
Abstract
Crohn’s disease (CD) is a chronic, relapsing inflammatory disorder of the gastrointestinal tract with limited curative therapies. Genomic drug repurposing offers a rapid strategy to identify existing drugs that could be redirected to treat CD. We retrieved 1,333 CD–associated single-nucleotide polymorphisms from the GWAS Catalog and expanded these to 15,590 variants using linkage disequilibrium analysis in HaploReg. Fourteen layers of functional annotation prioritized 45 high‐confidence risk genes. Drug‐gene interactions were screened against DGIdb, and tissue expression patterns were assessed using GTEx bulk RNA-seq data. Cellular associations were explored via HuBMAP network analysis. Denosumab, an FDA-approved Receptor Activator of NF-κB Ligand (RANKL) inhibitor, exhibited the highest interaction score with TNFSF11 among prioritized genes. TNFSF11 expression peaked selectively in the terminal ileum—the primary site of CD pathology—while other risk genes showed broader. Network analysis linked TNFSF11 to mast cells in the ileal mucosa, suggesting that RANKL blockade could attenuate local inflammatory cascades. Integrating GWAS, functional genomics, and drug-gene interaction data highlights denosumab as a promising candidate for CD repurposing. Targeting TNFSF11 may enable focused anti-inflammatory effects in the ileum while minimizing systemic toxicity, warranting further preclinical and clinical evaluation.
Similar content being viewed by others

Introduction
Crohn’s disease (CD) is a progressive, relapsing, chronic inflammatory disease of the gastrointestinal tract, commonly affecting the terminal ileum and proximal colon as discontinuous, patchy, segmental, and transmural inflammation1,2. With peak incidence in between the age of 20–30 years3, CD affects individuals of all ages and leads to significant morbidity and disability caused by progressive bowel damage1,3, as well as an increased risk of intestinal and colorectal cancer2. Chronic diarrhea is the most frequently reported symptom and often accompanied by other symptoms: abdominal pain, fever, and bloody or mucopurulent diarrhea4, as well as systemic symptoms: fatigue and weight loss, and extraintestinal manifestations: arthropathy, dermatological, and ocular features3. While CD is currently one of an unknown cause, the etiology is believed to be the interplay of environmental factors, such as smoking, medications, and lifestyle5, in genetically predisposed individuals, resulting in dysfunctional interaction between the immune system and the symbiotic intestinal commensal microbiota4.
As the precise etiology continues to be unclear, a curative therapy has yet to be discovered. The goal of the prevailing therapy is to manage inflammation using medications and symptom alleviation using surgical interventions6. Current specific medical therapy for CD aims to suppress a hyperactive intestinal immune system in two phases: (1) induction, which leads to rapid clinical remission by the involvement of higher dose of steroid-sparing medications, sometimes also involving steroids such as budesonide, and (2) maintenance, which aims to maintain the remission and prevents the disease from relapsing by the involvement of lower dose of steroid-sparing medications without steroid use5. Medications currently used for Crohn’s disease often fail to produce a clinically significant response, and among those who initially respond, some experience a decline in efficacy or develop intolerance with long-term use5,7. This is believed to be due to phenotypic and genetic variations between patients, as well as the development of antidrug antibodies8.
Steroid-free clinical remission remains the main objective of CD management9, but developing a curative therapy involving targeted and patient-personalized therapy based on genomic studies is attainable. In preference to traditional drug development, which is often resource-intensive and time-consuming10, this study aims to contribute in search of new drugs for CD by repurposing existing, approved drugs for novel indications, which is achieved by identifying genes associated with CD and analyzing the drug-gene interactions to determine drug candidates11.
Recent advances research in bioinformatics have facilitated the integration of genome-wide association studies (GWAS) with functional genomics resources to better understand disease mechanisms and prioritize druggable targets12,13,14,15,16,17,18,19,20,21. Incorporating such approaches into Crohn’s disease research enhances the translational relevance of genetic discoveries and supports the rational design of targeted therapies. Therefore, the outcomes of this study are expected to identify repurposed drugs for targeted therapy in Crohn’s disease.
Methods
Study design
A comprehensive workflow of the current drug repositioning study for CD is illustrated in Fig. 1. First, SNPs associated with CD were retrieved from the NHGRI-EBI GWAS Catalog, then a Manhattan plot was used to visualize the significant loci, identifying 1,333 SNPs (Supplementary 1). These SNPs were then analyzed using HaploReg, expanding the dataset to 15,590 SNP variants (Supplementary 2) mapped to 846 genes (Supplementary 3). To prioritize genes with strong associations with CD, a functional annotation approach was applied based on 14 biological risk criteria, resulting in 45 high-confidence risk genes. The 45 genes were subsequently analyzed using a drug-gene interaction database (DGIdb) to identify existing drugs that target them, leading to the identification of Denosumab as a potential repositioning candidate due to its specific interactions with TNFSF11 and highest interaction score among these genes. This integrative approach - combining GWAS data, functional annotations, and computational drug repurposing strategies - provides insights into novel therapeutic options for CD, with TNFSF11 serving as a gene target (Fig. 1).

A workflow for discovering potential drugs to repurpose for Crohn’s disease. This figure was created in Biorender with agreement number GN28E877CV.
Identification of Crohn’s disease-associated SNPs
Our investigation began with the identification of single-nucleotide polymorphisms (SNPs) that exhibit associations with CD. To achieve this, we utilized data from the GWAS Catalog, a curated repository of genome-wide association studies. A database search was conducted on April 28th 2025, via the GWAS Catalog platform (https://www.ebi.ac.uk/gwas/) to identify SNPs associated with the CD trait. Following data extraction, a Manhattan plot was generated to visualize the most significant SNPs. We then performed gene mapping by identifying the most significantly associated genes on each chromosome based on the GWAS data.
Identification of Crohn’s disease SNP variants in an Asian population
It is important to note that SNPs do not always act independently in gene expression. They can be influenced by neighboring variants, and complex interactions often occur both between genes and between proteins, contributing to disease mechanisms. To account for these factors, we employed HaploReg version 4.2, a well-established tool that provides comprehensive annotations of regulatory variants, linked genetic variants, and other functional insights into gene loci. This approach enabled a more detailed examination of the genomic landscape surrounding CD-associated SNPs from 1000 genome project, through linkage disequilibrium (LD) r² ≥ 0.8. Furthermore, we specifically analyzed genetic data from the Asian population, allowing for a more refined understanding of the genetic architecture of CD within this demographic.
Functional analysis of Crohn’s disease-risk genes
To improve the identification of genes potentially contributing to CD susceptibility, we applied a comprehensive functional annotation approach aimed at systematically prioritizing genes based on their biological relevance to the disease. This strategy involved assessing each candidate gene using 14 curated databases, each representing different biological dimensions pertinent to CD. Genes that attained annotation scores of eight or above were designated as CD-risk genes, with higher scores indicating stronger biological plausibility in contributing to disease pathogenesis. Genes with annotation scores ≥ 8 were classified as CD risk genes. The gene prioritization scoring system was adapted and refined from a previous study19,22,23,24.
Prioritization of repurposed drugs for Crohn’s disease treatment
To identify potential drug candidates for CD treatment, we systematically analyzed the set of biological CD risk genes using the Drug-Gene Interaction Database (DGIdb) (https://www.dgidb.org). DGIdb is a comprehensive online resource that integrates information from scientific literature, publicly available drug databases, and other biomedical resources to curate known drug-gene interactions and identify druggable genes. This database standardizes and categorizes drug-gene interaction data into structured conceptual groups, facilitating efficient drug repurposing strategy25.
In this study, we utilized DGIdb to screen for FDA-approved drugs that exhibit well-characterized interactions with the prioritized CD-associated genes. Candidate drugs were selected based on interaction score threshold of ≥ 3, ensuring a mechanistic link between the drug and target gene. This approach allowed us to systematically filter and prioritize existing pharmaceuticals that may be repurposed for CD treatment based on their established drug-gene interactions.
Statistical analysis
Statistical analyses were carried out using RStudio version 2024.09.0 + 375 (RStudio, Boston, MA, USA). Functional annotation was performed using the WEB-based GEne SeT AnaLysis Toolkit (WebGestalt; https://www.webgestalt.org), covering eleven different annotation categories. Missense variant genes were identified using the haploR package (https://cran.r-project.org/web/packages/haploR/index.html). Drug–gene interaction analysis was conducted using the DGIdb database (https://dgidb.org). Visualization of drug-gene interactions was performed using the circlize package (https://cran.r-project.org/web/packages/circlize/index.html) to generate a chord diagram. Gene network analysis was conducted via Enrichr (https://maayanlab.cloud/enrichr-kg) to explore interaction pathways. Additionally, gene expression profiles across specific human tissues and organs were examined using data retrieved from the GTEx Portal (https://gtexportal.org). All database searches were conducted on April 30, 2025.
Result
GWAS visualization of Crohn’s disease-associated SNPs using a Manhattan plot
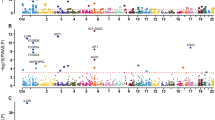
A total of 1,333 SNPs were identified from the GWAS data for CD. The distribution of their statistical significance across the genome was illustrated by a Manhattan plot (Fig. 2) that accentuates the loci with potential roles in the pathogenesis and progression of CD.

Manhattan plot of genome-wide association study (GWAS) results for Crohn’s disease. Each dot represents an SNP plotted according to its chromosomal position on the x-axis and its − log₁₀(p-value) on the y-axis. The horizontal black line indicates the genome-wide significance threshold (p = 5 × 10⁻⁸). The five most frequently appearing genes on each chromosome are annotated, highlighting candidate regions potentially involved in the pathogenesis of CD. This figure was created by R studio.
In this study, we performed gene mapping using genome-wide association data and visualized the results with a Manhattan plot. This plot allows us to identify loci with strong statistical associations with Crohn’s disease; additionally, we used it to determine the five most frequently appearing genes on each chromosome, thereby observing the genomic distribution of potential risk loci. Among them, IL23R, NOD2, and STAT3 are consistently reported as significant genetic contributors to disease susceptibility. IL23R plays a central role in immune signaling, with its variants linked to dysregulated inflammatory responses in the gut. NOD2, one of the earliest genes associated with CD, is involved in bacterial recognition; mutations in this gene impair microbial sensing, leading to chronic intestinal inflammation. STAT3, a key transcription factor in cytokine signaling, has also been implicated in the inflammatory process characteristic of CD. The consistent appearance of these genes across studies underscores their relevance in the genetic architecture and pathogenesis of CD 26–32.
While GWAS remains a valuable tool for identifying loci with strong statistical associations, it has inherent limitations. Relying solely on statistical thresholds may result in the omission of genes that, despite their modest association signals, contribute meaningfully to disease pathogenesis through functional interactions, such as gene regulation, signaling cascades, or epigenetic mechanisms. Moreover, our mapping revealed that candidate genes associated with CD are dispersed across nearly all chromosomes, with no SNP clustering confined to a specific chromosome. This observation supports the notion that CD is driven by a complex interplay of multiple genetic and biological factors. To address these limitations, functional annotations are required to uncover the full spectrum of biologically relevant genes. By moving beyond significance alone, researchers can gain a more comprehensive understanding of disease mechanisms and identify novel gene targets for therapeutic intervention.
Identification of genes associated with Crohn’s disease
Following the expansion of the initial 1,333 SNPs through linkage disequilibrium (LD) analysis using HaploReg (r² ≥ 0.8 in Asian populations), a total of 15,590 SNPs corresponding to 846 genes were identified as potentially associated with CD. To evaluate the functional relevance of these genes, a gene set enrichment analysis was performed using the DisGeNET database and openXGR database. As depicted in Fig. 3, the analysis revealed significant enrichment of genes associated with CD, inflammatory bowel disease, and ulcerative colitis.
This result supports the biological validity of the genes obtained from LD expansion and confirms that a substantial portion of these genes have known associations with CD and related inflammatory conditions. Notably, several top-enriched terms were highly specific to CD, strengthening the evidence that the identified genes contribute meaningfully to disease mechanisms. Furthermore, the presence of overlapping gene sets among multiple disease, suggests potential shared genetic pathways, particularly between CD, inflammatory bowel disease, and ulcerative colitis. These findings highlight the value of integrating GWAS with enrichment analysis to uncover not only statistically significant variants but also functionally coherent gene networks.

A scatter plot visualizing the enrichment analysis of 846 genes associated with Crohn’s disease using the DisGeNET database. Dot size indicates the number of overlapping genes, and color intensity represents enrichment significance. The top enriched terms—Crohn’s disease, inflammatory bowel disease, and ulcerative colitis—are closely related conditions, highlighting the strong disease relevance of the identified gene set. This figure was created by https://www.webgestalt.org.
To further validate the biological relevance of the identified SNP variants, we performed gene set enrichment analysis using disease ontology. The results are visualized in Fig. 4 as a bubble plot, where Crohn’s disease (CD) emerged as the most enriched trait with the highest Z-score (12) and odds ratio (OR = 6.93), compared to other related conditions. Inflammatory bowel disease, bone inflammation disease, rheumatoid arthritis, and arthritis also showed significant enrichment, but with lower Z-scores and OR values. These findings confirm that the expanded SNP set captures not only shared inflammatory pathways but also strongly prioritizes CD as the primary disease phenotype. The enrichment pattern underscores the robustness of our SNP expansion approach and supports the use of these variants for downstream functional annotation and drug repurposing analyses.

Gene set enrichment analysis of disease ontology associations using OpenXGR. Bubble plot showing enriched disease terms identified through gene set enrichment analysis. The x-axis represents the Z-score, and the y-axis represents –log10(FDR). Bubble size indicates the number of associated genes. Among the top five enriched diseases, Crohn’s disease (CD) exhibited the highest Z-score (12) and the strongest odds ratio (OR = 6.93, 95% CI [4.69–10.1], FDR = 2.20E–16), indicating the most robust association. Data were obtained from the OpenXGR platform (http://www.openxgr.com/OpenXGR).
Functional annotation prioritization reveals key genes implicated in Crohn’s disease
To refine gene prioritization linked to CD, we employed 14 distinct layers of functional annotation, each contributing one point per gene if applicable (Supplementary 3). These included (1) the functional impact of missense variants, which evaluates amino acid substitutions that may disrupt protein function; (2) expression quantitative trait loci (eQTL) in whole blood; (3) gene involvement in key biological processes; (4) localization of gene products within specific cellular components; (5) molecular functions, including enzymatic activity or receptor interaction; (6) inclusion in the 2022 Primary Immunodeficiency gene list; (7) associations cataloged in DisGeNET for gene-disease relationships; (8) cellular profiles across tissues from the Human Cell Landscape; (9) links to clinical phenotypes via the Human Phenotype Ontology; (10) pathway associations from the Kyoto Encyclopedia of Genes and Genomes (KEGG); (11) kinase target information; (12) regulation by microRNAs from miRNA databases; (13) interaction pathways curated in reactome pathway database; and (14) regulatory interactions involving transcription factors.
As summarized in Fig. 5A, DisGeNET-derived annotations were the most prevalent, confirming that the most frequently annotated genes are those with the strongest known associations to CD (Fig. 3). While most genes exhibited low annotation scores, a subset showed strong functional convergence (Fig. 5B). A threshold score of ≥ 8 was used to define high-priority genes, yielding 45 genes for downstream analysis. The threshold of annotation scores ≥ 8 was adapted from prior bioinformatics-driven drug repurposing studies that employed multi-layer functional annotation frameworks18,19,20,21,33. The top-ranked gene was NFKB1, with score of 12, followed by STAT3, SMAD3, and NFKBIA, each with scores of 11 (Fig. 5C). Many of these top candidates are involved in inflammatory signaling pathways, cytokine response, and transcriptional regulation, all of which are critical in the pathophysiology of CD26,34–41. Our study complements prior large-scale GWAS of Crohn’s disease42,43,44,45,46 by employing a different methodological framework. Unlike these studies that relied on meta-analysis and fine-mapping, we integrated functional annotation from 14 independent databases to systematically prioritize candidate genes. Despite methodological differences, our findings converged on key loci such as NOD2, IL23R and TNFSF15, reinforcing the robustness of our approach. This highlights the novelty of combining LD expansion with multi-layered functional annotation to provide deeper biological insight beyond statistical associations.

Functional annotation–based prioritization of genes associated with Crohn’s disease. (A) Number of genes identified in each of the 14 functional annotation categories. (B) Distribution of genes based on cumulative annotation scores. (C) Genes with scores ≥ 8 (n = 45) were considered high-priority.
Determination of drug candidates
We utilized the DGIdb to identify drug candidates targeting our 45 high-priority genes. A threshold of ≥ 3 was applied to capture drugs with strong interaction evidence, following the approach adapted from previous work19. However, when restricting the analysis to FDA-approved drugs with an interaction score ≥ 3, only five genes remained actionable. Furthermore, DGIdb provides only quantitative interaction scores without specifying whether a drug–gene interaction is agonistic or antagonistic, so the functional consequence of each hit must be inferred from theory or the literature. For example, one of the drugs, mifamurtide, was excluded from potential drug candidates due to its immunostimulatory effects mediated via NOD2 activation, whose receptors are expressed in macrophages and monocytes47. Therefore, its use would be counterproductive to the immunosuppressive goal of CD management. Among the remaining seven potential drugs, denosumab, showing specific interactions with TNFSF11, emerged as the drug with the highest interaction score of 43.5 (Fig. 6). Denosumab was approved and is currently used as a treatment for osteoporosis48. Other drugs with lower interaction scores (shown in grey links in Fig. 6) may still possess therapeutic potential and require extended research.

A chord diagram showing the interactions of drug candidates for Crohn’s disease with their target genes and their current indications. Denosumab’s interaction with TNFSF11 is highlighted in red to denote its highest interaction score (IS = 43.5), while all other drug–gene interactions are shown in grey. This figure was created by R studio.
Tissue expression and cellular interactions of TNFSF11
Clarifying the role of TNFSF11 in the molecular mechanisms underlying this disease may offer valuable insights for the development of targeted therapeutic strategies. The network diagram reveals how TNFSF11 which encodes receptor activator of NF-κB ligand (RANKL) connects with mast cells in the small intestine and various reticular cell types in lymph nodes (Fig. 7). At the gut mucosa, TNFSF11 appears to stimulate mast cells, setting off NF-κB pathways activation that lead to the release of inflammatory mediators. This response could help the body respond to bacterial threats but, if unchecked, may also drive the chronic inflammation seen in CD49–52. Such insights suggest that targeting TNFSF11 could offer a way to dial down harmful gut inflammation.

Network diagram illustrating the gene TNFSF11 and its associations with specific cell types. This figure obtained from https://maayanlab.cloud/enrichr-kg.
To analyze the tissue distribution of our five prioritized CD risk genes, we examined bulk RNA-seq data from 30 human tissues in the Genotype-Tissue Expression (GTEx) Portal database. Among these, TNFSF11 displayed a striking, organ-restricted expression peak in the terminal ileum of the small intestine—precisely the region most often afflicted in CD. In contrast, NOD2 and IL6R were predominantly expressed in whole blood, SH2B3 showed its highest expression in spleen but also high-level expression across multiple other organs. Meanwhile, TGFBR2 exhibited a fairly uniform, high-level expression across many tissues (Fig. 8).
These divergent expression profiles carry important therapeutic implications. The highly restricted localization of TNFSF11 suggests that RANKL blockade using an agent such as denosumab could deliver potent anti-inflammatory effects directly to the ileal mucosa while minimizing systemic exposure and off-target toxicity. By contrast, targeting broadly expressed receptors like TGFBR2 may increase the risk of systemic adverse events. Aligning drug-repurposing strategies with tissue-specific gene expression landscapes thus offers a precision-medicine framework to maximize both efficacy and safety in CD treatment.

This analysis shows that the target gene TNFSF11 is localized in gastrointestinal tracts, which differs to the other 4 genes (TGFBR2, NOD2, IL6R, SH2B3) that have variation in expression in each organ, proposing a more organ-targeted therapy in CD. This figure obtained from https://gtexportal.org.
Beyond denosumab, our analysis also revealed agents targeting IL6R, TGFBR2, SH2B3, and NOD2. However, most of these drugs had substantially lower interaction scores than denosumab and did not meet our prioritization criteria. For instance, mifamurtide, an immunostimulatory compound acting via NOD2, was excluded because it would likely exacerbate, rather than suppress, intestinal inflammation. Similarly, drugs targeting TGFBR2, SH2B3 or IL6R raised concerns regarding systemic toxicity possibility due to their broad expression patterns across multiple tissues.
Transcriptome-wide association evidence and therapeutic implications
To further explore the causal relationship between GWAS loci, gene expression, and CD risk, we performed a transcriptome-wide association study (TWAS) using reference prediction models obtained from the TWAS Hub (https://twas-hub.org/). These models were built upon the GTEx v8 reference panel encompassing 49 human tissues. For this analysis, we prioritized tissues most relevant to CD pathogenesis, including the small intestine (terminal ileum), transverse and sigmoid colon, and whole blood, while also incorporating immune-related tissues such as spleen and lymph node to capture systemic immune contributions.
The TWAS results indicated that genetically predicted expression of TNFSF11 showed a positive association with Crohn’s disease across two independent datasets—Crohn’s Disease 2012 and Crohn’s Disease 2017—with Z-scores of 2.9 and 4.4, respectively (Supplementary 4). These associations were most evident in intestinal and lymphoid tissues, consistent with the gene’s localized expression peak in the terminal ileum from the GTEx dataset (Fig. 8). This tissue-specific enrichment highlights TNFSF11’s potential role in mucosal immune regulation and its translational relevance to CD.
At the same time, TWAS identified other TNF-superfamily members: TNFSF8 and TNFSF15, as significantly associated with CD at the transcriptional level (Supplementary 5). This distinction suggests a complementary model: TNFSF8 and TNFSF15 may represent key genetic drivers of disease susceptibility, whereas TNFSF11 (RANKL) primarily contributes through therapeutically targetable signaling mechanisms that mediate immune–stromal crosstalk in the intestinal microenvironment.
To strengthen these transcriptomic findings, we integrated clinical and network-level validation layers. Evidence from a Phase 2 clinical trial (NCT02321280) demonstrated the safety and mechanistic plausibility of denosumab treatment in CD patients, while preclinical studies using a dinitrobenzenesulfonic acid (DNBS)-induced colitis mouse model showed that RANKL inhibition by denosumab markedly reduced mucosal cytokine expression and restored microbial diversity. In addition, network-based prioritization using Open Targets and OpenXGR confirmed that TNFSF11 resides within a highly enriched immune-signaling subnetwork relevant to Crohn’s disease. Taken together, these multi-layered lines of evidence—genetic, transcriptomic, clinical, and network-based—provide convergent support for TNFSF11 as a biologically relevant and pharmacologically tractable target. Consequently, denosumab, a monoclonal antibody against RANKL, emerges as a robust and rational repurposing candidate for Crohn’s disease.
Discussion
Crohn’s disease is a chronic, progressive, and immunologically mediated disease. CD could induce inflammation from entire GI tract, but most often affects terminal ileum and right colon. Gastrointestinal symptoms such as diarrhea and abdominal pain are common, accompanied by systemic manifestations including weight loss, fever, and fatigue. Without treatment, the inflammation could lead to severe complication for the patient55.
The immune dysregulation of CD involves defect in innate immunity and adaptive immunity56. Defects in innate immunity highlight the important role of mast cell as a major contributing factor, alongside gene variants that impair the mucous barrier. A study found increased mast cell accumulation in fibrotic areas, especially around strictures, indicating a possible role in promoting tissue fibrosis57. In CD, mast cell production of pro-inflammatory mediators—such as TNF-α, IL-16, histamine, tryptase, and substance P—is dysregulated, amplifying inflammation through autocrine and paracrine loops and contributing to disease chronicity58. Although the trigger of mast cell activation remains largely unknown, there is no evidence for IgE-dependent pathway, suggesting alternative mechanism such as stress-related factor59,60,61,62,63,64. Mast cells may be activated by bacterial products such as LPS through TLR-4, Fcγ receptors, and soluble CD14 from mucosal microenvironment, further promoting inflammation65. These findings underscore the importance of mast cells in Crohn’s disease pathophysiology. However, although adaptive immunity—driven by Th1 and Treg cell dominance via proinflammatory cytokines such as TNF, IL-12, IL-34, and IL-23—fuels ongoing inflammation, current therapies for Crohn’s disease, including anti-TNF agents and interleukin inhibitors, are not curative and often fail to achieve sustained remission66,67,68,69,70.
In our study, we discovered TNFSF11 as the highest potential gene target. TNFSF11, also known as RANKL, is a ligand for osteoprotegerin and plays a role in osteoclast differentiation and activation71. Further analysis of this gene was conducted using two methods. The first analysis utilized HuBMAP (the Human BioMolecular Atlas Program), which confirmed and expanded the immune role of TNFSF11 by showing its association with mast cell in the small intestine. The consistent appearance of mast cells highlights their potential significance in TNSFS11-mediated immune response and supports their important role in pathogenesis of CD. The second analysis shows the bulk tissue gene expression for TNFSF11 is dominantly in small intestine–specifically the terminal ileum, a region that is commonly affected by CD. Integrating these findings, we propose that upregulation of TNFSF11 will drive increased mast cell activation. This conclusion helps explain why mast cells in terminal ileum were found to be elevated in patient with IBS, according to a study72. This finding also suggests that targeting TNFSF11 with its inhibitor could exert the greatest effect in the terminal ileum–where it is most needed–while minimizing side effect in other organs. This approach exemplifies organ-specific targeted therapy.
TNFSF11 also shows an interaction with NFKB1 gene, which held the highest score of 12 in the functional annotation score (Fig. 5C). NF-κB is a transcription factor that becomes activated when the receptor activator of NF-κB (RANK) binds to its ligand (RANKL) IBD73,74,75,76. RANKL is originated in subepithelial stromal cells in the Peyer’s patch domes and mast cells that is situated at the same location77,78. Peyer’s patches are mostly concentrated in the distal ileum79, consistent with bulk-tissue gene expression showing high titer of TNFSF11 (the gene encoding RANKL) in the distal or terminal ileum (Fig. 8). During inflammation (e.g., bacterial infection), a study showed that microfold cells are upregulated via RANK–RANKL signaling, implying that RANKL expression and activity increase under inflammatory conditions. RANK on the other hand, is primarily expressed by intestinal epithelial cells such as follicle-associated epithelium which cover Peyer’s patches, villous and crypt epithelial cells, and epithelial precursor cells which situated in crypts adjacent to Peyer’s patches80.
The RANK-RANKL complex will activate TRAF6, which recruits TAK1/Table 2 complexes to initiate NF-κB and MAPK pathway, creating feed-forward loop. The loop continues on with NF-κB upregulating RANKL, amplifying NF-κB activity via TRAF6-TAK1-Table 2 that will sustain pro-inflammatory cytokine production, explaining why it is hyperactivated in IBD81,82. Therefore, using a RANKL inhibitor should stop the feed-forward loop and reduce inflammation in inflammatory bowel diseases, in this context, CD.
Utilizing DGIdb with a criterion of interaction scores greater than 3, the only drug that was identified to interact with TNSF11 is denosumab, a RANKL inhibitor. Denosumab is currently used as an anti-resorptive bone medication and is FDA-approved for conditions such as skeletal-related bone pain and fractures caused by multiple myeloma or bone metastases from solid tumors, giant cell tumors of the bone, hypercalcemia of malignancy, as well as osteoporosis in postmenopausal women at high risk of fractures and in men at increased risk due to osteoporosis or glucocorticoid-induced bone loss83. A study showed that in interleukin 2-deficient mouse model demonstrated that spontaneous osteopenia and colitis were driven by excessive RANKL production. Blocking the RANKL-RANK interaction reversed bone abnormalities and reduced colitis severity by lowering dendritic cell numbers in the colon84,85. These findings support the repurposing of RANKL inhibitors such as denosumab as a potential therapeutic strategy in inflammatory bowel diseases such as CD.

Proposed mechanism of TNFSF11 (RANKL)-mediated inflammatory signaling in Crohn’s disease and its inhibition by Denosumab. (1) Bacterial infection in the terminal ileum triggers upregulation and activation of TNFSF11 in subepithelial stromal cells and mast cells of Peyer’s patches. (2–3) Activated TNFSF11 is released as soluble RANKL into the subepithelial compartment. (4) RANKL binds to its receptor RANK on epithelial and mast cells, resulting in NF-κB activation. (5) NF-κB signaling drives activation of downstream immune cells. (6) Mast cell degranulation releases pro-inflammatory mediators (including TNF-α), fueling chronic intestinal inflammation characteristic of CD. (7) TNF-α further enhances RANKL expression and RANKL-induced NF-κB signaling, creating a pathogenic feed-forward loop.
Denosumab, a monoclonal anti-RANKL antibody, binds and neutralizes RANKL (dashed arrow), preventing RANK–RANKL interactions and downstream NF-κB activation to attenuate this inflammatory cascade. This figure was created in Biorender with agreement number GR28E87B3N.
Based on multiple analyses and several supporting studies, our study proposes a model to integrate and simplify the interplay of TNFSF11 with immune response and the role of denosumab (Fig. 9). Bacterial infection and inflammatory condition will upregulate TNFSF11 in terminal small intestine that originates from subepithelial stromal and mast cells in the Peyer’s patch that will encode RANKL. RANKL will then bind with RANK which is located in follicle-associated epithelium cells, villous and crypt epithelial cells, and epithelial precursor cells forming RANK-RANKL complex. The complex will activate NF-κB and MAPK pathways, causing a pro-inflammatory condition with a feed-forward mechanism, creating a loop causing a chronic condition in CD. Additionally, mast cells also play a role in this mechanism via TNF-α in their degranulation products. TNF-α increase expression of NFATc1, which will prime immune cells, specifically macrophages that cause an increased response in NF-κB pathway activation86,87,88. Moreover, TNF-α will sustain canonical NF-κB activation (p50/p65), while RANKL primarily activates the non-canonical NF-κB activation (p52/ReIB). The combined activation will drive chronic inflammation in the gut mucosa86,89,90. Besides that, the same cytokine will cause a feed-forward inflammatory loop, TNF-α will enhance RANKL effects, and RANKL induced NF-κB activation further stimulates production of pro-inflammatory cytokines, including TNF-α91,92,93,94. Denosumab plays a role in inhibiting RANKL by binding the RANKL, preventing the complex to form and stopping TNF-α production. Which means the drug will stop both RANKL-NF-κB and mast cell feed forward mechanisms and will reduce inflammation in CD. To strengthen the translational validity of our findings, we adopted a systematic multi-layer validation framework encompassing transcriptomic, clinical, and network-based approaches. First, TWAS results across independent cohorts confirmed a positive association between TNFSF11 expression and CD risk, especially in intestinal and lymphoid tissues. Second, clinical data from a Phase 2 trial (NCT02321280) provided direct human-based validation for denosumab’s mechanistic relevance in CD, supported by preclinical models of experimental colitis showing that RANKL blockade mitigates mucosal inflammation. Third, reanalysis using the Open Targets and OpenXGR platforms consistently positioned TNFSF11 within Crohn’s disease–enriched immune subnetworks, underscoring its integrative role in disease biology. These complementary validation layers collectively strengthen the evidence supporting denosumab as a promising repurposed agent for CD.
Denosumab, a fully human monoclonal antibody against RANKL, is currently approved for osteoporosis and administered subcutaneously every six months with a well-established safety and pharmacokinetic profile in over 10,000 patients from phase III trials and post-marketing data (ClinicalTrials.gov ID NCT02321280). Its mechanism is highly relevant to Crohn’s disease (CD), as RANKL not only mediates bone resorption but also regulates T cell–dendritic cell interactions integral to intestinal inflammation. Preclinical studies, including dinitrofluorobenzene sulfonic acid (DNBS) models of colitis, demonstrated that RANKL inhibition improved both bone mass and colitis, supporting translational relevance. Taken together, its favorable pharmacokinetics, safety record, and mechanistic overlap with CD pathogenesis provide a rational basis for evaluating denosumab as a novel therapeutic option in this setting.
This study highlights the role of TNFSF11 in the pathogenesis of Crohn’s disease and supports the potential repurposing of denosumab. Identified as a prioritized gene through GWAS data integration and functional annotation scoring, TNFSF11 shows strong associations with immune responses specific to the small intestine, particularly the terminal ileum. Therefore, targeting TNFSF11 with RANKL inhibitors such as denosumab represents a promising novel treatment strategy for Crohn’s disease. However, several limitations exist. First, our study may not capture all relevant genetic variants across diverse populations, as we only expanded SNP variants in the Asian population, which may affect the generalizability of our findings. Second, denosumab’s clinical safety and efficacy in Crohn’s disease must still be validated through preclinical and clinical studies. Third, although our findings are supported by clinical trial evidence (Phase III trial NCT02321280), we recognize that complementing our approach with other established methodologies from prior studies could enhance the robustness of our conclusions12,13,14,15,16,95. Such future research will be crucial to determine whether TNFSF11–RANKL blockade can ultimately be translated into an effective precision therapy for Crohn’s disease. Fourth, beyond denosumab, our analysis also identified agents targeting TGFBR2, SH2B3, and IL6R, which may hold therapeutic potential, although concerns regarding systemic toxicity and broad tissue expression remain. Therefore, further studies are warranted to explore the feasibility and safety of these alternative therapeutic strategies.
Conclusion
Genomic analysis identified five high-confidence targets for drug repurposing in CD. Among these, denosumab—targeting TNFSF11—was prioritized due to its highest interaction score and its selective action in the gastrointestinal tract, the primary site of CD pathology. This suggests that it may serve as a targeted therapeutic agent for CD.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Dolinger, M., Torres, J. & Vermeire, S. Crohn’s disease. Lancet 403, 1177–1191 (2024).
Pasternak, G. et al. Crohn’s disease: basic characteristics of the Disease, diagnostic Methods, the role of Biomarkers, and analysis of metalloproteinases: A review. Life 13, 2062 (2023).
Cockburn, E. et al. Crohn’s disease: an update. Clin. Med. 23, 549–557 (2023).
Karabulut, A. & Kaya, M. Crohn’s disease from past to present: research trends and global outcomes with scientometric analysis during 1980 to 2022. Medicine 102, e34817 (2023).
Cushing, K. & Higgins, P. D. R. Management of Crohn disease. JAMA 325, 69 (2021).
Gordon, H. et al. ECCO guidelines on therapeutics in crohn’s disease: medical treatment. J. Crohns Colitis. 18, 1531–1555 (2024).
Wang, Z. et al. Microbial and genetic-based framework identifies drug targets in inflammatory bowel disease. Theranostics 11, 7491–7506 (2021).
Scheurlen, K. M., Parks, M. A., Macleod, A. & Galandiuk, S. Unmet challenges in patients with crohn’s disease. J. Clin. Med. 12, 5595 (2023).
Cheifetz, A. S. Management of active Crohn disease. JAMA 309, 2150 (2018).
Grenier, L. & Hu, P. Computational drug repurposing for inflammatory bowel disease using genetic information. Comput. Struct. Biotechnol. J. 17, 127–135 (2019).
Garza-Hernandez, D. et al. A systematic review and functional bioinformatics analysis of genes associated with crohn’s disease identify more than 120 related genes. BMC Genom. 23, 302 (2022).
Kiryluk, K. et al. Genome-wide association analyses define pathogenic signaling pathways and prioritize drug targets for IgA nephropathy. Nat. Genet. 55, 1091–1105 (2023).
Brown, A. C. et al. Comprehensive epigenomic profiling reveals the extent of disease-specific chromatin States and informs target discovery in ankylosing spondylitis. Cell. Genomics. 3, 100306 (2023).
Fang, H. & Knight, J. C. Priority index: database of genetic targets in immune-mediated disease. Nucleic Acids Res. 50, D1358–D1367 (2022).
Fang, H. et al. A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat. Genet. 51, 1082–1091 (2019).
Buniello, A. et al. Open targets platform: facilitating therapeutic hypotheses Building in drug discovery. Nucleic Acids Res. 53, D1467–D1475 (2025).
Satria, R. D. et al. Identification of druggable genes for multiple myeloma based on genomic information. Genomics Inf. 21, e31 (2023).
Sukorini, U. et al. Genome-wide association study-driven identification of thrombomodulin and factor V as the best biomarker combination for deep vein thrombosis. Genomics Inf. 23, 11 (2025).
Satria, R. D. et al. Genome-wide association study – Driven drug repositioning for the treatment of insomnia. J. Genetic Eng. Biotechnol. 23, 100502 (2025).
Satria, R. D. et al. Bioinformatic and genomic analysis identifies C allele of APOE rs7412 as the most prominent variant limiting extreme human longevity. Sci. Rep. 15, 21688 (2025).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Irham, L. M., Adikusuma, W. & Perwitasari, D. A. Genomic variants-driven drug repurposing for tuberculosis by utilizing the established bioinformatic-based approach. Biochem. Biophys. Rep. 32, 101334 (2022).
Adikusuma, W. et al. Transcriptomics-driven drug repositioning for the treatment of diabetic foot ulcer. Sci. Rep. 13, 10032 (2023).
Adikusuma, W. et al. Integrated genomic network analysis revealed potential of a druggable target for hemorrhoid treatment. Saudi Pharm. J. 31, 101831 (2023).
Freshour, S. L. et al. Integration of the Drug-Gene interaction database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49, D1144–D1151 (2021).
Jostins, L. et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Khor, B., Gardet, A. & Xavier, R. J. Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 (2011).
Hugot, J. P. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to crohn’s disease. Nature 411, 599–603 (2001).
Pastras, P., Aggeletopoulou, I., Papantoniou, K. & Triantos, C. Targeting the IL-23 receptor gene: A promising approach in inflammatory bowel disease treatment. Int. J. Mol. Sci. 26, 4775 (2025).
Ashton, J. J. et al. Prediction of Crohn’s Disease Stricturing Phenotype Using a NOD2- derived Genomic Biomarker. Inflamm. Bowel Dis. 29, 511–521 (2023).
Samad, M. A. et al. STAT3 signaling pathway in health and disease. MedComm (Beijing) 6, (2025).
Robinson, P. et al. Genetic and Small-Molecule modulation of Stat3 in a mouse model of crohn’s disease. J. Clin. Med. 11, 7020 (2022).
Lu, H. F. et al. Meta-Analysis of Genome-Wide association studies identifies three loci associated with stiffness index of the calcaneus. J. Bone Miner. Res. 34, 1275–1283 (2019).
Chen, J., Bardes, E. E., Aronow, B. J. & Jegga, A. G. ToppGene suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311 (2009).
Borm, M. E. A., van Bodegraven, A. A., Mulder, C. J. J., Kraal, G. & Bouma, G. A NFKB1 promoter polymorphism is involved in susceptibility to ulcerative colitis. Int. J. Immunogenet. 32, 401–405 (2005).
Barrett, J. C. et al. Genome-wide association defines more than 30 distinct susceptibility loci for crohn’s disease. Nat. Genet. 40, 955–962 (2008).
Bouwman, W., Verhaegh, W. & van de Stolpe, A. Improved diagnosis of inflammatory bowel disease and prediction and monitoring of response to anti-TNF alpha treatment based on measurement of signal transduction pathway activity. Front Pharmacol 13, (2022).
Liu, H. et al. A broken network of susceptibility genes in the monocytes of crohn’s disease patients. Life Sci. Alliance. 7, e202302394 (2024).
Moneva-Sakelarieva, M. et al. The role of the transcription factor NF-kB in the pathogenesis of inflammation and carcinogenesis. Modulation capabilities. Pharmacia 72, 1–13 (2025).
Ji, Z., He, L., Regev, A. & Struhl, K. Inflammatory regulatory network mediated by the joint action of NF-kB, STAT3, and AP-1 factors is involved in many human cancers. Proc. Natl. Acad. Sci. 116, 9453–9462 (2019).
Guo, Q. et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal. Transduct. Target. Ther. 9, 53 (2024).
Liu, Z. et al. Genetic architecture of the inflammatory bowel diseases across East Asian and European ancestries. Nat. Genet. 55, 796–806 (2023).
de Lange, K. M. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 49, 256–261 (2017).
El Hadad, J., Schreiner, P., Vavricka, S. R. & Greuter, T. The genetics of inflammatory bowel disease. Mol. Diagn. Ther. 28, 27–35 (2024).
Huang, R. et al. Identifying immune cell infiltration and effective diagnostic biomarkers in crohn’s disease by bioinformatics analysis. Front Immunol 14, (2023).
Nguyen, V. Q. et al. Noncanonical NF-κB signaling upregulation in inflammatory bowel disease patients is associated with loss of response to Anti-TNF agents. Front Pharmacol 12, (2021).
Jiang, H. et al. Application of carbohydrates in approved small molecule drugs: A review. Eur. J. Med. Chem. 223, 113633 (2021).
Kendler, D. L., Cosman, F., Stad, R. K. & Ferrari, S. Denosumab in the treatment of osteoporosis: 10 years later: A narrative review. Adv. Ther. 39, 58–74 (2022).
Kim, H. Y., Kang, H. G., Nam, S. Y., Kim, H. M. & Jeong, H. J. Blockade of RANKL/RANK signaling pathway by Epigallocatechin gallate alleviates mast cell-mediated inflammatory reactions. Int. Immunopharmacol. 88, 106872 (2020).
Lleo, A. & Gershwin, M. E. Targeting the RANK/RANKL pathway in autoimmune disease and malignancy: future perspectives. Expert Rev. Clin. Immunol. 17, 933–936 (2021).
Mao, H., Zhao, X. & Sun, S. C. NF-κB in inflammation and cancer. Cell. Mol. Immunol. 22, 811–839 (2025).
Pagnini, C. & Cominelli, F. Tumor necrosis factor’s pathway in crohn’s disease: potential for intervention. Int J. Mol. Sci 22, (2021).
Dostert, C., Grusdat, M., Letellier, E. & Brenner, D. The TNF family of ligands and receptors: communication modules in the immune system and beyond. Physiol. Rev. 99, 115–160 (2019).
Khafipour, A. et al. Denosumab regulates gut microbiota composition and cytokines in dinitrobenzene sulfonic acid (DNBS)-Experimental colitis. Front Microbiol 11, (2020).
Ranasinghe, I. R., Tian, C., Ronald & Affiliations, H. Crohn Disease.
Petagna, L. et al. Pathophysiology of Crohn’s disease inflammation and recurrence. Biology Direct vol. 15 Preprint at (2020). https://doi.org/10.1186/s13062-020-00280-5
Li, Y. et al. Inflammation-fibrosis interplay in inflammatory bowel disease: mechanisms, progression, and therapeutic strategies. Front Pharmacol 16, (2025).
Albert-Bayo, M. et al. Intestinal Mucosal Mast Cells: Key Modulators of Barrier Function and Homeostasis. Cells 8, (2019).
Bischoff, S. C. Mast cells in Gastrointestinal disorders. Eur. J. Pharmacol. 778, 139–145 (2016).
Farhadi, A. et al. Reduced immunostaining for c-kit receptors in mucosal mast cells in inflammatory bowel disease. J. Gastroenterol. Hepatol. 22, 2338–2343 (2007).
Noto, C. N., Hoft, S. G. & DiPaolo, R. J. Mast cells as important regulators in autoimmunity and cancer development. Front Cell. Dev. Biol 9, (2021).
Castells, M. et al. Mast cell activation syndrome: current Understanding and research needs. J. Allergy Clin. Immunol. 154, 255–263 (2024).
von Beek, C. et al. A two-step activation mechanism enables mast cells to differentiate their response between extracellular and invasive enterobacterial infection. Nat. Commun. 15, 904 (2024).
Draberova, L., Tumova, M. & Draber, P. Molecular mechanisms of mast cell activation by Cholesterol-Dependent cytolysins. Front Immunol 12, (2021).
Tam, J. S. Y., Coller, J. K., Hughes, P. A., Prestidge, C. A. & Bowen, J. M. Toll-like receptor 4 (TLR4) antagonists as potential therapeutics for intestinal inflammation. Indian J. Gastroenterol. 40, 5–21 (2021).
Stidham, R. W. et al. Systematic review with network meta-analysis: The efficacy of anti-TNF agents for the treatment of Crohn’s disease. Alimentary Pharmacology and Therapeutics vol. 39 1349–1362 Preprint at (2014). https://doi.org/10.1111/apt.12749
Moja, L., Danese, S., Fiorino, G., Del Giovane, C. & Bonovas, S. Systematic review with network meta-analysis: comparative efficacy and safety of Budesonide and mesalazine (mesalamine) for crohn’s disease. Aliment. Pharmacol. Ther. 41, 1055–1065 (2015).
Dvornikova, K. A., Platonova, O. N. & Bystrova, E. Y. Inflammatory bowel disease: crosstalk between Histamine, Immunity, and disease. Int J. Mol. Sci 24, (2023).
Becker, J., Ott, D. & Diener, M. Impact of sensitization and inflammation on the interaction of mast cells with the intestinal epithelium in rats. Front Physiol 10, (2019).
Gudiño, V., Bartolomé-Casado, R. & Salas, A. Single-cell omics in inflammatory bowel disease: recent insights and future clinical applications. Gut 74, 1335–1345 (2025).
National Center for Biotechnology Information (NCBI). TNFSF11 TNF Superfamily Member 11 [ Homo Sapiens (Human) ]. (2025).
Van Remoortel, S., Hussein, H. & Boeckxstaens, G. Mast cell modulation: A novel therapeutic strategy for abdominal pain in irritable bowel syndrome. Cell. Rep. Med. 5, 101780 (2024).
Anderson, D. M. et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390, 175–179 (1997).
Escalante, C. R., Shen, L., Thanos, D. & Aggarwal, A. K. Structure of NF-κB p50/p65 heterodimer bound to the PRDII DNA element from the Interferon-β promoter. Structure 10, 383–391 (2002).
Chang, M., Lee, A. J., Fitzpatrick, L., Zhang, M. & Sun, S. C. NF-κB1 p105 regulates T cell homeostasis and prevents chronic inflammation. J. Immunol. 182, 3131–3138 (2009).
Souza, R. F., Caetano, M. A. F., Magalhães, H. I. R. & Castelucci, P. Study of tumor necrosis factor receptor in the inflammatory bowel disease. World J. Gastroenterol. 29, 2733–2746 (2023).
De Giovanni, M. et al. Mast cells help organize the peyer’s patch niche for induction of IgA responses. Sci Immunol 9, (2024).
Li, B. et al. Roles of the RANKL–RANK axis in Immunity—Implications for pathogenesis and treatment of bone metastasis. Front Immunol 13, (2022).
Kobayashi, N., Takahashi, D., Takano, S., Kimura, S. & Hase, K. The roles of peyer’s patches and microfold cells in the gut immune system: relevance to autoimmune diseases. Front Immunol 10, (2019).
Onji, M. et al. RANK drives structured intestinal epithelial expansion during pregnancy. Nature 637, 156–166 (2025).
Mizukami, J. et al. Receptor activator of NF-kappaB ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, tables 2, and TRAF6. Mol. Cell. Biol. 22, 992–1000 (2002).
De Leon-Oliva, D. et al. The RANK-RANKL-OPG system: A multifaceted regulator of Homeostasis, Immunity, and cancer. Medicina (Kaunas) 59, (2023).
Hildebrand, G. K., Patel, P. & Kasi, A. Denosumab. (2025).
Ashcroft, A. J. et al. Colonic dendritic Cells, intestinal Inflammation, and T Cell-Mediated bone destruction are modulated by Recombinant osteoprotegerin. Immunity 19, 849–861 (2003).
Yang, M., Zhu, L. & Osteoimmunology The crosstalk between T Cells, B Cells, and osteoclasts in rheumatoid arthritis. Int J. Mol. Sci 25, (2024).
Yarilina, A., Xu, K., Chen, J. & Ivashkiv, L. B. TNF activates calcium–nuclear factor of activated T cells (NFAT)c1 signaling pathways in human macrophages. Proc. Natl. Acad. Sci. 108, 1573–1578 (2011).
Kimura, S. et al. Osteoprotegerin-dependent M cell self-regulation balances gut infection and immunity. Nat. Commun. 11, 234 (2020).
Lai, N. Y. et al. Gut-Innervating nociceptor neurons regulate peyer’s patch microfold cells and SFB levels to mediate Salmonella host defense. Cell 180, 33–49e22 (2020).
Veerasubramanian, P. K., Wynn, T. A., Quan, J. & Karlsson, F. J. Targeting TNF/TNFR superfamilies in immune-mediated inflammatory diseases. Journal Experimental Medicine 221, (2024).
Prados, A. et al. Fibroblastic reticular cell lineage convergence in peyer’s patches governs intestinal immunity. Nat. Immunol. 22, 510–519 (2021).
Blaschke, M. et al. Crohn’s disease patient serum changes protein expression in a human mesenchymal stem cell model in a linear relationship to patients’ disease stage and to bone mineral density. J. Clin. Transl Endocrinol. 13, 26–38 (2018).
Papadaki, M. et al. New insights for RANKL as a Proinflammatory modulator in modeled inflammatory arthritis. Front Immunol 10, (2019).
Glasnović, A., O’Mara, N., Kovačić, N., Grčević, D. & Gajović, S. RANK/RANKL/OPG signaling in the brain: A systematic review of the literature. Front Neurol 11, (2020).
Wu, C. S. et al. The alterations of molecular repertoire of the RANKL-induced osteoclastogenesis in the M1 macrophage-derived inflammatory milieu. Sci. Rep. 15, 16137 (2025).
Bao, C. et al. OpenXGR: a web-server update for genomic summary data interpretation. Nucleic Acids Res. 51, W387–W396 (2023).
Funding
This original article did not receive any specific grant from funding agencies.
Author information
Authors and Affiliations
Contributions
RP and NR: study design, interpretation, and manuscript drafting. PB and FI: manuscript revision, and proofreading. NA, HR and ATW: experiment performing and data analyzing experiment instruction. WA, EM, VFH and GCS: data analyzing and material preparation. LMI, CG, and SSA: data analyzing and interpretation. CFL : supervise. RDS: study design, experiment instruction, data analyzing and interpretation, manuscript drafting, supervise and resource providing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This original article involved only the analysis of anonymized, publicly available data; ethical approval and informed consent were therefore not required.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pranata, R., Ratnasari, N., Bayupurnama, P. et al. Genomic analysis unlocks the potential of denosumab as a targeted therapy for Crohn’s disease. Sci Rep 15, 44713 (2025). https://doi.org/10.1038/s41598-025-28395-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28395-7
