
Abstract
The most common kind of kidney cancer, clear cell renal cell carcinoma (ccRCC), presents challenges in clinical management and prognosis. Although the roles of long non-coding RNAs (lncRNAs) and migrasomes in tumorigenesis and tumor development have gradually attracted attention, there is currently relatively little research on migrasomes in ccRCC. The objective of this research was to construct a migrasome-associated lncRNAs model and assess its predictive value for immune reactions and survival outcomes in patients with ccRCC. First, the Cancer Genome Atlas (TCGA) database was utilized to acquire transcriptome data and clinical information pertaining to ccRCC. Using this data, Pearson correlation identified migrasome-associated lncRNAs, and Cox regression analysis was utilized to build a prognostic model. Subsequently, the model’s utility was validated against clinical characteristics from multiple perspectives. Enrichment results, immune infiltration analysis, Tumor Mutational Burden (TMB), and Tumor Immune Dysfunction and Exclusion (TIDE) analysis delineated differences in the tumor immune microenvironment. Quantitative real-time polymerase chain reaction (qPCR) confirmed the expression of key lncRNAs in the model. Finally, a interference plasmid for UBE2Q1-AS1 was constructed and transfected into 786-O cells. The CCK-8 assay and Transwell assay were then used to verify its effects on cell proliferation and migration. A prognostic model incorporating 12 migrasome-associated lncRNAs with independent prognostic value was developed, demonstrating robust predictive power across various clinical features. Enrichment and immune infiltration analyses revealed significant disparities in immune responses between groups. TMB and TIDE analyses indicated that high TMB groups had lower survival rates compared to low TMB groups, and TIDE scores were greater for high-risk groups than for low-risk groups. Additionally, the quantities of expression for migrasome-associated lncRNAs were validated in human ccRCC cell line. Cell experiments revealed that interference of UBE2Q1-AS1 significantly inhibited the proliferation and migration of 786-O cells. The migrasome-associated lncRNA model accurately predicts ccRCC patient prognosis, offering new insights for immunotherapy and clinical applications.
Similar content being viewed by others

Introduction
The most common histological subtype of renal cell carcinoma (RCC), which makes up 70% to 80% of total renal malignancies, is clear cell renal cell carcinoma (ccRCC)1. Clinical approaches to ccRCC primarily encompass surgical treatment, targeted therapy, and immunotherapy. Surgical resection is vital in the early stages of ccRCC and achieves favorable outcomes for localized tumors. Nevertheless, recurrence post-surgery is observed in 20% to 40% of cases, with poor prognoses, especially for those with advanced or metastatic disease2. There is an urgent clinical need for novel predictive tools to assess the prognostic risks and immune responses in ccRCC patients.
Extracellular vesicles known as migrasomes, discovered in recent years3, form on the contractile fibers of migrating cells and carry a variety of biomolecules, including proteins, mRNA, and lncRNA, being a significant factor in signal transduction during active cell migration processes such as tumor metastasis, immune response, homeostasis maintenance, and developmental biology4. In the metastatic process of malignant tumors, cancer cells engage in migratory movements, interacting with extracellular matrix proteins and vascular endothelial cells, a process often accompanied by the release of numerous migrasomes5. Zhang et al. identified migrasomes as facilitators in Hepatocellular Carcinoma (HCC), promoting cancer cell invasion and metastasis and intensifying tumor progression by enhancing intercellular communication6. Cheng et al. reported that nanoparticles are able to diminish migrasome’s ability to promote cell migration by binding to contractile fibers and migrasomes, thereby inhibiting tumor cell metastasis7. These reports indicated that migrasomes might be viable targets for cancer therapy. Currently, research on migrasomes in RCC remains in a void. However, key molecules that regulate cell migration, vesicle secretion (such as exosomes), and epithelial-mesenchymal transition (EMT), including Bmi-1, SPOP, and Trim21, have been proven to be closely associated with the invasiveness of RCC8,9,10,11,12.
Long Non-coding RNAs (lncRNAs), which are non-coding RNAs with a length of more than 200 nucleotides13, play a pivotal role in the initiation and progression of ccRCC. According to existing research, there has been no report of direct evidence regarding migrasome-associated lncRNAs in RCC. However, numerous studies have demonstrated that lncRNAs regulating cell migration significantly influence the progression of renal cell carcinoma through mechanisms such as EMT and vesicle transport. These processes have potential associations with the functions of migrasomes14,15,16. Meng et al. discovered that SNHG17 facilitates the migration and invasion of ccRCC cells by recruiting E2F1 to the CENPE promoter and activating its transcription17. Wang et al. discovered that lncRNA OTUD6B-AS1 delays the growth of ccRCC by intervening in the Wnt/β-catenin signaling pathway and has a close connection with the prognosis of ccRCC18. Numerous studies have identified lncRNAs as predictive factors for ccRCC progression19,20. However, the impact of migrasome-associated lncRNAs on ccRCC patient prognosis remains undetermined. Within this research, we comprehensively analyzed the prospective function of migrasome-associated lncRNAs in ccRCC and constructed a novel prognostic model for migrasome-associated lncRNAs.
Materials and methods
Data acquisition
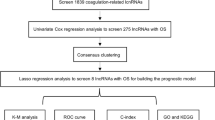
In previous studies, seven migrasome-associated genes were obtained, namely CPQ, EOGT, ITGA5, ITGB1, NDST1, PIGK, and TSPAN421,22,23. Subsequently, by searching for “migrasome” in the database at https://www.genecards.org/, another three migrasome-associated genes were obtained, namely EPCIP, PKD1, and PKD2. Transcriptomic, mutational, and clinical data from ccRCC were extracted from the Cancer Genome Atlas database (TCGA) on August 31, 2024. This study encompassed 72 normal tissue samples and 542 ccRCC samples. Subsequently, we utilized Strawberry Perl 5.30.0 software to amalgamate the acquired gene transcriptome data of ccRCC samples into a gene expression matrix and organized the downloaded clinical data to extract pertinent clinical information. A diagram of the study’s methodology was shown in Fig. 1.

Flowchart of this study.
Selection of migrasome-associated lncRNAs
LncRNAs were isolated from the acquired ccRCC transcript expression data. Pearson correlation analysis was employed to determine the relationship between lncRNAs and migrasome-associated genes; lncRNAs with |Pearson R|> 0.4 and p < 0.001 were classified as migrasome-associated lncRNAs19,24. Employing the “limma”, “dplyr”, “ggalluvial”, and “ggplot2” packages in R software, we visualized the outcomes of the correlation analysis in a Sankey diagram, thereby elucidating the up-regulation and down-regulation relationships associated with migrasomes25.
Establishment and validation of migrasome-associated lncRNA prognostic model
In this experimental segment, computations and analyses were performed with R language, segmenting the TCGA database’s KIRC dataset into testing and training sets with a 1:1 proportion in accordance with a stochastic approach. A prognostic model involving migrasome-associated lncRNAs was crafted with the training set and subsequently authenticated through the test set and the entire cohort. Table 1 describes the clinical traits of patients with ccRCC. The training dataset was utilized to select prognostic lncRNAs through univariate Cox regression analysis. To mitigate overfitting, the “glmnet” R package facilitated Least Absolute Shrinkage and Selection Operator (LASSO) regression analysis. Subsequently, a multivariate Cox regression analysis was conducted to establish a model with the identified lncRNAs. The Eq. (1) as follows was employed for calculating risk scores:
Within the formula, Coef(i) denotes the regression factor, and Expr(i) signifies lncRNA expression quantity. The median risk score value was used to stratify the patients into high-risk and low-risk groups. Survival disparities between these groups were examined with survival analysis, with corresponding Kaplan–Meier (KM) curves plotted. The variation in lncRNA expressions between the two groups was assessed via the “pheatmap” package and portrayed in a thermal map. The independence of the model in forecasting ccRCC prognosis, in conjunction with clinical information, was verified through Cox regression analysis. The model’s prognostic capabilities were appraised with the “timeROC” package in R software, by plotting receiver operating characteristic (ROC) curves and calculating the concordance index (C-index). Nomogram models were constructed with the “survival” and “rms” packages, with calibration curves employed to affirm the precision of the nomograms.
Principal component analysis (PCA) and enrichment analysis
We employed PCA for dimensionality reduction of all genes, migrasome-associated genes, migrasome-associated lncRNAs, and model data, thereby ascertaining the discernibility among different risk groups. The “clusterProfiler” R package was leveraged to perform gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses on genes with differential expression in the two groups26. Furthermore, gene set enrichment analysis (GSEA) was carried out for filtering functional pathways. Gene sets “c2.cp.kegg.Hs.symbols” and “c5.go.Hs.symbols” were retrieved from the Molecular Signatures Database (MSigDB) and evaluated via the “GSVA” R package to identify enriched pathways among different risk groups.
Immune infiltration and tumor microenvironment analysis
With the help of CIBERSORT algorithm, we quantified the infiltration status of immune cells in relation to risk scores, encompassing the measurement of tumor-infiltrating immune cell subsets in the two groups, and evaluated their immune-related functions27. Utilizing the ESTIMATE algorithm, we determined the tumor microenvironment (TME) score for each sample in the two groups, thereby further exploring the TME in ccRCC patients28.
Somatic mutation analysis and immunotherapy response prediction
Post-downloading somatic variation data from the TCGA site, we employed the R package “maftools” to amalgamate and dissect the correlation between survival rates and tumor mutational burden (TMB) across the two groups. Additionally, we utilized the Tumor Immune Dysfunction and Exclusion (TIDE) online database to grade all samples29, followed by a differential analysis of TIDE scores among risk groups to anticipate potential responses to immunotherapy.
Drug sensitivity prediction
In this analytical segment, we forecasted the half-maximal inhibitory concentrations of anticancer drugs with the R package “oncoPredict”30 and scrutinized disparities across the two groups to identify latent therapeutics for ccRCC.
Quantitative real-time polymerase chain reaction (qPCR)
Human renal cell line (HK-2, CL-0109, Pricella, China) was maintained in DMEM/F12 medium (PM150312B, Pricella, China) with 10% Fetal Bovine Serum (FBS, AC03L055, Life-iLab, China). Human ccRCC cell line (786-O, CL-0010, Pricella, China) was maintained in RPMI-1640 medium (PM150110B, Pricella, China) with 1% penicillin–streptomycin solution (C0222, Beyotime, China) and 10% FBS. The incubation conditions were set at 37℃, 95% humidity, and 5% CO2 concentration. For subsequent detection, cells in the logarithmic growth phase and in good condition should be selected.
Total RNA of the two types of cells was extracted via Trizon reagent (CW0580S, Cwbio, China). Complementary DNA (cDNA) was generated through the Goldenstar™ RT6 cDNA Synthesis Kit Ver.2 (TSK302M, Tsingke, China). qPCR was performed using a real-time fluorescence quantitative PCR instrument (IQ5, Bio-Rad, USA), employing SYBR Green I (TSE202, Tsingke, China). Expressions of UBE2Q1-AS1, NOP53-AS1, and FAM225B were ascertained with U6 as the reference gene. Expressions of lncRNAs were calculated with the 2-△△Ct method, with primers displayed in Table 2.
RNA interference
Interfering targets were designed based on the UBE2Q1-AS1, and the relevant sequences are listed in Table 3. The single-stranded oligos were annealed to form double-stranded molecules, which were then ligated into the linearized pLVX-shRNA vector. Subsequently, the ligation products were transformed into competent Escherichia coli cells (strain DH5α, TSV-A07, Tsingke, China). After that, positive transformants were selected for sequencing. Monoclonal colonies with verified correct sequences from the sequencing results were picked, and high-purity plasmid extraction was carried out.
Transfection
The 786-O cells were seeded into two culture dishes at a density of 5 × 106 cells per dish, and then the cells were divided into the sh-NC group and the sh-UBE2Q1-AS1 group. For the preparation of the transfection system, 10 μL of shRNA plasmid (the sh-NC empty vector control plasmid and the sh-UBE2Q1-AS1 plasmid) were separately taken and dissolved in 490 μL of Opti-MEM™ medium (31985070, Gibco, USA), and then incubated at room temperature for 5 min. Additionally, 30 μL of the transfection reagent Max (24765-1, BioMedicine, China) was mixed with 470 μL of Opti-MEM™ medium, and the mixture was left standing at room temperature for 5 min. Subsequently, the above two solutions were combined, gently pipetted, and then left standing at room temperature for 15 min. After the transfection system was ready, it was added to the cells of the sh-NC group and the sh-UBE2Q1-AS1 group respectively, and then the cells were continuously cultured. Six hours after transfection, the medium was replaced with complete medium. Forty-eight hours after transfection, the cells were collected for subsequent analysis.
Cell proliferation assay
The treated cell suspension was seeded into 96-well plates at a density of 5 × 103 cells per well. Then, 10 μL of CCK-8 solution (C0038, Beyotime, China) was added to each well, and the plates were incubated for 1 h. Zero-adjustment wells (containing medium + CCK-8 solution, without cells) and control wells (containing untreated cells + CCK-8 solution) were set up. Finally, the absorbance was measured at 450 nm using a microplate reader. The calculation formula for cell viability (in percentage) is: Cell viability (100%) = [(experimental well—zero-adjustment well) / (control well—zero-adjustment well)] × 100%.
Transwell migration assay
The cell density was adjusted to 10 × 104 cells/mL. 100 μL of the cell suspension was added to each well of the Transwell chamber (3422, Corning, China), followed by a 24-h incubation. After the incubation, the Transwell chamber was removed, and the culture medium in the wells was discarded. The cells were then washed twice with PBS. Subsequently, the cells were fixed with 4% paraformaldehyde for 20 min. After washing with PBS, the cells were stained with 0.1% crystal violet (G1062, Solarbio, China) for 20 min. Then, the non-migrated cells on the upper layer of the chamber were gently wiped off with a wet cotton ball and washed three times with PBS. Finally, the cells were counted in randomly selected fields under a 100 × microscope, and the average value was calculated.
Statistical analysis
We employed R version 4.3.3 and associated packages for data processing and analysis. Inter-group comparisons of survival durations were executed via KM analysis and log-rank testing. The Wilcoxon test was used to assess disparities between variables across the two groups, with a two-sided p < 0.05 considered statistically significant.
Results
Selection of migrasome-associated prognostic lncRNAs
In this experimental phase, Pearson correlation analysis was conducted with stringent thresholds (|Pearson R|> 0.4 and p < 0.001) to identify 624 statistically significant lncRNAs in ccRCC samples. The relationship between these 624 migrasome-associated lncRNAs and 10 migrasome-associated genes is depicted in a Sankey diagram (Fig. 2A), with details provided in Supplementary Table S1. Univariate Cox analysis of single features yielded 182 prognostic-associated lncRNAs with significant single-factor associations (Supplementary Table S2). We proceeded to perform LASSO regression analysis on the significantly associated lncRNAs from the univariate analysis, resulting in LASSO coefficient distribution plots (Fig. 2B) and LASSO regression cross-validation plots (Fig. 2C). Ultimately, 23 lncRNAs with significant LASSO regression associations were retained. Subsequently, a multivariable Cox regression analysis identified 12 lncRNAs that impact prognosis, leading to the development of a risk scoring model. Score calculation was as follows: Risk Score = AC009318.3 × (0.814122439132301) + NOP53-AS1 × (−0.456561952324909) + UBE2Q1-AS1 × (0.367735041542032) + AC016026.1 × (0.858563305420942) + AC021037.1 × (−1.26984968493847) + AC095055.1 × (−0.575578993672608) + AC018809.2 × (−0.732108004856524) + AC068279.2 × (−0.569571735667287) + FAM225B × (1.25783614786488) + AP001107.3 × (0.735037159086312) + CD27-AS1 × (0.364103444097015) + MALINC1 × (−0.615075383989782). A correlation heatmap (Fig. 2D) illustrated the connections between migrasome-associated genes and these 12 lncRNAs. It could be clearly seen from the numerical annotations added in Fig. 2D that the four most relevant associations were as follows: PIGK was positively correlated with AC009318.3, with a correlation coefficient of 0.510; PKD1 was positively correlated with MALINC1, with a correlation coefficient of 0.471; PKD1 was positively correlated with AC018809.2, with a correlation coefficient of 0.470; ITGA5 was positively correlated with AP001107.3, with a correlation coefficient of 0.455. These results indicated that there was a relatively high correlation between these migrasome-associated genes and lncRNAs.

Selection of migrasome-associated prognostic lncRNAs in ccRCC. (A) Sankey diagram of migrasome-associated lncRNAs and migrasome-associated genes. (B) Plot of LASSO coefficients’ distribution for migrasome-associated lncRNAs. (C) LASSO regression cross-validation. (D) Correlation between migrasome-associated genes and 12 migrasome-associated lncRNAs.
Validation of migrasome-associated lncRNA prognostic model
With the help of a risk nomogram coupled with KM survival analysis, we appraised the prognostic capacity of migrasome-associated lncRNA model in forecasting disease outcomes. Hazard curves, survival condition diagrams, and thermal maps for the 12 migrasome-associated lncRNAs were evaluated across the two groups (Fig. 3A–I). Observations from the scatter plots of prognostic survival status in both groups revealed an increase in mortality coincident with an escalation in risk scores. Moreover, the progression-free and total survival durations were markedly shorter in patients at high risk vs those at low risk (both with p < 0.05, Fig. 3J–O).

Risk model’s prognostic significance in the training, testing, and overall dataset. (A–C) Hazard curves across two groups. (D–F) Survival condition diagrams across two groups. (G–I) Thermal maps of risk scores across two groups. (J–L) KM curves for total survival across two groups. (M–O) KM curves for progression-free survival across two groups.
Furthermore, we stratified ccRCC patients into subgroups in accordance with clinical features including age, sex, staging, G grading, T classification, M classification, and N classification to assess the model’s capability to differentiate across the two groups in terms of various clinical and pathological features (Fig. 4A–N). Survival analysis allowed us to conclude that, concerning age, gender, staging, G grading, T classification, M classification, and N0 status, the survival rates were markedly shorter in high-risk patients compared to those at low risk (all with p < 0.001). Although the difference in survival probabilities across these two groups in the N1 category was statistically non-significant (p = 0.077), a similar trend was observed. These findings suggested that this migrasome-associated lncRNA model holds considerable potential for forecasting the outcomes of patients suffering from diverse clinical and pathological characteristics.

KM curves across the two groups stratified by various clinical features. (A) Individuals aged > 65 years. (B) Individuals aged ≤ 65 years. (C) Male individuals. (D) Female individuals. (E) Individuals at Stage I-II. (F) Individuals at Stage III-IV. (G) Individuals with Grade G1-2. (H) Individuals with Grade G3-4. (I) Individuals with Tumor stage T1-2. (J) Individuals with Tumor stage T3-4. (K) Individuals with Metastasis M0. (L) Individuals with Metastasis M1. (M) Individuals with Node negative N0. (N) Individuals with Node positive N1.
Subsequently, Cox analyses were carried out to ascertain the prognostic significance independently of this model (Fig. 5A,B). These findings indicated that age, cancer grading, cancer staging, risk score served as standalone prognostic elements for ccRCC (all with p < 0.001). Additionally, we plotted ROC curves for this model, with corresponding AUCs of 0.757, 0.784, and 0.773 for 1, 3, and 5 years (Fig. 5C). The ROC curve findings revealed that the AUC of the risk score (0.773) surpassed that of other clinical and pathological parameters (Fig. 5D). Concurrently, C-index curve indicated that the risk model holds a certain degree of reference value in clinical settings (Fig. 5E). Taking into account the risk score and clinical elements, we created a nomogram to forecast the 1-, 3-, and 5- year total survival rates of patients (Fig. 5F). Analysis of calibration plots indicated a high concordance between forecasted and observed outcomes (Fig. 5G).

Stand-alone prognostic assessment and verification of the model. (A) Univariate Cox analysis forest plot considering risk scores and additional features. (B) Multivariate Cox analysis forest plot. (C) ROC plots for this model at 1, 3, and 5 years. (D) Comparison of ROC curves between the risk model and additional features. (E) C-index curves for the risk model and additional features. (F) Nomogram for predicting overall survival. (G) Calibration curve for overall survival.
PCA and functional analysis
Upon the application of principal component analysis, distinct clustering characteristics were observed across the two groups in spatial distribution, signifying the precision of the migrasome-associated lncRNA model in discerning patients across two risk strata (Fig. 6A–D). Outcomes from GO analysis demonstrated significant concentration of differential genes in antigen binding, collagen-containing extracellular matrix, and leukocyte mediated immunity (Fig. 7A,B). KEGG enrichment analysis indicated substantial concentration of differential genes including Rheumatoid arthritis, Viral protein interaction with cytokine and cytokine receptor, and IL-17 signaling pathway (Fig. 7C,D). GSEA demonstrated divergent primary pathways enriched across the two groups (Fig. 7E,F).

Principal component analysis across two groups. (A) Principal component analysis of all genes. (B) Principal component analysis of migrasome-associated genes. (C) Principal component analysis of migrasome-associated lncRNAs. (D) Principal component analysis of the 12 lncRNAs used in model building.

Functional evaluation of the risk model. (A, B) GO analysis. (C, D) KEGG analysis. (E, F) GSEA across two groups.
Immunofunction analysis
In this section, we delineated the composition of immunological cells in ccRCC patients within the TCGA dataset (Fig. 8A). Immune cell analysis of differences highlighted notable disparities in the abundance of T cells CD8, Dendritic cells resting, T cells follicular helper, Macrophages M1, Macrophages M0, T cells regulatory (Tregs), Resting CD4+ memory T cells, Mast cells resting, and Mast cells activated across two risk groups (all with p < 0.05, Fig. 8B). Functional immune difference analysis indicated significant variations in various immune functions across these two groups (all with p < 0.05). Notably, scores for Type II IFN Response and iDCs were markedly reduced in the high-risk group (Fig. 8C). Furthermore, high-risk patients demonstrated elevated immune scores and ESTIMATE scores as compared to low-risk patients (both with p < 0.001), while the stromal scores between the two risk groups were not statistically significant (Fig. 8D).

Variations in tumor microenvironment across two groups. (A) Relative levels of immunological cells in ccRCC patients. (B) Differential analysis of immunological cells across two groups. (C) Differential analysis of immunological response capabilities across two groups. (D) TME scores across two groups.
TMB and TIDE analyses
We conducted an assessment of the somatic mutational landscape of ccRCC patients within the TCGA cohort. Cascade plots revealed that the top commonly mutated genes in both groups were VHL, PBRM1, TTN, SETD2, and BAP1 (Fig. 9A,B). Moreover, discrepancy analysis of TMB demonstrated no significant disparity between these two groups (Fig. 9C). Individuals diagnosed with ccRCC were stratified into high and low TMB groups according to their TMB. Subsequent survival analysis of the two groups indicated that the survival probabilities of high TMB group were markedly reduced vs low TMB group (p < 0.001, Fig. 9D). Joint survival analysis incorporating TMB and patient risk scores showed statistically meaningful variations in survival rates among those groups (p < 0.001, Fig. 9E). Lastly, we calculated the TIDE scores for both risk groups, with results indicating that high-risk patients exhibited increased TIDE values vs low-risk patients, suggesting a poorer response to immunotherapy (p < 0.001, Fig. 9F).

Correlation of model scores with TMB and TIDE. (A, B) Somatic mutation landscapes across two groups. (C) TMB across two groups. (D) Survival analysis of TMB. (E) Integrative survival assessment of TMB and risk scores. (F) TIDE scores across two groups.
Sensitive drug screening
By utilizing the “oncoPredict” software package, we identified 65 drugs with significantly varying sensitivities across two groups. Within this cohort, 19 drugs demonstrated heightened sensitivity in low-risk patients (Supplementary Fig. S1), whereas 46 drugs displayed greater sensitivity in high-risk patients (Supplementary Fig. S2). Notably, sorafenib was more sensitive in high-risk patients (p < 0.001, Fig. 10A), and cediranib was more sensitive in low-risk patients (p < 0.001, Fig. 10B).

Sensitive drugs. (A) Increased sensitivity to sorafenib in high-risk group. (B) Increased sensitivity to cediranib in low-risk group.
Model external verification
External validation of the risk model was conducted via NOP53-AS1, FAM225B and UBE2Q1-AS1 from the TCGA dataset. These findings indicated that NOP53-AS1, FAM225B and UBE2Q1-AS1 were more present in tumor tissues vs neighboring tissues (all with p < 0.001, Fig. 11A–C). Subsequent survival analysis indicated that higher expression of NOP53-AS1 was linked to significantly higher survival rates vs lower expression group (p < 0.01, Fig. 11D). And high expressions of FAM225B and UBE2Q1-AS1 were associated with significantly lower survival rates (both with p < 0.001, Fig. 11E,F), suggesting that elevated expression of these two lncRNAs is correlated with adverse prognosis. Additionally, ROC curves were plotted, yielding AUCs of 0.654 for NOP53-AS1, 0.784 for FAM225B and 0.827 for UBE2Q1-AS1 (Fig. 11G–I).

External verification of the model. (A–C) Levels of NOP53-AS1, FAM225B and UBE2Q1-AS1 in tumor and neighboring tissues. (D–F) Survival analysis of NOP53-AS1, FAM225B and UBE2Q1-AS1. (G–I) ROC curves for NOP53-AS1, FAM225B and UBE2Q1-AS1.
Verification of migrasome-associated lncRNAs expression levels
Our qPCR findings revealed that, as compared to normal renal cells, expressions of NOP53-AS1, FAM225B, and UBE2Q1-AS1 were markedly elevated in ccRCC cells (all with p < 0.01, Fig. 12A–C).

Differential expression of lncRNAs in normal renal cells and ccRCC cells. (A) NOP53-AS1 expression levels. (B) FAM225B expression levels. (C) UBE2Q1-AS1 expression levels (Inter-group comparison, **p < 0.01).
Interfering with the expression of UBE2Q1-AS1 significantly inhibits the proliferation and migration of ccRCC cells
To further validate the function of lncRNAs in the progression of ccRCC, we selected UBE2Q1-AS1, which had the highest AUC, for experimental verification. Firstly, we constructed the interference plasmid of UBE2Q1-AS1 and transfected it and the NC plasmid into 786-O cells respectively. The results of qPCR detection showed that, compared with the sh-NC group, the expression level of UBE2Q1-AS1 mRNA in the sh-UBE2Q1-AS1 group was significantly decreased (p < 0.01, Fig. 13A), indicating that the interference of UBE2Q1-AS1 lncRNA was successfully achieved in this cell model. Subsequently, we further employed the CCK-8 assay and Transwell migration assay to evaluate the effects of the interference of UBE2Q1-AS1 on the proliferation and migration abilities of ccRCC cells. The results showed that, compared with the sh-NC group, both the cell viability and the number of migrated cells in the sh-UBE2Q1-AS1 group were significantly decreased (all with p < 0.01, Fig. 13B,C). This suggests that the interference of UBE2Q1-AS1 can significantly inhibit the proliferation and migration of ccRCC cells.

Effects of UBE2Q1-AS1 on the proliferation and migration of ccRCC cells. (A) Detection of UBE2Q1-AS1 mRNA expression by qPCR. (B) Effects on cell proliferation. (C) Effects on cell migration (Inter-group comparison, **p < 0.01).
Discussion
ccRCC, a prevalent variant of renal cancer, is frequently detected in its later stages, where patients face a grim prognosis and the likelihood of distant metastases31. The intractability of malignant tumors stems not only from their swift proliferation but also their high susceptibility to metastasis, which is intricately connected to the migratory propensity of tumor cells. Migrasomes, a recently identified type of membranous organelle discovered in 2015, are vesicular structures that emerge at the termini of contractile filaments within migrating cells5. Subsequent investigations have demonstrated that migrasomes are ubiquitous in various somatic and oncogenic cells, beyond their presence in in vitro cultured cells. Migrasomes are replete with signaling and bioactive molecules, facilitating the transference of mRNA and other substances between disparate cells through endocytosis, thereby begetting a remarkable impact across a spectrum of physiological and pathological processes32. Research has established that migrasomes derived from mesenchymal stem cells (MSCs) are capable of directing the chemotaxis of primary CD34+ hematopoietic progenitor cells or KG-1a leukemia cells, suggesting an essential function in the regulation of immunity33. Following podocyte injury, a surge in migrasome production occurs during migration, with an increase in the quantity of podocyte-derived migrasomes in urine observed during renal impairment. This indicated the latent capacity of migrasomes as a non-disruptive biomarker for the diagnosis or prognosis of urinary system diseases, serving as an indicator of podocyte damage34.
LncRNAs are integral to the modulation of genetic expression, cellular proliferation and migration, and modulation of immune responses, and they significantly contribute to oncogenesis and tumor progression35. Zhang et al. documented that lncRNA NEAT1 is upregulated in gastric cancer cells, and its suppression can curtail gastric neoplastic cell expansion and migration by regulating the miR-495-3p/Sp1 axis36. Zhong et al. identified that lncRNA ARAP1-AS1 fosters the aggressive progression of ccRCC cells via the miR-361-3p/PGF37. Furthermore, Qu et al. established a ccRCC prognostic model derived from lipid metabolism-related lncRNAs, which displayed robust predictive capabilities38. Nevertheless, no studies have yet harnessed migrasome-associated lncRNAs to create a prognostic model for ccRCC patients.
The research employed bioinformatics to evaluate the prognostic potential of migrasome-associated lncRNAs in the progression of ccRCC. Initially, 624 migrasome-related lncRNAs were identified within ccRCC samples. Subsequently, univariate Cox and LASSO regression analyses pinpointed 23 lncRNAs with differential prognostic relevance. Ultimately, a multivariable Cox regression analysis delineated 12 signature lncRNAs, encompassing AC009318.3, NOP53-AS1, UBE2Q1-AS1, AC016026.1, AC021037.1, AC095055.1, AC018809.2, AC068279.2, FAM225B, AP001107.3, CD27-AS1, and MALINC1, to construct a prognostic prediction model. The heatmap analysis showed that PIGK was positively correlated with AC009318.3, PKD1 was positively correlated with MALINC1 and AC018809.2 respectively, and ITGA5 was positively correlated with AP001107.3. Previous studies have indicated that PKD plays a role in the process of tumor progression by regulating the connections between renal cancer cells and epithelial cells39. Che et al.40 found that in ccRCC patients, high expression of ITGA5 would lead to an unfavorable prognosis. This implies that MALINC1, AC018809.2, and AP001107.3 may participate in the development of ccRCC by regulating the expression or function of migrasome-associated genes PKD1 and ITGA5, and ultimately have an impact on the prognosis of patients. Patients were stratified into high and low-risk groups in accordance with the predictive scoring model, revealing superior survival rates in the low-risk group. Multivariable Cox analysis confirmed the risk score as a freestanding prognostic element. The ROC analysis and nomogram results demonstrated that the model’s AUC surpassed other clinical factors, with its integration of patient clinical characteristics accurately forecasting 1-, 3-, and 5-year survival probabilities. Furthermore, PCA demonstrated the model’s efficacy in distinguishing between the two risk groups, validating its predictive accuracy for ccRCC patient prognosis.
Enrichment analysis revealed important associations between migrasome-related lncRNAs and immune response pathways. Our study further investigated the correlation between prognostic model and immune infiltration. These findings indicated a substantial correlation between risk scores and CD8+ T cells, as well as Tregs. Known for their cytotoxic effects, CD8+ T cells efficiently identify and eliminate tumor cells41. However, an increase in CD8+ T cells in the high-risk group was observed, aligning with the findings of Wang et al19. Treg cell accumulation in the tumor microenvironment is typically linked to immune suppression and poor prognosis42. Consistent with previous reports, a radical increase in Treg cells was noted in the high-risk group. Additionally, differences in macrophage M0 and M1 subtypes between risk groups were observed. Macrophages have a complex impact on the neoplasmic microenvironment, with M1 macrophages generally representing anti-tumor activity and M0 levels being inversely correlated with tumor prognosis43,44,45. In line with previous reports, our study results highlighted the association of high-risk scores with low expressions of M1 macrophages and high expressions of M0 macrophages. Notably, the high-risk group showed greatly reduced scores for iDCs and Type II IFN responses. iDCs are crucial in antigen presentation and initiating immune responses46, while Type II IFN is related to antiviral immunity and tumor immune surveillance47. The reduction in these functions might be associated with the worse prognosis of individuals in the high-risk group. The study also conducted an analysis of tumor immunity scores. Individuals with high-risk values had elevated immune and ESTIMATE scores, suggesting lower tumor purity and potentially higher immune suppression in the tumor microenvironment of these patients. Furthermore, we applied the TIDE algorithm to estimate tumor immune responses between different risk groups, with results showing higher TIDE scores in high-risk vs low-risk group. This indicated a stronger immune escape capability in high-risk patients, enabling them to escape immune surveillance and attack, thus potentially responding poorly to immunotherapy. We also explored variations in sensitivity to anti-cancer drugs across these two risk groups. The research indicated that sorafenib was more effective in high-risk patients, whereas cediranib showed greater sensitivity in low-risk patients. Sorafenib, a common treatment for ccRCC, efficiently prolongs progression-free survival in advanced patients who have failed previous treatments48. Additionally, cediranib has shown good anti-tumor activity in untreated metastatic RCC and its efficacy is comparable to some approved drugs49. However, there is limited research on the use of cediranib for the treatment of ccRCC. These findings would provide more effective treatment options for clinical care. LncRNAs play complex roles in neoplasms and can be differently influenced by the tumor subtype, microenvironment, and disease stage. In this study, high expressions of FAM225B and UBE2Q1-AS1 were linked to unfavorable prognosis, while high level of NOP53-AS1 was correlated with good prognosis. Among these three lncRNAs, UBE2Q1-AS1 had the highest AUC, which means that UBE2Q1-AS1, as a biomarker, has relatively high accuracy and reliability for forecasting the prognostication of ccRCC. Notably, this study confirmed through external validation cohorts and cell functional assays that UBE2Q1-AS1 is highly expressed in ccRCC tissues, and its interferenece significantly inhibits the proliferation and migration of ccRCC cells, suggesting its potential as a therapeutic target.
Although the migrasome-associated lncRNA model constructed in this study demonstrates promising prognostic predictive efficacy, its clinical translation faces multiple challenges. First, in terms of technical feasibility, the model involves the expression detection of 12 lncRNAs, which relies on high-throughput sequencing or qPCR technology and imposes rigorous requirements on the testing equipment and standardized operating procedures of clinical laboratories. Second, cost-effectiveness is another critical issue: integrating bioinformatics analysis platforms for sample processing, data calculation, and risk score generation may increase diagnostic costs, particularly posing difficulties for promotion in resource-constrained medical settings. Additionally, establishing how to effectively integrate risk scores with existing clinical pathological indicators to form standardized diagnostic and treatment protocols requires multi-center collaboration to develop industry standards.
The retrospective analysis of this study based on the TCGA dataset has limitations. It has only been validated within the internal cohort and lacks external validation. Future research should conduct external validation through the GEO dataset or prospective clinical samples to further evaluate the generalization ability of the model and its clinical application value. Moreover, the precise mechanisms by which the lncRNAs in our model contribute to the genesis and progression of ccRCC necessitate further investigation.
Conclusion
To sum up, our study created a ccRCC prognostic model on account of 12 migrasome-associated lncRNAs, with its risk score serving as a freestanding prognostic element. This model correlates with survival rates in ccRCC patients, in with the high-risk group faces poorer prognoses. Furthermore, we explored our model’s utility in the immune infiltration and tumor microenvironment of ccRCC, offering novel insights to guide immunotherapeutic strategies.
Data availability
The datasets analyzed during the current study are available in The Cancer Genome Atlas (https:// portal. gdc. cancer.gov/ repository). All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Ran, L. et al. miRNAs in prediction of prognosis in clear cell renal cell carcinoma. Biomed. Res. Int. 2017, 4832931. https://doi.org/10.1155/2017/4832931 (2017).
Makhov, P. et al. Resistance to systemic therapies in clear cell renal cell carcinoma: mechanisms and management strategies. Mol. Cancer Ther. 17, 1355–1364. https://doi.org/10.1158/1535-7163.Mct-17-1299 (2018).
Yu, S. & Yu, L. Migrasome biogenesis and functions. FEBS J. 289, 7246–7254. https://doi.org/10.1111/febs.16183 (2022).
Dharan, R. & Sorkin, R. Biophysical aspects of migrasome organelle formation and their diverse cellular functions. BioEssays News Rev. Mol. Cell. Dev. Biol. 46, e2400051. https://doi.org/10.1002/bies.202400051 (2024).
Ma, L. et al. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 25, 24–38. https://doi.org/10.1038/cr.2014.135 (2015).
Zhang, K. et al. CD151-enriched migrasomes mediate hepatocellular carcinoma invasion by conditioning cancer cells and promoting angiogenesis. J. Exp. Clin. Cancer Res. CR 43, 160. https://doi.org/10.1186/s13046-024-03082-z (2024).
Cheng, Y. et al. Nanoparticulates reduce tumor cell migration through affinity interactions with extracellular migrasomes and retraction fibers. Nanoscale Horiz. 7, 779–789. https://doi.org/10.1039/d2nh00067a (2022).
Fu, Z. B. et al. Exosomes in diagnosis and treatment of renal cell carcinoma: recent progress. J. Clin. Urol. 35, 78–82. https://doi.org/10.13201/j.issn.1001-1420.2020.01.019 (2020).
Kim, K. et al. Determinants of renal cell carcinoma invasion and metastatic competence. Nat. Commun. 12, 5760. https://doi.org/10.1038/s41467-021-25918-4 (2021).
Cheng, Q. et al. Effect of Bmi-1 on invasion and metastasis of renal cancer cells and its related mechanism. Chin. Pharm. Bull. 36, 514–519. https://doi.org/10.3969/j.issn.1001-1978.2020.04.013 (2020).
Yan, G. et al. Effect of Trim21 on the proliferation, migration and invasion of renal carcinoma cells and its mechanisms. J. Xuzhou Med. Univ. 41, 703–709. https://doi.org/10.3969/j.issn.2096-3882.2021.10.001 (2021).
Liu, X. F. et al. The relationship between the expression of cell adhesion molecules and the invasion and metastasis of renal cell carcinoma. Lab. Med. 25, 67–70 (2010).
Puvvula, P. K. LncRNAs regulatory networks in cellular senescence. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20112615 (2019).
Lei, K. Y. et al. Expression of long non-coding RNA FER1L4 in renal cell carcinoma tissues and its effect on the proliferation, invasion, and migration of renal cell carcinoma cells. Chin. J. Clin. Oncol. 48, 882–886. https://doi.org/10.12354/j.issn.1000-8179.2021.20210842 (2021).
Xu, X. D. et al. The role of long non-coding RNAs in the development of renal cancer and prospect of clinical application. Progress Pharm. Sci. 45, 941–947 (2021).
Zhang, F. et al. Migrasome, a migration-dependent organelle. Front. Cell Dev. Biol. 12, 1417242. https://doi.org/10.3389/fcell.2024.1417242 (2024).
Meng, L. D. et al. SNHG17 activates CENPE by binding to transcription factor E2F1 to promote the proliferation, migration and invasion of clear cell renal cell carcinoma cells. J. Mod. Oncol. 32, 205–213. https://doi.org/10.3969/j.issn.1672-4992.2024.02.002 (2024).
Wang, G. et al. Novel long noncoding RNA OTUD6B-AS1 indicates poor prognosis and inhibits clear cell renal cell carcinoma proliferation via the Wnt/β-catenin signaling pathway. Mol. Cancer 18, 15. https://doi.org/10.1186/s12943-019-0942-1 (2019).
Wang, N. et al. Development of a novel disulfidptosis-related lncRNA signature for prognostic and immune response prediction in clear cell renal cell carcinoma. Sci. Rep. 14, 624. https://doi.org/10.1038/s41598-024-51197-2 (2024).
Wang, Y. N. et al. Long non-coding RNAs: A double-edged sword in aging kidney and renal disease. Chem. Biol. Interact. 337, 109396. https://doi.org/10.1016/j.cbi.2021.109396 (2021).
Zhao, X. et al. Identification of markers for migrasome detection. Cell Discov. 5, 27. https://doi.org/10.1038/s41421-019-0093-y (2019).
Qin, Y. et al. Pan-cancer analysis identifies migrasome-related genes as a potential immunotherapeutic target: A bulk omics research and single cell sequencing validation. Front. Immunol. 13, 994828. https://doi.org/10.3389/fimmu.2022.994828 (2022).
Zheng, Y., Lang, Y., Qi, B. & Li, T. TSPAN4 and migrasomes in atherosclerosis regression correlated to myocardial infarction and pan-cancer progression. Cell Adh. Migr. 17, 14–19. https://doi.org/10.1080/19336918.2022.2155337 (2023).
Guo, Z. et al. A novel disulfidptosis-related lncRNAs signature for predicting survival and immune response in hepatocellular carcinoma. Aging 16, 267–284. https://doi.org/10.18632/aging.205367 (2024).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53, D672–D677. https://doi.org/10.1093/nar/gkae909 (2025).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. https://doi.org/10.1038/nmeth.3337 (2015).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612. https://doi.org/10.1038/ncomms3612 (2013).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558. https://doi.org/10.1038/s41591-018-0136-1 (2018).
Maeser, D. et al. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. https://doi.org/10.1093/bib/bbab260 (2021).
Simonaggio, A. et al. Tumor microenvironment features as predictive biomarkers of response to immune checkpoint inhibitors (ICI) in metastatic clear cell renal cell carcinoma (mccRCC). Cancers https://doi.org/10.3390/cancers13020231 (2021).
Jiao, H. & Yu, L. Migrasomes: Biogenesis, physiological roles, and therapeutic potentials. J. Cell Biol. https://doi.org/10.1083/jcb.202403051 (2024).
Deniz, I. A. et al. Mesenchymal stromal cell-associated migrasomes: a new source of chemoattractant for cells of hematopoietic origin. Cell Commun. Signal 21, 36. https://doi.org/10.1186/s12964-022-01028-6 (2023).
Liu, Y. et al. Podocyte-released migrasomes in urine serve as an indicator for early podocyte injury. Kidney Dis. (Basel Switzerland) 6, 422–433. https://doi.org/10.1159/000511504 (2020).
Xiao, Y. et al. Long non-coding RNA-encoded micropeptides: functions, mechanisms and implications. Cell Death Discov. 10, 450. https://doi.org/10.1038/s41420-024-02175-0 (2024).
Zhang, X. R. et al. LncRNA NEAT1 modulates the effects of miR-495-3p/Sp1 axis on proliferation, migration and invasion of gastric cancer cells. Chin. J. Gerontol. 44, 3976–3981. https://doi.org/10.3969/j.issn.1005-9202.2024.16.032 (2024).
Zhong, L. & Zhong, X. Long non-coding RNA ARAP1-AS1 contributes to cell proliferation and migration in clear cell renal cell carcinoma via the miR-361-3p/placental growth factor axis. Bioengineered 12, 6629–6642. https://doi.org/10.1080/21655979.2021.1975019 (2021).
Qu, G. et al. Prognostic prediction of clear cell renal cell carcinoma based on lipid metabolism-related lncRNA risk coefficient model. Front. Genet. 13, 1040421. https://doi.org/10.3389/fgene.2022.1040421 (2022).
Brenner, W. et al. Adhesion of renal carcinoma cells to endothelial cells depends on PKCmu. BMC Cancer 10, 183. https://doi.org/10.1186/1471-2407-10-183 (2010).
Che, X. et al. Systematic multiomics analysis and in vitro experiments suggest that ITGA5 could serve as a promising therapeutic target for ccRCC. Cancer Cell Int. 24, 363. https://doi.org/10.1186/s12935-024-03546-4 (2024).
Zhou, Z. et al. Optimizing CD8(+) T cell-based immunotherapy via metabolic interventions: a comprehensive review of intrinsic and extrinsic modulators. Exp. Hematol. Oncol. 13, 103. https://doi.org/10.1186/s40164-024-00575-7 (2024).
Bozward, A. G. et al. Natural killer cells and regulatory T cells cross talk in hepatocellular carcinoma: exploring therapeutic options for the next decade. Front. Immunol. 12, 643310. https://doi.org/10.3389/fimmu.2021.643310 (2021).
Huang, L. et al. EFEMP2 indicates assembly of M0 macrophage and more malignant phenotypes of glioma. Aging 12, 8397–8412. https://doi.org/10.18632/aging.103147 (2020).
Li, W. et al. High expression of activated CD4(+) memory T cells and CD8(+) T cells and low expression of M0 macrophage are associated with better clinical prognosis in bladder cancer patients. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi Chin. J. Cell. Mol. Immunol. 36, 97–103 (2020).
Cao, J. & Liu, C. Mechanistic studies of tumor-associated macrophage immunotherapy. Front. Immunol. 15, 1476565. https://doi.org/10.3389/fimmu.2024.1476565 (2024).
Koumantou, D. et al. Specific requirement of the p84/p110γ complex of PI3Kγ for antibody-activated, inducible cross-presentation in murine type 2 DCs. Adv. Sci. (Weinheim Baden-Wurttemberg Germany) https://doi.org/10.1002/advs.202401179 (2024).
Chattopadhyay, A. et al. IFN-γ lowers tumor growth by increasing glycolysis and lactate production in a nitric oxide-dependent manner: implications for cancer immunotherapy. Front. Immunol. 14, 1282653. https://doi.org/10.3389/fimmu.2023.1282653 (2023).
Escudier, B. et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 356, 125–134. https://doi.org/10.1056/NEJMoa060655 (2007).
Sridhar, S. S. et al. A phase II study of cediranib (AZD 2171) in treatment naive patients with progressive unresectable recurrent or metastatic renal cell carcinoma. A trial of the PMH phase 2 consortium. Investig. New Drugs 31, 1008–1015. https://doi.org/10.1007/s10637-013-9931-1 (2013).
Acknowledgements
We are grateful to the personnel who have contributed to the public databases used in this study, and also to the medical writers, proof-readers and editors for their assistance to us.
Funding
This work was supported by the Science and Technology Innovation Development Project of Tai’an City, Shandong Province, China (Policy Guidance category) (No. 2022NS202).
Author information
Authors and Affiliations
Contributions
All authors participated in the conception and design of the study. L.Z. wrote the first draft of the paper. L.D. and J.L. collected and analyzed the data. T.T. and H.Z. provided suggestions for revising the previous versions of the manuscript. W.Z. guided the progress of this study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, L., Dong, L., Tian, T. et al. A novel migrasome-associated lncRNA model for clear cell renal cell carcinoma prognosis and immune response prediction. Sci Rep 16, 1895 (2026). https://doi.org/10.1038/s41598-025-31455-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-31455-7
