
Abstract
Aripiprazole (ARP), an atypical antipsychotic, suffers from poor aqueous solubility and extensive hepatic metabolism, resulting in limited brain bioavailability. This study aimed to develop and optimize nanostructured lipid carriers (NLCs) for intranasal delivery of ARP to enhance nose-to-brain transport and improve therapeutic outcomes for schizophrenia. ARP-loaded NLCs were formulated using a high-shear homogenization followed by ultrasonication. A 3² factorial design was employed to optimize the formulation variables. The prepared NLCs were characterized for particle size, polydispersity index, zeta potential, drug loading, and entrapment efficiency. Further evaluations included SEM, in-vitro drug release, ex-vivo permeation, hemolytic activity, nasal ciliotoxicity, and stability studies. The optimized formulation (A4), consisting of 2.5% stearic acid and oleic acid (solid lipid phase), Tween 80 (surfactant), and PEG 400 (co-surfactant), demonstrated favorable physicochemical properties: particle size of 99.6 nm, zeta potential of − 36.7 mV, PDI of 0.249, drug loading of 20.96%, and entrapment efficiency of 96.23%. The in-vitro release showed a biphasic pattern over 72 h, fitting the Fickian diffusion model. Ex-vivo studies confirmed enhanced drug permeation across nasal mucosa. Toxicity studies indicated excellent biocompatibility. Stability studies revealed that the formulation was more stable at 5 ± 0.3 °C compared to higher storage temperatures. The results indicated that the optimized intranasal ARP-NLCs developed in this study offer a novel and promising approach for improved brain targeting in schizophrenia, potentially enhancing therapeutic efficacy and patient adherence by enabling sustained drug release and avoiding first-pass metabolism.
Similar content being viewed by others
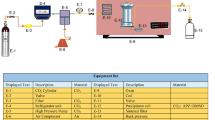


Introduction
Schizophrenia is a type of neuropsychiatric disease that causes severe effects on the patient’s life, as it poses a high risk of suicide and life-threatening behaviors1,2. After discontinuing medications, about 80% of patients experienced severe relapses within two years, resulting in negative, cognitive, aggressive, hostile, anxious, and depressive symptoms. Although all the biological mechanisms of the disease are not well-known, developing evidence shows that dopamine receptors play a key role in the prominence of the above symptoms3. The blood-brain barrier (BBB) limits the entry of drugs into the brain, resulting in an ineffective drug concentration reaching the brain/central nervous system (CNS)4,5. Various drugs have been developed to treat schizophrenia with the goals of curing the illness’s symptoms, lowering the frequency of relapses and their severity, and enhancing social interactions6.
Nose-to-brain drug administration has emerged as a potential non-invasive technique to cross the BBB and prevent first-pass metabolism, thereby improving brain targeting7,8,9. This might be accomplished by avoiding first-pass metabolism. In order to optimize the delivery of drugs to the CNS, this route makes use of the high permeability of the nasal mucosa as well as the direct transport channels provided by the olfactory and trigeminal neurons10,11. Among the many nanocarriers that have been investigated for this purpose, nanostructured lipid carriers (NLCs) provide a number of specific benefits, such as a high drug loading capacity, regulated release profiles, and increased stability12,13,14,15,16. Previous studies have demonstrated that NLCs are effective in transporting various antipsychotics such as clozapine and amisulpride17,18,19,20, as well as treatments for Parkinson’s disease21 and glioblastoma22. These results provide additional evidence for the potential that NLCs have for nose-to-brain delivery.
Aripiprazole (ARP), is an oral, second-generation antipsychotics agent used for the treatment of schizophrenia. It is a BCS class II drug (low solubility and high permeability), undergoes extensive first-phase metabolism and P-glycoprotein efflux, leading to a decrease in its in vivo efficacy and an increase in its dose-related side effects23,24. However, there has been little investigation into their effectiveness for ARP for the treatment of schizophrenia through nose-to-brain targeting. So, our main objective of the research was to enhance the therapeutic efficacy of ARP by nose-to-brain drug delivery by developing optimized NLCs that ensure the maximum drug absorption with effective targeted delivery and safe retention in the olfactory region.
Since nanostructured lipid carriers have been previously explored for nose-to-brain delivery of other antipsychotic drugs such as risperidone, and, olanzapine the application of these systems for ARP has not been adequately explored25,26. The novelty of this work lies in the systematic development and optimization of ARP-NLCs using a factorial design approach specifically tailored to overcome the dual challenges of ARP’s poor solubility and limited brain bioavailability. This study provides a comprehensive evaluation, including robust statistical optimization, detailed characterization, and ex vivo permeation studies, to establish a promising nose-to-brain delivery system for aripiprazole with enhanced therapeutic efficacy.
Materials and methods
Pure Aripiprazole was provided by Hetero Labs Limited (Andhra Pradesh, India). Stearic and Oleic acid, Castor oil, Olive oil, Span 20, Span 80, Tween 20, PEG 200, PEG 4000, PEG 6000, PEG 8000, Carnauba wax, Bees wax, Palmitic acid, Glyceryl monostearate, Castor wax, Sodium lauryl sulphate, Iso propyl alcohol, EDTA and Benzalkonium chloride (BKC) was obtained from Rajesh Chemicals, Mumbai, (India). Tween 80 and Polyethylene glycol (PEG-400) were procured from SD Fine Chemicals (Mumbai, India). Methanol, Ethanol, Acetonitrile, and Chloroform was obtained from Rajesh Chemicals Mumbai, (India). Capmul MCM, Labrafil M2125, Captex 200, Labrafac PG, Capryol 90, and Labrasol are obtained by Gattefosse. The instruments were used, UV spectrophotometer (UV 1800, Shimadzu Co., Japan), DSC apparatus (Model: Q200, Shimadzu Co, Japan), Scanning Electron Microscopy (SEM-EVO LS 15, Zeiss, Germany), Fourier-Transform Infrared Spectroscopy (FT-IR-8400-S, Shimadzu, Japan), Viscometer (Brookfield DV-E, USA) and Malvern zeta sizer (DTS Ver. 5.10, Sr. No. MAL1031371, Malvern Instruments Ltd., UK).
Solubility of drug in solid lipid and liquid lipid
Solid lipid solubility
The solid lipid solubility study was performed by taking a fixed amount of drug (5 mg) in a test tube, in which the solid lipid (stearic acid, palmitic acid, glycerol monostearate, carnauba wax, bees wax, and castor wax) was added with an increment of 100 mg, followed by the heating of the test tube at a temperature of 10 °C above the actual melting temperature of the solid lipid in a water bath with controlled temperature. The same process was repeated until a clear solution in the melted lipid was observed with the naked eye under normal light. This method was used to estimate the right lipid for drug solubility and NLC preparation27,28.
Liquid lipid solubility
To determine the drug solubility in liquid lipid (oleic Acid, capmul MCM, labrafil M 2125, olive oil, castor oil, captex 200, and labrafac PG), a fixed volume of liquid lipid (1 ml) was taken in a small Eppendorf (2 ml) tube, followed by the addition of drug with an increment of 2 mg to the individual liquid lipid and surfactant. Then solubilization was performed by using a vortex mixer. The same thing was repeated until saturation. After 72 h, centrifugation of each sample was performed at 5000 rpm for 10 min, from which a clear supernatant layer was obtained. The supernatant phase was separated, diluted with phosphate buffer (PB), pH 6.4 [which represents the pH of nasal mucosa], and analyzed by UV spectrophotometer (UV 1800, Shimadzu Co., Japan) at 216 nm using quartz cuvettes. The undissolved drug concentration was determined and subtracted from the total concentration of drug added to determine the amount of drug dissolved29,30.
Screening for surfactant and co-surfactant
Surfactants are chosen based on their ability to self-emulsify with the lipids and the drug. In order to choose the most suitable surfactant, their capacity to self-emulsify was tested. The solubility experiments determined that stearic acid and oleic acid were the most suitable lipid phases. Selected lipid phase, surfactant and co-surfactant (Span 80, Tween 80, Tween 20, Span 20, capryol 90, PEG 6000, PEG 4000, PEG 8000, labrasol, PEG 400, PEG 200, and propylene glycol) were mixed in 1:1 and 1:2 ratios and heated at 75 °C and vortexed to form homogenous mixture. From the resultant homogenous mixture, 50 mg was accurately weighed and dispersed in 50 ml of Millipore water with gentle stirring to yield a fine emulsion.
The self-emulsification of surfactants was evaluated visually using a grading system for eight hours in terms of dispersibility, ease of emulsification, physical appearance, and optical clarity (Table 1). Resulting systems were also evaluated for turbidity by using the turbidimetric method31.
Fourier-transform infrared (FT-IR) spectroscopy
The FT-IR spectra of aripiprazole pure drug, individual excipients, and their physical mixtures were recorded using an FT-IR spectrophotometer (FT-IR-8400-S, Shimadzu, Japan) by the KBr pellet method over a range of 4000 –400 cm⁻¹ to assess potential incompatibilities between them.
Crystalline behavior of drug and lipids by differential scanning calorimetry (DSC)
The thermal behavior of aripiprazole and the optimized formulation of NLCs was analyzed using a DSC (DSC Q200, Shimadzu Co, Japan). The samples were sealed in aluminum pans and heated from 25 °C to 200 °C at a constant rate of 10 °C/min under a nitrogen purge, it gives the information regarding the physical properties, like the crystalline or amorphous nature of the samples32.
Optimization of formulations33,34,35,36,37
Effect of liquid lipid concentration
Three different batches of NLCs were prepared with varying liquid lipid phase concentrations (1%, 2%, and 3%) to study the effect of the lipid phase on particle size34,35,36. The ratio of surfactant to co-surfactant was maintained at 2:1 by keeping the other parameters constant. Briefly, the lipid phase and surfactant mixture were weighed into glass vials, mixed by stirring on a Vortex mixer, and heated at 70 °C to get a homogeneous mixture. The particle size of each batch was assessed visually (Table 2) and determined to be delicately milky.
Effect of the amount of solid lipid on ease of emulsification
Three different batches of NLCs were prepared, varying the ratio of solid lipid used for the lipid phase, i.e., liquid lipid: solid lipid. 1:1, 1:2, and 1:3. Briefly, 500 mg of each formulation was added dropwise to 500 ml of purified water. Gentle agitation was provided by stirring with a magnetic stirrer, and the formulations were assessed visually based on the following aspects: optically clear solution or turbid solution, observance of lumps, or no presence of particles (as given in Table 2) and found to be finely milky.
Effect of homogenization speed
Selecting the best concentration of solid lipid and liquid lipid by the above screening method, four different batches of NLCs were prepared by varying the homogenization speed corresponding to 10,000, 15,000, 20,000, and 25,000 rpm and by keeping the other parameters constant; the particle size of each batch was determined visually and using a zeta sizer (Malvern, Worcestershire, UK) and found to be fine and milky.
Effect of homogenization time
The best particle size formulation was selected from the above test by keeping the lipid phase, aqueous phase concentration, and homogenization speed constant. Five different batches of NLCs were prepared by varying the homogenization time to correspond to 5, 10, 15, 20, and 25 min; the particle size of each batch was determined visually and using a zeta sizer (Malvern, Worcestershire, UK) and found to be fine and milky.
Effect of sonication amplitude
Ascertained the best particle-sized formulation from the above screening by keeping all other parameters constant. Again, four different batches of NLCs were prepared, varying the sonication amplitudes corresponding to 300, 375, 450, and 525-watt sonication amplitudes, using a probe sonicator (Vibra Cell Sonic and Materials, Newtown, CT, USA), and the particle size of each batch was determined visually and using zeta sizer (Malvern Worcestershire, UK) and found to be fine milky.
Effect of sonication time
Selected the best particle-sized formulation from the above screening by keeping all the above parameters constant. The four different batches of NLCs were prepared, corresponding to 5, 10, 15, and 20 min of sonication time, and the particle size of each batch was determined visually, and using a zeta sizer (Malvern, Worcestershire, UK); it was found to be fine and milky.
Construction of pseudo ternary phase diagrams (PTPDs)
PTPD was constructed through aqueous titrations involving lipid, surfactant, and co-surfactant mixtures. The lipid phase was subjected to gentle heating at a temperature of 75 °C for a duration of 5 min. Surfactant and co-surfactant (Smix/aqueous phase) were combined in various volume ratios (1:0, 1:1, 2:1, 3:1) and subjected to heating at a consistent temperature. The selected Smix ratios are designed to illustrate the increasing concentration of surfactant in relation to co-surfactant. This approach facilitates a comprehensive analysis of the phase diagrams pertinent to the formulation of NLCs. Lipid phase and Smix were combined in various volume ratios (1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, and 9:1) to create homogeneous mixtures. These mixtures were subsequently titrated slowly with the aqueous phase. After each addition of the aqueous phase, visual observations were made as follows: clear nanoemulsions, nanoemulsion gels, emulsions, or emulsion gels. Oil (liquid lipid), surfactants, and co-surfactants were grouped in two different combinations for phase studies38.
PTPDs were generated using TriPlot Software, version 4.1.2. The addition of the aqueous phase was determined by calculating the percentage of each component within the nanoemulsion. In a pseudo-three-component phase diagram, the physical state is represented with one axis denoting the aqueous phase, the second axis representing the lipid phase, and the third axis illustrating a combination of surfactant and co-surfactant (Smix) at a predetermined volume ratio. A distinct phase diagram was developed for each Smix ratio. In the phase diagrams, only NE points were plotted within the shaded area to prevent overcrowding of the phases. For formulation development, the focus is exclusively on the nanoemulsion area.
Formulation and optimization of NLCs containing Aripiprazole was done by homogenization followed by ultrasonication by 32 factorial design. The formulation design of these Aripiprazole NLCs is shown in Table 3. Design-Expert® software (Version 13, Stat-Ease Inc., Minneapolis, MN) was used to perform the 3² factorial design, statistical analysis, ANOVA, and generation of response surface plots.
Algebraic expression can be defined as the dependence of a response variable on the independent variables. The first-order linear model for the three components is given in Eq. (1).
Where Y is the response, B0 is the intercept (the response when the level of all factors is low), X1, X2 and X3 are the levels (concentrations) of the 1, 2, 3 factors, respectively, B1, B2 and B3 are the polynomial coefficient, ε represents the experimental error. Statistical methods (such as the design of experiments (DoE)) can be applied to increase the efficacy of the experiment. DoE method is widely used in chemical research and the pharmaceutical industry for the development of new processes and optimizing the performance of existing ones. The empirical model of DoE includes the following orders: zero order: intercept; first order: main effects: A, B, C, D; second order: 2FI: AB, AC, BC, AD; and third order: 2FI: AB, AC, BC, AD and Third order: 3FI - ABC, ABD, ACD….
The NLCs were prepared by homogenization followed by ultrasonication18. The solid lipid and the liquid lipid were chosen based on the solubility of the drug in the lipids. Both solid lipids and liquid lipids were heated to 10 °C above the melting point of the solid lipid, and the drug was dissolved in the liquid mixture. Simultaneously, and at the same temperature. The aqueous phase; composed of the surfactant, BKC 0.001%, the co-surfactant, and purified water, was heated and mixed. The lipid phase was poured into the aqueous phase and homogenized using a homogenizer (Polytron Pt 1600e, Switzerland) at 10,000 rpm for 15 min. The pre-emulsion obtained was sonicated using a probe sonicator (Vibra Cell Sonic and Materials, Newtown, CT, USA), with an amplitude of 450 watts for 15 min. Subsequently, the NLCs formed were transferred to light resistant glass vials (sterilized) and cooled at room temperature. The different prepared formulations of Aripiprazole NLCs are shown in Table 4.
Characterization of nanostructured lipid carriers39,40,41
Particle size (PS), zeta potential (ZP), and polydispersity index (PDI)
PS, ZP, and PDI of the NLCs were measured by using a Malvern zeta sizer (DTS Ver. 5.10, Sr. No. MAL1031371, Malvern Instruments Ltd., UK) using the principle of the dynamic light scattering (DLS) method. All PS, ZP, and PDI measurements were carried out at room temperature (n = 3) by disposable plain folded zeta cells and polystyrene cells, respectively, following suitable dilution of 1:200 with the original dispersion preparation medium using Millipore water.
Drug loading and entrapment efficiency (EE%)
A measured volume of NLC solution was centrifuged at 18,000 rpm for 20 min, and the resulting solution was filtered and separated. The supernatant fraction was diluted using pH 6.4 phosphate buffer and was analyzed using a UV spectrophotometer (Model 1800, Shimadzu Co., Japan) at 216 nm for the quantification of unbound medication. The drug loading (DL%) and entrapment efficiency (%EE) were determined using the subsequent formulae.
Where, CT: total drug concentration in NLCs, CAP: free drug concentration in aqueous phase, WDL: weight of drug loaded in nanoparticles, WNP: weight of nanoparticles, pH: The pH of the NLC formulations was measured using a digital pH meter (n = 3) (Eutech Instruments, Mumbai).
Surface morphology by scanning electron microscopy (SEM)
SEM measurements were performed using SEM-EVO LS 15 (EVO LS 15, Zeiss, Germany) to study the external morphology of the prepared NLCs. Samples were mounted on an aluminum mount using double-sided adhesive tape and sputtered with gold under vacuum, then scanned at an accelerating voltage of 5 KV before observation42.
Viscosity and rheological studies
The optimized NLC formulation (A4) viscosity was evaluated at 25 °C using a rotating viscometer (Brookfield DV-E, USA) with spindle number 40. The viscosity was measured in triplicate (n = 3) and averaged in millipascal-seconds (mPa·s). The flow behavior was evaluated by measuring shear stress at different shear rates.
In vitro drug release by using Franz diffusion cell
In vitro drug release was carried out by dialysis membrane using the Franz diffusion cell [surface area of 1.79 cm2 and volume of 12 ml] for 72 h. The medication was released from the produced NLC formulations and drug solution using a pH 6.4 phosphate buffer that was agitated at 600 rpm and kept at 37 ± 0.5 °C43,44. The dialysis membrane (Dialysis membrane 150, Hi-Media, Mumbai, India) was utilized; its pore size was 2.4 nm, and its molecular weight ranged from 12,000 to 14,000. Before use, the dialysis membrane was immersed in a pH 6.4 phosphate buffer for a whole day. 1 mL of the pure drug solution (1 mg/mL of ARP in PB pH 6.4) or the ARP-NLC formulation (equivalent to 1 mg of ARP) was placed in the donor compartment onto the dialysis membrane and sealed it. Aliquots 0.5 mL were withdrawn from the receptor compartment at predetermined time intervals (0.5, 1, 2, 4, 6, 8, 12, 24, 48, and 72 h). In order to maintain sink condition replaced with an equal volume of fresh pre-warmed dissolution medium.
The samples were analyzed for drug content by a UV-visible spectrophotometer (UV 1800, Shimadzu Co., Japan) at 216 nm. The drug release profile was constructed by plotting the cumulative percent drug release versus time (h). The kinetic analysis of the release data was modelled using several kinetic equations, including zero order, first order, Hixon, Peppas, and Higuchi’s equation. The Franz diffusion cell technique was tested for accuracy, precision, and linearity prior to the investigation. The analytical approach showed excellent linearity (R² > 0.999) throughout concentration ranges of 1–50 µg/mL. The intra-day and inter-day precision, reported as coefficient of variation (CV), were found to be less than 2%, with an average percentage recovery of aripiprazole of more than 98% (n = 3).
Ex vivo diffusion/ permeation using sheep nasal mucosa and kinetic studies
Ex vivo permeation studies were conducted utilizing the Franz diffusion cell, which has a surface area of 1.79 cm² and a capacity of 12 ml. Sheep nasal mucosa was sourced from a local slaughterhouse, exempt from ethical approval per institutional guidelines, as no live animals were involved. The recently excised sheep nasal mucosa was obtained from the abattoir and immersed in PBS with a pH of 6.4. The excised superior nasal membrane was trimmed to an acceptable size and thickness (0.2 mm), detached from adherent tissues, and positioned between the donor and receptor compartments of the Franz diffusion cell, with the mucosal side oriented towards the donor compartment45. We used a digital micrometer to make sure that the mucosal membrane was 0.2 mm thick. Before the experiment, a light microscope was used to check the tissue’s viability and integrity to make sure there was no obvious damage or epithelial sloughing.
The mounted tissue was given 15 min to stabilize and stir in PBS pH 6.4 (the diffusion media). The body temperature of the sheep was maintained at 39 ± 0.5 °C in the diffusion medium. After fifteen minutes, the solution from both compartments was withdrawn, and new diffusion medium was added to the receptor compartment. The nasal membrane was installed on the rim of the receptor compartment; the donor compartment of the diffusion cell was positioned over it and fastened with a clamp to prevent diffusion media leakage. Placing 0.5 ml onto stabilized sheep nasal membranes in the donor compartment and constantly magnetically stirring at 600 rpm allowed permeation investigations of pure drug solution and drug-loaded optimized NLCs to be performed. Media aliquots (0.5 ml) were taken from the receptor compartment at specified intervals and examined for drug content under a UV-Visible spectrophotometer (UV 1800, Shimadzu Co., Japan) at 216 nm. To keep the same volume at every time interval, each removed sample was promptly replaced with an equivalent volume of fresh, warm diffusion media. Each trial was conducted over a duration of 7 h, during which the quantity of drug permeating through the sheep’s nasal mucosal membrane was quantified at each sampling point utilising a UV-Visible spectrophotometer (UV 1800, Shimadzu Co., Japan). The permeation profile was generated by graphing the percentage of medication penetrated against time (hours). The ex vivo permeation research method was verified with linearity, accuracy (CV < 2%), and recovery (> 98%) for aripiprazole measurement in permeation samples.
The steady-state flux (Jss, µg/cm²/h) was determined by calculating the slope of the linear segment of the curve derived from plotting the cumulative amount of drug permeated per unit area (µg/cm²) vs. time (h) (n = 3).
In vitro haemolytic toxicity study
The haemolytic activity was conducted to assess in vitro toxicity and also functions as a straightforward and dependable indicator of membrane damage induced by the formulation in vivo. The fresh blood from sheep was collected in a vial containing ethylene diamine tetra acetic acid (EDTA) as an anticoagulant. It was stored at 4 °C for no longer than 4 h before processing to prevent spontaneous hemolysis. The blood taken from slaughterhouse following routine slaughter. This study had no live animals, so ethical approval was not needed. The blood was centrifuged for 20 min at 3000 rpm to remove white blood cell debris and suspended RBCs. The RBCs were prepared for an erythrocyte stock dispersion by first diluting them with buffer after three washes in an isotonic saline solution (0.15 M NaCl and pH 7.4). 50% haematocrit was achieved by adjusting the RBC cell suspension. Haemolysis experiments were carried out for both blank and drug-loaded formulations of NLCs at a concentrations of 0, 2, 3, 4, 5, 10, 20, 40, 80, and 160 µg/ml. The incubation process lasted one hour at 37 °C. Following a shaking incubation period, the debris and undamaged red blood cells were extracted using centrifugation, and 100 µL of the obtained supernatant was mixed with 2 ml of a 39:1, or 99%, ethanol and HCl, w/v ethanol/hydrochloric acid mixture. All of the ingredients were dissolved in this combination, preventing haemoglobin from precipitating46.
The absorbance of the mélange was ascertained at 216 nm by monitoring it with a blank sample using a UV spectrophotometer. A control sample of 0% lysis (in buffer) and 100% lysis (in Triton X 100) was utilized in the experiment. The following equation was used to determine the percentage of haemolysis caused by the test sample (n = 3)
Nasal ciliotoxicity studies
The nasal mucosa of freshly isolated sheep was obtained from a slaughterhouse and subjected to ciliotoxicity studies in a phosphate buffer solution (PBS) having a pH of 6.4. As a negative control, PBS pH 6.4 was used on each mucosal sample. Isopropyl alcohol (IPA) was used as a positive control to test its ability to damage nasal mucociliary lining. All of the samples were treated for two hours, then washed well with distilled water and kept safe with 10% formalin until they could be analyzed further (n = 3). Parts of each sample that were used for histopathology were cut out and stained with H and E. The mucosa was then cut apart, and an optical trinocular microscope was used to look at the mucocilia47.
Stability studies
Optimized NLC formulation (A4) was filled into 10 mL phosphate buffer (0.1 M, pH 6.4) amber glass vials, sealed with rubber stoppers and aluminum crimps to protect against light and moisture, and subjected to different thermodynamic stability studies for short term (3 months) at temperatures of 5 ± 3°, 25 ± 2 °C /60 ± 5% RH and 40 ± 2 °C /75 ± 5% RH according to ICH Q1A(R2) Guidelines48. The samples were withdrawn from amber glass vials periodically and were analyzed for various parameters [drug loading, entrapment efficiency, % transmittance, pH, particle size (nm), zeta potency (mV), and in vitro release studies of the preparation] against the initial results of formulation A4 (n = 3).
Results and discussion
Solubility study
The solubility studies conducted during pre-formulation were critical for selecting excipients that maximize drug loading. The solubility of Aripiprazole exhibited the highest in stearic acid (200 mg/mL) among the solid lipids and in oleic acid (43.660 mg/mL) among the liquid lipids. This significant affinity for stearic and oleic acid provided a strong rationale for their selection as the primary lipid components, forming a matrix capable of solubilizing a significant payload of the drug, which directly contributed to the high entrapment efficiency observed in the final NLCs. However, mixing the solid lipid (stearic acid) with the liquid lipid (oleic acid) reduced the solubility of aripiprazole to 38.36 mg/mL. We found that Tween 80 (85.506 mg/ml) and Span 80 (109.603 mg/ml) had higher drug solubility compared to other surfactants. Aripiprazole was more soluble in PEG 400 (63.935 mg/mL) and Labrasol (55.190 mg/mL) when compared to other co-surfactants, and hence they are considered for further studies. The detailed solubility data of aripiprazole in solid lipids, liquid lipids, surfactants, and co-surfactants were given in Supplementary Tables 1 and 2. These supplementary tables support selection of excipients with maximum ARP solubilization, which formed the basis for factorial design.
Emulsification studies with surfactant
According to the results of the emulsification screening, Tween 80 was able to successfully produce a stable microemulsion with the chosen lipid system (stearic and oleic acid). This was demonstrated by the fact that it had a Grade MEa rating and a low turbidity of 21.6 NTU. In comparison, Span 80 resulted in a less stable, watery emulsion. Tween 80’s better performance can be ascribed to its high HLB value (15) and molecular structure, which promoted the formation of a stable interfacial coating over the lipid droplets, a critical feature in the resulting formation of stable NLCs with tiny particle sizes. The evaluation of surfactants’ relative emulsification clearly demonstrates their ability to emulsify certain oily phases such as stearic acid and oleic acid. The grading for relative emulsification of surfactants and co-surfactants is shown in Table 5.
Compatibility study by Fourier transform infrared (FT-IR) spectroscopy
The compatibility of the pure Aripiprazole drug with the excipients was evaluated. The study comprised different ratios of blends of drug with excipients, and their compatibility was analyzed using FT-IR spectroscopy by the KBR pellet method. The results found that the excipients used in the study were not affecting the major peaks of the pure drug, either individually or in the mixture49. It clearly shows that there was no interaction between the drug and the excipients used in the study, as shown in Fig. 1.

FT-IR spectra of aripiprazole and with excipients in the study.
Crystalline behavior of drug and lipids by DSC
DSC is used to investigate the melting temperature and recrystallization behavior of crystalline materials. Aripiprazole was analyzed by DSC, and a major peak was observed at 144.45 °C. This peak confirmed the characteristic melting point of aripiprazole. DSC thermograms were recorded for the pure drug aripiprazole and optimized aripiprazole NLCs (A4). As shown in thermograms, a single sharp endothermic peak at 144.45 °C and 143.39 °C, respectively, was observed, and the melting peaks were at the relative temperatures of the drug and lipid used (Fig. 2). This conclusion suggests that the drug is in a crystalline state. Though the NLCs were prepared by the homogenization followed by ultra-sonication method, drug molecules dispersed in the lipid phase can crystallize. There are a few authors who have demonstrated that the substance does not exhibit an endothermic peak in the nanoparticles that are formed: it is found to be in the amorphous state50. Furthermore, the crystallization of the drug is unaffected by the presence of surfactants and lipids.

DSC data of pure aripiprazole and formulation.
Optimization of formulation
The formulation was prepared by varying all the parameters, such as liquid lipid concentration, solid lipid concentration, homogenization time and speed, sonication time and amplitude, and it was found to be finely milky in the optimum formulation.
Construction of pseudo-ternary phase diagrams
A pseudo-ternary phase diagram is one of the methods that is used for studying the boundaries of different phases as a function of their structure. The pseudo ternary phase diagrams can determine the structure and nature of the nanoemulsions, and they are widely used in selecting the optimum formulation. Oil, surfactant, and co-surfactant properties determine the type of nanoemulsion that is produced51.
As indicated by previous studies, the surfactant must have an HLB less than 10 to produce an oil-in-water emulsion. However, in this study, Tween 80 with an HLB value of 15 was selected based on the results found in the solubility studies. This is because Tween 80 is a non-ionic surfactant, and it was previously used due to its low toxicity compared to other ionic surfactants. In addition, pH and ionic strength had low influence on the performance of Tween 8052. Most of the surfactants used in the manufacture of nanoemulsions were single-chain surfactants, which were unable to sufficiently reduce the interfacial tension to produce nanoemulsion. To resolve this issue, a second amphiphile, known as a “co-surfactant,” was incorporated into the system. Accordingly, the PEG 400, with an HLB value of 11, was included. It revealed that the use of co-surfactants had the effect of differentiating between surfactant molecules and diminishing polar head group interactions. In addition, they have the potential to enhance the elasticity of the interfacial film surrounding the nanoemulsion droplets53.
PTPDs were constructed for each group, and each Smix ratio and nanoemulsion area was plotted. The three axes of the phase diagram represented the oil phase, surfactant mixture (Smix), and aqueous phase. The remaining area on the phase diagram represented the turbulent, milky, and conventional emulsion zones.
According to the pseudo-ternary phase diagram, nanoemulsions composed of oleic acid and stearic acid (1:1) as the oil phase, Tween 80 and PEG 400 (2:1) (Smix) as the surfactant and co-surfactant, and different weight ratios of the oil phase to the Smix (9:1, 8:2, 7:3, and 6:4) were selected for further optimization. No nanoemulsion region was obtained when the surfactant and co-surfactant mixture (Smix) ratio was decreased from 9:1 to 1:9 as shown in Supplementary Fig. 1.
When 1.96% v/v of Tween 80 was used alone (Smix 1:0), the maximum amount of oil that needed to be solubilized was 17.64% v/v. Upon the addition of co-surfactant (Smix 1:1), an increase in nanoemulsion area was observed and approximately 18.75% v/v oil was solubilized. Further increase in the co-surfactant (1:2 and 1:3) had led to an increase in the nano emulsion area to 21.42% v/v and 20.45% v/v of the solubilized oil, respectively. The increases in surfactant to co-surfactant ratio had the effect of increasing micelle formation, which enhanced the solubilization capacity of the nanoemulsion. When surfactant concentration was increased with respect to co-surfactant (2:1), the nanoemulsion area was greater compared to a 1:1 ratio; however, a nanogel area was obtained due to high viscosity. When surfactant concentration was increased further (3:1), the nanoemulsion area was reduced in comparison to 2:1, and an increase in the nanogel area was obtained54. When 1.85% v/v Tween 80 was used alone (Smix 1:0), a maximum oil of 16.66% v/v could be solubilized. With the addition of co-surfactant (Smix:1), an increase in the nanoemulsion area was observed and 21.42% v/v of the oil was solubilized. Further increases in co-surfactant (1:2 and 1:3) had led to a decrease in nano nanoemulsion area55, and 14.45% v/v and 13.23% v/v of the oil were solubilized, respectively.
Accordingly, the highest nanoemulsion formation area was obtained at a 2:1 Smix of (Liquid lipid (oil): solid lipid = 1:1) and it was considered as the optimized ratio for further formulations. This ratio used were to determine the NLCs region in the pseudo-ternary phase diagram; the upper and lower limits of the oil phase and Smix were set at 5–10% and 20–50% respectively.
Formulation of ARP-NLCs by 32 factorial design
Investigating the result of formulation variables on various parameters, a total of nine experiments were conducted. Formulation A4 was considered the optimized formulation. The values of responses y1 (Drug loading), y2 (entrapment efficiency), and y3 (particle size) were in the range of 2.245 to 20.96% (y1), 10.4 to 96.218% (y2) and 98.6 to 219.6 nm (y3) respectively.
The optimization of ARP-NLCs was conducted using a 32 factorial design, which is a robust statistical tool for evaluating the effect of independent variables on dependent responses. In this study, two factors were selected: the lipid phase (solid lipid and liquid lipid) and the surfactant-co-surfactant mixture (Smix). Each factor was tested at three levels: low (-1), medium (0), and high (+ 1). The lipid phase varied between 5 and 10%, while the Smix concentration ranged from 20 to 50%. The responses evaluated included drug loading (%), entrapment efficiency (%), and particle size (nm). The selection of these factors and levels was based on preliminary solubility and emulsification studies, which indicated that stearic acid (solid lipid) and oleic acid (liquid lipid) provided optimal drug solubility, while Tween 80 and PEG 400 (Smix) ensured stable emulsification. The factorial design allowed for the identification of significant interactions between factors, as evidenced by the ANOVA results, which showed that both the lipid phase and Smix significantly influenced drug loading and entrapment efficiency (p < 0.05). This systematic approach ensured the development of an optimized formulation with enhanced drug delivery properties. The ratios of maxima to minima for y1, y2 and y3 were 9.3, 9.2 and 2.22, respectively. Accordingly, there was no need for a power conversion of the values. The change in the response was a significant component for data analysis. The change was necessary if the degree of the conversion—residual—was determined by the size of the response—predicted values56. When the highest-to-lowest response ratio exceeded 10, the power change of the reactions became essential. The adjustment has minimal effect for a ratio smaller than three. The adequacy of the model was examined on the basis of experimental data to determine whether the hypothesized model would yield poor or misleading results. Polynomial and linear models were fit to the experimental data. The F value of P < 0.0001, low standard deviation, high R2, and low-range approximate residual suggested the selection of polynomial models for residual sum responses. The 32 Factorial design and responses for ARP-NLCs are shown in Table 6.
An ANOVA of the data verified that the model was significant (model probability > F less than 0.05). The model F values for responses y1, y2, and y3 were 113.35, 5.691, and 68.368, respectively, indicating the model’s significance. The ANOVA data for responses y1, y2, and y3 indicate significant factors that affect these responses31,47. The measured values of y1, y2, and y3 were determined to be 20.478%, 96.436%, and 99.369 nm for drug loading, entrapment efficiency, and particle size, respectively, and closely aligned with the expected values. Analysis of variance as shown in Table 7.
Regression equations for the responses: R1= -28.892 + 3.894*A + 0.993*b-0.079*A*B; R2= -11.741 + 2.899*A + 1.728*B; R3= -41.711 + 30.586*A + 1.388*B-0.325*A*B.
Residual plots were examined to verify the model’s adequateness, and they demonstrated random scatter. The Overlay contour plot and surface plots drug loading, entrapment efficiency and particle size were shown in Supplementary Fig. 2). Additionally, a numerical optimization technique that utilizes a desirability function was implemented to optimize drug delivery and entrapment efficiency while simultaneously decreasing particle size. The optimized formulation (A4) had an overall desirability of 0.92. the desirability Plot as shown in Fig. 3.

Desirability plot of oil phase and Smix.
The study used an actual vs. predicted plot for formulation and a response surface graph to compare the theoretical values (predicted by the model) with the experimental values (obtained from actual experiments) shown in Supplementary Figs. 3 and 4 respectively. The fact that the two results are consistent with one another is evidence that the predictions made by the model are correct. The relationship between variables was further studied using contour plots and 3D plots to show the effect of factors A and B on responses y1, y2, and y3, helping to better understand the system’s behavior57. The ssummary of design of experiment (DoE) responses is shown in Supplementary Table 3.
Statistical significance between individual groups was determined using student t-test. A Student’s t-test was performed to compare drug loading (as the independent variable) and entrapment efficiency across formulations to assess their interdependence. The comparison was carried out between drug loading and entrapment efficiency (considering drug loading as the reference independent variable). In addition, Levene’s Test was applied to confirm the equality of variances between drug loading and entrapment efficiency, ensuring the validity of the t-test assumptions. [determined using IBM Software, statistics version 20]. The student’s t-test and Levine’s test for equality of variance comparing drug loading and entrapment efficiency across formulations are shown in Table 8.
Characterization of nanostructured lipid carriers
Particle size, zeta potential and polydispersity index
The optimized formulation (A4) was found to have the particle size of 99.6 nm. Supplementary Fig. 5 (a)), indicating it is small enough for effectively absorbed through the nasal route, a zeta potential of -36.7mV (Supplementary Fig. 5 (b) and a Polydispersity index (PDI) of 0.249, showing good stability because of strong repulsion between the particles, avoiding repulsion and uniform particle distribution respectively58, making the formulation best for intranasal administration. The transmittance (%), pH, ZP, PDI, and cumulative release after 72 h for all formultions (A1-A9) were given in supplemetary Table 4.
Drug loading and entrapment efficiency (%)
The drug loading and entrapment efficiency of optimized formulation (A4) were found to be high at 20.96% and 96.218% respectively, compared to other formulation. The results indicated that the drug can be dissolved to a greater extent in liquid lipids than in solid lipids. The higher entrapment efficiency was noticed due to most lipophilic drugs, such as aripiprazole, solubilizing mainly in liquid lipids rather than in solid lipids, which facilitates improved encapsulation inside the lipid matrix59.
Surface morphology by scanning electron microscopy (SEM)
The SEM were studies used to investigate the morphology and particle size of the NLCs. The SEM revealed that the particles were spherical in shape and homogeneous with a smooth surface. The prepared NLCs could be re-dispersed in water easily with dilutions of 1:200 using Millipore water. The nanoparticle size observed by SEM correlated well with the particle size measured by the particle size analyzer 60. The SEM images of the optimized formulation (A4), are shown in Fig. 4.

SEM photograph of optimized ARP-NLCs (A4) Formulation showing spherical, homogeneous particles. The scale bar represents 100 nm.”
In vitro release by using Franz diffusion cell
The in vitro release profile of aripiprazole from the prepared NLCs was evaluated using Franz diffusion cells and compared against a pure drug solution. The drug solution exhibited rapid and extensive release, reaching 91.78 ± 0.28% within 24 h. In stark contrast, all NLC formulations (A1-A9) demonstrated a significant sustained release pattern (p < 0.05), confirming their ability to prolong drug delivery. The drug release from the NLCs exhibited a distinct biphasic pattern, characterized by an initial burst release followed by a sustained, controlled phase. This profile is attributed to the immediate release of drug molecules adsorbed onto the nanoparticle surface. Subsequently, the sustained release phase is governed by the slower diffusion of the drug encapsulated within the lipid matrix core, ensuring prolonged drug availability, which can reduce dosing frequency and enhance therapeutic efficacy60.
Among all formulations, A4 showed the most favorable release profile, achieving 45.01 ± 0.20% cumulative release at the end of 72 h. This significantly higher release (p < 0.05) is indicative of an optimized ratio of solid to liquid lipids and surfactants within the formulation. The observed burst release can be further explained by the microstructure of the NLCs. The difference in melting characteristics between the liquid and solid lipids can lead to the formation of a liquid lipid-rich outer shell. This shell has high solubility for the lipophilic drug, facilitating rapid diffusion in the initial phase. The subsequent sustained release is primarily due to the slower diffusion of the drug from the solid lipid core of the matrix61. The in vitro drug release profiles for the pure aripiprazole solution and all NLC formulations (A1-A9) are presented in Fig. 5. Comprehensive in vitro characterization data for all prepared ARP-NLC formulations are provided in Supplementary Table 5. Collectively, these results illustrate the significant influence of formulation variables on modulating the drug release kinetics over time.
The obtained drug release data were fitted to release kinetic models such as the Higuchi’s equation, zero and first order, Korsmeyer-Peppas, and Hixon-Crowell equation. Drug release from NLCs was found to follow the Peppas model. It was determined that the release exponent (n) is less than 0.45, indicating that the release mechanism is governed by Fickian diffusion. Table 9 summarizes the model parameters, which include the correlation coefficient (R²), release rate constant (k), and release exponent (n) for the zero-order, first-order, Higuchi, and Korsmeyer-Peppas models62.

In vitro drug release profile of pure aripiprazole solution and NLCs formulation (A1-A9) in phosphate buffer (PB pH 6.4). Data represents mean ± SD (n = 3).
Ex vivo diffusion by sheep nasal mucosa and release kinetics
The ex vivo diffusion study carried out by using sheep nasal mucosa. The study revealed a significantly slower diffusion rate for the (A4) NLCs (27.154 ± 0.037%) compared to the pure drug solution (88.230 ± 0.157%) in 6 h. This behavior is strategically advantageous for targeting nose-to-brain. The NLCs act as a drug reservoir at the nasal mucosal site, facilitating sustained release. This prolonged residence time increases the window for direct uptake via the olfactory and trigeminal nerves, thereby enhancing direct brain delivery while minimizing rapid systemic absorption and associated side effects. Conversly, the rapid permeation of the drug solution likely leads to swift entry into the systemic circulation, subjecting it to first-pass metabolism and reducing the fraction available for targeting the brain. The Ex-vivo drug release of pure drug and optimized formulation (A4) NLCs in PB pH 6.4 as shown in Fig. 6. The ARP a lipophilic drug exhibits a greater affinity for the lipid matrix, resulting in a reduced quantity of drug diffusion from the NLCs compared to the drug solution. The NLCs could permeate through the receptor compartment, holding the drug within the matrix42. Kinetic analysis was done on the drug’s diffusion. When the regression coefficients of the aggregates’ various orders were examined, it became clear that the NLCs release pattern adhered to the Peppas model, as indicated by a greater R2 value (0.9637) than those of the other models. Based on the calculated ‘n’ value, it was found the mechanism followed the Fickian-diffusion.

Ex vivo drug release plot of pure drug and A4 NLCs in PB pH 6.4.
In vitro haemolytic toxicity
The NLCs may be transported to systemic circulation upon intranasal administration and can cause damage to RBC’s. In order to assess the safety of the formulated products, in vitro haemolytic toxicity experiments were conducted. The findings indicated that both the blank and drug-loaded NLCs were tested in a concentration range of 0–160 µg/ml in phosphate buffer at pH 6.4.showed negligible haemolytic effects (0.509–1.528%) and were within an acceptable range (< 5%). In this study, a haemolytic agent, Triton X 100, was used as a positive control, showing 100% haemolysis of RBC’s. The interpretation of the in vitro haemolytic study may not exactly predict the in vivo behavior, but it reveals the haemolytic potential. Therefore, the estimated in vitro RBC’s toxicity may be largely reduced in vivo8.
Nasal ciliotoxicity studies
Nasal ciliotoxicity studies were performed to investigate the toxicity of excipients employed in the formulation. When the nasal membrane was treated with isopropyl alcohol, there was nasociliary and epithelial layer damage observed. On the contrary, no nasociliary and epithelial layer damage was observed when treated with PB pH 6.4. Treatment with blank and drug loaded NLCs formulations showed no-toxicity on nasal mucosa, which implied that the formulation and excipients were non-toxic for nasal administration63, the haematoxylin and Eosin stained histopathological sections of sheep nasal mucosa as shown in Fig. 7.

Haematoxylin and Eosin (H & E) stained Histopathological sections of sheep nasal mucosa treated with (A) PB pH 6.4, (B) IPA, and (C1, C2) ARP-NLCs at magnification of 100x to study the nasal ciliotoxicity. (1: Venules; 2: Mucous glands; 3: Serous glands; 4: Pseudostratified columnar ciliated epithelial cells)
Stability studies
The physicochemical parameters, formulations were visually examined throughout the investigation. The optimized formulation (A4) NLCs held at 5 ± 3 °C remained homogeneous with no obvious phase separation or color change over time. In contrast, samples held at higher temperatures (25 ± 2 °C and 40 ± 2 °C) showed minor creaming after the first month, which increased over time. This visual finding is consistent with the reported increase in particle size and reduction in zeta potential at these temperatures, indicating physical instability and possible particle aggregation under stress conditions.
The results of stability studies indicated that optimized formulation of NLCs (A4) were stable with respect to drug loading, % transmittance, entrapment efficiency, in vitro release, pH, particle size, and zeta potential. The increase in particle size of NLCs was observed both at 25 ± 2 °C and 40 ± 2 °C stability stations. However, it was still in the nano range, making it optimal for nasal delivery. In contrast, the particle size growth was slower when NLCs were stored at 5 ± 2 °C. The system’s kinetic energy was found to increase at temperatures of 25 ± 2 °C and 40 ± 2 °C, which allowed for the acceleration of particle collisions64. Consequently, the potential of nanoparticle aggregation was increased. In addition, a decrease in DL (%), EE (%), in vitro release, and an upsurge in particle size were observed when NLCs were stored at 25 ± 2 °C and 40 ± 2 °C, compared to 5 ± 2 °C. The lipids undergo polymorphic transitions upon storage, which may cause drug expulsion. This indicated that the prepared NLCs were more stable at 5 ± 2 °C than other temperatures, so it was recommended to store the NLCs at 5 ± 2 °C. These findings confirm the temperature-dependent behavior of NLCs and support storage recommendations.
The optimized formulation (A4) exhibited Newtonian flow behavior with a viscosity of 32.5 ± 1.2 mPa·s, which is suitable for nasal administration and consistent with good mucosal spreadability and retention. During the stability investigation, the optimized formultion A4 viscosity remained steady at 32–35 mPa·s across all storage settings, demonstrating no substantial changes in its rheological characteristics over time. The Summary of stability study data for optimized formulation as shown in Supplementary Table 5.
Clinical implications of ARP-NLCs for schizophrenia treatment
The sustained release profile, enhanced nasal permeation, and stability of the optimized A4 ARP-NLCs suggest significant potential for improving schizophrenia therapy. The biphasic release pattern, with an initial burst followed by controlled release up to 72 h, could reduce the frequency of administration compared to conventional oral aripiprazole tablets, which require daily dosing due to a 75-hour half-life and extensive hepatic metabolism. This prolonged drug availability may enhance patient compliance, a critical factor given that approximately 80% of schizophrenia patients experience severe relapses within two years of discontinuing medication. Furthermore, the nose-to-brain delivery approach bypasses the blood-brain barrier and first-pass metabolism, potentially increasing drug bioavailability at the target site and minimizing systemic side effects, such as extrapyramidal symptoms or weight gain, commonly associated with oral aripiprazole therapy. The absence of significant haemolytic or ciliotoxic effects further supports the safety of this formulation for intranasal use. However, while these in vitro and ex vivo findings are promising. The future studies should explore in vivo pharmacokinetic profiling, brain biodistribution, and long-term safety assessments of ARP-NLCs in schizophrenia models.
Conclusions
ARP-loaded NLCs, an interesting tool as a drug carrier, were prepared successfully by homogenization followed by the ultrasonication method by a 32-factorial design using stearic acid, oleic acid, Tween 80, and PEG 400. The optimized formulation of NLCs (F4) (stearic acid and oleic acid 2.5% each) showed spherical-shaped particles with a narrow size distribution of particle size < 150 nm, a drug loading capacity of 20.96%, and an entrapment efficiency of 96.22% and sustained the drug release followed by Fickian diffusion. Toxicity studies confirmed that the prepared NLCs formulations had no significant hemolysis or mucosal damage. Thus, the developed ARP-NLCs improves the solubility, stability, and bioavailability of aripiprazole. The results suggest that ARP-loaded NLCs are a promising intranasal drug delivery system for targeting the brain, with improved solubility, stability, and bioavailability in vitro and ex vivo. In the future studies will focus on in vivo pharmacokinetics and pharmacodynamic evaluations in rodent models to conclusively validate the enhanced therapeutic efficacy, biodistribution, and brain targeting of this intranasal ARP-NLC method.
Data availability
All data analyzed or generated during this study are included in this article.
References
Paranjpe, M. & Müller-Goymann, C. C. Nanoparticle-mediated pulmonary drug delivery: A review. Int. J. Mol. Sci. 15, 5852–5873. https://doi.org/10.3390/ijms15045852 (2014).
Petric, P. S., Ifteni, P., Miron, A. A., Sechel, G. & Teodorescu, A. Brain abnormalities in schizophrenia: A comparative imagistic study. Medicina 60, 564. https://doi.org/10.3390/medicina60040564 (2024).
Bosun, A. et al. Dopaminergic epistases in schizophrenia. Brain Sci. 14, 1089. https://doi.org/10.3390/brainsci14111089 (2024).
Gongcheng, X. et al. Test-retest reliability of fNIRS in resting-state cortical activity and brain network assessment in stroke patients. Biomed. Opt. Exp. 14, 4217–4236. https://doi.org/10.1364/BOE.491610 (2023).
Luo, H., Gu, X., Tong, G. & Han, L. Research progress of apelin in acute ischemic brain injury. Am. J. Transl Res. 14, 7260–7267 (2022).
Jiaquan, L. et al. Unraveling the prefrontal cortex-basolateral amygdala pathway’s role on schizophrenia’s cognitive impairments: A multimodal study in patients and mouse models. Schizophr. Bull. 50, 913–923. https://doi.org/10.1093/schbul/sbae063 (2024).
Mistry, A., Stolnik, S. & Illum, L. Nose-to-brain delivery: Investigation of the transport of nanoparticles with different surface characteristics and sizes in excised porcine olfactory epithelium. Mol. Pharm. 12, 2755–2766. https://doi.org/10.1021/acs.molpharmaceut.5b00088 (2015).
Keller, L. A., Merkel, O. & Popp, A. Intranasal drug delivery: Opportunities and toxicologic challenges during drug development. Drug Deliv Transl Res. 12, 735–757. https://doi.org/10.1007/s13346-020-00891-5 (2022).
Alam, M. I. et al. Strategy for effective brain drug delivery. Eur. J. Pharm. Sci. 40, 385–403. https://doi.org/10.1016/j.ejps.2010.05.003 (2010).
Bonaccorso, A. et al. Optimization of curcumin nanocrystals as promising strategy for nose-to-brain delivery application. Pharmaceutics 12, 476. https://doi.org/10.3390/pharmaceutics12050476 (2020).
Shadab; Bhattmisra, S. K. et al. Nano-carrier enabled drug delivery systems for nose to brain targeting for the treatment of neurodegenerative disorders. J. Drug Deliv Sci. Technol. 43, 295–310. https://doi.org/10.1016/j.jddst.2018.03.022 (2018).
Korade, Z. et al. Chronic aripiprazole and trazodone polypharmacy effects on systemic and brain cholesterol biosynthesis. Biomolecules 13, 1321. https://doi.org/10.3390/biom13091321 (2023).
Preda, A. & Shapiro, B. B. A safety evaluation of aripiprazole in the treatment of schizophrenia. Expert Opin. Drug Saf. 19, 1529–1538. https://doi.org/10.1080/14740338.2020.1832990 (2020).
Banks, W. A. Physiology and pathology of the blood-brain barrier: Implications for microbial pathogenesis, drug delivery and neurodegenerative disorders. J. Neurovirol. 5, 538–555. https://doi.org/10.3109/13550289909021284 (1999).
Rahman, M. A. et al. Fabrication of sustained release curcumin-loaded solid lipid nanoparticles (Cur-SLNs) as a potential drug delivery system for the treatment of lung cancer: Optimization of formulation and in vitro biological evaluation. Polymers 15, 542. https://doi.org/10.3390/polym15030542 (2023).
Shivananjegowda, M. G. et al. Development and evaluation of solid lipid nanoparticles for the clearance of Aβ in Alzheimer’s disease. Pharmaceutics 15, 221. https://doi.org/10.3390/pharmaceutics15010221 (2023).
Patel, H. P. et al. Clozapine loaded nanostructured lipid carriers engineered for brain targeting via nose-to-brain delivery: Optimization and in vivo pharmacokinetic studies. J. Drug Deliv Sci. Technol. 64, 102533. https://doi.org/10.1016/j.jddst.2021.102533 (2021).
Gadhave, D. et al. Nose-to-brain delivery of amisulpride-loaded lipid-based poloxamer-gellan gum nanoemulgel: In vitro and in vivo pharmacological studies. Int. J. Pharm. 607, 121050. https://doi.org/10.1016/j.ijpharm.2021.121050 (2021).
Stoilova, S., Georgieva, D., Mihaylova, R., Petrov, P. D. & Kostova, B. Nanogels based on N,N-dimethylacrylamide and β-cyclodextrin triacrylate for enhanced solubility and therapeutic efficacy of aripiprazole. Gels 10, 217. https://doi.org/10.3390/gels10040217 (2024).
Abo El-Enin, H. A. et al. Evaluation of brain targeting and antipsychotic activity of nasally administrated ziprasidone lipid–polymer hybrid nanocarriers. Pharmaceuticals 16, 886. https://doi.org/10.3390/ph16060886 (2023).
Dimiou, S. et al. Particulate levodopa nose-to-brain delivery targets dopamine to the brain with no plasma exposure. Int. J. Pharm. 618, 121658. https://doi.org/10.1016/j.ijpharm.2022.121658 (2022).
Qu, Y. et al. Nose-to-brain delivery of disulfiram nanoemulsion in situ gel formulation for glioblastoma targeting therapy. Int. J. Pharm. 597, 120250. https://doi.org/10.1016/j.ijpharm.2021.120250 (2021).
U.S. Food and Drug Administration. Aripiprazole label. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/021436s038,021713s030,021729s022,021866s023lbl.pdf (2014). Accessed 17 Jan 2023.
Sawant, K., Pandey, A. & Patel, S. Aripiprazole loaded poly(caprolactone) nanoparticles: Optimization and in vivo pharmacokinetics. Mater. Sci. Eng. C. 66, 230–243. https://doi.org/10.1016/j.msec.2016.04.089 (2016).
Patel, S. et al. Brain targeting of risperidone-loaded solid lipid nanoparticles by intranasal route. J. Drug Target. 19, 468–474. https://doi.org/10.3109/1061186X.2010.523787 (2011).
Gadhave, D. G., Tagalpallewar, A. A. & Kokare, C. R. Agranulocytosis-protective olanzapine-loaded nanostructured lipid carriers engineered for CNS delivery: Optimization and hematological toxicity studies. AAPS PharmSciTech. 20 (1), 22. https://doi.org/10.1208/s12249-018-1213-y (2019). Published 2019 Jan 2.
Kumbhar, S. A., Kokare, C. R., Shrivastava, B., Gorain, B. & Choudhury, H. Antipsychotic potential and safety profile of TPGS-based mucoadhesive aripiprazole nanoemulsion: Development and optimization for nose-to-brain delivery. J. Pharm. Sci. 110, 1761–1778. https://doi.org/10.1016/j.xphs.2021.01.021 (2021).
Nguyen, T. T. L. & Duong, V. A. Solid lipid nanoparticles. Encyclopedia 2, 952–973. https://doi.org/10.3390/encyclopedia2020063 (2022).
Elmowafy, M. & Al-Sanea, M. M. Nanostructured lipid carriers (NLCs) as drug delivery platform: advances in formulation and delivery strategies. Saudi Pharm. J. 29, 999–1012. https://doi.org/10.1016/j.jsps.2021.07.015 (2021).
Gilani, S. J. et al. Formulation and optimization of nano lipid based oral delivery systems for arthritis. Coatings 11, 548. https://doi.org/10.3390/coatings11050548 (2021).
Hirun, N. et al. Fabrication and characterization of pectin films containing solid lipid nanoparticles for buccal delivery of fluconazole. Int. J. Mol. Sci. 25, 5413. https://doi.org/10.3390/ijms25105413 (2024).
Patil, K. et al. Quality-by-design-driven nanostructured lipid scaffold of apixaban: Optimization, characterization, and pharmacokinetic evaluation. Pharmaceutics 16, 910. https://doi.org/10.3390/pharmaceutics16070910 (2024).
Zaky, M. F. et al. Tailoring apixaban in nanostructured lipid carrier enhancing its oral bioavailability and anticoagulant activity. Pharmaceutics 15, 80. https://doi.org/10.3390/pharmaceutics15010080 (2023).
Zingale, E. et al. Optimization of lipid nanoparticles by response surface methodology to improve the ocular delivery of diosmin: Characterization and in-vitro anti-inflammatory assessment. Pharmaceutics 14 (1961). https://doi.org/10.3390/pharmaceutics14091961 (2022).
Clemente, N. et al. Solid lipid nanoparticles carrying temozolomide for melanoma treatment. Preliminary in vitro and in vivo studies. Int. J. Mol. Sci. 19, 255. https://doi.org/10.3390/ijms19020255 (2018).
Qi, C., Chen, Y., Jing, Q. Z. & Wang, X. G. Preparation and characterization of catalase-loaded solid lipid nanoparticles protecting enzyme against proteolysis. Int. J. Mol. Sci. 12, 4282–4293. https://doi.org/10.3390/ijms12074282 (2011).
Ozsoy, Y., Gungor, S. & Cevher, E. Nasal delivery of high molecular weight drugs. Molecules 14, 3754–3779. https://doi.org/10.3390/molecules14093754 (2009).
Siafaka, P. I., Üstündağ Okur, N., Karavas, E. & Bikiaris, D. N. Surface modified multifunctional and stimuli responsive nanoparticles for drug targeting: Current status and uses. Int. J. Mol. Sci. 17, 1440. https://doi.org/10.3390/ijms17091440 (2016).
Vlaia, L. et al. Topical biocompatible fluconazole-loaded microemulsions based on essential oils and sucrose esters: Formulation design based on pseudo-ternary phase diagrams and physicochemical characterization. Processes 9, 144. https://doi.org/10.3390/pr9010144 (2021).
Gandhi, S., Shastri, D. H., Shah, J., Nair, A. B. & Jacob, S. Nasal delivery to the brain: Harnessing nanoparticles for effective drug transport. Pharmaceutics 16 (481). https://doi.org/10.3390/pharmaceutics16040481 (2024).
Kiss, E. L. et al. Design and optimization of nanostructured lipid carrier containing dexamethasone for ophthalmic use. Pharmaceutics 11, 679. https://doi.org/10.3390/pharmaceutics11120679 (2019).
Lu, W. et al. Cationic albumin-conjugated pegylated nanoparticles as novel drug carrier for brain delivery. J. Control Release. 107, 428–448. https://doi.org/10.1016/j.jconrel.2005.03.027 (2005).
Joshi, M. & Patravale, V. Nanostructured lipid carrier (NLC) based gel of celecoxib. Int. J. Pharm. 346, 124–132. https://doi.org/10.1016/j.ijpharm.2007.05.060 (2008).
Jagaran, K., Habib, S. & Singh, M. Bio-Inspired polymeric solid lipid nanoparticles for SiRNA delivery: Cytotoxicity and cellular uptake in vitro. Polymers 16, 3265. https://doi.org/10.3390/polym16233265 (2024).
Pund, S., Rasve, G. & Borade, G. Ex vivo permeation characteristics of venlafaxine through sheep nasal mucosa. Eur. J. Pharm. Sci. 48, 195–201. https://doi.org/10.1016/j.ejps.2012.10.029 (2013).
Golubeva, O. Y., Alikina, Y. A., Brazovskaya, E. Y. & Vasilenko, N. M. Hemolytic activity and cytotoxicity of synthetic nanoclays with montmorillonite structure for medical applications. Nanomaterials 13, 1470. https://doi.org/10.3390/nano13091470 (2023).
Pires, A., Fortuna, A., Alves, G. & Falcão, A. Intranasal drug delivery: How, why and what for? J. Pharm. Pharm. Sci. 12, 288–311. https://doi.org/10.18433/J3NC79 (2009).
Béduneau, A. et al. Brain targeting using novel lipid nanovectors. J. Control Release. 126, 44–49. https://doi.org/10.1016/j.jconrel.2007.11.001 (2008).
Queiroz, M. C. V. & Muehlmann, L. A. Characteristics and preparation of solid lipid nanoparticles and nanostructured lipid carriers. J. Nanotheranostics. 5, 188–211. https://doi.org/10.3390/jnt5040012 (2024).
Singh, A., Dubey, P., Pandey, R., Verma, R. & Singh, S. Preparation and characterization of aripiprazole-cyclodextrin inclusion complexes: A study on molecular interactions using FTIR and DSC. Molecules 28, 1125. https://doi.org/10.3390/molecules28041125 (2023).
Gupta, R., Sharma, M. & Patel, A. Analysis of aripiprazole formulations using DSC and FTIR techniques for enhanced drug delivery. Pharmaceutics 15, 1450. https://doi.org/10.3390/pharmaceutics15061450 (2023).
Mura, P., Maestrelli, F., D’Ambrosio, M., Luceri, C. & Cirri, M. Evaluation and comparison of solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs) as vectors to develop hydrochlorothiazide effective and safe pediatric oral liquid formulations. Pharmaceutics 13, 437. https://doi.org/10.3390/pharmaceutics13040437 (2021).
Wolska, E. & Brach, M. Distribution of drug substances in solid lipid microparticles (SLM)—Methods of analysis and interpretation. Pharmaceutics 14, 335. https://doi.org/10.3390/pharmaceutics14020335 (2022).
Patil, G. B., Patil, N. D., Deshmukh, P. K., Patil, P. O. & Bari, S. B. Nanostructured lipid carriers as a potential vehicle for carvedilol delivery: Application of factorial design approach. Artif. Cells Nanomed. Biotechnol. 44, 12–19. https://doi.org/10.3109/21691401.2014.909820 (2016).
Shinde, C. G. et al. Intra-articular delivery of a methotrexate loaded nanostructured lipid carrier based smart gel for effective treatment of rheumatic diseases. RSC Adv. 6, 12913–12924. https://doi.org/10.1039/C5RA22672D (2016).
Samiun, W. S., Ashari, S. E., Salim, N. & Ahmad, S. Optimization of processing parameters of nanoemulsion containing aripiprazole using response surface methodology. Int. J. Nanomed. 15, 1585–1594. https://doi.org/10.2147/IJN.S198914 (2020).
Patel, R. B., Patel, M. R., Bhatt, K. K. & Patel, B. G. Formulation consideration and characterization of microemulsion drug delivery system for transnasal administration of carbamazepine. Bull. Fac. Pharm. Cairo Univ. 51, 243–253. https://doi.org/10.1016/j.bfopcu.2013.07.002 (2013).
Prajapati, J. B. & Patel, G. C. Nose to brain delivery of rotigotine loaded solid lipid nanoparticles: Quality by design based optimization and characterization. J. Drug Deliv Sci. Technol. 63, 102377. https://doi.org/10.1016/j.jddst.2021.102377 (2021).
Radtke, M., Müller, R. & Souto, E. Nanostructured lipid carriers: A novel generation of solid lipid drug carriers. Pharm. Technol. Eur. 17, 45–50 (2005).
Silki; Sinha, V. R. Enhancement of in vivo efficacy and oral bioavailability of aripiprazole with solid lipid nanoparticles. AAPS PharmSciTech. 19, 1264–1273. https://doi.org/10.1208/s12249-017-0944-5 (2018).
K M, A. S. et al. Box-Behnken design-based optimization and evaluation of lipid-based nano drug delivery system for brain targeting of bromocriptine. Pharmaceuticals 17, 720. https://doi.org/10.3390/ph17060720 (2024).
Ghadiri, M., Young, P. & Traini, D. Strategies to enhance drug absorption via nasal and pulmonary routes. Pharmaceutics 11 (113). https://doi.org/10.3390/pharmaceutics11030113 (2019).
Kumar, A. & Jain, S. K. Preliminary studies for the development of intranasal nanoemulsion containing CNS agent: emphasizing the utilization of cut and weigh method. Artif. Cells Nanomed. Biotechnol. 45, 515–521. https://doi.org/10.3109/21691401.2016.1160405 (2017).
Illum, L. Nasal drug delivery—Possibilities, problems and solutions. J. Control Release. 87, 187–198. https://doi.org/10.1016/S0168-3659(02)00363-2 (2003).
Acknowledgements
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Large Groups [RGP. 2/233/46]. The authors thank JSS College of Pharmacy, Mysuru and JSS Academy of Higher Education and Research, Mysuru for providing all the facilities for carrying out the research work.
Funding
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Large Groups [RGP. 2/233/46].
Author information
Authors and Affiliations
Contributions
Conceptualization, M.P.V., and M.R.; methodology, M.P.V., M.R. and B.R.; software, S.A.F., and S.S.B.; validation, I.P. and M.P.V.; formal analysis, B.R., D.R.P., and H.H.S.; investigation, B.R., and H.H.S.; resources, M.R. and M.M.A.; data curation, S.S.B., M.G. and M.M.A.; writing—original draft preparation, M.P.V., B.R., and D.R.P.; writing—review and editing, S.A.F., M.R., and M.G.; visualization, M.R., I.P., and M.G.; supervision, M.P.V.; project administration, M.P.V; funding acquisition, M.R.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Institutional review board statement
was not required as no live animals or humans were used. Fresh sheep blood and nasal mucosa was obtained from a local slaughterhouse following standard ethical guidelines for tissue sourcing.” However, we can affirm that all procedures and experimental protocols were carried out in accordance with the relevant institutional guidelines and regulations, and the reporting of methods complies with the ARRIVE guidelines, no animals were sacrificed for this study, and that this aligns with the ‘3Rs’ principle (Replacement). so animal ethical approval is “Not applicable”.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rahamathulla, M., Bindiya, R., Venkatesh, M.P. et al. Design and optimization of intranasal aripiprazole-loaded nanostructured lipid carriers for enhanced brain targeting in schizophrenia: in vitro and ex vivo evaluation. Sci Rep 15, 45118 (2025). https://doi.org/10.1038/s41598-025-32985-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-32985-w
