
Abstract
Psoriasis features keratinocyte hyperproliferation. This study aimed to identify disulfidptosis-related key genes in psoriasis for potential diagnostic biomarkers or therapeutic targets. Using the GSE30999 dataset, WGCNA was applied to identify key gene modules. GO, KEGG were used for analysis. IHC, Western blot, PCR and free thiol tests verified results. WGCNA clustered differentially expressed genes into 18 modules, with the blue module being significantly associated with psoriasis (Pearson’s r = 0.82, P < 0.001). Six hub genes (FRK, GYS1, HECW2, MYH10, S100A12, SLC7A11) were identified. GYS1, S100A12, and SLC7A11 showed pronounced dysregulation (FDR < 0.01) and upregulation in psoriasis epidermis (P < 0.05). The double-stranded death core pathogenic gene SLC7A11 can regulate the level of free sulfhydryl groups and the expression levels of molecules in the NF-κB pathway.(P < 0.05). GYS1, S100A12, and SLC7A11 are disulfidptosis-related driver genes in psoriasis, aiding molecular diagnosis and treatment.
Similar content being viewed by others

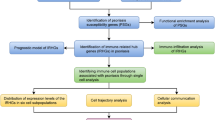

Introduction
Psoriasis is a common, chronic, recurrent and immune-mediated inflammatory skin disease. Its pathogenesis is complex and involves interactions among multiple factors such as genetics, immunity and environmental factors. Its pathological feature is the clonal proliferation of keratinocytes in the epidermis1. The disease’s metabolic nexus emerges from nutrient competition dynamics, wherein hyperproliferative keratinocytes deplete local glucose reservoirs, disrupting cutaneous metabolic homeostasis2. Psoriasis is a common, chronic, recurrent, and immune-mediated inflammatory skin disease. Its pathogenesis is complex and involves multiple interactions among genetic, immune, and environmental factors. The core pathological physiology of psoriasis is the abnormal activation of the innate and acquired immune systems, as well as the excessive proliferation and abnormal differentiation of keratinocytes. The main mechanisms can be summarized into the following key steps: genetic susceptibility including key regulatory factors such as HLA-C, IL23R, and NF-κB pathways; immune dysregulation related to the IL-23/Th17 axis, Th17 cell differentiation and activation, and dendritic cell activation; environmental triggers such as infection, trauma, drugs, and mental stress1,2. Psoriasis affects a large number of people worldwide, and the prevalence varies by region and ethnicity. Studies have shown that the global prevalence of psoriasis is approximately 503.6/100,0003. Moreover, psoriasis, especially in severe cases, is an important independent risk factor for multiple systemic diseases. These comorbidities include psoriatic arthritis, metabolic syndrome, cardiovascular diseases, inflammatory bowel disease, and depression, etc. Its high prevalence, chronic course, significant impact on quality of life, and high risk of severe systemic comorbidities make it a major public health problem.
However, the metabolic competition and pathological mechanisms that lead to excessive epidermal proliferation in psoriasis have not yet been fully elucidated. A newly discovered programmed cell death mode, induced by increased disulfide levels, is termed disulfidoptosis.. This mode is driven by the oxidative-reductive imbalance (disulfide stress) caused by excessive accumulation of disulfides (such as cysteine) within the cells and triggers cell death through abnormal disulfide bond formation. Studies have shown that the solute carrier protein SLC7A11 is crucial in the process of disulfide stress-induced cell death4.Under glucose deprivation conditions, SLC7A11 becomes the “death switch”: when this protein is highly expressed, it promotes cysteine intake, and SLC7A11 mediates the entry of a large amount of cysteine into the cytoplasm; during metabolic stability, NADPH reduces cysteine to cystine; but if NADPH supply is insufficient (such as during glucose starvation), the reduction process is blocked, and cysteine cannot be effectively reduced to cystine, resulting in the accumulation of disulfide toxicity, which leads to abnormal disulfide bonds forming between actin cytoskeletal proteins, cytoskeleton contraction and cell death5. Currently, research on disulfidptosis in disease focuses on cancer6. In a variety of cancers, such as liver and lung adenocarcinoma7,8,9, changes in SLC7A11 expression are closely related to tumourigenesis, progression, metastasis and response to treatment, and cancer cells are prone to disulfidptosis under conditions such as glucose deprivation6. This provides a new direction for cancer treatment. However, disulfide death has been relatively little studied in other diseases. Within the pathologically remodeled epidermal microenvironment of psoriasis, hyperproliferative keratinocytes have a strong glucose uptake capacity, resulting in reduced glucose uptake by suprabasal cells and glucose starvation, leading to a decrease in the intracellular NADPH/NADP + ratio10. Therefore, it is reasonable to speculate that there is a specific process of disulfidptosis in psoriatic skin tissue.
Thanks to the significant advancements in bioinformatics over the past few decades, researchers are now able to analyze large-scale complex genomic sequencing data, a method that is widely recognized as a key approach in life science research. Weighted Gene Co-expression Network Analysis (WGCNA) is a cutting-edge computational framework that systematically studies the correlations between gene clusters and the associations between genomic features and clinical phenotypes by dynamically constructing co-expression module systems11. These modules are based on differences in expression profiles and the key driver genes that play an important role in critical cellular signaling pathways, especially in specific cell types12.WGCNA can identify highly correlated gene modules and their characteristics, reveal the driving genes within the modules, establish associations between gene modules and samples, and calculate the module membership13.
Although the current direct studies on the role of farnesoid X receptor (FXR) death in psoriasis are still in the exploratory stage, its multiple roles in immune metabolism and cell apoptosis suggest that it may become an important molecular target to connect metabolic abnormalities with psoriasis. This study aims to identify the potential associations between FXR death-related genes and psoriasis, with the expectation of providing a new theoretical basis for metabolic intervention strategies for this disease.
Methods
Data sources and pre-processing
The expression datasets GSE30999 [22,763,790] and GSE13355 [19, 169, 254] of skin biopsy samples from patients with moderate-to-severe psoriasis were downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo). GSE30999 incorporated gene expression levels from 85 healthy controls, 85 patients with moderate to severe psoriasis. Disulfidoptosis-related genes were retrieved from the GeneCard database (https://www.genecards.org/) and relevant literature, and the results obtained was further validated using the external dataset GSE13355.
Construction of WGCNA
During the process of using the WGCNA R package, we make use of the fashClust function for hierarchical clustering tree drawing (den-program). The soft threshold selected via the pick Soft Threshold function ensures that the network is standard in the scale-free topology, which makes the constructed network exhibit power-law distribution characteristics. The choice of this soft threshold effectively reduces the error by strengthening strong correlations and weakening weak ones in the scale-free network, thus making the results more consistent with the characteristics of the biological data. The scale-free topology fitting index showed exponential variation, further indicating that the data network conformed to the scale-free topology distribution. Therefore, the good correlation verifies the scale-free property of the data network.
Clinical significance module
During the analysis, we identified key modules by evaluating the association strength between module eigengenes and clinical features. Specifically, we employed effect sizes (Pearson correlation coefficients) between gene expression profiles and clinical traits as the measure of Gene Significance (GS), as this provides a more robust and biologically interpretable metric of association strength compared to p-values alone.The GS for each gene was calculated as the absolute value of its correlation coefficient with the clinical trait of interest, reflecting the magnitude of association regardless of direction. For each module, we then computed the Module Significance (MS) by averaging the GS values (i.e., absolute correlation coefficients) of all genes within that module.The module with the highest MS value was identified as the key module most strongly associated with the clinical features, ensuring our selection was based on biological effect magnitude rather than statistical significance alone.
Functional enrichment analysis
In order to deeply explore the biological functions of the genes in the key modules, we performed Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of the modules using the clusterProfiler package. By uploading the gene lists derived from the module, enrichment results for Biological Process (BP) and KEGG pathways were obtained. The significance threshold was set at an adjusted p-value of < 0.05 to ensure the reliability of the analysed results.
Candidate gene selection and validation
To assess the diagnostic potential of candidate genes in discriminating disease states, we systematically compared expression levels between case and control groups in an independent validation cohort (GSE13355). The discriminative performance was subsequently assessed by subject operating characteristic (ROC) curve analysis, and corresponding curves were generated for the training and validation sets, respectively. Differential expression patterns were presented visually by box-and-line plots. Diagnostic accuracy was quantitatively assessed by calculating the area under the curve (AUC) metrics via the pROC calculation package (v1.18.5), thus ensuring rigorous statistical validation of biomarker effectiveness.
Identification of differentially expressed genes
In order to validate the differentially expressed genes (DEGs) between healthy control samples and psoriasis samples in the dataset GSE30999, we used the ‘limma’ R package to screen for differentially expressed genes. We used the ‘limma’ R package to perform the screening with a cut-off value of log2|FC|> 1 and an adjusted p-value < 0.05. Volcano plots were drawn by the R package ggplot2 to show the distribution of differentially expressed genes, and hierarchical clustering analyses were performed using pheatmap to visualise the gene expression patterns.
Construction of the column chart
In order to predict the risk of psoriasis, a column chart was constructed based on the differentially expressed driver genes. Next, calibration curves and decision curve analysis (DCA) curves were plotted to determine the performance of the column maps.
Experimental validation of driver genes
Immunohistochemical verification
To validate the expression of driver genes, this study included normal controls (obtained from normal skin adjacent to lesions in patients with surgically resected nevi) and patients with moderate-to-severe psoriasis (derived from previously archived pathological sections and fresh pathological tissues). The study adhered to the ethical guidelines outlined in the Declaration of Helsinki and was approved by the Medical Ethics Committee of Dalian Dermatology Hospital (Approval No.: KY2025-009). Immunohistochemistry (IHC) was employed to detect the expression levels of GYS1, S100A12, and SLC7A11. Skin tissue samples for IHC were fixed using 4% paraformaldehyde, embedded in paraffin, and sectioned into 4-μm thick slices according to a standardized protocol. Primary antibodies used included GYS1 polyclonal antibody (1:100, Cat. 10,566–1-AP, Proteintech), S100A12 polyclonal antibody (1:100, Cat. 16,630–1-AP, Proteintech), and SLC7A11 polyclonal antibody (1:200, Cat. 26,864–1-AP, Proteintech). The secondary antibody was HRP-conjugated sheep anti-rabbit IgG (1:200, GB23303, Servicebio). Protein expression levels were quantified using ImageJ win64 software, while data visualization and graph creation were performed using GraphPad Prism 10.
Western blotting verification
The tissue was minced into 1–3 mm3 pieces and washed in pre-cooled PBS to remove residual blood. Strong lysis buffer (RIPA) and proteinase inhibitors were added, and the tissue was homogenized using a mechanical homogenizer on ice for 30 min. The lysate was centrifuged and the supernatant (12,000–14,000 rpm, 4℃, 15 min) was collected to obtain the protein sample. The primary antibodies used included GYS1 polyclonal antibody (1:2000, Cat. 10,566–1-AP, Proteintech), S100A12 polyclonal antibody (1:2000, Cat. 16,630–1-AP, Proteintech), and SLC7A11 polyclonal antibody (1:2000, Cat. 26,864–1-AP, Proteintech). (1:500–1:2000), incubated at 4℃ overnight; the secondary antibody was HRP-labeled goat anti-rabbit IgG (1:15,000, GB23303, Seville Biotech) incubated at room temperature for 1 h, and the chemiluminescence method was used for imaging, with the gray value of the band analyzed using Image Lab software and normalized with the internal reference GAPDH. Statistical analysis was performed using GraphPad Prism 10.
Detection of free sulfhydryl groups in psoriatic skin tissue
The above-mentioned source organization used 0.9% NaCl containing 0.16 mg/ml heparin to wash away the residual blood on the tissue and homogenized and centrifuged the cells using cell lysis buffer at approximately 4ºC. The supernatant was collected and measured using the Free Sulfhydryl Assay Kit with DTNB. Using the Biyung-S0138S-Free Sulfhydryl Detection Kit (DTNB method), prepare the DTNB storage solution and Ellman’s Reagent Solution according to the kit instructions. Add 180 μl Ellman’s Reagent Solution to each well of the 96-well plate and add 20ul of the sample to each well. Set 3 duplicate wells for each group. Incubate at room temperature (approximately 20–25ºC) for 15 min. Measure the absorbance at 412 nm using an enzyme reader. The absorbance values of the samples or standards are recorded as Sample. Use the absorbance value of the blank control (ABlank) with a concentration of 0 in the standard curve. Subtract ABlank from Sample to calculate the average absorbance value, which is recorded as ASample. Substitute ASample into the standard curve to calculate the sulfhydryl concentration (recorded as B). Directly calculate C (mM) = B × n (dilution factor). GraphPad Prism 10 is used for data visualization and graphic creation.
Effect of SLC7A11 overexpression and knockdown on free sulfhydryl groups in keratinocytes
For the gene overexpression group, the SLC7A11 cDNA was cloned into a plasmid and transfected into HaCaT cells using a liposome-based method. Conversely, the knockdown group was established by transfecting cells with siRNA targeting SLC7A11 mRNA. HaCaT cells transfected with an empty plasmid vector served as the control group. The method for measuring free sulfhydryl groups was identical to that described above.
Alterations in genes associated with the NF-κB pathway in a psoriatic in vitro cell model
HaCaT cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin until they reached adherence. Subsequently, the cells were treated with the M5 cytokine cocktail (IL-17A, IL-22, TNF-α, IL-1α, and Oncostatin M) for 24 h. The concentrations used were 50 ng/mL for IL-17A and IL-22, and 5 ng/mL for TNF-α.Following the 24-h cytokine stimulation, total RNA was isolated from HaCaT cells using a commercial RNA extraction kit according to the manufacturer’s instructions. The concentration and purity of the extracted RNA were determined spectrophotometrically. Complementary DNA (cDNA) was then synthesized from 1 µg of total RNA using a reverse transcription kit with oligo(dT) primers.The mRNA expression levels of NFKB1, RELA, IKBKB, and TNF-α were quantified by quantitative real-time polymerase chain reaction (qRT-PCR). PCR amplification was performed using SYBR Green master mix on a real-time PCR detection system. The relative expression of each target gene was normalized to the endogenous reference gene GAPDH and calculated using the 2^–ΔΔCt method.
Statistical analysis
The results of immunohistochemistry, Western blotting, PCR and free thiol detection were presented in the form of the average value ± standard deviation obtained from at least three independent experiments. By regulating the expression of SLC7A11(independent variable), the levels of free sulfhydryl groups and the expression levels of genes in the NF-κB pathway (dependent variable) were detected. And the statistical significance of the differences was determined by the unpaired two-tailed Student’s t-test (*P < 0.05; **P < 0.01; ***P < 0.001). In addition, all other statistical analyses in this study were conducted using the R-4.4.3 version of the Windows software.
Results
Microarray data expression value analysis
Background correction and normalization of GSE30999 raw data were performed using the R package. Probe and gene symbols were matched using R package annotations. For multi-matched genes the median was selected as the expression value. A total of 6980 genes were identified, and the number of eligible genes screened by a standard deviation greater than 0.5 totaled 6409, classifying a total of 170 samples (Fig. 1).

Sample clustering was based on GSE3099 expression data.
Construction of WGCNA and identification of key modules
A suitable soft threshold is selected so that the built network presents a scale-free distribution. Network topology analysis was performed for the first 20 threshold powers, and a soft threshold (β = 7) was finally selected such that the fit index R2 > 0.9 (Figs. 2A and B), while maintaining a high average connectivity between genes to produce a hierarchical clustering tree containing 6409 genes (Fig. 2C). The neighbour-joining matrix was converted to a topological overlap matrix (TOM) to measure gene co-expression similarity, and the neighbour-joining matrix was converted to a topological overlap matrix to construct the network and build the hierarchical clustering tree. Using the dynamic shear tree algorithm (deepSplit = 2, minClusterSize = 50) to identify biologically significant gene modules and merge highly similar modules (MEDissThres = 0.25), a total of 18 modules were generated (Fig. 2D). Specifically, the black, blue, brown, cyan, green, grey, grey60, lightcyan, lightgreen, lightyellow, magenta, midnightblue, pink, purple, royalblue, salmon, tan and yellow modules, have 441, 2943, 648, 128, 672, 163, 76, 105, 69, 61, 222, 112, 266, 191, 57, 129, 146, 551 genes, respectively.

(A) Analysis of the scale-free fit index for various soft-thresholding powers (β). (B) Analysis of the mean connectivity for various soft-thresholding powers. In all, 7 was the most fit power value. Co-expression module construction using the WGCNA package in R. (C-D) (C) The cluster dendrogram of genes. Each branch in the Fig. represents one gene, and each color represents one coexpression module. (D) The cluster dendrogram of module eigengenes.
Correlation between modules and identification of key modules
Pearson correlation analyses were performed between modules and clinical traits, where each row represents a module (e.g. MEblue, MEbrown, etc.) and each column corresponds to a clinical trait (e.g. Normal and Psoriasis). The value in each cell represents the correlation coefficient (ranging from −1 to 1) and its corresponding p-value, reflecting the strength and significance of the correlation between the module and the trait. The BLUE module showed the strongest positive correlation with psoriasis, and the CYAN module showed the strongest negative correlation (Fig. 3), suggesting that the BLUE module induces progression in the course of psoriasis, and so we chose the BLUE module for the following analyses.

Heatmap of the correlation between module eigengenes and the disease status of psoriasis. The blue module was the most negatively correlated with status, and the cyan module was the most positively correlated with status.
Functional enrichment analysis
Since the blue module was positively correlated with disease progression, the genes in the blue module were included in further GO and KEGG analyses14, through which we identified multiple significantly enriched biological processes and pathways. Among the biological processes, nuclear division, chromosome segregation, inflammatory response regulation and leukocyte migration, which are related to cell cycle and immune regulation, were significantly enriched. In the pathway analysis, a total of 30 significantly enriched pathways were identified, of which the most significant ones included cytokine-cytokine receptor interactions, chemokine signalling pathway, TNF signalling pathway, IL-17 signalling pathway and NF-κB signalling pathway (Fig. 4A and B), which are mainly closely related to immune regulation, inflammatory response and cell signalling. The p-values of all enrichment results were less than 0.05, which were highly statistically significant and provided important clues for further functional analyses and biological mechanism studies.

GO and KEGG analysis of blue module. (A) GO analysis of the enriched genes in the blue module. (B) The KEGG pathways of the enriched genes in the blue module.
Gene intersection analysis and visualisation
Constructing Venn diagrams for blue module genes and bisulfide death-related genes (Fig. 5), we successfully screened key genes jointly identified by LASSO regression and SVM methods. These genes (FRK, GYS1, HECW2, MYH10, S100A12, SLC7A11) may be core regulators in the pathogenesis of psoriasis and may be associated with disulfidptosis, which is worthy of further functional validation and experimental studies.

Identification of common genes between disulfidptosis genes and the blue module by overlapping them.
External dataset validation biomarkers
In the external datasetGSE13355 [19, 169, 254], the expression differences of the target gene (MYH10, S100A12, SLC7A11, FRK, GYS1 and HECW2) in the skin between healthy individuals and psoriasis patients were analyzed.) We found that the expression levels of S100A12, SLC7A11, FRK, and GYS1 genes in the experimental group (Treat) of the external dataset were significantly higher than those in the control group (Con), and the differences were highly statistically significant (p < 0.05) (Fig. 6A-F). Further analysed by ROC curves, S100A12 (AUC = 1.000) and SLC7A11 (AUC = 0.999) exhibited near-perfect classification ability, MYH10 (AUC = 0.940) and GYS1 (AUC = 0.905) also showed excellent diagnostic performance, while the diagnostic performance of HECW2 (AUC = 0.829) and FRK (AUC = 0.699) were good and moderate, respectively (Fig. 7A-F). Taken together, these results suggest that GYS1, S100A12 and SLC7A11 are not only significantly up-regulated in the disease state, but may also serve as potential biomarkers for distinguishing experimental and control groups, providing an important molecular basis for disease diagnosis and treatment.

A-F:Box plot of target gene expression differences.The p values of all genes were less than 0.05, but the expression levels of SLC7A11 (A), S100A12 (B), GYS1 (C) and FRK (D) genes in the experimental group were higher than those in the control group; and the expression levels of MYH10 (E) and HECW2 (F) genes in the experimental group were lower than those in the control group.

F:Gene ROC curve. SLC7A11 (A) and S100A12 (B) are potential ideal biomarkers, followed by GYS1 (C) and FRK (D), and MYH10 (E) and HECW2 (F) can be used as complementary indicators.
Identification of disease-related gene expressions in the blue module
Use the ‘limma’ R package to screen for differentially expressed genes (DEGs) between healthy control samples and moderate-to-severe psoriasis samples in dataset GSE30999 for validation. Genes with |logFC|> 1 and adj.P.Val < 0.05 were screened as differentially expressed genes. Presented by heatmap and volcano plot (Fig. 8A and B), the analysis results screened three genes, GYS1, S100A12 and SLC7A11, which were the same as the validation results of the external dataset.

Identification of driving genes in GSE30999. (A) Volcano plot visualizing DEGs in GSE30999 (85 with psoriasis and 85 healthy controls (HC)). The green nodes are downregulated genes, and the red nodes are upregulated genes (|fold change|> 1, p < 0.05). (B) Heatmap hierarchical clustering reveals dysregulated genes in the psoriasis groups compared with the healthy controls.
Construction of column plots
After screening for characterized genes, column line plots were created to predict the likelihood of disease in patients with the three diseases. The accuracy of the column maps was relatively high, and the calibration curve and decision curve analyses showed that SLC7A11, GYS1, and S100A12 were valuable in psoriasis risk prediction, and their translation to the clinic could be promoted through multidisciplinary cross-validation in the future (Fig. 9A-C). However, the direction of GYS1 coefficients was reversed, which may be due to the high correlation between GYS1 and other genes, resulting in the masking of its independent effect. It is also possible that the small sample size makes the multivariate model parameter estimation unstable, which needs to be further verified in combination with immunohistochemical experiments.

Nomogram model construction and evaluation of candidate feature genes involved in psoriasis. (A) the nomogram model construction on the basis of feature genes to predict the likelihood of disease in patients with psoriasis (p > 0.05; mae < 0.05). (B) Calibration curve showed the accuracy of the nomogram was relatively high and validated the model performance of psoriasis(n = 170,Mean absolute error = 0.016,Mean squared error = 0.00121,0.9 Quantile of absolute error = 0.019). (C) DCA curve suggested net profitof the constructed model is better than the default method.
Experimental validation of driver genes
To validate our identification, we examined the expression of the three genes in skin pathology biopsy specimens from psoriasis patients and healthy controls (Figs. 10 and 11), with the brown representation representing positive staining. The intensity was read for identification using Image J software, and the value was then divided by the group mean to obtain the fold change in the healthy control. Immunohistochemistry results showed up-regulation of GYS1, S100A12, and SLC7A11 protein levels in skin pathology biopsies from psoriasis patients compared to healthy controls, consistent with our bioinformatics analysis.

The expression of 3 driver genes in psoriasis samples by IHC(n = 3,p < 0.01).

The expression of 3 driver genes in psoriasis samples by western blot.(n = 3).
Detection of free sulfhydryl groups in psoriatic skin tissue
The results of free thiol detection showed that, compared with the normal group, the relative expression level of free thiol in psoriasis skin tissue was significantly decreased (Fig. 12), indicating that during the onset of psoriasis, the free thiol showed a decreasing trend and there was an increase in disulfide bond formation. The experimental results indirectly suggested that disulfide death might occur in psoriasis skin tissue.

The expression of free sulfhydryl assay by DTNB.(n = 4).
The impact of SLC7A11 overexpression and knockdown on free thiol levels in keratinocytes
To investigate the role of SLC7A11 in free thiol groups of HaCaT cells, we overexpressed and knocked down the SLC7A11 gene in HaCaT cells respectively. The SLC7A11 overexpression plasmid was introduced into the cells by liposome transfection as the overexpression group, and the shRNA plasmid was introduced by liposome transfection as the knockdown group, while the empty vector was used as the control group. After 48 h of transfection, the intracellular free thiol levels were detected by DTNB method, and the total protein concentration was standardized by BCA method.The results demonstrated that SLC7A11-overexpressing cells exhibited lower levels of free thiols compared to the control group, the level of free sulfhydryl groups in SLC7A11-knockdown cells showed no significant difference compared to the control group(Fig. 13). These findings suggest that SLC7A11 overexpression may disrupt intracellular redox homeostasis by depleting antioxidant molecules such as glutathione (GSH), thereby promoting abnormal disulfide bond accumulation and triggering cell death. This mechanism aligns with the characteristics of disulfide-induced cell death, indicating that SLC7A11 may contribute to the pathogenesis of psoriasis through modulation of thiol homeostasis.

The expression of free sulfhydryl assay by DTNB.(n = 3).
The PCR experiment validated the gene expression of the NF-κB pathway
To validate the KECG result of the NF-κB signaling pathway from bioinformatics analysis, we detected the mRNA expression levels of key genes in the NF-κB pathway (NFKB1,RELA,IKKβ,TNF-α,IL-6) in the skin of psoriasis patients and healthy individuals using the RT-qPCR experiment (n = 3).The results show that, compared with the skin of healthy individuals, the expression of genes in the A pathway was significantly upregulated in the skin tissues of psoriasis patients (see Fig. 9A) Specifically, the expression levels of mRNA in NFKB1 (p < 0.001),RELA (p < 0.01),IKKβ (p < 0.001) and TNF-α (p < 0.01) were all increased (Fig. 14).

The relative expression levels of NF-κB pathway-related genes. (n = 3).
Discussion
This study, by integrating weighted gene co-expression network analysis (WGCNA) for the first time revealed the collaborative regulatory network of genes related to disulfide death in psoriasis, and identified three core driving genes: SLC7A11, GYS1 and S100A12. Among them, SLC7A11, as the pathogenic gene of the double sulfur death core, we further investigated the potential mechanism of SLC7A11 in psoriasis by regulating its expression.These genes, SLC7A11, GYS1 and S100A12, by regulating metabolic reprogramming, inflammatory responses, and cell death pathways, form the core molecular mechanism of the "metabolism-inflammation-cell death" axis in psoriasis, providing a new perspective for disease diagnosis and treatment. The following discusses in depth the molecular mechanism, methodological innovation, clinical translational potential, and research limitations, and proposes future research directions.
The association between gene function and the mechanism of disulfide bond degradation.
SLC7A11: a hub of cystine metabolic imbalance
The significant up-regulation of the cystine/glutamate reverse transporter (system xc-) encoded by SLC7A11 (solute carrier family 7 member 11) in psoriasis lesions (Fig. 7) suggests that it may drive the disease process by regulating cystine metabolism15. Under glucose-sufficient conditions, SLC7A11-mediated cystine uptake generates cysteine via NADPH-dependent reduction reactions, maintains intracellular redox homeostasis and inhibits ferroptosis16,17,18. However, in psoriatic epidermis, aberrant proliferation of keratin-forming cells leads to a chronic glucose starvation of basal epithelial cells19, NADPH synthesis is blocked and cystine cannot be efficiently reduced to cysteine, leading to an abnormal accumulation of disulphides (e.g. cystine and cystamine)20, these disulfides trigger cytoskeletal contraction, actin network collapse and eventual disulfidptosis by inducing the formation of aberrant disulfide bonds between actin skeleton proteins21. This mechanism is similar to disulfidptosis in cancer5, However, psoriasis is unique in that it may co-mediate the maintenance of a cytopathic state in psoriasis, and this metabolic competition plays a role in skin tissue homeostasis19.
GYS1: a bridge between glycogen metabolism and immune activation
GYS1 (glycogen synthase 1) catalyzes the addition of glucose monomers to growing glycogen molecules through the formation of α—1,4—glycosidic bonds and participates in glycogen metabolism as the rate-limiting enzyme for glycogen synthesis, and its aberrant up-regulation in psoriasis was found to be of dual significance22. GYS1 (glycogen synthase 1) affects cellular energetic homeostasis by regulating cellular metabolism23,24, especially in energy-demanding immune cells and keratinocytes25,26,27. In psoriasis, the metabolic activity of immune cells and keratin-forming cells is significantly enhanced28, and GYS1 can exacerbate the inflammatory response by regulating glycogen metabolism and affecting immune cell activation, proliferation, and the release of inflammatory factors (e.g., IL-17, IL-23, TNF-α)29,30. In addition, GYS1 promotes epidermal thickening and scale formation by regulating the proliferation and differentiation of keratinocytes. Meanwhile, abnormal GYS1 function may lead to an imbalance in intracellular redox status, triggering disulfide stress-related disulfidptosis31, further exacerbating cellular damage and inflammatory responses. Although these mechanisms are not yet fully understood, studies have demonstrated the importance of metabolic reprogramming in psoriasis. Future studies could further explore the specific role of GYS1 and disulfidptosis in psoriasis and its potential as a therapeutic target.
S100A12: a cross-regulator of inflammation and metabolism
The high expression of S100A12 (calcium-binding protein A12), a pro-inflammatory alarmin, in psoriasis lesions (Fig. 7) reveals a deep interaction between inflammation and metabolism. S100A12 activates the NF-κB32 and MAPK signalling pathways33 by binding to RAGE (receptor for advanced glycosylation end products), driving the release of pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IL-1β)34, creating a positive feedback loop of inflammation and cell death35,36. S100A12 has been previously observed to be overexpressed in inflamed tissues with various diseases such as IBD, psoriatic arthropathy, juvenile idiopathic arthritis and rheumatoid arthritis37,38,39. Li et al. also found a strong positive correlation between S100A12 and monocytes40. Notably, S100A12 also functions to regulate calcium homeostasis41, and fluctuations in calcium ion concentration may directly affect the activity of disulfide-metabolising enzymes (e.g., glutathione reductase)42, and thus indirectly the susceptibility to disulfidptosis. The discovery of this dual action in psoriasis is rare, suggesting that targeting S100A12 may intervene in both the inflammatory cascade response and disulfide bond stress, providing a theoretical basis for the development of multi-targeted therapeutic strategies.
Methodological innovation supports the reliability of the conclusion
This study captured the strong correlation between the blue module (2943 genes) and psoriasis through WGCNA. The enriched IL-17, NF-κB, etc. pathways are highly consistent with the known mechanisms of psoriasis43,44,45,46,47,48. The advantage of WGCNA lies in its ability to identify highly co-expressed gene clusters and to analyze the association between the modules and clinical phenotypes, which is particularly applicable to the molecular classification of complex diseases (such as psoriasis)11. Further, by combining external datasets for validation, it ensured that the selection of key genes was not data noise but had universal biological characteristics. Immunohistochemical experiments and Western blotting experiments confirmed at the protein level that the genes were upregulated in the skin basal epithelial cells of psoriasis patients (Fig. 8), which compensated for the limitations of transcriptomic data and provided a multi-dimensional evidence chain for the conclusion. We determined the causal relationship between these three genes and psoriasis through bar charts. Additionally, we detected the level of free sulfhydryl groups in psoriasis skin tissues, which indirectly reflected the abnormal accumulation of disulfide bonds in psoriasis skin tissues. This "module-gene-function-validation" research framework provides a methodological paradigm for the molecular mechanism analysis of complex diseases.
Clinical transformation potential and strategies
This study found that the diagnostic efficacy of genes such as SLC7A11 (with an AUC > 0.9) far exceeded that of traditional markers (such as IL-17), suggesting their potential as non-invasive diagnostic markers48. More importantly, by drawing on the targeted strategies used in cancer for SLC7A11, intervention measures targeting the metabolic microenvironment of psoriasis can be developed: by balancing the "pro-death—antioxidant" dual roles of SLC7A11, or by jointly inhibiting glycogen synthesis mediated by GYS1, it may break the "metabolism—inflammation" vicious cycle49. Additionally, the high specificity expression of S100A12 provides a basis for the development of new biological agents targeting the RAGE signal.
Limitations of the study
Despite the breakthroughs in the research, the following issues still need to be addressed: (1) Direct pathological evidence of disulfide bond death (such as in vivo imaging of disulfide bond accumulation) has not yet been obtained; (2) The existing data rely on public cohorts and require validation of the clinical universality of the genes through multicenter prospective studies; (3) The specific interaction mechanism between the SLC7A11 regulatory network and the IL-17 pathway still needs to be analyzed through animal experiments.
Future direction
Considering the above limitations, this study can be strengthened in the future by conducting more comprehensive mechanistic studies: constructing a psoriasis-like organoid model, combining spatial transcriptome technology, and dynamically observing the metabolic heterogeneity of cells in different epidermal layers and the spatiotemporal characteristics of disulfidptosis under glucose deprivation; through the joint analysis of metabolomics and proteomics, to resolve the metabolite network regulated by SLC7A11, GYS1, S100A12 and its regulation mechanism on inflammatory signaling pathway.
Conclusion
This study reveals for the first time the synergistic role of disulfidptosis-related genes in psoriasis, which provides a new perspective for understanding the ‘metabolism-inflammation-cell death’ interactive network of the disease. Taking into account the advances in cancer research on the SLC7A11 pathway, it is necessary to develop targeting strategies for the metabolic microenvironment of psoriasis and to balance the dual roles of SLC7A11 in antioxidant and pro-death therapy to achieve precise treatment in the future. This study lays an important foundation for mechanism elucidation and therapeutic innovation in psoriasis.
Data availability
The datasets analyzed during the current study are available through the GEO repositories (GSE30999,GSE13355)[https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE30999https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE13355].
Code availability
This study relevant data can be obtained through the following links from jianguoyun: https://www.jianguoyun.com/#/sandbox/1aa7167/5103b67bae7f2624/%2F/.
Abbreviations
- AUC:
-
Area under the ROC curve
- FDR:
-
False discovery rate
- FRK:
-
Fyn related Src family tyrosine kinase
- GLM:
-
Generalized linear model
- GO:
-
Gene ontology
- GYS1:
-
Glycogen synthase 1
- HECW2:
-
HECT, C2 and WW domain containing E3 ubiquitin protein ligase 2
- IHC:
-
Immunohistochemistry
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- MYH10:
-
Myosin, heavy polypeptide 10, non-muscle
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate, reduced form
- ROC:
-
Receiver operating characteristic
- S100A12:
-
Calcium-binding protein A12
- SLC7A11:
-
Solute carrier family 7 member 11
- SVM:
-
Support vector machine
- WGCNA:
-
The weighted gene co-expression network analysis
- XGBoost:
-
EXtreme gradient boosting
References
Griffiths, C. E. & Barker, J. N. Pathogenesis and clinical features of psoriasis. Lancet 370, 263–271. https://doi.org/10.1016/s0140-6736(07)61128-3 (2007).
Chen, C. et al. Metabolomic profiling reveals amino acid and carnitine alterations as metabolic signatures in psoriasis. Theranostics 11, 754–767. https://doi.org/10.7150/thno.51154 (2021).
Damiani, G. et al. The Global, Regional, and National Burden of Psoriasis: Results and Insights From the Global Burden of Disease 2019 Study. Front Med (Lausanne) 8, 743180. https://doi.org/10.3389/fmed.2021.743180 (2021).
Zheng, T., Liu, Q., Xing, F., Zeng, C. & Wang, W. Disulfidptosis: a new form of programmed cell death. J Exp Clin Cancer Res 42, 137. https://doi.org/10.1186/s13046-023-02712-2 (2023).
Liu, X. et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol 25, 404–414. https://doi.org/10.1038/s41556-023-01091-2 (2023).
Zheng, P., Zhou, C., Ding, Y. & Duan, S. Disulfidptosis: a new target for metabolic cancer therapy. J Exp Clin Cancer Res 42, 103. https://doi.org/10.1186/s13046-023-02675-4 (2023).
Wang, T. et al. Disulfidptosis classification of hepatocellular carcinoma reveals correlation with clinical prognosis and immune profile. Int Immunopharmacol 120, 110368. https://doi.org/10.1016/j.intimp.2023.110368 (2023).
Ni, L., Yang, H., Wu, X., Zhou, K. & Wang, S. The expression and prognostic value of disulfidptosis progress in lung adenocarcinoma. Aging (Albany NY) 15, 7741–7759 (2023).
Zhu, J. et al. Disulfidptosis-related gene SLC7A11 predicts prognosis and indicates tumor immune infiltration in lung adenocarcinoma. Transl. Cancer Res. 13, 5064–5072 (2024).
Ma, J. et al. Inhibition of PI3K/AKT/GLUT1 Signaling Pathway by Quercetin in the Treatment of Psoriasis. Front Biosci (Landmark Ed) 30, 26884 (2025).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Lin, W. et al. Role of Calcium Signaling Pathway-Related Gene Regulatory Networks in Ischemic Stroke Based on Multiple WGCNA and Single-Cell Analysis. Oxid Med Cell Longev 2021, 8060477. https://doi.org/10.1155/2021/8060477 (2021).
Song, C. et al. DUSP6 protein action and related hub genes prevention of sepsis-induced lung injury were screened by WGCNA and Venn. Int J Biol Macromol 279, 135117. https://doi.org/10.1016/j.ijbiomac.2024.135117 (2024).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res 51, D587-d592. https://doi.org/10.1093/nar/gkac963 (2023).
Liu, X., Zhuang, L. & Gan, B. Disulfidptosis: disulfide stress-induced cell death. Trends Cell Biol 34, 327–337. https://doi.org/10.1016/j.tcb.2023.07.009 (2024).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. https://doi.org/10.1016/j.cell.2012.03.042 (2012).
Badgley, M. A. et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. https://doi.org/10.1126/science.aaw9872 (2020).
Cramer, S. L. et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med 23, 120–127. https://doi.org/10.1038/nm.4232 (2017).
Liu, W. et al. Energy competition remodels the metabolic glucose landscape of psoriatic epidermal cells. Theranostics 14, 3339–3357. https://doi.org/10.7150/thno.93764 (2024).
Gu, Q. et al. Disulfidptosis, A Novel Cell Death Pathway: Molecular Landscape and Therapeutic Implications. Aging Dis 16, 917–945 (2024). https://doi.org/10.14336/ad.2024.0083
Chen, J. et al. Disulfidptosis decoded: a journey through cell death mysteries, regulatory networks, disease paradigms and future directions. Biomark Res 12, 45. https://doi.org/10.1186/s40364-024-00593-x (2024).
Li, Y. et al. Identification of novel immune subtypes and potential hub genes of patients with psoriasis. J Transl Med 21, 182. https://doi.org/10.1186/s12967-023-03923-z (2023).
Fastman, N. M. et al. The structural mechanism of human glycogen synthesis by the GYS1-GYG1 complex. Cell Rep 40, 111041. https://doi.org/10.1016/j.celrep.2022.111041 (2022).
Xirouchaki, C. E. et al. Impaired glucose metabolism and exercise capacity with muscle-specific glycogen synthase 1 (gys1) deletion in adult mice. Mol Metab 5, 221–232. https://doi.org/10.1016/j.molmet.2016.01.004 (2016).
Sadiku, P. et al. Neutrophils Fuel Effective Immune Responses through Gluconeogenesis and Glycogenesis. Cell Metab 33, 411-423.e414. https://doi.org/10.1016/j.cmet.2020.11.016 (2021).
Qian, L. et al. MK8617 inhibits M1 macrophage polarization and inflammation via the HIF-1α/GYS1/UDPG/P2Y(14) pathway. PeerJ 11, e15591. https://doi.org/10.7717/peerj.15591 (2023).
Cassidy, F. C. et al. Glycogen-fuelled metabolism supports rapid mucosal-associated invariant T cell responses. Proc Natl Acad Sci U S A 120, e2300566120. https://doi.org/10.1073/pnas.2300566120 (2023).
Griffiths, C. E. M., Armstrong, A. W., Gudjonsson, J. E. & Barker, J. Psoriasis. Lancet 397, 1301–1315. https://doi.org/10.1016/s0140-6736(20)32549-6 (2021).
Thwe, P. M. et al. Cell-Intrinsic Glycogen Metabolism Supports Early Glycolytic Reprogramming Required for Dendritic Cell Immune Responses. Cell Metab 30, 225. https://doi.org/10.1016/j.cmet.2019.05.017 (2019).
Robinson, J. M., Karnovsky, M. L. & Karnovsky, M. J. Glycogen accumulation in polymorphonuclear leukocytes, and other intracellular alterations that occur during inflammation. J Cell Biol 95, 933–942. https://doi.org/10.1083/jcb.95.3.933 (1982).
Xie, J. et al. (2024) Multi-omics analysis of disulfidptosis regulators and therapeutic potential reveals glycogen synthase 1 as a disulfidptosis triggering target for triple-negative breast cancer. MedComm 5, e502. https://doi.org/10.1002/mco2.502 (2020).
Nazari, A. et al. S100A12 in renal and cardiovascular diseases. Life Sci 191, 253–258. https://doi.org/10.1016/j.lfs.2017.10.036 (2017).
Tanaka, N. et al. S100A12 inhibits fibroblast migration via the receptor for advanced glycation end products and p38 MAPK signaling. In Vitro Cell Dev Biol Anim 55, 656–664. https://doi.org/10.1007/s11626-019-00384-x (2019).
Chen, J. et al. Characterization of S100A12 from nile tilapia (Oreochromis niloticus) and its roles on inflammatory responses. Fish Shellfish Immunol 131, 401–407. https://doi.org/10.1016/j.fsi.2022.10.011 (2022).
Lira-Junior, R. et al. S100A12 Expression Is Modulated During Monocyte Differentiation and Reflects Periodontitis Severity. Front Immunol 11, 86. https://doi.org/10.3389/fimmu.2020.00086 (2020).
Kessel, C., Holzinger, D. & Foell, D. Phagocyte-derived S100 proteins in autoinflammation: putative role in pathogenesis and usefulness as biomarkers. Clin Immunol 147, 229–241. https://doi.org/10.1016/j.clim.2012.11.008 (2013).
Foell, D. et al. Expression of the pro-inflammatory protein S100A12 (EN-RAGE) in rheumatoid and psoriatic arthritis. Rheumatology (Oxford) 42, 1383–1389. https://doi.org/10.1093/rheumatology/keg385 (2003).
Foell, D. et al. Monitoring neutrophil activation in juvenile rheumatoid arthritis by S100A12 serum concentrations. Arthritis Rheum 50, 1286–1295. https://doi.org/10.1002/art.20125 (2004).
Orczyk, K. & Smolewska, E. A Granulocyte-Specific Protein S100A12 as a Potential Prognostic Factor Affecting Aggressiveness of Therapy in Patients with Juvenile Idiopathic Arthritis. J Immunol Res 2018, 5349837. https://doi.org/10.1155/2018/5349837 (2018).
Li, L. et al. Regulation mechanisms of disulfidptosis-related genes in ankylosing spondylitis and inflammatory bowel disease. Front Immunol 15, 1326354. https://doi.org/10.3389/fimmu.2024.1326354 (2024).
Wang, Q. et al. Calcium mediated static and dynamic allostery in S100A12: Implications for target recognition by S100 proteins. Protein Sci 33, e4955. https://doi.org/10.1002/pro.4955 (2024).
Ma, Y., Ren, X. & Liang, C. Exogenous Ca(2+) enhances antioxidant defense in rice to simulated acid rain by regulating ascorbate peroxidase and glutathione reductase. Planta 254, 41. https://doi.org/10.1007/s00425-021-03679-0 (2021).
Bugaut, H. & Aractingi, S. Major Role of the IL17/23 Axis in Psoriasis Supports the Development of New Targeted Therapies. Front Immunol 12, 621956. https://doi.org/10.3389/fimmu.2021.621956 (2021).
Mu, X. et al. IL-17 in wound repair: bridging acute and chronic responses. Cell Commun Signal 22, 288. https://doi.org/10.1186/s12964-024-01668-w (2024).
Maschalidi, S. et al. Targeting SLC7A11 improves efferocytosis by dendritic cells and wound healing in diabetes. Nature 606, 776–784. https://doi.org/10.1038/s41586-022-04754-6 (2022).
Zhao, C. et al. Sulfasalazine promotes ferroptosis through AKT-ERK1/2 and P53-SLC7A11 in rheumatoid arthritis. Inflammopharmacology 32, 1277–1294. https://doi.org/10.1007/s10787-024-01439-6 (2024).
Hu, K. et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J Clin Invest 130, 1752–1766. https://doi.org/10.1172/jci124049 (2020).
Gupta, A. K., Ellis, C. N., Siegel, M. T. & Voorhees, J. J. Sulfasalazine: a potential psoriasis therapy?. J Am Acad Dermatol 20, 797–800. https://doi.org/10.1016/s0190-9622(89)70092-x (1989).
Wang, Y. et al. Transcriptional regulation of pig GYS1 gene by glycogen synthase kinase 3β (GSK3β). Mol Cell Biochem 424, 203–208. https://doi.org/10.1007/s11010-016-2856-1 (2017).
Funding
This study was approved by the Major Collaborative Innovation Program of Medical and Health Science and Technology Innovation Project of the Chinese Academy of Medical Sciences (2021-I2M-1–059).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript. Qingshan Li, Ping Xu, Yongjun Piao, Zhiyi Wang, and Chengzhi Lv performed research and analyzed data; Qingshan Li, Ping Xu and Zhiyi Wang designed research and wrote the paper; Yongjun Piao, and Chengzhi Lv responsed to the reviewers.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Our study followed the ethical guidelines outlined in the Declaration of Helsinki and received ethical approval from the Medical Ethics Committee of Dalian Dermatological Hospital (KY2025-009).
Informed consent of human participants
We confirm that all human participants (and/or their legal guardians) in this study provided written informed consent before participating. The consent clearly states: Scope and methods of data collection and use. Protect the privacy and confidentiality of participants. Participants have the right to quit at any time without being fined.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Q., Xu, P., Piao, Y. et al. Screening of key genes related to disulfidptosis in psoriasis based on the analysis of WGCNA. Sci Rep 15, 45579 (2025). https://doi.org/10.1038/s41598-025-28457-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28457-w
