited p65 translocation into the nucleus (Fig. 4a). Furthermore, LPS-stimulated NF-κB DNA-binding activity (4.1 ± 0.3-fold) was impaired by 100 µg mL−1 of the S. horneri 70% EtOH extract (2.38 ± 0.2-fold), 100 µg mL−1 of its CH2Cl2-soluble subfraction (1.94 ± 0.27-fold), and 200 µg mL−1 of its water-soluble subfraction (2.35 ± 0.28-fold), as shown in Fig. 4b.

Effects of the S. horneri 70% EtOH extract and its CH2Cl2- and water-soluble subfractions on (a) the activated NF-κB-p65 expressions, (b) NF-κB DNA-binding activity in LPS-stimulated RAW264.7 cells. Immunofluorescence assay for the nuclear translocation of NF-κB p65. The DAPI-stained or non-stimulated RAW264.7 group displayed blue color and no intense green fluorescence in the nucleus. In lipopolysaccharide (LPS) stimulated cells with p65 staining displayed intense green fluorescence in the nucleous. The S. horneri (c) 70% EtOH extract, (d) CH₂Cl₂-soluble, and (e) water-soluble fractions decreased nuclear localization of NF-κB p65 and reduced green fluorescence intensity within the nucleus in LPS-treated RAW264.7 cells; quantification of NF-κB p65 nuclear localization is displayed for (f) 70% EtOH extract, (g) CH₂Cl₂-soluble, and (h) water-soluble fractions. The white arrows indicate the expression of p65 with intense green fluorescence. Immunoblots of the detected NF-κB-p65 expressions was quantified using ImageJ software, and specific band intensity was normalized to DAPI. Data are expressed as means ± SD (n = 4). *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the LPS-stimulated groups.
In addition, we ascertained whether the anti-inflammatory effects of the S. horneri 70% EtOH extract (100 µg mL−1) and its subfractions (100 or 200 µg mL−1) are mediated by the NF-κB signaling pathway. In this study, we observed the nuclear localization of NF-κB p65 transcription factors in LPS-stimulated (1 µg mL−1) RAW264.7 cells using an FITC-labeled anti-p65 secondary antibody via immunofluorescence with DAPI as the nuclear stain. The results indicate that the control or non-stimulated RAW264.7 cells exhibited p65 expression in the cytosol, suggesting low constitutive nuclear translocation of NF-κB. However, after 24 h LPS stimulation, green-colored fluorescence signals increased in response to both DAPI and p65 staining owing to p65 accumulation in the nuclei, as shown in the merged images of Fig. 4c–e, and the quantification of NF-κB p65 nuclear localization was significantely decreased with various fractions of S. horneri treatments was shown in Fig. 4f–h. These results indicate that NF-κB p65 accumulated in the nucleus because of translocation from the cytoplasm, thus activating RAW264.7 macrophage immunomodulation and promoting the NO, TNF-α, and IL-6 production. Three-hour pretreatment with the S. horneri 70% EtOH extract and its subfractions before LPS stimulation resulted in markedly weaker green fluorescence in the macrophages compared with that in the individual LPS-stimulated cells; this might have resulted from p65 expression throughout the cytosol. Therefore, the S. horneri extract and its subfractions presumably regulated iNOS and COX-2 protein expressions by preventing NF-κB and inhibitor of nuclear factor kappa B alpha (IκB-α) phosphorylation in RAW264.7 cells.
Effects of the S. horneri EtOH extract and its subfractions on HO-1 protein expression and Nrf2 nuclear translocation in RAW264.7 macrophages
We conducted western blotting analysis to determine whether the S. horneri 70% EtOH extract (100 µg mL−1) and its n-Hexane- (100 µg mL−1), CH2Cl2- (100 µg mL−1), and water-soluble (200 µg mL−1) subfractions exert an anti-inflammatory effect by regulating HO-1 protein expression in LPS-treated RAW264.7 macrophages. In this study, we used cobalt protoporphyrin (CoPP) to induce HO-1 expression as a positive control. The results reveal that the CH2Cl2-soluble subfraction of S. horneri treatment significantly increased HO-1 protein expression compared with other S. horneri extracts (Fig. 5a). This indicates that the S. horneri CH2Cl2-soluble subfraction potentially suppresses the inflammatory response in macrophages by initiating the Nrf2/HO-1 pathway. Accordingly, we examined the effect of CH2Cl2-soluble subfraction treatment on the activation of Nrf2 nuclear translocation. During this process, RAW264.7 macrophages were treated with the CH2Cl2-soluble subfraction (100 µg mL−1) over different time durations (0.5–1.5 h), and the Nrf2 protein expression levels were subsequently analyzed via western blotting. The results shown in Fig. 5b indicate that CH2Cl2-soluble subfraction treatment induced Nrf2 nuclear translocation in a time-dependent manner.

Effects of the (a) S. horneri 70% EtOH extract and its CH2Cl2-, Hexane-, and water-soluble subfractions on HO-1, as well as (b) the effects of the CH2Cl2-soluble subfraction on Nrf2 activation in RAW264.7 macrophages. HO-1 mediates the CH2Cl2-soluble subfraction-induced suppression of LPS-stimulated (c) nitrite content, (d) TNF-α, and (e) NF-κB DNA-binding activity in RAW264.7 cells. Data are expressed as means ± SD (n = 4). **p < 0.01, ***p < 0.001, #p < 0.05, ##p < 0.01, and ###p < 0.001 compared with the non-treated group.
Furthermore, we determined whether the S. horneri CH2Cl2-soluble subfraction has an anti-inflammatory effect by stimulating the Nrf2/HO-1 pathway. Accordingly, NO and TNF-α concentrations and the NF-κB-binding activity were examined in the presence of tin protoporphyrin (SnPP) as an HO-1 inhibitor. In this study, the cells were treated with the CH2Cl2-soluble subfraction (100 µg mL−1) for 3 h, with or without SnPP (50 μM), and subsequently stimulated with 1 µg mL−1 LPS treatment for 24 h. The results in Fig. 5c–e indicate that exclusive treatment with the S. horneri CH2Cl2-soluble subfraction significantly inhibited NO and TNF-α production, including NF-κB DNA-binding activity, in LPS-treated RAW264.7 macrophages. However, these inhibitory effects attenuated after SnPP addition. In contrast, SnPP treatment did not affect the NO and TNF-α levels and NF-κB DNA-binding activity of LPS-stimulated macrophages. These results suggest that the S. horneri CH2Cl2-soluble subfraction exhibited the anti-inflammatory effects of LPS-treated macrophages by inducing HO-1 expression.
Effects of S. horneri on physiological, serological, and histological examination in HFD-fed obese mice
We assessed the food efficiency ratio (FER) and body weight (BW) of ICR mice across all experimental groups in order to investigate the impact of S. horneri on body weight increase. Compared to the other groups, the obese CON group fed a high-fat diet (HFD) gained more weight (Fig. 6a, b). However, when compared to the HFD-induced obese CON group, there was a substantial (p < 0.05) reduction in body weight gain of 4.59% and 8.28% in the S. horneri treated SH1 and SH2 groups, respectively. There was no significant difference in food intake among the HFD-fed groups, despite a significant difference in food intake between the NOR and HFD-fed groups. However, a similar trend in the FER was noted in conjunction with the increase in body weight (Fig. 6b). Furthermore, compared to the NOR group, the CON group’s weight of the liver and all other adipose tissues (perirenal, retroperitoneal, mesenteric, and epididymal) increased significantly. Additionally, the S. horneri therapy significantly decreased the weight of the liver and fat tissues in a dose-dependent manner. Figure 6c, d shows that the weight changes in the kidney and heart were not statistically significant (p < 0.05). The HFD-fed obese mice showed signs of hyperlipidemia, as seen by the higher levels of blood TC, TG, and LDL cholesterols as well as the decreased HDL-cholesterol in the CON group of mice. Furthermore, in HFD-fed obese mice, S. horneri treatment (15 and 30 g kg−1 body weight) markedly decreased the serum cholesterols (TC, TG, and LDL), and elevated HDL levels (Fig. 6e, f). Moreover, S. horneri treatment significantly lowered the arteriosclerosis index (AI), and cardiovascular risk factor (CVRF) values in HFD-fed obese mice (Fig. 6f).

Effects of dietary S. horneri on (a-d) initial body weight, food intake, body and organs weight changes including adipose tissues, (e) triglycerides and LDL-cholesterol levels, (f) total and HDL cholesterol levels with AI and CVRF. Effect of S. horneri on histology of (g) mouse liver and (h) epididymal fat in all experimental groups, with (i) adipocyte area. Data are expressed as means ± SD (n = 6). Values not sharing a common letter are significantly different at p < 0.05 by Tukey’s multiple comparison test.
The histological alterations in the liver and adipose tissue of HFD-fed mice treated with S. horneri are summarized in Fig. 6g–i. The liver tissues of NOR and obese mice were stained with H&E, and any structural damage was then observed. The obese CON group exhibited signs of inflammation, hepatocyte cytoplasmic vacuoles, and severe micro- and macro-steatosis with hepatocyte atrophy. Supplementation with 1.5% and 3% S. horneri reduced or eliminated fat droplet production in the liver tissues of HFD-fed mice. The hepatocyte architecture and hepatic tissue morphology of mice treated with S. horneri were similar to those of mice in the NOR group. The treatment of S. horneri reduced these histological changes in the liver tissues of the HFD group in a dose-dependent manner (Fig. 6g). The adipocytes in the CON obesity group were significantly larger, according to histological alterations of the epididymal tissues. Nevertheless, compared to the CON group, the adipocytes in the groups of ICR mice treated with S. horneri (at low and high dosages) were much smaller (Fig. 6h). In comparison to the NOR group, the CON group’s total adipocyte area also rise (Fig. 6i). However, the overall adipocyte area was dramatically reduced in a dose-dependent manner after the S. horneri therapy (Fig. 6i).
By examining biochemical markers in serum samples from experimental animals given S. horneri treatment, we assessed liver and kidney functioning (Table 3). The NOR group’s protein and albumin levels were noticeably higher than those of the HFD-fed animals. When compared to the CON group, the activity of the marker enzymes ALT and AST was considerably lower in the S. horneri-treated groups. No significant change was noted in the BUN levels as an indicator of renal function across the HFD-diet groups, while creatinine levels in the NOR group were not substantially different from those in the S. horneri treated SH1 and SH2 groups.
Changes in the activities of antioxidant enzymes in liver tissues
The purpose of this work is to determine whether administering S. horneri to HFD-fed ICR mice increases the activity of liver antioxidant enzymes. The GPx, GST, GR, CAT, SOD enzyme activities and GSH levels were reduced in the obesity CON group as compared to the NOR group. Figure 7a–f illustrates the increased activity of these antioxidant enzymes in the groups supplemented with S. horneri powder. The NOR, CON, and SH1 groups did not significantly differ in their GST antioxidant activity; however, the GST antioxidant activity was highest in the SH2 group. A high-fat diet has been shown to reduce the antioxidant activity of the liver. These findings imply that adding S. horneri to a supplement increases antioxidant activity, which in turn inhibits free radicals.

Effects of dietary S. horneri treatment with low (SH1) and high (SH2) dose on hepatic antioxidant enzyme activities in HFD induced obese ICR mice. Each value represents the mean ± SD (n = 6). Values not sharing a common letter are significantly different at p < 0.05 by Tukey’s multiple comparison test.
S. horneri activates AMPK signaling pathway in liver tissues
We investigated whether S. horneri affects the expression of genes and proteins involved in the metabolism of fatty acids in the liver tissues of obese mice. Western blot analysis results revealed that, in comparison to control HFD-fed obese mice, S. horneri therapy significantly enhanced both total AMPK and phosphorylated AMPK (p-AMPK) levels (Fig. 8a, b). Specifically, the effect of S. horneri treatment led to a significant increase the phosphorylation ratio of AMPK (p-AMPK/AMPK) when compared to the obese control group, indicating enhanced AMPK activation. Additionally, S. horneri treatment markedly raised the expression of genes involved AMPK activation (Fig. 8d), suggesting that S. horneri enhances the AMPK signaling pathway. Furthermore, in a dose-dependent way, S. horneri treatment significantly suppressed the protein expression of key lopogenic transcription factors and enzymes, including SREBP1, FAS, and ACC (Fig. 8c). In addition, S. horneri treatment markedly raised the expression of genes involved in fatty acid oxidation (Fig. 8e; CPT) and dramatically decreased the expression of genes involved in fatty acid synthesis (Fig. 8f–h; FAS, ACC, and SCD). These findings revealed that S. horneri treatment exerts anti-obesity effects by activating the AMPK signaling pathway, which subsequently inhibits lipogenic enzymes activity and promotes fatty acid oxidation.

S. horneri treatment modulates lipid metabolism related proteins and gene expressions in HFD-induced obese mice. In vivo effect of S. horneri treatment on (a) Representative blots for AMPK, p-AMPK, SREBP-1, FAS and ACC. (b) Quantitaive analysis of AMPK, p-AMPK and p-AMPK/ AMPK protein expression levels in liver tissues. (c) Quantitaive analysis of SREBP-1, FAS and ACC protein expression levels in liver tissues. Results are expressed as the ratio to β-actin. Image J was applied for the quantification of the protein bonds. Effect of S. horneri on relative mRNA expression of lipogenic genes in HFD-induced obese mice: (d) AMPK, (e) CPT, (f) FAS, (g) ACC, and (h) SCD gene expressions was determined using RT-PCR. Each value represents the mean ± SD (n = 4). Values not sharing a common letter are significantly different at p < 0.05 by Tukey’s multiple comparison test.
Discussion
Phlorotannins, fucoxanthin, alginic acid, fucoidans, laminarin, loliolide, and other bioactive substances are abundant in marine algae, which are also valuable sources of food and functional components20,36,37,38,39. Bioactive substances found in S. horneri, such as benzophenone, denatonium, phenolic acids, pheophytin, and vitamin D3, have been shown to possess advantageous physiological actions, including anti-inflammatory, anti-obesity, and antioxidant properties40,41,42,43,44. By lowering body weight and adiposity, the phenolic compounds have been demonstrated to decrease obesity by improving lipid metabolism. Moreover, they mitigate metabolic problems linked to obesity, including hepatic steatosis, hyperlipidemia, and cardiometabolic diseases45. Moreover, the bioactive compound fucosterol (trans-24-ethylidene cholesterol) is belonging to the sterol group. It contains steroidal rings, and exhibits hydrophobic interaction with alanine (Ala), methionine (Met), phenylalanine (phe) via pi-alkyl interactions. The fucosterol side chain also interacts various hydrophobically with leucine (Leu), isoleucine (ilu), phenylalanine (phe), tryptophane (Trp), among others. Although these bioactive chemicals have been shown to have a number of positive effects, no research has looked into how they affect inflammatory disorders, particularly obesity brought on by a high-fat diet.
According to the experimental results, the antioxidant properties of S. horneri extract and its subfractions displayed similar trends for both ABTS and DPPH radical-scavenging ability. This indicates that the S. horneri extract and its subfractions may contain hydroxyl groups that potentially provide an electron-donating ability to prevent free radical toxicity. In addition, Sargassum species and other brown seaweeds are known to contain a variety of bioactive compounds, including phenols, fucosterol, fucoxanthin, and phlorotannins19,20,39. The activated RAW264.7 cells by LPS-treatment can produce NO, TNF-α, PGE2, and IL-6, among others, leading to several complications, such as migraines, rheumatoid arthritis, multiple sclerosis, and other autoimmune diseases20,39. The aforementioned cytokines upregulate COX-2 and iNOS enzyme activities, increasing pro-inflammatory mediator (NO and PGE2) production.
Therefore, mitigating inflammatory disorders by inhibiting COX-2 and iNOS expression is a promising method for analyzing anti-inflammatory drugs. In our study, the experimental results (Fig. 3a–f) indicate that the LPS treatment group induced iNOS and COX-2 expression compared with the non-LPS treatment group. Nonetheless, pretreatment with the S. horneri extract and its CH2Cl2- and water-soluble subfractions inhibited COX-2 and iNOS protein expression, including cytokine (IL-6 and TNF-α) production. It indicates the CH2Cl2- and water-soluble subfraction having ability to extract moderately polar bioactive compounds from S. horneri with selectivity is responsible for its bioactivity. According to Lefebvre et al.46, the CH2Cl2 solvent system is very good in extracting semi-polar substances, such as particular phlorotannins and sterol derivatives with the best lipophilicity for biological activity. Previous studies have reported that phlorotannins, fucosterol and other sterol compounds are commonly found in brown seaweed CH2Cl2 fractions, also exhibit strong antioxidant, anti-inflammatory effects by modulating the NF-κB and Nrf2 pathways47,48,49,50. Moreover, CH2Cl2-soluble subfraction is especially valuable for therapeutic applications due to its bioactive property with low yield (12.1%), which indicates a concentrated presence of these potent bioactive compounds. These results reveal that S. horneri effectively suppresses PGE2 and NO production by inhibiting COX-2 and iNOS protein expression in LPS-treated RAW264.7 macrophages, without affecting cell viability. They also indicate that S. horneri is a promising source of bioactive compounds that exhibit anti-inflammatory potential by inhibiting LPS-induced inflammatory mediator and cytokine production through blocking NF-κB translocation into the nucleus.
During the inflammatory response, an NF-κB dimer containing REL-associated proteins (p65) is initiated and translocated from the cytosol into the nucleus to activate inflammatory gene transcriptions and adhesion factors, directly leading to inflammation7,51,52. This implies that p65 protein expression is a prospective target for the development of specific NF-κB-pathway-blocking drugs or reagents. Therefore, we measured NF-κB p65 protein expression in LPS-treated RAW264.7 cells using western blot analysis, and quantification of NF-κB p65 nuclear localization using immunofluorescence. The results demonstrate that LPS alone enhanced NF-κB-p65 protein translocation into the nucleus, and pretreatment with the S. horneri 70% EtOH extract and its CH2Cl2- and water-soluble subfractions downregulated NF-κB protein expression in the nuclei, including p65 DNA-binding activity, as shown in Fig. 4a–h. This indicates that a number of bioactive coumpounds caontained S. horneri extract and its subfractions, may responsible for the anti-inflammatory mechanism, which may entail blocking the NF-κB pathway. According to the earlier research, S. horneri extracts significantly reduces the NF-κB DNA binding activity in LPS-induced RAW264.7 macrophages53. However, S. horneri is abundant in fucoxanthin, fucosterol, and other bioactive sterols that have been attenuate the inflammatory responses by inhibiting the nuclear translocation of NF-κB p65 and reducing the phosphorylation of IκB-α in macrophages53,54,55,56. Our findings that S. horneri extract regulates iNOS and COX-2 protein expressions by inhibiting NF-κB and IκB-α phosphorylation are supported by the synergistic effects of these several bioactive substances, which probably contribute to the potent anti-inflammatory activity we observed in our investigation.
In this study, we found that the antioxidant and anti-inflammatory effect of S. horneri regulate Nrf2/HO-1 expression in RAW264.7 macrophages. Nrf2 is a “master” controller of the cytoprotective program against inflammation and related diseases, and it is usually located in the cytoplasm. Nrf2/HO-1 pathway activation inhibits pro-inflammatory mediator and cytokine production via antioxidant activity in LPS-stimulated macrophages8,9. To maintain the cell’s microenvironment-dependent homeostasis, HO-1 is an important enzyme for producing antioxidants via heme group degradation57. It is an inducible and rate-limiting enzyme that catalyzes and degrades the heme group into CO, Fe2+, and biliverdin, which is subsequently transformed into bilirubin by biliverdin reductase. The CO and bilirubin generated via inflammatory heme group degradation possess antioxidant and anti-inflammatory properties that play an important role in providing an HO-1 anti-inflammatory effect in LPS-stimulated macrophages57,58. Meanwhile, CO suppresses LPS-induced NO, TNF-α, PGE2, and IL-6 formation by suppressing the NF-κB pathway in macrophages (Fig. 9). Similarly, this study’s results also reveal that S. horneri treatment induced HO-1 expression and was responsible for the antioxidant and anti-inflammatory effects (Fig. 5). Hence, our findings suggest that S. horneri extract and its subfractions exert anti-inflammatory and antioxidant effects via Nrf2/HO-1 pathway activation.

Proposed mechanism for the anti-inflammatory effect of S. horneri through the NF-κB and Nrf2/HO-1 signaling pathway.
Chronic obesity is characterized by elevated TC and TG levels, which can lead to a number of metabolic disorders because of aberrant pancreatic and liver function. In addition to causing aberrant lipid metabolism, obesity brought on by a high-fat diet also inflames the body. Since the HFD-induced obesity mice model in our study closely resembles the underlying etiology of obesity in humans, it is regarded as highly translational45. Furthermore, the HFD-induced obesity model offers a trustworthy way to investigate obesity since it is easy to make animals gain weight when they are fed a high-fat diet. We now studied the effects of giving mice an oral dose of S. horneri for six weeks together with a high-fat diet. The outcomes showed that the S. horneri treatment dramatically decreased hepatotoxicity indicators and restored alterations in serum lipids brought on by obesity. Moreover, treatment with S. horneri markedly reduced the expression of lipogenic genes in the liver tissue.
According to our findings, taking supplements containing S. horneri may help stop excess weight gain without reducing overall food intake, which is linked to weight increase. An excessive increase in adipose tissue mass and the liver can be characterized as obesity45. Thus, we measured the mass of adipose tissue and various organs in this study. We found that S. horneri (1.5% and 3%) treatment prevented a significant increase in the weight of organs and mesenteric, epididymal, retroperitoneal, perirenal, and adipose tissues among HFD-fed mice compared to the NOR group. In this investigation, supplementing HFD with S. horneri was found to considerably attenuate the blood TC, TG, LDL, AI, and CVRF, in addition to reducing body and adipose tissue weight. The primary process underlying the lower levels of TC, TG, LDL, and AI may be the extract’s constituents breaking down lipids and restoring normal lipid metabolism59. The decrease in serum levels of TC, TG, and LDL suggests a removal of circulating lipids, which may shield cells from harm caused by free radicals. AI is thought to be a marker for a number of cardiovascular diseases, and the value of AI is closely correlated with the development of the cardiovascular risk factor (CVRF)45. As a result, the AI and CVRF values of the groups treated with S. horneri seem advantageous, showing lipid-lowering characteristics linked to a putative cardioprotective effect. Furthermore, a six-week high-fat diet (HFD) in mice was shown to increase hepatic steatosis with lipid accumulation and increase adipocyte cell size13, while treatment with S. horneri significantly decreased hepatic steatosis with lipid accumulation in liver and adipose tissues (Fig. 6g–i). These findings were supported by histopathological observations of liver and adipose tissues. Moreover, S. horneri treatment dramatically reduced serum ALT, BUN, creatinine, and AST levels in relation to renal and liver metabolic markers in obese mice13.
According to this study, the liver tissue of HFD-induced obese mice had increased oxidative stress and inflammation, which led to a decrease in the activity of antioxidant enzymes like GPx, GR, GST, SOD, and CAT as well as a drop in GSH levels. The rapid and exhausting use of antioxidant enzymes stored to combat the production of free radicals in obesity may be the reason for this decrease in antioxidant enzyme activity; obesity-associated oxidative stress involves mechanisms that can exacerbate the deposition of fat in organs and the generation of free radicals60. In groups treated with S. horneri, the decrease in hepatic antioxidant enzyme activity was effectively reversed in a dose-dependent manner. Western blotting and RT-PCR were used to assess alterations in the expression of genes linked to fat metabolism in the liver tissue in order to clarify the mechanism by which S. horneri carries out its anti-obesity activity (Fig. 7).
Liver and adipose tissue have significant levels of the pro-adipogenic transcription factor SREBP1, which has the ability to directly control the expression of the lipogenic genes FAS and ACC, which are involved in lipid metabolism45,61. In obesity, AMPK function is dysregulated, leading to elevated ACC and SREBP1 expressions, which in turn blocks the β-oxidation62. According to the current in vivo investigation, HFD group had considerably higher levels of SREBP-1, FAS, ACC protein expressions, as well as increased mRNA expression of FAS, ACC, and SCD. However, treatment with S. horneri decreased these proteins and gene expressions by activated AMPK phosphorylation in the liver tissues while enhancing CPT expressions (Fig. 8). These findings suggest that, rather than a decreased hepatic lipid absorption, the anti-obesity activity of S. horneri may be due to a diminished fatty acid metabolism and a decrease in lipogenesis genes. Furthermore, S. horneri has the ability to regulate the activities of malonyl-CoA and CPT1, which are essential for the β-oxidation of fatty acids, via the AMPK signaling pathway. Notably, S. horneri treatment activating AMPK while simultaneously reducing the expression of FAS in the liver tissue of obese mice to reduce the accumulation of fat (Fig. 10).

Proposed mechanism for the effect of S. horneri on lipid accumulation in hepatic and adipocytes. S. horneri treatment suppresses fatty acid synthesis by activating AMPK, which leads to the down regulation of lipogenesis-related transcription factor SREBP-1, and downregulated the lipogenic enzymes such as ACC, and FAS. In addition, the activation of AMPK may also enhance the fatty acid oxidation by increasing the CPT-1 expressions.
Recent studies suggested that S. horneri is rich in fucoxanthin, fucosterol and other sterol compounds that enhances AMPK phosphorylation through the LKB1-AMPK or AMPK signaling pathway in hepatocytes, leading to increased fatty acid oxidation and decreased lipogenesis63,64,65. Moreover, S. horneri containing flavonoids, polysaccharides, fucoidan, alginate, polyphenolic compounds, including phlorotannins, which activates AMPK by inhibiting mitochondrial ATP synthesis, thereby increasing the AMP/ATP ratio and promoting AMPK activation22,66,67,68. It clarifies how the activation of AMPK by the bioactive compounds of S. horneri provides a comprehensive mechanism underlying the observed anti-obesity effects and metabolic improvements in our high-fat diet-induced obesity model22,68). It was clear from the biochemical and gene expression characteristics that S. horneri prevented obesity brought on by a high-fat diet.
Conclusion
Our findings confirm that S. horneri and its extracts containing bioactive compounds including phlorotannins, fucoxanthin, fucosterol, and other phenolic compounds exhibits anti-inflammatory and anti-obesity properties. The S. horneri extracts potentially reduces NO and PGE2 levels by suppressing iNOS and COX-2 protein expressions. They also inhibit the production of pro-inflammatory cytokines, such as TNF-α and IL-6, by suppressing NF-κB p65 nuclear translocation in LPS-treated RAW264.7 macrophages. In addition, S. horneri significantly attenuates inflammation by suppressing the initiation of the NF-κB/iNOS pathway and promoting Nrf2/HO-1 expression. It is evidence that S. horneri extracts particularly CH2Cl2-soluble fraction effectively reduced inflammation and other related disorders. The anti-obesity effect of S. horneri in HFD-induced obese mice involves a significant inhibition of weight gain, fat accumulation, and hepatic steatosis. Moreover, S. horneri’s treatment regulates the lipid metabolism by activating AMPK mechanism, and decreases the expression of hepatic lipogenic enzymes, such as SREBP-1, FAS, and ACC while enhancing CPT experession, thereby promoting fatty acid β-oxidation and inhibiting lipogenesis. In addition, S. horneri’s treatment significantly reduced hepatotoxic serum markers (ALT, AST) and restored antioxidant enzyme activities (GPx, GST, GR, CAT, SOD) including GSH levels in liver tissue. The histopathological analysis also provided a morphological evidence to supporting the biochemical findings by reducing lipid accumulation in liver and adipose tissues. Importantly, S. horneri and its extracts did not exhibit any cytotoxic effects, rendering it a safe resource for nutraceutical applications. This study is the first to demonstrate that the bioactive compounds in S. horneri exhibit strong antioxidant, anti-inflammatory, and anti-obesity properties and providing scientific evidence for its potential development a natural therapeutic agent for preventing inflammation and obesity-related diseases. These findings suggested that the S. horneri can be used as a comparatively safe and effective drug for the treatment of inflammation and obesity related diseases.
Data availability
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
References
Shen, J. et al. Regulating effect of baicalin on IKK/IKB/NF-kB signaling pathway and apoptosis-related proteins in rats with ulcerative colitis. Int. Immunopharmacol. 73, 193–200 (2019).
Choi, Y.-H. & Myung, N.-Y. The anti-inflammatory mechanism of the peel of Zanthoxylum piperitum DC is by suppressing NF-κB/Caspase-1 activation in LPS-induced RAW264.7 cells. Korean J. Plant Resourc. 2019, 256 (2019).
Fang, Y., Yang, L. & He, J. Plantanone C attenuates LPS-stimulated inflammation by inhibiting NF-κB/iNOS/COX-2/MAPKs/Akt pathways in RAW 264.7 macrophages. Biomed. Pharmacother. 143, 112104 (2021).
Han, B. H. et al. Hwangryunhaedoktang exerts anti-inflammation on LPS-induced NO production by suppressing MAPK and NF-κB activation in RAW264.7 macrophages. J. Integr. Med. 15, 326–336 (2017).
Wang, Q. et al. Targeting NF-kB signaling with polymeric hybrid micelles that co-deliver siRNA and dexamethasone for arthritis therapy. Biomaterials 122, 10–22 (2017).
Druszczyńska, M., Godkowicz, M., Kulesza, J., Wawrocki, S. & Fol, M. Cytokine receptors—regulators of antimycobacterial immune response. Int. J. Mol. Sci. 23, 1112 (2022).
Hwangbo, H. et al. Anti-inflammatory effect of auranofin on palmitic acid and LPS-induced inflammatory response by modulating TLR4 and NOX4-mediated NF-κB signaling pathway in RAW2647 Macrophages. Int. J. Mol. Sci. 22, 5920 (2021).
Saha, S., Buttari, B., Panieri, E., Profumo, E. & Saso, L. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules 25, 5474 (2020).
Wen, Y.-L., He, Z., Hou, D.-X. & Qin, S. Crocetin exerts its anti-inflammatory property in LPS-induced RAW2647 cells potentially via modulation on the crosstalk between MEK1/JNK/NF-κB/iNOS pathway and Nrf2/HO-1 pathway. Oxid. Med. Cell. Longev. 2021, 2563 (2021).
Esser, N., Legrand-Poels, S., Piette, J., Scheen, A. J. & Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 105, 141–150 (2014).
Sanyal, A. et al. Interplay between obesity-induced inflammation and cGMP signaling in white adipose tissue. Cell Rep. 18, 225–236 (2017).
Li, J., Wu, H., Liu, Y. & Yang, L. High fat diet induced obesity model using four strains of mice: Kunming, C57BL/6, BALB/c and ICR. Exp. Anim. 69, 326–335 (2020).
Meng, X. et al. Effects of Jowiseungki-tang on high fat diet-induced obesity in mice and functional analysis on network pharmacology and metabolomics analysis. J. Ethnopharmacol. 283, 114700 (2022).
Khateeb, S., Albalawi, A. & Alkhedaide, A. Diosgenin modulates oxidative stress and inflammation in high-fat diet-induced obesity in mice. Diabetes Metabol. Syndrome Obes. Targets Therapy 2022, 1589–1596 (2022).
Panchal, S. K. et al. High-carbohydrate, high-fat diet–induced metabolic syndrome and cardiovascular remodeling in rats. J. Cardiovasc. Pharmacol. 57, 611–624 (2011).
Upadhyay, R. K. Emerging risk biomarkers in cardiovascular diseases and disorders. J. Lipids 2015, 256 (2015).
Hedayatnia, M. et al. Dyslipidemia and cardiovascular disease risk among the MASHAD study population. Lipids Health Dis. 19, 1–11 (2020).
Hyung, J.-H., Ahn, C.-B., Kim, B. I., Kim, K. & Je, J.-Y. Involvement of Nrf2-mediated heme oxygenase-1 expression in anti-inflammatory action of chitosan oligosaccharides through MAPK activation in murine macrophages. Eur. J. Pharmacol. 793, 43–48 (2016).
Sanjeewa, K. K. A. et al. Anti-inflammatory activity of a sulfated polysaccharide isolated from an enzymatic digest of brown seaweed Sargassum horneri in RAW 264.7 cells. Nutr. Res. Practice 11, 3–10 (2017).
Han, E.-J. et al. Sargachromenol purified from Sargassum horneri inhibits inflammatory responses via activation of Nrf2/HO-1 signaling in LPS-stimulated macrophages. Mar. Drugs 19, 497 (2021).
Ko, W. et al. The anti-oxidative and anti-neuroinflammatory effects of sargassum horneri by heme oxygenase-1 induction in BV2 and HT22 cells. Antioxidants 10, 859 (2021).
Lee, E. J., Lee, S., Jang, H.-J. & Yoo, W. Loliolide in Sargassum horneri alleviates ultrafine urban particulate matter (PM 01)-induced inflammation in human RPE cells. Int. J. Mol. Sci. 25, 162 (2023).
Kim, M. J. et al. Anti-obesity and Anti-hyperglycemic Effects of Meretrix lusoria Protamex Hydrolysate in ob/ob mice. Int. J. Mol. Sci. 23, 4015 (2022).
Berridge, M. V. & Tan, A. S. Characterization of the cellular reduction of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch. Biochem. Biophys. 303, 474–482 (1993).
Titheradge, M. A. Nitric Oxide Protocols 83–91 (Springer, 1998).
Ko, W. et al. Inhibitory effects of alternaramide on inflammatory mediator expression through TLR4-MyD88-mediated inhibition of NF-кB and MAPK pathway signaling in lipopolysaccharide-stimulated RAW264.7 and BV2 cells. Chemico-Biol. Interact. 244, 16–26 (2016).
Jeon, H. L. et al. Anti-inflammatory and antioxidant actions of N-arachidonoyl serotonin in RAW264.7 cells. Pharmacology 97, 195–206 (2016).
Kim, D.-C. et al. Prenylated flavonoids from Cudrania tricuspidata suppress lipopolysaccharide-induced neuroinflammatory activities in BV2 microglial cells. Int. J. Mol. Sci. 17, 255 (2016).
Underwood, W. & Anthony, R. AVMA guidelines for the euthanasia of animals: 2020 edition. Retrieved on March 2013, 2020–2021 (2020).
Zhang, M. et al. Beneficial effects of taurine on serum lipids in overweight or obese non-diabetic subjects. Amino Acids 26, 267–271 (2004).
Park, S. Y. et al. In vivo hepatoprotective effects of a peptide fraction from krill protein hydrolysates against alcohol-induced oxidative damage. Mar. Drugs 17, 690 (2019).
Minakawa, M., Miura, Y. & Yagasaki, K. Piceatannol, a resveratrol derivative, promotes glucose uptake through glucose transporter 4 translocation to plasma membrane in L6 myocytes and suppresses blood glucose levels in type 2 diabetic model db/db mice. Biochem. Biophys. Res. Commun. 422, 469–475 (2012).
Wang, Y., Nair, S. & Gagnon, J. Herring Milt and Herring Milt protein hydrolysate are equally effective in improving insulin sensitivity and pancreatic beta-cell function in diet-induced obese-and insulin-resistant mice. Mar. Drugs 18, 635 (2020).
Kim, K.-N. et al. 2, 4, 6-Trihydroxybenzaldehyde, a potential anti-obesity treatment, suppressed adipocyte differentiation in 3T3-L1 cells and fat accumulation induced by high-fat diet in C57BL/6 mice. Environ. Toxicol. Pharmacol. 39, 962–968 (2015).
Ren, J. et al. Anti-inflammatory effects of Aureusidin in LPS-stimulated RAW264.7 macrophages via suppressing NF-κB and activating ROS-and MAPKs-dependent Nrf2/HO-1 signaling pathways. Toxicol. Appl. Pharmacol. 387, 114846 (2020).
Li, Y. et al. Brown algae carbohydrates: structures, pharmaceutical properties, and research challenges. Mar. Drugs 19, 620 (2021).
Silva, A. et al. Recent advances in biological properties of brown algae-derived compounds for nutraceutical applications. Crit. Rev. Food Sci. Nutr. 64, 1283–1311 (2024).
Fernando, I. P. S. et al. (−)-Loliolide isolated from Sargassum horneri abate UVB-induced oxidative damage in human dermal fibroblasts and subside ECM degradation. Mar. Drugs 19, 435 (2021).
Han, E.-J. et al. (–)-Loliolide isolated from Sargassum horneri suppressed oxidative stress and inflammation by activating Nrf2/HO-1 signaling in IFN-γ/TNF-α-stimulated HaCaT keratinocytes. Antioxidants 10, 856 (2021).
Khanum, S. A., Shashikanth, S. & Deepak, A. Synthesis and anti-inflammatory activity of benzophenone analogues. Bioorg. Chem. 32, 211–222 (2004).
Jack, B. U. et al. Adipose tissue as a possible therapeutic target for polyphenols: a case for Cyclopia extracts as anti-obesity nutraceuticals. Biomed. Pharmacother. 120, 109439 (2019).
Zagorchev, P. I., Kokova, V. Y., Apostolova, E. G. & Draganova-Filipova, M. N. Denatonium benzoate decreases the effect of histamine in vitro and in rats. Trop. J. Pharm. Res. 19, 1879–1885 (2020).
Bento-Silva, A. et al. Factors affecting intake, metabolism and health benefits of phenolic acids: do we understand individual variability?. Eur. J. Nutr. 59, 1275–1293 (2020).
Kusmita, L., Puspitaningrum, I. & Limantara, L. Identification, isolation and antioxidant activity of pheophytin from green tea (Camellia sinensis (L.) Kuntze). Procedia Chem. 14, 232–238 (2015).
Boby, N. et al. Microbiota modulation and anti-obesity effects of fermented Pyrus ussuriensis Maxim extract against high-fat diet-induced obesity in rats. Biomed. Pharmacother. 154, 113629 (2022).
Lefebvre, T., Destandau, E. & Lesellier, E. Selective extraction of bioactive compounds from plants using recent extraction techniques: a review. J. Chromatogr. A 1635, 461770 (2021).
Omoarelojie, L. O. & van Staden, J. Perspectives on the potentials of phlorotannins in enhancing phytoremediation performance. J. Plant Growth Regul. 43, 2972–2992 (2024).
Sohn, S.-I. et al. Phytosterols in seaweeds: an overview on biosynthesis to biomedical applications. Int. J. Mol. Sci. 22, 12691 (2021).
de Jong, D. L., Timmermans, K. R., de Winter, J. M. & Derksen, G. C. Effects of nutrient availability and light intensity on the sterol content of Saccharina latissima (Laminariales, Phaeophyceae). J. Appl. Phycol. 33, 1101–1113 (2021).
Santos, S. A. et al. Lipophilic fraction of cultivated Bifurcaria bifurcata R. Ross: detailed composition and in vitro prospection of current challenging bioactive properties. Mar. Drugs 15, 340 (2017).
Chen, H.-B. et al. Anti-inflammatory activity of coptisine free base in mice through inhibition of NF-κB and MAPK signaling pathways. Eur. J. Pharmacol. 811, 222–231 (2017).
Geng, L., Hu, W., Liu, Y., Wang, J. & Zhang, Q. A heteropolysaccharide from Saccharina japonica with immunomodulatory effect on RAW 264.7 cells. Carbohydr. Polymers 201, 557–565 (2018).
Han, E.-J. et al. Sargachromenol isolated from Sargassum horneri attenuates glutamate-induced neuronal cell death and oxidative stress through inhibition of MAPK/NF-κB and activation of Nrf2/HO-1 signaling pathway. Mar. Drugs 20, 710 (2022).
Tan, P. X. et al. Algae-derived anti-inflammatory compounds against particulate matters-induced respiratory diseases: a systematic review. Mar. Drugs 19, 317 (2021).
Fonseca-Barahona, I., Shahbaz, K. & Baroutian, S. Bioactives from brown algae: antioxidant, anti-inflammatory, anticancer, and antimicrobial potential. ChemBioEng Rev. 2025, e70007 (2025).
Ye, Y. et al. Isolation and purification of fucoxanthin from brown seaweed Sargassum horneri using open ODS column chromatography and ethanol precipitation. Molecules 26, 3777 (2021).
Zhang, J. et al. MicroRNA-24 inhibits the oxidative stress induced by vascular injury by activating the Nrf2/Ho-1 signaling pathway. Atherosclerosis 290, 9–18 (2019).
Ahmed, S. M. U., Luo, L., Namani, A., Wang, X. J. & Tang, X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim. Biophys. Acta BBA Mol. Basis Dis. 1863, 585–597 (2017).
Juhel, C. et al. Green tea extract (AR25®) inhibits lipolysis of triglycerides in gastric and duodenal medium in vitro. J. Nutr. Biochem. 11, 45–51 (2000).
Vincent, H., Powers, S., Dirks, A. & Scarpace, P. Mechanism for obesity-induced increase in myocardial lipid peroxidation. Int. J. Obes. 25, 378–388 (2001).
Payne, V. A. et al. C/EBP transcription factors regulate SREBP1c gene expression during adipogenesis. Biochem. J. 425, 215–224 (2010).
Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 48, e245–e245 (2016).
Chang, Y.-H., Chen, Y.-L., Huang, W.-C. & Liou, C.-J. Fucoxanthin attenuates fatty acid-induced lipid accumulation in FL83B hepatocytes through regulated Sirt1/AMPK signaling pathway. Biochem. Biophys. Res. Commun. 495, 197–203 (2018).
Feng, J., MengHuan, L., TingTing, Y., XueJie, Y. & HaiNing, G. Research progress on AMPK in the pathogenesis and treatment of MASLD. Front. Immunol. 16, 1558041 (2025).
Li, M. et al. Natural products targeting AMPK signaling pathway therapy, diabetes mellitus and its complications. Front. Pharmacol. 16, 1534634 (2025).
Herzig, S. & Shaw, R. J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135 (2018).
Kim, J.-Y. et al. Sargassum horneri extract fermented by Lactiplantibacillus pentosus SH803 mediates adipocyte metabolism in 3T3-L1 preadipocytes by regulating oxidative damage and inflammation. Sci. Rep. 14, 15064 (2024).
Lee, H.-G., Jeon, Y.-J. & Kang, M.-C. Fucoidan from Sargassum thunbergii obtained via step gradient ethanol precipitation indicate potential anti-obesity and anti-hepatic steatosis in vitro 3T3-L1 and HepG2 cells and in vivo high-fat diet-induced obesity mice. Food Chem. Toxicol. 174, 113686 (2023).
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea government (MSIT) (No. RS-2023-00217471). This study was financially supported by University Industry Liaison of Chonnam National University (Grant number:2025-0929).
Author information
Authors and Affiliations
Contributions
Ramakrishna Chilakala: Conceptualization, Methodology, Software, Formal analysis, investigation, Writing- original draft preparation. Hyeon Jeong Moon, Min Ju Kim, Kang Ho Ko, Jong Won Han and Min Seouk Jung: Formal analysis, Investigation, Software, Data curation. Sun Hee Cheong: Writing—review and editing, Visualization, Supervision, Project administration, Funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Ethical approval
This study was conducted in accordance with the ARRIVE guidelines (Animal Research: Reporting of In Vivo Experiments) for reporting animal research. Animal experiments were conducted in accordance with the Guidelines for Care and Use of Laboratory Animals of Chonnam National University and were approved by the Animal Ethics Committee of Chonnam National University (No: CNU IACUC-YS-2020-9); the approval date is 7 October 2020. Mice were euthanized humanely using isoflurane inhalation anesthesia following AVMA guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chilakala, R., Moon, H.J., Kim, M.J. et al. Bioactive compounds from Sargassum horneri attenuates inflammation and obesity regulating by Nrf2/HO-1 and AMPK signaling pathways. Sci Rep 15, 43519 (2025). https://doi.org/10.1038/s41598-025-27131-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-27131-5
