
Abstract
Skin cutaneous melanoma (SKCM) is a highly aggressive and malignant neoplasm. A wealth of evidence highlights the critical role of LINC01116 in tumorigenesis and progression. Additionally, numerous small molecules within the FK506-binding protein (FKBP) family have been implicated in cancer development. Herein, we interrogated the functional significance of lncRNA LINC01116 and FKBP7/14 in melanoma pathogenesis. This study aim to the expression of long non-coding RNA (lncRNA) LINC01116 in melanoma and its mechanistic role in promoting de novo protein synthesis and cancer stemness through the miR-432-5p/FKBP7/14 axis. A competing endogenous RNA (ceRNA) network for LINC01116 was constructed through bioinformatics analysis. LINC01116 expression levels were assessed in clinical melanoma tissues, adjacent normal tissues, and melanoma cell lines. In vitro experiments included dual-luciferase reporter assays to validate miR-432-5p targeting of FKBP7/14, quantitative PCR (qPCR) and western blotting (WB) to quantify gene/protein expression, and sphere-forming assays to assess protein synthetic capacity and stemness. In vivo, xenograft mouse models were employed to evaluate tumor growth. LINC01116 was significantly upregulated in melanoma tissues and correlated with adverse clinical outcomes. Overexpression of LINC01116 enhanced melanoma cell proliferation, spheroid formation, and invasive potential in vitro. Mechanistically, LINC01116 acted as a sponge for miR-432-5p, thereby upregulating FKBP7/14. In vivo, LINC01116 overexpression accelerated tumor growth in xenograft models. Our findings unveil a novel LINC01116/miR-432-5p/FKBP7/14 axis that drives melanoma stemness and protein synthesis addiction. These insights identify promising therapeutic targets for melanoma intervention.
Similar content being viewed by others

Introduction
SKCM is a highly aggressive malignant tumor arising from melanocytes and predominantly occurs in the skin1. Epidemiological studies indicate that SKCM has the highest mortality rate among all cutaneous malignancies2,3. The use of systemic immunotherapy has improved outcomes for patients with advanced melanoma (stage III and IV), increasing 5-year survival rates to over 70% and 30%, respectively4,5. The melanomas were divided into those etiologically related to sun exposure and those that are not, based on their mutational signatures, anatomic site, and epidemiology6. The tumor‒node‒metastasis (TNM) staging system established by the American Joint Committee on Cancer (AJCC)7categorizes melanomas into clinical stages I through IV8. However, current TNM-based staging systems demonstrate limited prognostic precision for melanoma outcomes. This highlights the urgent need to identify biomarkers and therapeutic targets with enhanced diagnostic, staging, and predictive value.
Extensive investigations into the molecular mechanisms underlying SKCM invasion, proliferation, and metastasis have yielded significant advances9. Nevertheless, the precise molecular pathways driving SKCM pathogenesis and progression remain incompletely understood. Current evidence confirms that the proliferative phenotype of melanoma is regulated by SOX10/MITF signalling, whereas the invasive phenotype is governed by AP1/TEAD transcription factors9. Although several protein-coding genes involved in the phenotypic switching of melanoma have been characterized, the involvement of noncoding genes in this process remains poorly characterized. An increasing number of studies have demonstrated that noncoding RNAs (ncRNAs) may perform tumor-suppressive or oncogenic functions during malignant transformation, invasion, and metastasis10,11,12. lncRNA, a subclass of nonprotein-coding RNA molecules, were historically regarded as “transcriptional noise” during DNA transcription13. However, emerging research has revealed their multifaceted roles in epigenetic regulation, chromatin remodelling, and cellular differentiation14,15,16. LINC01116 was first identified in 2017 as an oncogenic lncRNA that is overexpressed in hepatocellular and prostate carcinomas, where it enhances colony formation and invasive capacity in prostate cancer cells17,18. A meta-analysis19 incorporating 12 original studies (809 patients) across diverse malignancies—including gastric, breast, and colorectal cancers; osteosarcoma; glioma; oral squamous cell carcinoma; and ovarian cancer—revealed that consistent LINC01116 upregulation was correlated with poor prognosis. It has been demonstrated that LINC01116 promotes melanoma progression by sequestering miR-3612 and targeting GDF11/SSD3 signalling20. While bioinformatics analyses revealed LINC01116-associated differentially expressed genes and pathway enrichment in melanoma, the precise regulatory mechanisms through which LINC01116 drives melanomagenesis and progression await further experimental validation.
LINC01116 may directly regulate downstream genes or signalling pathways to influence tumor cell behavior. In our study, a ceRNA regulatory network for LINC01116 was constructed via the ENCORI/starBase (https://rnasysu.com/encori/) for miRNA prediction, combined with RNA-seq/miRNA-seq data and bioinformatics analyses. This approach identified FKBP7 and FKBP14 as key targets showing strong positive correlations with LINC01116 expression. Both FKBP7 and FKBP14 are critical regulators of posttranslational protein folding, with documented roles in tumorigenesis, progression, and drug resistance21,22. The FKBP family comprises highly conserved proteins present across diverse eukaryotes, from yeast to humans23. Multiple FKBP family members, including prostate cancer, colorectal cancer, breast cancer, myeloma, non-small cell lung cancer, and melanoma, have been implicated in cancer pathogenesis and therapeutic resistance, positioning them as promising therapeutic targets24,25.
Our findings demonstrate that miR-432-5p exerts antitumour effects on melanoma by targeting FKBP7/14. Consistently, melanoma cell lines presented elevated expression of LINC01116, FKBP7/14 and reduced miR-432-5p levels. During oncogenesis, active genes frequently reprogram cancer cells to activate stem cell-like properties26. Further in vivo experiments revealed that LINC01116 knockdown significantly attenuated tumor growth, proliferation, and apoptosis while impairing nascent protein synthesis and melanoma cell stemness.
Materials and methods
Bioinformatics construction of the CeRNA network
All genomic and transcriptomic data utilized in this study were sourced from publicly available repositories to ensure reproducibility and ethical compliance. First, melanoma-related gene expression data, utilized data from previously published studies, were downloaded from The Cancer Genome Atlas (TCGA) Data Portal (https://portal.gdc.cancer.gov/) on [2023-12-12] and the Gene Expression Omnibus (GEO) under accession GSE15605 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE15605). The dataset with ID GSE15605 (Platform: GPL570 [HG-U133_Plus_2] included 16 normal and 58 tumor samples. Differential expression analysis was performed using the limma package in R 4.1.3. Genes with |logFC| > 1 and an adjusted p-value < 0.05 were considered significantly differentially expressed. The TCGA melanoma expression profile (RNA-Seq) was downloaded and processed from UCSC Xena. A total of 473 samples were initially included, from which patients lacking survival information were excluded. Transcriptomic data from skin tissues were also obtained from the GTEx database. After merging and batch effect removal, differential expression analysis was conducted using limma in R 4.1.3, comparing 557 normal and 472 tumor samples, with the same thresholds (|logFC| > 1, adjusted p-value < 0.05).
Using the TCGA transcriptomic data, samples were divided into two groups based on the median expression level of LINC01116. Differential analysis was carried out with limma (R 4.1.3), applying thresholds of |logFC| > 1 and adjusted p-value < 0.05. Spearman correlation coefficients between LINC01116 and all other genes were calculated. Genes with |cor| > 0.2 and p-value < 0.05 were selected. A circos plot was generated for the top 10 positively correlated genes with LINC01116. Functional enrichment analyses, including Gene Ontology (GO) enrichment analysis annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, were performed.
Potential downstream miRNAs of LINC01116 were predicted using ENCORI/starBase (https://rnasysu.com/encori/). Downstream target mRNAs of these miRNAs were predicted using miRDB, mirDIP, and TargetScan. The intersection of these predicted mRNAs with the co-expressed genes yielded 87 overlapping genes, identified as targeted co-expressed genes.
A ceRNA network was constructed using Cytoscape (v3.9.0). From the 87 targeted co-expressed genes, 17 upregulated differentially co-expressed genes were identified by intersection with the GEO#GSE15605, and 44 were obtained from the intersection with the combined TCGA and GTEx data. The overlap between these two sets of upregulated genes resulted in 13 common target genes. Spearman correlation analysis was performed on these 13 genes, and a correlation heatmap was generated. The heatmap revealed a strong positive correlation between FKBP7/14 (cor = 0.96). Finally, a ceRNA network centered on the LINC01116-miR-432-5p-FKBP7/14 axis was systematically constructed.
Specimen collection and experimental materials
Tissue samples
Primary malignant melanoma tissues (n = 20), normal tissues(n = 20), and melanocytic nevi (n = 20) were collected from melanoma patients at the Affiliated Hospital of Zunyi Medical University and the Second Affiliated Hospital of Zunyi Medical University. All specimens were histopathologically confirmed, with diagnoses independently verified by two pathologists. Patients had not undergone prior chemotherapy or radiotherapy, and written informed consent was obtained from all participants. This study was approved by the Ethics Committee of the Second Affiliated Hospital of Zunyi Medical University (Approval No. KYLL-2022-007). Confirmation of ethical approval from the Animal Welfare and Ethics Committee of the Zunyi Medical University review board (Approval No. ZMU21-2405-001).
Cell lines
The human melanoma cell lines A375 (Procell, CL-0014) and SK-MEL-28 (Procell, CL-0717) were purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin‒streptomycin (Gibco) and maintained at 37 °C in a humidified incubator with 5% CO₂.
RT‒qPCR analysis
Total RNA was extracted using TRIzol reagent (15596026, Invitrogen, Carlsbad, CA, USA). For miRNA analysis, reverse transcription was performed with the miRNA First Strand cDNA Synthesis Kit (B532451, Sangon Biotech, Shanghai, China). cDNA synthesis for mRNA analysis was carried out using the PrimeScript RT reagent kit (RR037A, Takara, Shiga, Japan). Quantitative real-time PCR was conducted using SYBR Green PCR master mix (4309155, Thermo Fisher Scientific, Waltham, MA, USA) on a Roche LightCycler 480 system (Roche, Basel, Switzerland). miRNA (with 10 ng input) and mRNA (with 5 µg input) expression levels were normalized to U6 and ACTB, respectively. The relative gene expression was calculated using the 2^(−ΔΔCT) method, where ΔCT = CT(target gene) − CT(reference gene) and ΔΔCT = ΔCT(experimental group) − ΔCT(control group). All samples were tested in triplicate. Primer sequences are provided in Table S1.
WB analysis
Protein separation was performed via the BioSci™ NewFlash Protein AnyKD PAGE Gel Kit (Dakewe, Cat# 8012011). Protein quantification was performed using the BCA assay. Proteins were extracted using RIPA buffer containing a protease inhibitor cocktail and phosphatase inhibitors. Following extraction, protein concentration was determined using the BCA assay. Equal amounts of protein (30 µg) were separated by 10% SDS-PAGE and transferred to PVDF membranes.Antibodies FKBP7 (Rabbit polyclonal antibody; Thermo Fisher Scientific, PA5-30629; 1:2000), FKBP14 (Rabbit monoclonal antibody [EPR16769]; Abcam, ab251703; 1:10000), Nestin (Mouse monoclonal antibody [2C1.3A11]; Abcam, ab18102; 1:5000), ABCB5 (Rabbit polyclonal antibody; Thermo Fisher Scientific, PA5-112068; 1:1000), CD133 (Rabbit monoclonal antibody [EPR16508]; Abcam, ab222782; 1:2000), ALDH1A1 (Rabbit monoclonal antibody [EP1933Y]; Abcam, ab52492; 1:5000), β-Actin (Rabbit monoclonal antibody [EPR16769]; Abcam, ab179467; 1:5000)were used for normalization.
Dual-luciferase reporter assay
To validate the relationship between LINC01116 and miR-432-5p, dual-luciferase reporter vectors containing either the wild-type (Wt) or mutant type (Mut) binding sites of LINC01116 were constructed (Hanheng Biotechnology Co., Ltd.). Each vector contained a respective 3’UTR (LINC01116, FKBP7, or FKBP14) cloned downstream of the firefly luciferase gene. Wild-type(Wt) constructs preserved the native sequence containing a single predicted miR-432-5p binding site, Mutant type(Mut) were generated by site-directed mutagenesis (QuikChange method) to disrupt the seed region pairing:
LINC01116-mut(CUCCAAG→GCGAUUC), FKBP7-mut(CUCCAAG→GAGGUUC), FKBP14-mut (CUCCAAG→GAGGUUC). These reporter plasmids were cotransfected with miR-432-5p mimics or inhibitors into A375 and SK-MEL-28 cells, followed by measurement of luciferase activity via a dual-luciferase detection system. The same methodology was used to confirm the interaction between miR-432-5p and the mRNA sequences of FKBP7/14.The experiment was independently conducted in triplicate.
Viral transfection
HEK293 cells were co-transfected with lentiviral transfer plasmid, packaging plasmid (pSPAX2), and envelope plasmid (pMD2.g) at a ratio of 4:3:1 using Lipofectamine 3000. Eighteen hours post-transfection, the culture medium containing plasmids and transfection reagents was replaced with fresh complete DMEM. After 48 h of transfection, supernatants containing lentiviral particles were harvested and used to infect A375 and SK-MEL-28 cells in the presence of polybrene (8 µg/mL). To determine viral titer, cell debris was removed by filtration through a 0.45 μm membrane. Subsequently, 293T cells were seeded into 96-well plates with serial dilutions (≥ 5 gradients) of viral suspensions, and viral titer was quantified using qPCR.
Functional assays
Click chemistry-based nascent protein synthesis detection
Nascent protein synthesis levels were assessed in A375 and SK-MEL-28 cells across four experimental groups (sh-NC, sh-LINC01116, sh-LINC01116 + Inh-NC, and sh-LINC01116 + miR-432-5p inhibitor) via a protein synthesis assay kit (red) (Abcam, Cat# ab235634). The assay was performed in strict accordance with the manufacturer’s protocol. The fluorescent signals were observed and documented via a fluorescence microscopy system.
CCK-8 proliferation assay
Cell suspensions were prepared and seeded into 5 × 10³ cells/well (96-well plate) culture plates for preculturing under standard conditions (37 °C, 5% CO2). Following cell adherence, 10 µL of CCK-8 solution (Dojindo Laboratories) was added to each well, followed by incubation for 2 h, and continuous detection at 24 h intervals. The absorbance at 450 nm (optical density, OD) was quantified via a microplate reader (BioTek Instruments), after which the IC50 was calculated via dose‒response curve analysis.
Colony formation assay 30 min
Cells in the logarithmic growth phase were trypsinized, resuspended in complete medium supplemented with 10% fetal bovine serum (FBS; Gibco), and quantified. The cell suspensions were seeded into 5 × 10⁴ cells/well (8 μm pore size, 24-well plate) culture plates and cultured until most individual colonies contained > 50 cells, 30 min at 37 °C for Matrigel polymerization. Colonies were photographed via phase-contrast microscopy (Leica Microsystems), fixed with 1 mL of 4% paraformaldehyde (PFA; Solarbio) for 15 min and stained with 0.1% crystal violet solution (Solarbio). After three washes with PBS, the plates were air-dried, and colonies were documented via a digital imaging system (Nikon).
Flow cytometric cell cycle analysis
The cells were trypsinized, centrifuged, and washed, followed by RNase treatment and propidium iodide (PI) staining. The cell cycle distribution was analysed via a Cell Cycle and Apoptosis Detection Kit (Beyotime Biotechnology, Cat# C1052) according to the manufacturer’s protocol. DNA content quantification was performed with FlowJo software (v10.8.1, BD Biosciences) through histogram deconvolution analysis.
3D tumor spheroid growth assay
A 24-well culture plate was prechilled on ice for 30 min. Following cell counting, the cells (2 × 10⁵–2 × 10⁷) were resuspended in 0.5 mL of 37 °C prewarmed culture medium and mixed with 0.5 mL of 37 °C basement membrane matrix (Corning Matrigel) at a 1:1 ratio, achieving a final density of 1 × 10⁵–1 × 10⁷ cells/mL. The mixture (20–40 µL) was aliquoted onto a prechilled plate. Following hydrogel polymerization, 1 mL of ice-cold stabilization buffer was added for 15 min of fixation. The cell-embedded hydrogels were subsequently cultured at 37 °C in a 5% CO2 incubator for 7–14 days, and spheroid formation was monitored daily via phase-contrast microscopy (Olympus).
Xenograft tumor assay
Balb/c-nu/nu male nude mice (6-week-old, 15–20 g) were purchased from Zunyi Medical University (Zunyi, China). SK-MEL-28 cells were divided into two experimental groups: the sh-NC group (negative control) and the sh-LINC01116 group (target gene silencing). For each group, 5 × 106 cells were mixed with Matrigel at a 2:1 volume ratio to prepare a 100 µL cell suspension per mouse. This suspension was then subcutaneously inoculated into the left flank of the mice. Tumor growth was monitored by measuring tumor volume every 3 days using the formula 0.5 × length × width², and growth curves were plotted accordingly. No animals were excluded from the final analysis. After 28 days, mice were euthanized via inhalation of isoflurane followed by cervical dislocation, tumor tissues were harvested for subsequent analyses, including Ki-67 immunohistochemistry, TUNEL staining, and WB analysis to evaluate stemness markers (Nestin, ABCB5, CD133, and ALDH1). All methods were performed in accordance with the relevant guidelines and regulations.
Statistical analysis
The data are presented as the means ± standard deviations (SDs). Statistical analyses were performed via SPSS software (version 26.0; IBM). Between-group comparisons were evaluated via Student’s t test or one-way analysis of variance (ANOVA). Spearman’s correlation analysis and multivariate analyses were conducted with MATLAB (version R2023a; MathWorks). Kaplan‒Meier survival curves were generated to assess the association between LINC01116 expression and melanoma patient survival. Melanoma tissues were stratified into groups on the basis of LINC01116 expression levels, with between-group differences analysed via the log-rank test. Survival curves were plotted with GraphPad Prism software (version 9.0; GraphPad Software). Statistical significance was defined as *P < 0.05 and **P < 0.01.
Results
LINC01116-associated differentially expressed genes and pathways
Analysis of the melanoma dataset (GEO#GSE15605) revealed significant upregulation of LINC01116 in melanoma tissues (Fig. 1A), which was further validated via data from TCGA (Fig. 1B). Integration of TCGA clinical data (survival time and status) with transcriptomic profiles revealed that high LINC01116 expression was correlated with poor 8-year overall survival (OS) in melanoma patients (Fig. 1C). K‒M analysis of our melanoma cohort confirmed significantly reduced survival rates in patients with elevated LINC01116 levels.
Using TCGA transcriptomic data, samples were stratified by median LINC01116 expression. Spearman correlation analysis further identified LINC01116-associated genes (Fig. 1D). A Venn diagram of the intersection of LINC01116-coexpressed genes, differential melanoma genes, and LINC01116-correlated genes revealed 129 differentially coexpressed genes (Fig. 1E). KEGG27,28enrichment analysis of these genes revealed significant associations with cancer-related pathways (Fig. 1F).
mRNAs positively correlated with LINC01116
Potential downstream miRNAs of LINC01116 were predicted via ENCORI/starBase (https://rnasysu.com/encori/), resulting in candidates: hsa-miR-3614-5p, hsa-miR-432-5p, hsa-miR-6512-3p, hsa-miR-3690, hsa-miR-6720-5p. Downstream mRNA targets of these miRNAs were further identified. Intersection analysis between the predicted mRNAs and coexpressed genes yielded 87 overlapping target coexpressed genes (Fig. 2A). A ceRNA network comprising LINC01116, 4 miRNAs, and 87 mRNAs was constructed via Cytoscape (v3.9.0) (Fig. 2B). Venn diagram analysis of the DEGs from the GEO#GSE15605 and TCGA datasets revealed 13 intersecting genes (Fig. 2C). Correlation heatmap analysis of the upregulated targets revealed that FKBP7 /14 exhibited strong positive correlations (Fig. 2D).
LINC01116 is significantly overexpressed in melanoma
The qPCR analysis of 60 clinical tissue samples—including normal, melanocytic nevus, and melanoma samples—revealed significantly elevated mRNA levels of LINC01116, FKBP7/FKBP14 in melanoma tissues compared with normal controls (P < 0.01). Conversely, miR-432-5p expression was markedly reduced in melanoma tissues (P < 0.01) (Fig. 3A). WB analysis further confirmed greater protein expression of FKBP7/14 in melanoma tissues than in both normal skin tissues and melanocytic nevi, (P < 0.01)(Fig. 3B). These findings collectively indicate that LINC01116, FKBP7/14 are overexpressed in melanoma and may drive tumorigenesis and progression.
LINC01116 modulates FKBP7/14 expression via miR-432-5p
Bioinformatic analysis predicted binding sites between LINC01116 and miR-432-5p, as well as complementary targeting regions in the 3’-UTRs of FKBP7/14. Dual-luciferase reporter assays confirmed the direct binding of miR-432-5pto the WT 3′-UTR of LINC01116, with MUT of the predicted binding sites abolishing this interaction (Fig. 4A, B). Dual-luciferase assays demonstrating that miR-432-5p targets the 3′-UTR regions of FKBP7/14. Mutations in the miR-432-5p seed-matching sequence (MUT) eliminated luciferase suppression, confirming sequence-specific binding (Fig. 4C, D). qRT‒PCR analysis of LINC01116, miR-432-5p, FKBP7/FKBP14 mRNA levels in A375 and SK-MEL-28 melanoma cell lines transfected with sh-LINC01116 versus sh-NC was performed (Fig. 4E). Rescue experiments evaluating the effects of miR-432-5p inhibition (via a miR-432-5p inhibitor) on FKBP7/14 expression in sh-LINC01116-transfected cells. Cotransfection with Inh-NC served as a baseline. The results demonstrated that miR-432-5p suppression reversed the downregulation of FKBP7/14 caused by LINC01116 knockdown (Fig. 4F). WB analysis of FKBP7/14 protein levels in the same experimental groups. β-actin was used as a loading control. Densitometric quantification confirmed that LINC01116 silencing reduces FKBP7/14 protein expression, which is rescued by miR-432-5p inhibition (Fig. 4G). Consistently, melanoma cell lines presented high LINC01116/FKBP7/14 expression and low miR-432-5p levels, and these patterns were reversed upon LINC01116 knockdown or miR-432-5p inhibition. These findings establish a LINC01116/miR-432-5p/FKBP7/14 regulatory axis driving melanoma progression.
LINC01116 promotes nascent protein synthesis
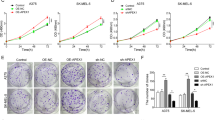
Fluorescence intensity analysis of the A375 and SK-MEL-28 cell lines across four experimental groups—sh-NC, sh-LINC01116, sh-LINC01116 + Inh-NC, and sh-LINC01116 + miR-432-5p inhibitor—revealed distinct protein synthesis levels (Fig. 5A). Posttransfection CCK-8 assays revealed that the sh-NC group had the highest proliferation rate, followed by the sh-LINC01116 + miR-432-5p inhibitor group, whereas the sh-LINC01116 and sh-LINC01116 + Inh-NC groups had the lowest proliferation rate (Fig. 5B). Colony formation assays further confirmed reduced clonogenic capacity in the sh-LINC01116 group, with partial rescue observed in the sh-LINC01116 + miR-432-5p inhibitor group compared with the sh-LINC01116 + Inh-NC group (Fig. 5C).
Cell cycle analysis revealed that LINC01116 knockdown induced G1/S phase arrest in melanoma cells, and this effect was reversed by miR-432-5p inhibition (Fig. 5D). In 3D tumor spheroid assays, the sh-NC and sh-LINC01116 + miR-432-5p inhibitor groups exhibited significantly enhanced spheroid-forming capacity (Fig. 5E). These results collectively indicate that LINC01116 drives nascent protein synthesis and malignant progression in melanoma through the miR-432-5p axis.
LINC01116 influences melanoma cell stemness
Cancer stem cells (CSCs) exhibit malignant biological properties, including self-renewal, multilineage differentiation, and unlimited proliferative capacity.The presence of CSCs in melanoma has been extensively demonstrated. We included four proteins related to melanoma cell stemness, and further investigated the impact of LINC01116 on these populations. WB analysis of cancer stemness markers, inclouding Nestin, ATP-binding cassette, sub-family B5(ABCB5), CD133(Prominin-1), Aldehyde dehydrogenase 1 (ALDH1) in A375 and SK-MEL-28 cell lines across four experimental groups—sh-NC, sh-LINC01116, sh-LINC01116 + Inh-NC, and sh-LINC01116 + miR-432-5p inhibitor—revealed significantly lower protein levels of Nestin, CD133, and ALDH1 (but not ABCB5) in the sh-LINC01116 and sh-LINC01116 + Inh-NC groups than in the sh-NC and sh-LINC01116 + miR-432-5p inhibitor groups. miR-432-5p inhibitor treatment partially restored the expression of these markers, confirming that the LINC01116/miR-432-5p axis regulates melanoma stemness (Fig. 5F). These results demonstrate that LINC01116 enhances melanoma stemness by promoting tumor cell self-renewal and differentiation.
Functional validation using LINC01116-knockdown and miR-432-5p inhibitor-treated A375 and SK-MEL-28 cells revealed that miR-432-5p inhibition inhibits the effects of LINC01116 knockdown on proliferation, cell cycle progression, clonogenicity, and 3D spheroid formation. Furthermore, LINC01116 depletion impairs nascent protein synthesis and attenuates melanoma stemness properties.
In vivo validation of the protumorigenic role of LINC01116
Finally, we investigated the impact of LINC01116 on melanoma growth in vivo. SK-MEL-28 cells stably expressing LINC01116 shRNA or lentiviral control were subcutaneously injected into six BALB/c nude mice per group. The mice were euthanized 28 days postinoculation, and the tumor growth curves revealed significantly slower progression in the sh-LINC01116 group than in the sh-NC group (Fig. 6A). Gross tumor morphology and volumetric analysis revealed markedly reduced tumor size in the sh-LINC01116 group (P < 0.0001; Fig. 6B).Immunohistochemical (IHC) staining for Ki-67 showed decreased proliferative activity in the sh-LINC01116 group (P < 0.001; Fig. 6C), whereas TUNEL staining indicated significantly elevated apoptosis in the same group (P < 0.001; Fig. 6D). WB analysis of tumor tissues revealed downregulated expression of cancer stem cell markers, including Nestin, ABCB5, CD133, and ALDH1, in the sh-LINC01116 group (Fig. 6E). These findings collectively demonstrate that LINC01116 knockdown attenuates melanoma progression in vivo by suppressing tumor growth, reducing proliferation, increasing apoptosis, and impairing cancer stemness and nascent protein synthesis.
Discussion
Melanoma is the most aggressive and dangerous form of skin cancer, It is crucial to identify melanoma at its early stages29,30. Management of advanced melanoma is complex, and multidisciplinary care is essential. In recent years, the introduction of BRAF/MEK tyrosine kinase inhibitors and immune checkpoint inhibitors, have substantially increased melanoma survival31,32,33. The role of LINC01116 in tumorigenesis and cancer progression has garnered increasing attention. LINC01116 can directly regulate downstream genes or signalling pathways to influence tumor cell behavior while also functioning as a ceRNA to modulate microRNA (miRNA) expression, thereby indirectly regulating cancer-associated genes or pathways19,20. Aberrantly expressed mRNAs are frequently implicated in melanoma pathogenesis and clinical prognosis34,35.
In this study, we first constructed a bioinformatics-predicted LINC01116-miRNA target gene network and speculate a potential ceRNA interaction network involving LINC01116, miR-432-5p, FKBP7/14 in melanoma. Among nevus-associated melanomas, which overall account for 20%-30% of all melanomas36. Since studies have shown that identical oncogenic driver mutations are found in benign nevi and melanoma37. and we choose primary malignant melanoma tissues(n = 20), normal tissues(n = 20), and melanocytic nevi (n = 20) as clinical tissue sample. Analysis revealed significantly higher protein expression levels of these molecules in the melanoma group than in the normal and the melanocytic nevi group, indicating that LINC01116, FKBP7/14 are overexpressed in melanoma and may contribute to tumorigenesis and progression.
Dual-luciferase reporter assays confirmed the direct binding betweenmiR-432-5p and LINC01116, while FKBP7/14 were confirmed as downstream targets of this regulatory axis. Notably, LINC01116 siRNA significantly suppressed the luciferase activity of the FKBP7/14 3’UTR reporter construct, and this suppression was reversed by the miR-432-5p inhibitor. Collectively, these findings demonstrate that LINC01116 promotes FKBP7/14 expression in melanoma by acting as a miRNA sponge for miR-432-5p, thereby driving oncogenic signalling.
Our study revealed a discrepancy in ABCB5 expression between in vivo and in vitro settings (Fig. 5F shows no change in ABCB5, whereas Fig. 6E shows its downregulation). This inconsistency may be attributed to the influence of the complex in vivo tumor microenvironment on the regulation of ABCB5 expression, which differs from that under in vitro conditions. ABCB5 has been previously demonstrated to be regulated by an interleukin (IL)1β/IL8/CXCR1 cytokine signaling pathway, through which IL8 mediates paracrine interactions between ABCB5-positive (ABCB5⁺) cells. Furthermore, certain isoforms within the ABCB5 family do not confer multidrug resistance38. Additionally, the differential sensitivity or expression kinetics of the ABCB5 marker compared to other stemness markers such as Nestin, CD133, and ALDH1 in response to LINC01116 knockdown may also contribute to the observed discrepancies.
As a tumor-suppressive miRNA, miR-432-5p is downregulated in multiple malignancies and participates in tumor cell proliferation, migration, and invasion39,40. Our study revealed that miR-432-5p exerts anti-proliferative and anti-metastatic effects in melanoma by targeting FKBP7/14 expression. FKBP7/14, as immune-associated proteins, may play critical roles in tumorigenesis and progression.The FKBP family belongs to the immunophilin superfamily, with its prototype FKBP12 first isolated in 1989, representing the minimal structural domain required for FKBP family membership41,42. Previous studies reported that FKBP51 promotes melanoma metastasis in vivo43, FKBP7 is upregulated in melanoma44, FKBP10 may serve as a therapeutic target45, and FKBP38 is selectively downregulated in highly metastatic B16-F10-plc melanoma clones46. Among the FKBP family members, FKBP38, FKBP51, FKBP52, and FKBP65 have also been implicated in cancer etiology and chemoresistance47. This study is the first to report the regulatory role of the LINC01116/miR-432-5p/FKBP7/14 axis in melanoma pathogenesis. However, the precise mechanistic roles of individual FKBP family members require further investigation.
We observed that LINC01116 promotes cell proliferation, cell cycle progression, clonogenicity, 3D spheroid formation, as well as tumor growth and proliferation, while inhibiting apoptosis in both in vivo and in vitro experiments. Conversely, knockdown of LINC01116 was found to suppress de novo protein synthesis and reduce stemness properties in melanoma cells. Additionally, melanoma cell lines were found to exhibit high expression levels of LINC01116, FKBP7/14. FKBP7/14, which function as peptidyl-prolyl cis-trans isomerases (PPIases), facilitate de novo protein synthesis. These proteins serve as common receptors for the natural products FK506 and rapamycin. By binding to these drugs, they form complexes that protect cancer cells and induce immunosuppressive effects24. In contrast, FKBP5, another member of the FKBP family, can form complexes with heat shock proteins Hsp90 and Hsp70. It plays a significant role in tumorigenesis and responses to anti-cancer therapies by influencing steroid receptor maturation as well as modulating the NF-κB and AKT signaling pathways.
Genes that are active during embryonic development play pivotal roles in reprogramming cancer cells to activate stem-like properties26. The first documentation of Nestin expression in melanoma dates to 199448, with subsequent studies demonstrating its expression in melanocytic hyperplasia—particularly in vascular endothelial cells adjacent to advanced melanomas—but not in normal melanocytes49,50. Its expression intensity and distribution correlate with tumor progression and reduced patient survival51. ABCB5 is critically involved in the chemoresistance of melanoma stem cells52. ABCB5, a member of the ATP-binding cassette (ABC) transporter superfamily comprising 48 transporters53, and it is ranks among the most frequently mutated genes in melanoma54,55.CD131, a pleiotropic cytokine receptor, regulates hematopoietic cell proliferation, differentiation, and self-renewal homeostasis56. ALDH1 overexpression is strongly correlated with poor prognosis and high therapeutic resistance rates57,58,59,60. Targeted inhibition of ALDH1 in cancer stem cells enhances drug sensitivity while suppressing proliferation, differentiation, and metastasis in solid tumors61. WB revealed differential protein expression levels of ABCB5 compared to other cancer stemness genes, suggesting this discrepancy may be attributed to the fact that our melanoma patients have not yet undergone chemotherapy.
This study demonstrated that, compared with sh-NC, sh-LINC01116 significantly reduced the expression of stemness-associated markers (Nestin, CD133, and ALDH1) and that cotreatment with a miR-432-5p inhibitor (sh-LINC01116 + miR-432-5p inhibitor) partially reversed this suppression. These findings indicate that LINC01116 enhances melanoma cell stemness by promoting self-renewal and differentiation, thereby driving tumor progression. Substantial research has been conducted on the treatment of patients with advanced melanoma, with available therapeutic agents including nivolumab and ipilimumab4,5. Additionally, adoptive cell therapy with tumor-infiltrating lymphocytes (TIL-ACT) has shown certain efficacy in advanced melanoma62. While the development of novel drugs has extended the overall survival (OS) rate in some patients, drug resistance remains a challenge for others. Targeting LINC01116 or restoring miR-432-5p function holds potential as a therapeutic strategy for melanoma, either as a monotherapy or in combination with existing regimens. However, clinical translation may face several challenges, particularly related to nucleic acid-based therapies—such as antisense oligonucleotides (ASOs) and miRNA mimics—including delivery efficiency, stability, tumor specificity, potential off-target effects, as well as underlying mechanisms of drug resistance. This study provides a solid theoretical foundation for subsequent translational research, identifies promising candidate targets, and supports preclinical investigations into combination therapies and the development of improved drug delivery systems targeting this axis.
Conclusion
In summary, this study integrates bioinformatics analysis, experimental validation, and functional investigations to uncover the oncogenic role of LINC01116 in melanoma. Our findings demonstrate that LINC01116 is significantly overexpressed in melanoma tissues and drives tumor progression by activating the miR-432-5p/FKBP7/14 axis, which promotes neonatal protein synthesis and augments cancer stem cell-like properties. These findings not only expand the understanding of melanoma pathogenesis but also provide a preclinical rationale for developing therapeutic strategies targeting LINC01116.

LINC01116-Associated Differentially Expressed Genes and Pathways (A) Volcano plot of differentially expressed genes (DEGs) in melanoma from the GEO#GSE15605, highlighting LINC01116 upregulation. (B) Validation of LINC01116 overexpression in melanoma using TCGA-derived DEG volcano plot. (C) Kaplan-Meier survival curve demonstrating reduced 8-year overall survival in melanoma patients with high LINC01116 expression. (D) Spearman correlation Scatter Plot of LINC01116 co-expressed genes (|cor| >0.2, P < 0.05). (E) Venn diagram intersecting LINC01116-correlated genes, melanoma co-expressed genes, and DEGs, identifying 129 overlapping genes. (F) KEGG pathway enrichment analysis of the 129 overlapping genes, highlighting cancer-related signaling pathways.

LINC01116-associated mRNA co-expression and regulatory networks. (A) Venn diagram illustrating the intersection of predicted LINC01116-targeting miRNAs (identified via the Starbase database, http://starbase.sysu.edu.cn) and co-expressed mRNAs. (B) ceRNA (competing endogenous RNA) network constructed using Cytoscape 3.9.0, comprising LINC01116, four miRNAs, and 87 mRNAs. (C) Heatmap analysis of overlapping targets derived from the GEO dataset GSE15605 and TCGA database, revealing 13 shared genes in the Venn diagram. (D) Correlation heatmap of overlapping targets, demonstrating a strong positive correlation between FKBP7 /14 expression (r > 0.8, p < 0.001).

LINC01116 is significantly overexpressed in melanoma. (A) qPCR analysis of LINC01116, miR-432-5p, FKBP7/14 in clinical tissue samples (20 normal skin tissues, 20 melanocytic nevi, and 20 melanomas). (B) WB analysis of FKBP7/14 protein levels in the same cohort of clinical tissues. Data were expressed as the mean ± SD, **P < 0.01, ***P < 0.001,****P < 0.0001.

LINC01116 Modulates FKBP7/14 Expression via miR-432-5p. (A-D) Dual-luciferase reporter assays demonstrating the direct binding of miR-432-5p to the 3′-UTR regions of LINC01116, FKBP7/14. (E) qRT-PCR analysis of LINC01116, miR-432-5p, FKBP7/14 expression in A375 and SK-MEL-28 melanoma cell lines transfected with sh-LINC01116 versus sh-NC. (F) Rescue experiments evaluating the effects of miR-432-5p inhibition (via miR-432-5p inhibitor) on FKBP7/14 expression in sh-LINC01116-transfected cells. Co-transfection with inhibitor negative control (Inh-NC) served as a baseline. (G) WB analysis of FKBP7/14 protein levels.
Data were expressed as the mean ± SD, *P < 0.05,**P < 0.01, ***P < 0.001,****P < 0.0001.

LINC01116 Promotes Nascent Protein Synthesis and Influences Melanoma Cell Stemness. (A) Fluorescence intensity analysis demonstrated successful transfection efficiency across experimental groups. (B) CCK-8 proliferation assay revealed distinct growth dynamics: h-NC group exhibited the highest proliferation rate. sh-LINC01116 and sh-LINC01116 + Inh-NC groups showed the suppression of proliferation. (C) Clonogenic assay results demonstrated: sh-NC group displayed superior clonogenic potential. sh-LINC01116 + miR-432-5p inhibitor group exhibited partial restoration of clonogenicity compared to sh-LINC01116 and sh-LINC01116 + Inh-NC groups. (D) Cell cycle analysis via flow cytometry identified: G1/S phase arrest in sh-LINC01116-transfected cells, miR-432-5p inhibitor rescued this arrest. (E) 3D tumor spheroid assay:sh-NC and sh-LINC01116 + miR-432-5p inhibitor groups formed larger, more compact spheroids. (F) WB analysis was performed to detect the protein levels of stemness genes Nestin, ABCB5, CD133, and ALDH1 in the A375 and sk-mel-28 cell lines across the following groups: sh-NC, sh-LINC01116, sh-LINC01116 + Inh-NC, and sh-LINC01116 + miR-432-5p inhibitor. The results showed that, with the exception of ABCB5, the expression levels of the other genes were significantly lower in the sh-LINC01116 and sh-LINC01116 + Inh-NC groups compared to the sh-NC and sh-LINC01116 + miR-432-5p inhibitor groups.

In vivo validation of the pro-tumorigenic role of LINC01116. (A) Tumor growth curves in BALB/c nude mice subcutaneously injected with SK-MEL-28 melanoma cells (sh-NC vs. sh-LINC01116 groups). (B) Gross tumor morphology and volumetric analysis at endpoint. Tumor volumes were calculated using the formula: Volume = 0.5×length×width2. The sh-LINC01116 groupshowed markedly smaller tumors compared to the sh-NC group(****P < 0.0001). (C) IHC analysis of Ki67in tumor tissues. The sh-LINC01116 group displayed significantly fewer Ki67-positive cells than the sh-NC group (***P < 0.001). (D ) TUNEL staining to assess apoptosis in tumor sections. The sh-LINC01116 group exhibited a higher percentage of TUNEL-positive cells (green fluorescence) compared to the sh-NC group (***P < 0.001). (E) WB analysis of stemness-associated markers (Nestin, ABCB5, CD133, ALDH1) in tumor lysates. Data were expressed as the mean ± SD, ***P < 0.001,****P < 0.0001.
Data availability
The RNA-seq data analyzed in this study were downloaded from TCGA (https://portal.gdc.cancer.gov/) and the GEO under accession GSE15605 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE15605).
References
Perniciaro, C. Dermatopathologic variants of malignant melanoma. Mayo Clin. Proc. 72, 273–279 (1997).
D’Ambrosio, A. E. et al. Pleural melanoma metastasis. Ann. Thorac. Surg. 112, e103–e105 (2021).
Garbe, C. et al. Epidemiology of cutaneous melanoma and keratinocyte cancer in white populations 1943–2036. Eur. J. Cancer. 152, 18–25 (2021).
Tawbi, H. A. et al. Three-Year overall survival with nivolumab plus relatlimab in advanced melanoma from RELATIVITY-047. Journal Clin. Oncology 43, (2025).
Wolchok, J. D. et al. Final, 10-Year outcomes with nivolumab plus ipilimumab in advanced melanoma. New Engl. J. Med 392, (2025).
Bobos, M. Histopathologic classification and prognostic factors of melanoma: a 2021 update. Ital. J. Dermatol. Venerol. 156, 300–321 (2021).
Balch, C. M. et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 27, 6199–6206 (2009).
Keung, E. Z. & Gershenwald, J. E. The eighth edition American joint committee on cancer (AJCC) melanoma staging system: implications for melanoma treatment and care. Expert Rev. Anticancer Ther. 18, 775–784 (2018).
Verfaillie, A. et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 6, 6683 (2015).
Dong, R. et al. Targeting long non-coding RNA-TUG1 inhibits tumor growth and angiogenesis in hepatoblastoma. Cell. Death Dis. 7, e2278 (2016).
Lambrou, G. I., Hatziagapiou, K. & Zaravinos, A. The Non-Coding RNA GAS5 and its role in tumor Therapy-Induced resistance. Int J. Mol. Sci 21, (2020).
Xue, M. et al. Hypoxic exosomes facilitate bladder tumor growth and development through transferring long non-coding RNA-UCA1. Mol. Cancer. 16, 143 (2017).
Herman, A. B., Tsitsipatis, D. & Gorospe, M. Integrated LncRNA function upon genomic and epigenomic regulation. Mol. Cell. 82, 2252–2266 (2022).
Bridges, M. C., Daulagala, A. C. & Kourtidis, A. LNCcation: LncRNA localization and function. J Cell. Biol 220, (2021).
Li, T. et al. LncRNA MNX1-AS1: A novel oncogenic propellant in cancers. Biomed. Pharmacother. 149, 112801 (2022).
Zhang, P. et al. LncRNA-Mediated adipogenesis in different adipocytes. Int J. Mol. Sci 23, (2022).
Beaver, L. M. et al. Long noncoding RNAs and sulforaphane: a target for chemoprevention and suppression of prostate cancer. J. Nutr. Biochem. 42, 72–83 (2017).
Xu, C. et al. LncNetP, a systematical LncRNA prioritization approach based on CeRNA and disease phenotype association assumptions. Oncotarget 8, 114603–114612 (2017).
Fan, S. T. et al. High linc01116 expression May contribute to a poor prognosis in various cancers based on systematic reviews and meta-analyses. BMC Cancer. 24, 1566 (2024).
Wang, K. et al. LINC01116 facilitates melanoma 1 progression via sequestering miR-3612 and Up-regulating GDF11 and SDC3. Arch. Med. Res. 53, 44–50 (2022).
Garrido, M. F. et al. Regulation of eIF4F translation initiation complex by the peptidyl Prolyl isomerase FKBP7 in Taxane-resistant prostate cancer. Clin. Cancer Res. 25, 710–723 (2019).
Sun, Z., Qin, X., Fang, J., Tang, Y. & Fan, Y. Multi-Omics analysis of the expression and prognosis for FKBP gene family in renal cancer. Front. Oncol. 11, 697534 (2021).
Romano, S. et al. FK506 binding proteins as targets in anticancer therapy. Anticancer Agents Med. Chem. 10, 651–656 (2010).
Kolos, J. M., Voll, A. M., Bauder, M. & Hausch, F. FKBP Ligands-Where we are and where to go? Front. Pharmacol. 9, 1425 (2018).
Zhu, W. et al. FKBP3 promotes proliferation of Non-Small cell lung cancer cells through regulating Sp1/HDAC2/p27. Theranostics 7, 3078–3089 (2017).
Vittorioso, P., Cowling, R., Faure, J. D., Caboche, M. & Bellini, C. Mutation in the Arabidopsis PASTICCINO1 gene, which encodes a new FK506-binding protein-like protein, has a dramatic effect on plant development. Mol. Cell. Biol. 18, 3034–3043 (1998).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–462 (2016).
Eddy, K. & Chen, S. Overcoming immune evasion in melanoma. Int J. Mol. Sci 21, (2020).
Mogensen, M. et al. [Melanoma]. Ugeskr Laeger 187, (2025).
Long, G. V., Swetter, S. M., Menzies, A. M. & Gershenwald, J. E. Scolyer, cutaneous melanoma. Lancet 402, 485–502 (2023).
Teixido, C., Castillo, P., Martinez-Vila, C., Arance, A. & Alos, L. Molecular markers and targets in melanoma. Cells 10, (2021).
Timar, J. & Ladanyi, A. Molecular pathology of skin melanoma: Epidemiology, differential diagnostics, prognosis and therapy prediction. Int J. Mol. Sci 23, (2022).
Chen, S., Luo, L., Chen, H. & He, C. The current state of research regarding the role of Non-Coding RNAs in cutaneous squamous cell carcinoma. Onco Targets Ther. 13, 13151–13158 (2020).
Zhang, J. et al. Identification of lncRNA-mRNA regulatory module to explore the pathogenesis and prognosis of melanoma. Front. Cell. Dev. Biol. 8, 615671 (2020).
Alos, L. et al. Melanoma on congenital melanocytic nevi. Pathol. Res. Pract. 256, 155262 (2024).
Zhang, Y., Ostrowski, S. M. & Fisher, D. E. Nevi and melanoma. Hematol. Oncol. Clin. North. Am. 38, 939–952 (2024).
Gerard, L. et al. Identification of two novel heterodimeric ABC transporters in melanoma: ABCB5β/B6 and ABCB5β/B9. J. Biol. Chem. 300, (2024).
Deng, Y., Zhang, L. & Luo, R. LINC01783 facilitates cell proliferation, migration and invasion in non-small cell lung cancer by targeting miR-432-5p to activate the Notch pathway. Cancer Cell. Int. 21, 234 (2021).
Liu, Y., Lu, C., Zhou, Y., Zhang, Z. & Sun, L. Circular RNA hsa_circ_0008039 promotes breast cancer cell proliferation and migration by regulating miR-432-5p/E2F3 axis. Biochem. Biophys. Res. Commun. 502, 358–363 (2018).
Harding, M. W., Galat, A., Uehling, D. E. & Schreiber, S. L. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 341, 758–760 (1989).
Siekierka, J. J., Hung, S. H., Poe, M., Lin, C. S. & Sigal, N. H. A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from Cyclophilin. Nature 341, 755–757 (1989).
Romano, S. et al. FK506 binding protein 51 positively regulates melanoma stemness and metastatic potential. Cell. Death Dis. 4, e578 (2013).
Crijns, A. P. et al. Survival-related profile, pathways, and transcription factors in ovarian cancer. PLoS Med. 6, e24 (2009).
Hagedorn, M., Siegfried, G., Hooks, K. B. & Khatib, A. M. Integration of zebrafish fin regeneration genes with expression data of human tumors in Silico uncovers potential novel melanoma markers. Oncotarget 7, 71567–71579 (2016).
Kashani-Sabet, M. et al. Identification of gene function and functional pathways by systemic plasmid-based ribozyme targeting in adult mice. Proc. Natl. Acad. Sci. U S A. 99, 3878–3883 (2002).
Solassol, J., Mange, A. & Maudelonde, T. FKBP family proteins as promising new biomarkers for cancer. Curr. Opin. Pharmacol. 11, 320–325 (2011).
Nowogrodzka, K. & Jankowska-Konsur, A. Emerging biomarker in carcinogenesis. Focus on Nestin. Postepy Dermatol. Alergol. 39, 1001–1007 (2022).
Brychtova, S., Fiuraskova, M., Hlobilkova, A., Brychta, T. & Hirnak, J. Nestin expression in cutaneous melanomas and melanocytic nevi. J. Cutan. Pathol. 34, 370–375 (2007).
Florenes, V. A., Holm, R., Myklebost, O., Lendahl, U. & Fodstad, O. Expression of the neuroectodermal intermediate filament Nestin in human melanomas. Cancer Res. 54, 354–356 (1994).
Sellheyer, K. & Krahl, D. Spatiotemporal expression pattern of neuroepithelial stem cell marker Nestin suggests a role in dermal homeostasis, neovasculogenesis, and tumor stroma development: a study on embryonic and adult human skin. J. Am. Acad. Dermatol. 63, 93–113 (2010).
Frank, N. Y. et al. Regulation of progenitor cell fusion by ABCB5 P-glycoprotein, a novel human ATP-binding cassette transporter. J. Biol. Chem. 278, 47156–47165 (2003).
Liu, W. et al. The BET inhibitor JQ1 suppresses tumor survival by ABCB5-mediated autophagy in uveal melanoma. Cell. Signal. 125, 111483 (2025).
Schatton, T. et al. Identification of cells initiating human melanomas. Nature 451, 345–349 (2008).
Duvivier, L. & Gillet, J. P. Deciphering the roles of ABCB5 in normal and cancer cells. Trends Cancer. 8, 795–798 (2022).
Donovan, C. Beyond single cytokines: targeting Co-Receptor CD131 in Asthma-COPD overlap. Respirology 30, 274–275 (2025).
Ciccone, V. et al. ALDH1A1 overexpression in melanoma cells promotes tumor angiogenesis by activating the IL–8/Notch signaling cascade. Int J. Mol. Med 50, (2022).
Lu, Y. et al. ALDH1A3-acetaldehyde metabolism potentiates transcriptional heterogeneity in melanoma. Cell. Rep. 43, 114406 (2024).
Nguyen, A. L., Facey, C. O. B. & Boman, B. M. The significance of aldehyde dehydrogenase 1 in cancers. Int J. Mol. Sci 26, (2024).
Zanoni, M., Bravaccini, S., Fabbri, F. & Arienti, C. Emerging roles of aldehyde dehydrogenase isoforms in Anti-cancer therapy resistance. Front. Med. (Lausanne). 9, 795762 (2022).
Wei, Y. et al. ALDH1: A potential therapeutic target for cancer stem cells in solid tumors. Front. Oncol. 12, 1026278 (2022).
Martín-Lluesma, S. et al. Efficacy of TIL therapy in advanced cutaneous melanoma in the current immuno-oncology era: updated systematic review and meta-analysis. Ann Oncol 35, (2024).
Author information
Authors and Affiliations
Contributions
Author Contributions Statement Template This study adheres to the CRediT (Contributor Roles Taxonomy) guidelines to transparently delineate each author’s contributions. Asterisks (*) indicating co-corresponding authors. [Author Ying Sun] Conceptualization: Designed the overarching research strategy and experimental framework. Methodology: Developed the xenograft model protocol and validated the LINC01116 knockdown efficiency assay. Formal Analysis: Performed statistical analyses of tumor growth kinetics and stemness marker expression. Writing – Original Draft: Authored Sections 2 (Methods) and 3 (Results), including figure legends. Data Analysis: Interpreted RNA-seq differential expression results. Writing – Review & Editing: Revised the Introduction and Discussion for clinical relevance. Writing – Original Draft: Authored Section 4 and Supplementary Materials. [Author Lin Li and Hua Yang] Visualization: Generated Figures 1–3 (tumor growth curves, Ki-67 IHC, and Western blot panels). [Author Ying Yang and Wei Yuan] Resources: Provided critical reagents (LINC01116 siRNA constructs, melanoma cell lines). Bioinformatics: Annotated stemness-related gene signatures using MSigDB databases. [Author HuiHuang Liu and YuTing Zhu] Data Curation: Managed raw datasets, including tumor volume measurements and immunohistochemical scoring. Investigation: Conducted in vivo experiments (animal handling, tumor inoculation, euthanasia). [Author ZhenJuan Wang and LiSheng Ying] Software: Programmed the R script for statistical modeling (two-way ANOVA, survival analysis). Validation: Verified reproducibility of TUNEL assay results across biological replicates. [Author Wen Yan and GengJian Zhang] Supervision: Oversaw ethical compliance (protocol submission to IACUC, welfare audits). Project Administration: Coordinated interdisciplinary collaboration between oncology and bioinformatics teams. *Correspondence Statement *Correspondence should be addressed to [Primary Corresponding Author Ying Yang] ([mailto:yangying8672@163.com]) and [Secondary Corresponding Author Wei Yuan] ([mailto:dryuanweihua@sina.com]).
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
All procedures involving human participants were conducted in accordance with the Declaration of Helsinki. This study was approved by the Ethics Committee of the Second Affiliated Hospital of Zunyi Medical University (Approval No. KYLL-2022-007).Written informed consent was obtained from all participants. Animal experiments were performed following the ARRIVE guidelines and approved by the Animal Care and Use Committee of Zunyi Medical University (Approval No. ZMU21-2405-001). Euthanasia was carried out via isoflurane inhalation followed by cervical dislocation to minimize suffering.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, Y., Li, L., Yang, H. et al. LINC01116 promotes nascent protein synthesis and cellular stemness through the miR-432-5p/FKBP7/14 regulatory axis in melanoma. Sci Rep 15, 44395 (2025). https://doi.org/10.1038/s41598-025-28087-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28087-2
