
Abstract
Nitrogen deprivation is a well-established strategy to enhance lipid accumulation in microalgae, yet the transcriptomic mechanisms underlying a gradient of nitrogen deprivation remain poorly understood. This study investigated the oleaginous microalga Auxenochlorella pyrenoidosa under a sodium nitrate (NaNO₃) gradient (1.5, 1, 0.5, and 0 g L⁻¹). RNA-seq analysis revealed distinct molecular responses tailored to nitrogen availability. Under mild limitation (1 g L⁻¹), nitrate assimilation and nitrogen metabolism were predominantly suppressed. Moderate deprivation (0.5 g L⁻¹) triggered extensive downregulation of the photosynthetic apparatus, encompassing chloroplast function and porphyrin/chlorophyll metabolism. In contrast, severe deprivation (0 g L⁻¹) induced nucleolar stress and upregulated ribosome biogenesis, indicating a fundamental shift in gene expression regulation. Common adaptive mechanisms across all stress levels included the consistent upregulation of the TCA cycle, MAPK signaling, and specific transporters. Furthermore, series cluster analysis identified monotonically expressed genes implicating tRNA processing, the COP9 signalosome, glycolysis, gibberellin response, and lysosomal hydrolases as potential rate-limiting steps governing lipid accumulation. Our findings provide a comprehensive transcriptomic landscape, revealing both tiered and shared regulatory networks, and propose novel targets for engineering lipid production in microalgae under precision nitrogen regulation.
Similar content being viewed by others

Introduction
Microalgal lipids mostly consist of saturated and monounsaturated fatty acids, which are promising replaceable sources for biodiesel manufacture1. Meanwhile, a certain species of microalgae produces polyunsaturated fatty acids (PUFAs) including eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Recently, their positive impacts on the health of humans and animals have made these PUFAs a subject of intense interest2. Therefore, the versatility of microalgal lipids as resources for energy, health, and industrial products gives them significant economic and practical value. Nowadays, the main challenge in the mass production of microalgal lipids remains the improvement of lipid accumulation3,4. A valuable approach to solving the problem is metabolic engineering, which is based on the comprehensive understanding of the regulatory mechanisms in the lipid biosynthesis of microalgae5. Thus, it is significant to use transcriptome analysis to elucidate the molecular mechanisms underlying lipid accumulation in microalgae. This is beneficial for the development of metabolic engineering, thereby facilitating the technological progress of microalgal lipid production6.
Deprivation of environmental nitrogen can effectively affect lipid contents in microalgae7. Many studies have shown that partial or complete nitrogen deprivation in the medium induces an increase in lipid content in microalgae. This suggests that different levels of nitrogen deprivation may trigger similar mechanisms to enhance lipid biosynthesis8. Moreover, some studies reported that the extent of lipid accumulation varies depending on the degree of nitrogen limitation, suggesting that distinct nitrogen deprivation levels may involve different regulatory mechanisms for lipid biosynthesis9,10,11. If these similarities and differences can be identified, it will deepen our understanding of the relevant mechanisms to facilitate the use of metabolic engineering to enhance lipid accumulation in microalgae. However, previous studies focused mainly on the molecular mechanism of lipid accumulation in microalgae under nitrogen starvation conditions (i.e. without nitrogen). These studies reported that transcriptional inductions of nitrogen assimilation, tricarboxylic acid (TCA) cycle, pyruvate kinase and a subset of diacylglycerol acyltransferases were important in increasing lipid biosynthesis. At the same time, the transcriptional processes of cellular photosynthesis, gluconeogenesis and translation (ribosomes) were inhibited12. However, transcriptomic information regarding lipid biosynthesis under moderate or limited nitrogen conditions remains scarce. Therefore, it remains unclear what commonalities and differences exist in the regulatory mechanisms across different levels of nitrogen deprivation13.
Auxenochlorella pyrenoidosa, known for its high tolerance to abiotic stresses, is a common and promising species for lipid production14. Therefore, it was chosen as the subject of this study and exposed to sodium nitrate (NaNO3) concentrations of 1.5, 1, 0.5 and 0 g L− 1 for 5 days. The lipid contents were analyzed and RNA-seq was applied to mRNA libraries to investigate the roles of key biological processes in the lipid accumulation of A. pyrenoidosa under varying nitrogen deprivation. This study aims to deepen our understanding of how varying levels of nitrogen deprivation regulate lipid biosynthesis in microalgae.
Methods
Microalgal strain and culture
A. pyrenoidosa SHOU-1002, obtained from a microalgal seed bank at Shanghai Ocean University, was cultured in BG-11 medium (containing 1.5 g L− 1 NaNO3, analytical grade) for two weeks. The algal culture was harvested during the exponential growth phase and centrifuged (4℃, 6000 rpm, 3 min) to remove the supernatant. The obtained algal pellet was washed twice with sterile water and then inoculated into 2.5 L BG-11 media with 1.5, 1, 0.5 and 0 g L− 1 NaNO3 in 3 L Erlenmeyer flasks. These flasks were labeled as N1.5 (Ncon), N1, N0.5 and N0 groups, respectively (Table 1). Three replicates were performed for each group, with initial biomass concentrations of approximately 0.8 g·L− 1. All cultures were incubated in an incubator with photoperiod 12:12 (L: D), temperature 25 °C, light intensity 70 µmol photons·m− 2·s− 1. They were shaken 3 times a day manually.
Measurement of exogenous sodium nitrate in media
The nitrate nitrogen was measured using a spectrophotometric method15. The absorbance of a series of standard solutions at 220 nm and 275 nm (denoted as A₍₂₂₀₎ and A₍₂₇₅₎, respectively) was measured using a fluorescence-spectrophotometer (Shimadzu RF-540, Japan) to perform the linear regression and construct the standard curve. The absorbance of the sample was then measured, and the nitrate nitrogen concentration was calculated according to the regression equation. Finally, the concentration of NaNO3 (g/L) was calculated using the regression equation obtained from the standard curve.
Biochemical analysis
The samples were collected every other day to analyze lipid contents. Cell biomass was harvested by centrifugation at 8000 × g for 10 min from 250 mL culture and washed with sterile water. Then, the algal cells were frozen at − 20 °C overnight, followed by vacuum drying (− 46 °C, 24 h), then weighed to determine the biomass (B, g L− 1). Then, the contents of total lipids were detected as described by Huang et al.9. In brief, 0.2 g dried microalgae were extracted using 50 mL chloroform-methanol for 24 h. The extraction process involved pulsed sonication (5 s on, 5 s off) at 60% amplitude (of a 500 W maximum output) and a frequency of 70 Hz intensity and room temperature. After two extraction cycles of 30 min each, the suspension was filtered and the obtained filtrate was washed twice with 0.88% KCl solutions. Subsequently, the lower organic phase was transferred into pre-weighed glass vials, which were dried at 40℃ under vacuum until they reached constant weights. The total lipid contents were assayed in gravimetrical (in % dry weight).
Total RNA Preparation and quality test
Three independent biological replicates were used for each treatment group. After 5-days of exposure, the NaNO3 in the N0.5 group was nearly depleted (Fig. 1). Then, 0.6 g fresh algal cells of each group were harvested to extract total RNA by TRIzol reagent (Invitrogen, USA) according to the manufacturer’s protocol. The genomic DNA was removed using RNase-free DNase I (Fermentas, Canada). Total RNA was quantified and assessed for quality as follows. 1% agarose gels were used to monitor the degradation and contamination of total RNA. The NanoPhotometer® (IMPLEN, USA) was used to measure the concentration of total RNA. The integrity was assessed through RNA Nano 6000 Assay Kit on the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). The criteria used to select total RNA for subsequent analyses were OD260/280 ≥ 1.9, 28 S/18S rRNA ratio ≥ 1.6, RIN value ≥ 7.0, and a minimum required amount and concentration of total RNA were 3 µg and 50 ng/µL, respectively.

The concentration of sodium nitrate in media. N1.5, 1.5 g/L NaNO3 group; N1, 1 g/L NaNO3 group; N0.5, 0.5 g/L NaNO3 group; N0, 0 g/L NaNO3 group. A one-way ANOVA was performed to compare the effects of different treatments within each day. The values with the same or no little letter superscripts mean no significant difference (P > 0.05), while those with different little letter superscripts mean significant difference (P < 0.05).
mRNA-seq library construction and sequencing
Twelve libraries from Ncon (Ncon-1, Ncon-2 and Ncon-3), N1 (N1-1, N1-2 and N1-3), N0.5 (N0.5-1, N0.5-2 and N0.5-3) and N0 (N0-1, N0-2 and N0-3) were constructed. Briefly, oligo (dT) magnetic beads (Yeasen, China) were added to purify mRNA from total RNA. The fragmenting of mRNA and the synthesis of double stranded cDNA were completed using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA). The samples obtained were purified using PCR purification kits (Takara, Japan). After adenylation and end-filling by Hieff NGS® Ultima™ Endprep Mix (Yeasen, China), the adapters were ligated to the cDNA by Hieff NGS® Ultima® DNA Ligation Module (Yeasen, China) and fragment sizes were verified by agarose gel electrophoresis. 150 bp fragments were selected using the AMPure XP system (Beckman Coulter, Beverly, USA) for PCR amplification: 1 cycle of 98 °C for 30 s and then 10 cycles of 98 °C for 10 s each, followed by 65 °C for 30 s, and 72 °C for 30 s. Finally, a 5-min extension step was performed at 72 °C. After that, all libraries were sequenced on an Illumina HiSeqTM 2500 platform. The raw data of the transcriptome have been submitted to the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/search/all/?term=GSE233967).
Bioinformatic analysis
The read filtering, unigene counting, Venn analysis, Gene ontology (GO) analysis, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, gene co-expression network and series cluster were carried out according to Zhang et al.16. Raw sequencing data in FASTQ format were first processed using Trimmomatic. Reads containing poly-N regions and low-quality reads were removed to obtain the clean reads. After removing adapters and low-quality sequences, the clean reads were assembled de novo into contigs and transcripts by Trinity (version 2.4) using the paired-end sequencing approach. The longest transcript was chosen as a unigene based on the similarity and length for subsequent analyses.
In particular, differentially expressed genes (DEGs) were identified using Baggerley’s test with default filtering conditions. FPKM and read counts value of each unigene were calculated using bowtie2 and eXpress. DEGs were identified using the DESeq2 functions estimateSizeFactors and nbinomTest. The thresholds were set as a false discovery rate (FDR) ≤ 0.05 and an absolute Log2 fold change (FC) ≥ 1. Hierarchical cluster analysis of DEGs was performed to explore transcripts expression pattern. GO enrichment and KEGG pathway enrichment analysis of DEGs were respectively performed using R based on the hypergeometric distribution. A p-value ≤ 0.05 was considered statistically significant for hypergeometric tests in GO analysis, KEGG analysis, and series clustering. Gene pairs showing significant correlation (p-value ≤ 0.05) were used to construct the co-expression network17,18.
Quantitative real-time PCR (qRT-PCR) verification
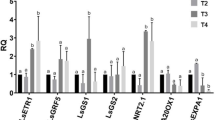
Six genes from each treatment were chosen for qRT-PCR. The same RNA extracts utilized for RNA-seq were also employed to analyze the target genes. The primers were designed using the Primer 5 software (Supplementary data Table S1) and the actin (ACT) gene was selected as the internal reference gene. DEPC water was used to substitute for cDNA in the negative control groups. The qRT-PCR was performed using Agilent AriaMx qRT-PCR System. The reaction systems contained 10 µL SYBR Green Master Mix (YEASEN Biotech, Shanghai, China), 0.4 µL of each forward and reverse primer (10 nmol), 1 µL cDNA, and 8.2 µL DEPC-treated water. The qRT-PCR protocol included an initial denaturation at 95 °C for 2 min, followed by 30 cycles of 95 °C for 10 s and 60 °C for 20 s. Finally, a melting curve analysis was conducted. 2−ΔΔCT method was applied to calculate the average threshold cycle (Ct) of gene expression (Supplementary data Table S2), which was then normalized16.
Statistical analysis
The lipid contents were presented as mean ± standard error. Significant differences among groups were assessed using one-way ANOVA in SPSS software (Version 11.0), after confirming normality and homogeneity of variances. Post-hoc multiple comparisons were performed using Duncan’s test and a significant level of p-value ≤ 0.05 was selected.
Results
Changes in lipid contents of microalgae under varying nitrogen deprivation
Over time, the effects of varying nitrogen deprivation on lipid contents in microalgae gradually became apparent. After 5 days, A. pyrenoidosa cultured in the 1.5 g L⁻¹ NaNO₃ group exhibited the lowest lipid content (11.82%). In contrast, higher lipid contents were observed in cultures supplemented with 1 g L⁻¹, 0.5 g L⁻¹, and 0 g L⁻¹ NaNO₃, with values of 14.18%, 18.95%, and 22.81%, respectively (Fig. 2). These results demonstrate a clear inverse relationship between nitrate concentration and lipid accumulation, with more severe nitrogen deprivation leading to greater lipid production.

Effects of different nitrogen concentrations on lipid contents in A. pyrenoidosa. N1.5, 1.5 g/L NaNO3 group; N1, 1 g/L NaNO3 group; N0.5, 0.5 g/L NaNO3 group; N0, 0 g/L NaNO3 group. A one-way ANOVA was performed to compare the effects of different treatments within each day. The values with the same or no little letter superscripts mean no significant difference (P > 0.05), while those with different little letter superscripts mean significant difference (P < 0.05).
Transcriptomic statistics
A total of 46.65–51.70 million raw reads were obtained. After removing low quality reads, approximately 46.61~51.66 million clean reads were generated in total. Quality analysis indicated that valid base percentages and Q30 values exceeded 98.37% and 90.48%, respectively, in all samples (Supplementary data Table S3). There was no significant difference in the data among the different treatment groups. Finally, 16,415 unigenes were assembled, with a total length of 28,388,667 bp and an average length of 1,729 bp. Specifically, Groups N0.5 and N0 produced the highest and statistically similar unigene counts, which were significantly greater than that of Group N1.5 (p < 0.05) (Supplementary data Table S4). The proportion of complete and single-copy BUSCOs reached 85.48%, indicating a high-quality transcriptome assembly.
DEGs in response to varying nitrogen deprivation
In the 1 g L− 1 NaNO3 group, total 352 DEGs were detected in microalgae, consisting of 168 significantly upregulated genes and 184 significantly downregulated genes. There were 1,976 significantly upregulated genes and 1,245 significantly down-regulated genes in the 0.5 g L− 1 NaNO3 group. At 0 g L⁻¹ NaNO₃, 1,716 genes were significantly upregulated and 1,650 genes were downregulated in microalgae. Venn diagram analysis revealed that 133 genes exhibited differential expression across the three nitrogen deprivation groups, including 70 co-upregulated genes and 26 co-downregulated genes (Figure S1). Figure 3A and C illustrates the distribution and levels of DEGs, highlighting changes in gene expression. Dot plots of gene ontology analysis identified nucleolus, chloroplast, and nitrate assimilation as the most represented pathways (Fig. 3D and F). A key finding is that the number of DEGs increased dramatically with the severity of nitrogen limitation, and a core set of 133 genes responded consistently across all deprivation levels, suggesting their fundamental role in the nitrogen stress response.

Transcriptome sequencing analysis of A. pyrenoidosa SHOU-1002. The different panels represent: (A-C) a volcano plot displaying the differentially expressed genes and (D-F) a dot plot displaying the gene ontology for differentially expressed genes of A. pyrenoidosa SHOU-1002 under varying nitrogen deprivation.
GO and KEGG classification of DEGs under varying nitrogen deprivation
The top ten significantly enriched GO terms in microalgae treated with 1 g L⁻¹, 0.5 g L⁻¹, and 0 g L⁻¹ NaNO₃ are shown in Table 2. Nitrate assimilation (GO:0042128) was the most significantly enriched term in the 1 g L⁻¹ NaNO₃ treated group. For the 0.5 g L⁻¹ NaNO₃ treated group, the most significantly enriched GO term was chloroplast (GO:0009507). In the nitrogen-deprived group (0 g L⁻¹ NaNO₃), the most significantly enriched GO term was nucleolus (GO:0005730).
The top ten significantly enriched KEGG pathways are presented in Table 3. Nitrogen metabolism (ko00910) was the most significantly enriched pathway in the 1 g L⁻¹ NaNO₃ treated group. Under 0.5 g L⁻¹ NaNO₃ treatment, porphyrin and chlorophyll metabolism (ko00860) emerged as the most significantly enriched pathway. Notably, ribosome biogenesis in eukaryotes (ko03008) was ranked second among the top ten significantly enriched KEGG pathways in the 0 g L⁻¹ NaNO₃ treated group.
These analyses revealed that the molecular response to nitrogen deprivation is highly dependent on its severity, shifting from nitrogen salvage and metabolism under mild deprivation to fundamental reorganization of chloroplast function and ribosomal assembly under severe deprivation.
Differential gene expression in key metabolic pathways under varying nitrogen deprivation
Transcriptome analysis of A. pyrenoidosa SHOU-1002 under varying nitrogen deprivation revealed significant differential expression of genes involved in central carbon and lipid metabolism. Key findings are summarized in Table 4.
In the Calvin cycle, genes encoding the ribulose bisphosphate carboxylase large chain (rbcL), ribulose bisphosphate carboxylase small chain (rbcS), phosphoribulokinase (PRK) and sedoheptulose-1,7-bisphosphatase (SBPase) were significantly downregulated. Conversely, genes encoding key glycolytic enzymes, including hexokinase (HK), phosphofructokinase (PFK) and pyruvate kinase (PK), were upregulated.
Within the TCA cycle, the gene expression of the pyruvate dehydrogenase (aceE) and malate dehydrogenase (MDH) was increased, while the gene expression of citrate synthase (CIS) and isocitrate dehydrogenase (IDH) was downregulated. For lipid metabolic pathways, the expression of the glycerol-3-phosphate acyltransferase (GPAT), lysophosphatidic acid acyltransferase (LPAAT), diacylglycerol acyltransferase (DGAT), phosphatidic acid phosphatase (PAP), acetyl-CoA carboxylase (ACC), malonyl-CoA: ACP transacylase (fabD), and β-Ketoacyl-ACP synthase (fabF) was significantly upregulated.
Collectively, these expression shifts indicate a major metabolic rewiring where carbon flux is redirected from photosynthesis and partial TCA cycle activity towards glycolysis and the robust upregulation of the entire lipid biosynthesis machinery.
qRT-PCR verification
Although the fold-change values differed between qRT-PCR and RNA-seq, the expression trends of the selected genes were consistent across both methods (Supplementary data Figure S2). Generally, the results of these platforms were in agreement, indicating the reliability and validity of the RNA-seq results.
Co-expression network of DEGs under varying nitrogen deprivation
The co-expression network of DEGs included total 45 genes in this study (Fig. 4). SFC1 (mitochondrial succinate-fumarate transporter 1 T10O8.50), CISY3 (citrate synthase 3, peroxisomal F7D19.21), CYSZ (citrate synthase, glyoxysomal) and MASY (Malate synthase, glyoxysomal NCU10007) had the highest degree of 37, followed by EDR1 (serine/threonine-protein kinase EDR1 F22O13.20) and YHA8 (Uncharacterized transporter YHL008C) with a degree of 27. Network analysis shows that these genes play pivotal roles in coordinating the global transcriptional response to nitrogen deprivation.

Co-expression network of DEGs from N1, N0.5 and N0 groups. Different color from 3–37 represents varying degree centrality, where the higher degree centrality stands for the more importance of a gene. AMT15: Putative ammonium transporter 1 member 5 K7M2.6; AB36G: ABC transporter G family member 36 P0410E03.7; ADH4: Alcohol dehydrogenase 4 SPAC5H10.06c; ACEA: Isocitrate lyase CC1G_07122; ACEA: Isocitrate lyase ICL; ABA2: Zeaxanthin epoxidase, chloroplastic ABA2; AMT12: Ammonium transporter 1 member 2 F13O11.9; ALEU: Thiol protease aleurain; BASS1: Probable sodium/metabolite cotransporter BASS1, chloroplastic T30F21.11; BALDH: Benzaldehyde dehydrogenase, mitochondrial BALDH; CSLC9: Probable xyloglucan glycosyltransferase 9 OSJNBa0072F13.8; CISY3: Citrate synthase 3, peroxisomal F7D19.21; CYSZ: Citrate synthase, glyoxysomal; DPOE1: DNA polymerase epsilon catalytic subunit A T23G18.21; DUR1: Allophanate hydrolase YBR1448; DHE4: NADP-specific glutamate dehydrogenase; EDR1: Serine/threonine-protein kinase EDR1 F22O13.20; FRO2: Ferric reduction oxidase 2 F22L4.12; FAEA: Probable feruloyl esterase A AFLA_066140; GLE: Autolysin; GL813: Germin-like protein 8–13 OsJ_026468; HUP2: H(+)/hexose cotransporter 2 HUP2; H4: Histone H4 MMN10.22; LAMA: Putative urea carboxylase AN0887; MU157: Meiotically up-regulated gene 157 protein SPAC12B10.16c; MASY: Malate synthase, glyoxysomal NCU10007; NRT21: High-affinity nitrate transporter 2.1 OJ1399_H05.5; NIR: Ferredoxin–nitrite reductase, chloroplastic NIR; NRT24: High affinity nitrate transporter 2.4 MAE1.2; NIA: Nitrate reductase [NADH] NITA; P2C05: Probable protein phosphatase 2 C 5 T12M4.15; PDC1: Pyruvate decarboxylase 1 OsI_019612; PRI1: DNA primase small subunit SPAC6B12.10c; RITF1: Protein RGF1 INDUCIBLE TRANSCRIPTION FACTOR 1 T4E5; RM1DH: L-rhamnose-1-dehydrogenase PICST_50944; STAD5: Stearoyl-[acyl-carrier-protein] 9-desaturase 5, chloroplastic OsJ_14516; SFC1: Mitochondrial succinate-fumarate transporter 1 T10O8.50; SPL10: Squamosa promoter-binding-like protein 10 B1047G05.25; SUB3: Subtilisin-like protease 3 SUB3; TRHN1: Group 1 truncated hemoglobin LI637 LI637; TRPB1: Tryptophan synthase beta chain 1, chloroplastic MBG8.7; TRYP: Trypsin; UBP1: Oligouridylate-binding protein 1 UBP1; Y232: Glycosyltransferase family 92 protein RCOM_0530710 RCOM_0699480; YHA8: Uncharacterized transporter YHL008C YHL008C.
Series cluster of DEGs under varying nitrogen deprivation
Total 50 unique model expression tendencies were found (Supplementary data Figure S3). Profile 9 and profile 42 had P-values less than 0.01, and their trends changed monotonically (Fig. 5). The gene cluster in profile 9 was downregulated while the gene cluster in profile 42 was upregulated. These changes were induced by 1 g L− 1, 0.5 g L− 1 and 0 g L− 1 NaNO3. Moreover, the genes in profile 9 played a significant role in various biological processes, including tRNA processing (GO:0008033), hydrolase activity (GO:0016787), NAD+ diphosphatase activity (GO:0000210), protein deneddylation (GO:0000338) and COP9 signalosome (GO:0008180). Genes in profile 42 mostly enriched in response to gibberellin (GO:0009739), thiamine pyrophosphate binding (GO:0030976), TCA cycle (GO:0006099), succinate dehydrogenase activity (GO:0000104), pyruvate decarboxylase activity (GO:0004737), mitochondrial electron transport, succinate to ubiquinone (GO:0006121), hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds (GO:0016810) related GO terms. Two gene clusters with statistically significant and monotonically changing expression patterns were identified, representing a suite of co-regulated genes that are systematically suppressed or activated in direct proportion to the degree of nitrogen deprivation.

Gene clusters and top ten GO terms of profile 9 (A) and profile 42 (B). The horizontal axis shows N1.5, N1, N0.5 and N0 groups, and the vertical axis represents the expression levels for genes after Log normalized transformation.
Discussion
We conducted 5-days exposure of A. pyrenoidosa under a series of 1.5, 1, 0.5 and 0 g·L− 1 NaNO3. With the decrease in nitrogen concentrations, the final lipid contents showed a stepwise increase (Fig. 2). The significantly higher number of unigenes in the N0.5 and N0 groups indicates a greater transcriptome complexity, which likely reflects a more active and diverse gene expression profile under these specific conditions. Moreover, the Venn diagram revealed both unique and shared DEGs among microalgae under different nitrogen concentrations (Supplementary data Figure S1). Based on the results of GO analysis and KEGG analysis, we aimed to uncover distinct regulatory mechanisms in the lipid accumulation of A. pyrenoidosa under varying nitrogen deprivation. Furthermore, the co-expression network was used to reveal common regulatory mechanisms. Furthermore, potential rate-limiting steps in these regulatory mechanisms were identified based on the series cluster analysis.
Distinct characteristics of regulatory mechanisms at different nitrogen concentrations
In this study, our GO enrichment analysis revealed that the most significantly altered biological processes under varying nitrogen concentrations were nitrogen assimilation (Profile N1), chloroplast metabolism (Profile N0.5), and nucleolar stress (Profile N0) (Table 2). These processes collectively regulate lipid biosynthesis by modulating the availability of carbon skeletons and energy. Specifically, the carbon skeleton alpha-ketoglutarate and adenosine triphosphate (ATP) are essential for nitrogen assimilation. Inhibition of nitrogen assimilation leads to accumulation of carbon skeletons and ATP, which can be redirected toward lipid biosynthesis19. Similarly, chloroplast metabolism affects lipid accumulation by altering the production of ATP and reducing nicotinamide adenine dinucleotide phosphate (NADPH)20. Moreover, nucleolar stress activates a distinct lipogenesis pathway to redirect excessive energy toward lipid storage21.
Complementarily, KEGG pathway analysis further confirmed these findings at the metabolic network level, identifying significantly enriched pathways including nitrogen metabolism (across all groups), photosynthesis and porphyrin/chlorophyll metabolism (N0.5), and the TCA cycle and ribosome biogenesis (N0) (Table 3). The consistent alteration in nitrogen metabolism pathways across gradients underscores its central role, while the distinct pathway enrichments specific to each nitrogen level—such as the downregulation of photosynthetic components in N0.5 and the activation of the TCA cycle in N0—reflect a graded metabolic reprogramming. This coordinated shift at the pathway level supports the notion that carbon and energy fluxes are fine-tuned to optimize lipid accumulation under different stress intensities22.
Common characteristics of regulatory mechanisms at different nitrogen concentrations
As shown in Fig. 4, the genes of the TCA cycle (i.e. SFC1, CISY3, CYSZ and MASY) had the highest degree centrality, followed by genes encoding MAPK and transporters (i.e. EDR1 and YHA8). These results suggest that the TCA cycle, MAPK cascade, and transporters play conserved roles in lipid accumulation of microalgae under varying nitrogen deprivation. Under the condition of nitrogen starvation, membrane and intracellular transporters play roles in cellular homeostasis. For example, the redistribution of intracellular carbon fluxes occurs, ultimately yielding acetyl-CoA or succinyl-CoA as intermediates in the TCA cycle. These intermediates enter the TCA cycle, which channels carbon flux toward lipid biosynthesis23. Mitogen-activated protein kinase (MAPK) cascade activates lipogenic genes through transcription factors to enhance lipid biosynthesis in microalgae24. These regulatory processes should also apply to lipid biosynthesis in microalgae under other nitrogen deprivation, which is supported by the co-expression network in this study. The centrality of these genes in the network suggests that the TCA cycle, MAPK cascade, and transporters play conserved roles in lipid accumulation of microalgae under varying nitrogen deprivation. The activity of these pathways may be modulated by reactive oxygen species (ROS) generated under nitrogen deprivation. It is known that varying nitrogen deprivation can affect electron transport chains in mitochondria and chloroplasts to increase ROS. When ROS levels reach a certain threshold, essential molecules can accumulate through the TCA cycle and transporters. Additionally, gene expression can be upregulated through MAPK cascades to enhance lipid accumulation25.
Critically, our findings showed that the consistent activation of the MAPK cascade across nitrogen regimes establishes it not merely as a common feature, but as a pivotal, conserved signaling hub and a master regulatory switch for stress-induced lipid biosynthesis, representing a prime target for future metabolic engineering24. This perspective extends recent advances in multi-omics and metabolic flux analysis, which have significantly improved our understanding of lipid accumulation in oleaginous microalgae under nitrogen stress. Under nitrogen deficiency, Nannochloropsis oceanica exhibits proteome-level adaptations—such as sustained photosynthesis, enhanced TCA cycle activity, and Kennedy pathway activation—that promote TAG biosynthesis26. In Nannochloropsis gaditana, nitrogen stress leads to marked downregulation of photosynthetic and protein synthesis machinery, while upregulating key lipid-assembly proteins like DGAT27. Similarly, nitrogen deprivation elevates TAG production in Chlamydomonas reinhardtii, with multi-omics reanalysis underscoring the central role of arginine and ornithine metabolism. Moreover, the involvement of phosphorylation, nitrosylation, and peroxidation signaling events was highlighted in the regulatory network28. Thus, while prior omics studies have cataloged the metabolic components of the response, our findings position the MAPK cascade as the central signaling integrator that commands this lipid biosynthesis program under varying nitrogen stress.
Gene expression patterns underpinning growth and lipid biosynthesis under varying nitrogen deprivation
The most pronounced transcriptional response was the general downregulation of key genes involved in the Calvin cycle, including rbcL, rbcS, and PRK. This suppression likely reduces carbon fixation into carbohydrates and redirects the carbon flux towards the synthesis of storage lipids, a common metabolic shift under nutrient stress when growth is arrested. Concomitantly, we observed a significant upregulation of several genes in the glycolytic pathway, such as HK, PFK, and PK. This enhanced glycolytic activity ensures an increased supply of phosphoenolpyruvate and pyruvate, which are crucial precursors for the synthesis of acetyl-CoA, the fundamental building block for de novo fatty acid synthesis13.
The fate of pyruvate appears to be strategically redirected. While the gene aceE was upregulated, facilitating the conversion of pyruvate into acetyl-CoA, key enzymes in the TCA cycle (CIS, IDH) were repressed. This pattern suggests a bypass of the full TCA cycle, which typically supports biomass production and energy generation during active growth. Instead, the cycle may operate in a non-cyclic, “broken” mode to primarily generate citrate for export to the cytosol, where it can be cleaved to provide acetyl-CoA for lipogenesis. The upregulation of MDH could further support this process by generating NADH, which is essential for redox balance in lipid biosynthesis12.
The most critical evidence for lipid hyperaccumulation comes from the dramatic upregulation of genes encoding enzymes in the Kennedy pathway and fatty acid synthesis. The key triacylglycerol (TAG) assembly enzymes—GPAT, LPAAT, PAP, and most notably DGAT—were all significantly upregulated. This concerted action channels fatty acids into TAG, the primary storage lipid. Furthermore, the rate-limiting step in fatty acid synthesis, catalyzed by ACC, was transcriptionally enhanced, along with other genes in the FAS pathway (e.g., fabD, fabF). This indicates a robust activation of the entire machinery for converting acetyl-CoA into long-chain fatty acids7.
In summary, our transcriptomic data delineate a coherent metabolic model (Fig. 6) for nitrogen deprivation-induced lipid accumulation : (1) carbon fixation is attenuated; (2) carbon flow is shunted through enhanced glycolysis to generate precursors; (3) The TCA cycle is remodeled to supply acetyl-CoA and reducing equivalents; and (4) The Kennedy pathway and FAS are transcriptionally activated to efficiently synthesize and store TAGs2.

The overall process of metabolic reprogramming from carbon fixation to TAG synthesis in A. pyrenoidosa SHOU-1002 under nitrogen deprivation.
Rate-limiting steps of regulatory mechanisms at different nitrogen concentrations
Genes with similar expression patterns were classified using Short Time-series Expression Miner (STEM). The trend characteristics of gene sets in profile 9 and profile 42 were found to be related to the monotonic trend of lipid accumulation (Fig. 5). These monotonic gene sets may indicate potential rate-limiting steps in microalgal lipid accumulation under different levels of nitrogen deprivation29.
The GO terms associated with tRNA processing and the COP9 signalosome showed monotonic trends in profile 9, suggesting that varying nitrogen deprivation could suppress protein biosynthesis and promote protein degradation through these processes. tRNA processing modifies the canonical L-shaped structure of tRNA through post-transcriptional modifications, which is necessary for protein synthesis15. COP9 signalosome suppresses the activity of E3 ubiquitin ligase by facilitating the cleavage of NEDD8-CUL1 conjugate (i.e. deneddylation), which prevents protein degradation30. It is known that changes in the biosynthesis and degradation of proteins contribute to lipid accumulation in microalgae. In particular, when protein biosynthesis is inhibited, excess photosynthetic carbons are redirected toward triglyceride production. In addition, protein breakdown occurs in response to environmental stresses, and nitrogen salvaged is diverted to the TCA cycle for lipid biosynthesis31. Therefore, these protein biosynthesis and degradation processes (tRNA processing and COP9 signalosome) may serve as rate-limiting steps in lipid accumulation under varying nitrogen deprivation.
The consistent patterns of GO terms correlated with oxidized nicotinamide adenine dinucleotide (NAD+), thiamine pyrophosphate (TPP), succinate dehydrogenase (SDH), and pyruvate dehydrogenase (PDH) in profile 9 and profile 42 indicate that varying nitrogen deprivation may affect the TCA cycle and glycolysis through the processes associated with these crucial molecules. Oxidized nicotinamide adenine dinucleotide (NAD+), as a hydrogen receptor, plays important roles in the TCA cycle and glycolysis32. Thiamine pyrophosphate (TPP) serves as an essential cofactor for catalytic enzymes, including α-ketose transketolase (TK, EC. 2.2.1.1) in the pentose phosphate (PPP) pathway, α-ketoglutarate dehydrogenase (KGDH, EC. 1.2.4.2) and pyruvate dehydrogenase (PDH, EC. 1.2.4.1) in TCA cycle etc33. Succinate dehydrogenase (SDH, EC. 1.3.5.1) supplies electrons to mitochondrial electron transport chains, whose activity can generally be used as an indicator to evaluate the operational degree of the TCA cycle34. Pyruvate decarboxylase (PDH, EC. 1.2.4.1) catalyzes the irreversible reaction of pyruvic acid to acetyl-CoA, which links glycolysis and TCA cycle31. In short, they are crucial molecules in the TCA cycle and glycolysis. Moreover, both TCA cycle and glycolysis are associated with energy metabolism and provide carbon skeletons for lipid biosynthesis31. Therefore, the processes described above, which participate in the TCA cycle and glycolysis, may become rate-limiting steps of lipid accumulation in microalgae under varying nitrogen deprivation.
In this study, the response to gibberellin (GO:0009739) was the most significantly enriched GO term in profile 42. Its trend was similar to that of lipid contents. This result suggests that endogenous gibberellins may respond to varying nitrogen deprivation and affect lipid accumulation in microalgae. Gibberellins are diterpene hormones that respond to environmental stresses and affect various physiological metabolisms in plants. Upon exposure to stresses, the homeostasis of gibberellins is altered through the expression of regulatory genes35. Besides, gibberellins have been reported to enhance the utilization of glucose and metabolites in the biosynthesis of fatty acids in microalgae36. Therefore, it is an interesting topic to identify and explore the roles of gibberellins in the lipid accumulation of microalgae under nitrogen deprivation. However, there are few related reports available. This situation may be attributed to the immaturity of endogenous gibberellin assays in microalgae. Fortunately, Stirk et al.37 first used appropriate methods to determine endogenous gibberellins in microalgae. This breakthrough may promote research on the regulation of lipid accumulation in microalgae by endogenous gibberellins.
The GO terms related to hydrolases were significantly enriched in the top 10 of profile 9 and profile 42, and their trends were monotonic. This result indicates that hydrolases in lysosomes may play a role in enhancing lipid accumulation in microalgae under varying nitrogen deprivation. Lysosomes contain hydrolases that participate in the digestion of proteins, nucleic acids and polysaccharides. This process helps achieve the turnover of substances necessary for lipid accumulation in microalgae38,39. Nitrogen deprivation has been reported to induce the storage of amino acids and intermediate molecules31. This process may activate multiple homeostatic signaling pathways through lysosomes, leading to changes in various cellular behaviors40. However, the roles of lysosomes and their internal hydrolases in lipid accumulation and the underlying mechanisms seem to be overlooked. Additional research is needed.
Conclusions
The distinct characteristics of lipid accumulation in A. pyrenoidosa under varying nitrogen deprivation were changes in nitrogen assimilation, chloroplast metabolism and nucleolar stress. In addition, the common characteristics were the variations in the TCA cycle, MAPK cascade and transporter. Moreover, the processes related to tRNA processing, COP9 signalosome, TCA cycle, glycolysis, gibberellins and lysosomes were possible rate-limiting steps. Notably, the pivotal role of the consistently activated MAPK cascade makes its exploration a priority for future metabolic engineering.
Data availability
The raw data of transcriptome in this study has been submitted to the National Center for Biotechnology Information (Accession number: GSE233967). Other data is available from the corresponding author on request.
References
Greene, J. M., Quiroz, D., Limb, B. J. & Quinn, J. C. Geographically-Resolved Techno-Economic and life cycle assessment comparing Microalgae-Based renewable diesel and sustainable aviation fuel in the united States. Environ. Sci. Technol. 59, 3472–3483 (2025).
Singh, R. P. et al. Unlocking the adaptation mechanisms of the oleaginous microalga Scenedesmus sp. BHU1 under elevated salt stress: a physiochemical, lipidomics and transcriptomics approach. Front. Microbiol. 15, 1475410 (2024).
Usman, H. M. et al. Comparative analysis assessing growth dynamics of locally isolated Chlorella Sorokiniana and Chlorella vulgaris for biomass and lipid production with biodiesel potential. Bioresour Technol. 403, 130868 (2024).
Pandey, S., Narayanan, I., Selvaraj, R., Varadavenkatesan, T. & Vinayagam, R. Biodiesel production from microalgae: a comprehensive review on influential factors, transesterification processes, and challenges. Fuel 367, 131547 (2024).
Kossalbayev, B. D. et al. Strategies for genetic modification of microalgae to improve the production efficiency of liquid biofuel. Int. J. Hydrogen Energ. 100, 1301–1314 (2025).
Yan, C. X. et al. Advances in multi-omics technologies for identifying metabolic engineering targets and improving lipid production in microalgae. Bioresour Technol. 429, 132501 (2025).
de Jesús-Campos, D. et al. Chemical composition, fatty acid profile and molecular changes derived from nitrogen stress in the diatom Chaetoceros muelleri. Aquac Rep. 16, 100281 (2020).
Kiran, B., Pathak, K., Kumar, R., Deshmukh, D. & Rani, N. Influence of varying nitrogen levels on lipid accumulation in Chlorella Sp. Int. J. Environ. Sci. Te. 13, 1823–1832 (2016).
Huang, X. X., Huang, Z. Z., Wen, W. & Yan, J. Q. Effects of nitrogen supplementation of the culture medium on the growth, total lipid content and fatty acid profiles of three microalgae (Tetraselmis subcordiformis, Nannochloropsis oculata and Pavlova viridis). J. Appl. Phycol. 25, 129–137 (2013).
Sibi, G., Shetty, V. & Mokashi, K. Enhanced lipid productivity approaches in microalgae as an alternate for fossil fuels-a review. J. Energy Inst. 89, 330–334 (2016).
Wang, X. X., Fosse, H. K., Li, K., Chauton, M. S. & Reitan, K. I. Influence of nitrogen limitation on lipid accumulation and EPA and DHA content in four marine microalgae for possible use in Aquafeed. Front. Mar. Sci. 6, 95 (2019).
Jaeger, D. et al. Time-resolved transcriptome analysis and lipid pathway reconstruction of the oleaginous green microalga Monoraphidium neglectum reveal a model for triacylglycerol and lipid hyperaccumulation. Biotechnol. Biofuels. 10, 197 (2017).
Bonnefond, H. et al. Coupling and uncoupling of triglyceride and beta-carotene production by Dunaliella Salina under nitrogen limitation and starvation. Biotechnol. Biofuels. 10, 25 (2017).
Liu, J. Y., Qiu, W. & Song, Y. M. Stimulatory effect of auxins on the growth and lipid productivity of Chlorella pyrenoidosa and Scenedesmus quadricauda. Algal Res. 18, 273–280 (2016).
Wei, W., Zhuang, H., Zhang, Y. & Wang, L. Crustacean zooplankton community in relation to physicochemical factors and phytoplankton of 13 waterbodies located in the Yangtze river delta. Ann. Limnol-Int J. Lim. 55, 16 (2019).
Zhang, Y. et al. Dose-dependent effects of polystyrene nanoplastics on growth, photosynthesis, and Astaxanthin synthesis in Haematococcus pluvialis. Environ. Pollut. 359, 124574 (2024).
o Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
o Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Gupta, N., Khare, P. & Singh, D. P. Nitrogen-dependent metabolic regulation of lipid production in microalga Scenedesmus vacuolatus. Ecotoxicol. Environ. Saf. 174, 706–713 (2019).
Stephenson, P. G., Moore, C. M., Terry, M. J., Zubkov, M. V. & Bibby, T. S. Improving photosynthesis for algal biofuels: toward a green revolution. Trends Biotechnol. 29, 615–623 (2011).
Wu, J. Y. et al. PHA-4/FoxA senses nucleolar stress to regulate lipid accumulation in Caenorhabditis elegans. Nat. Commun. 9, 1195 (2018).
Woodson, J. D. & Chory, J. Coordination of gene expression between organellar and nuclear genomes. Nat. Rev. Genet. 9, 383–395 (2008).
Yang, Z. K., Niu, Y. F., Ma, Y. H. & Jiao, X. Molecular and cellular mechanisms of neutral lipid accumulation in diatom following nitrogen deprivation. Biotechnol. Biofuels. 6, 67–67 (2013).
Wang, L. F., Ming, D., Pan, Y. Y., Shi, L. Q. & Wei, W. Z. Nitrogen limitation and hydrogen peroxide act synergistically to enhance lipids accumulation via ROS/Ca2 + dependent mechanism in Chlorella Sorokiniana. Algal Res. 70, 102974 (2023).
Zhang, S., He, Y., Sen, B. & Wang, G. Reactive oxygen species and their applications toward enhanced lipid accumulation in oleaginous microorganisms. Bioresour Technol. 307, 123234 (2020).
You, W. et al. Integration of proteome and transcriptome refines key molecular processes underlying oil production in Nannochloropsis oceanica. Biotechnol. Biofuels. 13, 109 (2020).
Hulatt, C. J. et al. Proteomic and transcriptomic patterns during lipid remodeling in Nannochloropsis Gaditana. Int. J. Mol. Sci. 21, 6946 (2020).
Monteiro, L. F. R., Giraldi, L. A. & Winck, F. V. From feasting to fasting: the arginine pathway as a metabolic switch in nitrogen-deprived Chlamydomonas reinhardtii. Cells 12, 1379 (2023).
Ernst, J. & Bar-Joseph, Z. Stem: a tool for the analysis of short time series gene expression data. BMC Bioinform. 7, 191 (2006).
Qin, N. X., Xu, D. Q., Li, J. G. & Deng, X. W. COP9 signalosome: Discovery, conservation, activity, and function. J. Integr. Plant. Biol. 62, 90–103 (2020).
Chen, H., Zheng, Y. L., Zhan, J., He, C. & Wang, Q. Comparative metabolic profiling of the lipid-producing green microalga Chlorella reveals that nitrogen and carbon metabolic pathways contribute to lipid metabolism. Biotechnol. Biofuels. 10, 153 (2017).
Noctor, G., Hager, J. & Li, S. Biosynthesis of NAD and its manipulation in plants. Adv. Bot. Res. 58, 153–201 (2011).
Rosado-Souza, L. et al. Appropriate Thiamin pyrophosphate levels are required for acclimation to changes in photoperiod. Plant. Physiol. 180, 185–197 (2019).
Affourtit, C., Krab, K., Leach, G. R., Whitehouse, D. G. & Moore, A. L. New insights into the regulation of plant succinate dehydrogenase. On the role of the protonmotive force. J. Biol. Chem. 31, 276, 32567–32574 (2001).
Yamaguchi, S. Gibberellin metabolism and its regulation. Annu. Rev. Plant. Biol. 59, 225–251 (2008).
Yu, X. J., Sun, J., Sun, Y. Q., Zheng, J. Y. & Wang, Z. Metabolomics analysis of phytohormone Gibberellin improving lipid and DHA accumulation in Aurantiochytrium Sp. Biochem. Eng. J. 112, 258–268 (2016).
Stirk, W. A. et al. Hormone profiles in microalgae: gibberellins and brassinosteroids. Plant. Physiol. Bioch. 70, 348–353 (2013).
De Duve, C. & Wattiaux, R. Functions of lysosomes. Annu. Rev. Physiol. 28, 435–492 (1966).
Nagappan, S. et al. Metabolomics integrated with transcriptomics and proteomics: evaluation of systems reaction to nitrogen deficiency stress in microalgae. Process. Biochem. 91, 1–14 (2020).
Zhang, Z. Q. et al. Role of lysosomes in physiological activities, diseases, and therapy. J. Hemato Oncol. 14, 79 (2021).
Acknowledgements
We thank Jingyuan Ge and Lihui Gao in OEbiotech.co.ltd for technical support in the transcriptome analysis of this study.
Funding
This research was primarily supported by grants from the Central Public-interest Scientific Institution Basal Research Fund, Freshwater Fisheries Research Center, CAFS (NO. 2025JBFR06). Additional support was provided by the Natural Science Foundation of China (31800435).
Author information
Authors and Affiliations
Contributions
Writing-original draft preparation, W.L.F. Conceptualization, W.L.F. Methodology, M.X.Y. Investigation, H.X.X. Validation, W.H.B. Formal analysis, J.W. Resources, C.W.W. Data Curation, W.Y.F. Writing-review and editing, X.P. Visualization, W.L.F. Supervision, W.H.B. and H.X.X. Project administration, W.H.B. and H.X.X. Funding acquisition, W.L.F. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liufu, W., Xueyan, M., Xuxiong, H. et al. A nitrogen deprivation gradient triggers transcriptional reprogramming for lipid biosynthesis in Auxenochlorella pyrenoidosa. Sci Rep 15, 43179 (2025). https://doi.org/10.1038/s41598-025-28963-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28963-x
