
Abstract
Multi-targeting drug design has become a prevalent attitude in exploring effective potent anticancer agents. In this regard, cyclooxygenase II (COX 2), tubulin, and focal adhesion kinase (FAK) are emerging as highly effective targets in the battle against cancer. Pyrazoles AS-(1–16) were prepared, and their structures were confirmed using NMR and IR tools. The designed compounds AS-(1–16) were investigated for their COXs inhibition and antiproliferative activities. Compounds AS-1, 2, and 14 showed high activities as COX inhibitors and antiproliferatives. Compounds AS-1, 2, and 14 were further investigated for their inhibitory activities towards tubulin polymerization, FAK, EGFR, BRAF and AS-14 showed the highest pronounced activities. AS-14 was subjected to cell cycle and showed activity at cell growth arrest G2/M. Likely the apoptotic activity of AS-14 in this cell line is triggered by pathways involving up-regulation of the proapoptotic proteins p53, Bax, and caspase-7 and down-regulation of the anti-apoptotic protein, Bcl-2. These findings illustrate the role of AS-14 in triggering the apoptotic pathway and shed light on a novel therapeutic drug in breast cancer. Docking studies were performed for the potent ligands to confirm the mechanism of action and showed good binding and a low energy score compared with the standard, especially for AS-14 as COX-2 inhibitor (-11.3 kcal/mol). Its molecular dynamics simulation with the target enzyme revealed high stability over the 200 ns trajectory.
Similar content being viewed by others
Introduction
In 2020, cancer surpassed the rates of stroke and coronary heart disease as one of the main causes of mortality, accounting for about 10 million deaths globally. His aggressiveness has exacerbated the most severe and lethal diseases during the past four years, particularly Covid-19, over the previous four instances. Breast cancer (BC) is a significant health issue marked by the unregulated proliferation of glandular breast tissue, leading to malignant tumors with considerable proliferative, invasive, and metastatic capabilities. Globally, BC has overtaken lung cancer as the most prevalent cancer in women (11.7%), with lung cancer (11.4%) becoming the fifth-ranking disease-causing death in women according to GLOBOCAN 2020 data1. Utilizing molecular biology approaches and gene expression profiles, BC can be categorized into five clinical characteristics and treated according to the presence of estrogen receptor (ER), progesterone receptor (PR), or human epidermal growth factor receptor (HER)−2 positivity2. Breast cancer (BC) arises from substantial modifications in genetic and epigenetic mechanisms, affecting multiple signaling pathways involved in growth and malignant progression towards an incurable and deadly condition3. Although the most effective strategy in clinical practice is chemotherapy, it suffers from several drawbacks, including poor drug selectivity, acquired drug resistance, recurrence, and metastasis after drug therapy4,5. Different approaches have been followed to overcome these limitations. Among these approaches, the combination strategy therapy, has proven its effectiveness for BC treatment and prevention of its progression more than the single-cell signaling pathway strategy6. Unfortunately, it faced other challenges due to its high cost, toxicity, and high potential for drug–drug interactions7,8,9. Additionally, the multi-target approach became an attractive competitor to the combination therapy approach because of its lower toxicity and fewer drug–drug interaction drawbacks.
One of the imperious targets for anticancer drugs is microtubules. They are cytoskeletal components vital for cell division, which are composed of α- and β-tubulin heterodimer assembly10. Microtubule targeting agents (MTA) act through binding with tubulin, leading to interruption of microtubule dynamics, triggering mitotic arrest, and rapidly growing cancer cells apoptosis11. There are at least four binding sites via which MTAs are known to interact with tubulin: the laulimalide, taxane/epothilone, vinca alkaloid, and colchicine sites. Significant scientific focus has been placed on the colchicine site, sometimes referred to as the colchicine-binding site (CBS), among these MTAs. Examples of the colchicine binding site inhibitors (CBSIs) that are currently known include colchicine (COL; I), combretastatin A-4 (CA4; II), and its equivalents III, and Crolibulin (IV), as shown in Fig. 1. The limited therapeutic windows and lack of oral bioavailability are two of the major challenges in creating this class of compounds as anticancer medications. Up to now, their therapeutic performance as single treatments has been unimpressive, and side effects such as brain toxicity, cardiovascular, and thromboembolic events continue to be serious concerns. So, a combination or multi-target protocol of CBSIs with other anticancer agents suggests a profitable direction to compensate for these obstacles.

The chemical structures of some known tubulin inhibitors.
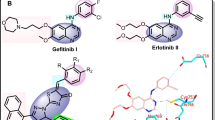
Multi-target strategy became an essential tool to counteract cancer drug resistance12. Non-receptor tyrosine protein kinase Focal adhesion kinase (FAK) is overexpressed in various cancer cells, like prostate, lung, breast, mesothelioma, cervical and kidney cancer cells13,14,15. FAK mostly acts through promotion the adhesion, R2 proliferation and angiogenesis of tumors16,17. Although numerous FAK inhibitors have been discovered, 6 compounds have reached anticancer clinical trials, among them VS-4718 (Phase I), GSK2256098 (Phase II), and defactinib (Phase II) (Fig. 2a.)18,19,20. Moreover, the decisive role of the epidermal growth factor receptor (EGFR) in innumerable cancers, including pancreatic cancer, non-small-cell lung cancer (NSCLC), colorectal cancer, squamous cell carcinoma of the head and neck, and breast cancer21. Plentiful EGFR inhibitors had been approved by FDA such as roclietinib, alflutinib, and osimertinib as shown in Fig. 222,23,24. Additionally, there is a hypothesis that the resistance mechanism occurred after EGFR treatment is due to acquired BRAFV600E mutation25,26. Moreover, EGFR is activated when BRAF is suppressed, which consequently leads to ongoing tumor proliferation27. Therefore, there are various FDA-approved drugs acting through dual inhibition of BRAFV600E and VEGFR-2 such as sorafenib and Regorafenib (Fig. 2a.).

(a) The chemical structures of some known FAD-approved FAK, EGFR and dual EGFR/BRAF inhibitors. (b) The chemical structures of some known FDA pyrazole-based anticancer drugs.
Additionally, on the other hand, the recent studies demonstrated that chronic inflammation raises the risk of developing a number of malignancies28,29. Overexpression of inflammatory cytokines, including TNF-α, IL-6, and IL-8, as well as some inflammatory pathways like COX-2 have critical roles in promoting tumorigenesis30. Depletion of COX-2 or prostaglandin E synthases changes the tumor’s inflammatory character to anticancer immunological pathways31,32,33,34. Furthermore, long-term treatment with NSAIDs has been shown to reduce the development of some malignancies and lower mortality rates in patients who received combination therapy with NSAIDs35,36. Additionally, non-steroidal anti-inflammatory drugs (NSAIDs) are utilized as complementary therapy with chemotherapeutics to lessen cancer’s pain or its treatment37,38. Selective COX-2 inhibitors have been identified as potential anti-proliferative agents for colorectal, breast, and prostate cancers, particularly in cases where there is an up-regulation of the COX-2 enzyme39,40. For example, celecoxib; a selective COX-2 inhibitor; provides anti-proliferative activity against several cancer cells, in addition to its administration with conventional chemotherapeutics, due to its chemo sensitizing effect41,42,43,44,45.
Collectively, in breast cancer, there is a strong linking between EGFR and COX-2. Activation of EGFR can lead to COX-2 upregulation through signaling pathways like PI3K/Akt and MAPK. This connection is noteworthy because it pushes invasiveness and a self-perpetuating loop. On the other hand, COX-2 can also activate EGFR, generating a closed cycle that encourages cancer growth. This relationship recommends that inhibition of both EGFR and COX-2 could be a favorable therapeutic strategy. Additionally, there is a strong link between EGFR/BRAF where blocking one can led to the other’s activation. So, a combination therapy that simultaneously targets both EGFR and BRAF may be more effective in inhibiting tumor growth and preventing resistance46,47,48,49.
From all the aforementioned data, it was hypothesized that targeting both the EGFR/BRAF pathway and COX-2 activity could be a probable therapeutic strategy in breast cancer, as single-agent often overcome resistance due to the signaling redundancies. Hereafter, it was expected that the synthesis of the novel multi-target compounds may wrestle the resistance obstacle and provide a more effective therapeutic agent.
Rationale and design
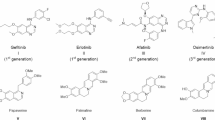
A literature survey of several potent FDA antineoplastic drugs containing a pyrazole ring have been reported against diverse cancer types. For example: XalkoriTM (crizotinib) and LorbrenaTM (lorlatinib) against non-small cell lung carcinoma (NSCLC); BraftoviTM (encorafenib), BalversaTM (erdafitinib), and Ruxolitinib against skin cancer, advanced urothelial cancer, and bone marrow cancer50,51,52,53,54. Additionally, NubeqaTM (darolutamide) has been approved to be used against prostate cancer55 as shown in Fig. 2b. So, our strategy depends on the utilization of a celecoxib, and SC-558-based pharmacophore as the major part for targeting the COX-2 enzyme due to the presence of the essential COX-2 pharmacophoric moieties: a Y-shaped structure (diaryl-heterocycles), a sulfonamide group at the p-position of one of the two phenyl rings, and the presence of a pyrazole ring, which has been critical for the activity.
Subsequently, other anticancer fragments are added to the pyrazole scaffold to improve its anti-proliferative activity. For example, the hydrazide-hydrazones linkage is well known to afford superfluous pharmacophoric points via including H-bond sites56. Additionally, it provides a fascinating option over hydrazides for prodrug synthesis through blocking of the NH2 group in the hydrazide linker. Also, hydrazone fragments show favorable anticancer activity due to their ability to be hydrolyzed in both in vitro and in vivo, providing less toxic metabolites57,58. Compound V affords high anticancer activity through interfering with DNA synthesis and angiogenesis inhibition59.
Furthermore, nitrogen-containing heterocyclic compounds such as oxadiazole have been profitably sighted for their efficacy as anti-tumor agents. Its important activity referred to its stability in aqueous medium, and its capability to form variable interactions with the different amino acids, including π –π interactions or strong hydrogen bonds60. 1,3,4-Oxadiazole derivatives provide their antitumor activity through various mechanisms including inhibition of epidermal growth factor receptor (EGFR) or tubulin polymerization, and histone deacetylases (HDAC), as illustrated in compounds VI-VIII61,62,63,64,65,66 Moreover, thiosemicarbazone linkers exhibit potential anticancer activity as reported in Triapine through growth inhibition as shown in Fig. 367,68,69,70.

The chemical structures of celecoxib and some known anticancer drugs containing different fragments.
From the abovementioned scientific literature results, the current work suggested a schematic design for the synthesis of novel diarylphenylpyrazole hybrids AS-1:AS-16. That is carried out by hybridization of the diphenylpyrazole core with different fragments, including hydrazones, thiosemicarbazones, pyrazole, and oxadiazoles, targeting optimization the antiproliferative ability through amalgamation of the pyrazole scaffold yielding hydrazone derivatives as in AS-1:AS-7, or with thiosemicarbazide derivatives yielding thiosemicarbazone derivatives as shown in compounds AS-8:AS-10. Moreover, it can be unifying with dicarbonyl compounds like succinic anhydride or phthalic anhydride, producing AS-11 and AS-12. Additionally, merging the pyrazole scaffold with a heterocyclic five-member ring like an isoxazole or dihydropyrazole ring affords AS-12:AS-16, as illustrated in Fig. 4.
The newly synthesized derivatives AS-1:AS-16 were assessed for their COXs, 5-LOX, IL-1β, IL-6, and TNF-α enzyme inhibitory activities, followed by their antiproliferative activity against human pancreatic cancer (PaCa-2), human non-small cell lung cancer (A-549), human breast (MCF-7), prostate cancer (PC-3), and human colon cancer (HT-29). Subsequently, the most favorable cytotoxic compound against breast cell lines was detected to enable us to assay its ability for suppression of tubulin, focal adhesion kinase (FAK), epidermal growth factor receptor (EGFR), and B-Raf Proto-Oncogene, Serine/Threonine Kinase (BRAF). Moreover, induction of apoptosis was detected via cell cycle analysis and exploration of caspase-3, caspase-7, caspase-9 activation, and Bax, and Bcl-2 protein levels. Finally, the binding modes within the enzyme active site were detected through the molecular modeling.

The designed strategy of target compounds.
Experimental
Chemistry
Material and methods
All chemicals and reagents were obtained from commercial suppliers without further purification. Melting points were uncorrected and were executed by open capillary tube methods using the IA 9100MK-Digital melting point apparatus. Elemental analysis was achieved at the micro- analytical center at the regional center for mycology and biotechnology, Al-Azhar University. Infrared spectra were conveyed on a Bruker FT-IR spectrophotometer Vector 22 and documented in wave number (cm− 1) using KBr discs at the micro-analytical center, Faculty of Science, Cairo University. 1H NMR and 13C NMR spectra were implemented with a Bruker APX400 spectrometer at 400 MHz and 101 MHz, respectively in the specified solvent at the Faculty of Pharmacy, Beni-Suef University. The J constant is given in Hz. Mass spectra were detected on a Finnegan MAT, SSQ7000 Mass spectrometer, at 70 eV (EI) at the micro-analytical center, Faculty of Science, Cairo University. Thin layer chromatography (TLC) was carried out using Kieselgel 60 F254 sheets (Merck, Darmstadt, Germany) and petroleum ether-ethyl acetate (6:4) as the eluting system.
Compounds 1a, 2a, 3 and 4 were prepared according to the reported procedures71,72,73,74,75.
General procedure A for synthesis of compounds (AS-1: AS-7)
A mixture of hydrazide intermediate 4 (0.357 g, 1 mmol) and various aldehydes/ketones (1.2 mmol) was dissolved in absolute ethanol in the presence of drops of glacial acetic acid. Heat the mixture under reflux for 3–24 h. The precipitate solid was collected by filtration and washed with ethanol. The crude products were purified by recrystallization from ethanol to obtain the target compounds.
4-(3-(2-(4-hydroxybenzylidene)hydrazine-1-carbonyl)−5-phenyl-1 H-pyrazol 1yl)benzenesulfonamide (AS-1): It was prepared as reported76.
4-(3-(2-(2-hydroxybenzylidene)hydrazine-1-carbonyl)−5-phenyl-1H-pyrazol-1-yl)benzenesulfonamide (AS-2): General procedure A, as buff solid (80%); m.p. 238℃. IR (cm− 1): 3416 (OH), 3127 (NH), 3061 (CH aromatic), 2923 (CH aliphatic), 1614 (CO), 1343, 1154 (SO2NH2).1H NMR (400 MHz, DMSO- d6): δ 12.15 (s, 1H, OH exchangeable with D2O), 11.32 (s, 1H, NH exchangeable with D2O), 8.74 (s, 1H, N = CH), 7.91 (d, J = 8.4 Hz, 2 H Ar-H), 7.60 (d, J = 8.4 Hz, 2 H, Ar-H), 7.52 (d, J = 6.9 Hz, 3 H, Ar-H), 7.43 (m, 2 H, Ar-H), 7.34 (s, 2 H, SO2NH2 exchangeable with D2O), 7.31 (d, J = 7.8 Hz, 1H, Ar-H), 7.20 (s, 1H, Ar-H), 6.95 (d, J = 8.6 Hz, 2 H, Ar-H). 13C NMR (101 MHz, DMSO- d6): δ 157.92 (C1), 157.81 (C2), 149.38 (C3), 146.64 (C4), 145.31(C5), 144.18 (C6), 141.98 (C7), 131.97 (C8), 130.09 (C9), 129.65 (C10), 129.34 (C11), 129.25 (C12), 127.19 (C13), 126.46 (C14), 119.90 (C15), 119.14 (C16), 116.92 (C17), 109.39 (C18). Anal. Calcd. for C23H19N5O4S: C, 59.86; H, 4.15; N, 15.18. Found: C, 59.85; H, 4.16; N, 15.19.
4-(3-(2-(3,4-dimethoxybenzylidene)hydrazine-1-carbonyl)−5-phenyl-1H-pyrazol-1-yl)benzenesulfonamide (AS-3): General procedure A, as white solid (65%); m.p. 197℃. IR (cm− 1): 3242 (NH), 3049 (CH aromatic), 2949 (CH aliphatic), 1694 (CO), 1321, 1165 (SO2NH2).1H NMR (400 MHz, DMSO- d6): δ 11.76 (s, 1H, NH exchangeable with D2O), 8.50 (s, 1H, N = CH), 7.92 (d, J = 8.5 Hz, 2 H, Ar-H), 7.61 (d, J = 8.5 Hz, 2 H, Ar-H), 7.54 (m, 2 H, Ar-H), 7.47–7.40 (m, 3 H, Ar-H), 7.39 (m, 1H, Ar-H), 7.35 (s, 2 H, SO2NH2 exchangeable with D2O), 7.20 (d, J = 4.2 Hz, 2 H, Ar-H), 7.04 (m, 2 H, Ar-H), 7.03 (d, J = 8.6 Hz, 2 H, Ar-H), 3.84 (s, 3 H, OCH3), 3.82 (s, 3 H, OCH3). 13C NMR (101 MHz, DMSO d6): δ 157.77 (C1), 151.30 (C2), 149.58 (C3), 148.96 (C4), 147.25 (C5), 145.16 (C6), 144.15 (C7), 142.03 (C8), 129.58 (C9), 129.37 (C10), 129.32 (C11), 129.25 (C12), 127.50 (C13), 127.19 (C14), 126.42 (C15), 122.55 (C16), 111.94 (C17), 109.35 (C18), 108.67 (C19), 56.04 (C20). Anal. Calcd. for C25H23N5O5S: C, 59.40; H, 4.59; N, 13.85. Found: C, 59.42; H, 4.60; N, 13.90.
4-(5-phenyl-3-(2-(3,4,5-trimethoxybenzylidene)hydrazine-1-carbonyl)−1 H-pyrazol-1-yl)benzenesulfonamide (AS-4) General procedure A, as reported76.
4-(5-phenyl-3-(2-(pyridin-3-ylmethylene)hydrazine-1-carbonyl)−1H-pyrazol-1-yl)benzenesulfonamide (AS-5): General procedure A, as white solid (75%); m.p. 207℃. IR (cm− 1): 3076 (CH aromatic), 2923 (CH aliphatic), 1626 (CO), 1339, 1194 (SO2NH2). 1H NMR (400 MHz, DMSO- d6): δ 12.05 (s, 1H, NH exchangeable with D2O), 8.85 (s, 1H, N = CH), 8.62 (s, 2 H, Pyridyl-H), 8.16 (d, J = 7.6 Hz, 1H, Pyridyl-H), 7.91 (d, J = 8.4 Hz, 2 H, Ar-H), 7.61 (d, J = 8.3 Hz, 2 H, Ar-H), 7.57–7.47 (m, 3 H, 2 H of Ar-H and 1H of Pyridyl-H), 7.43 (d, J = 3.0 Hz, 3 H, Ar-H), 7.35 (s, 2 H, SO2NH2 exchangeable with D2O), 7.22 (s, 1H). 13C NMR (101 MHz, DMSO- d6): δ 158.03 (C1), 151.21 (C2), 149.20 (C3), 146.93 (C4), 145.98 (C5), 145.27 (C6), 144.21 (C7), 142.35 (C8), 141.98 (C9), 134.02 (C10), 130.72 (C11), 129.62 (C12), 129.33 (C13), 129.26 (C14), 127.19 (C15), 126.47 (C16), 124.52 (C17), 109.44 (C18). MS (EI): m/z 446 (M+). Anal. Calcd. for C22H18N6O3S: C, 59.18; H, 4.06; N, 18.82. Found: C, 59.17; H, 4.07; N, 18.84.
4-(3-(2-(1-(4-bromophenyl)ethylidene)hydrazine-1-carbonyl)−5-phenyl-1H-pyrazol-1-yl)benzenesulfonamide (AS-6): General procedure A, as yellowish white solid (79%); m.p. 189℃. IR (cm− 1): 3341(NH), 3078 (CH aromatic), 2925 (CH aliphatic), 1685 (CO), 1331, 1173 (SO2NH2). 1H NMR (400 MHz, DMSO- d6): δ 10.59 (s, 1H, NH exchangeable with D2O), 7.92 (d, J = 8.5 Hz, 2 H, Ar-H), 7.83 (d, J = 8.4 Hz, 2 H, Ar-H), 7.63 (dd, J = 17.5, 8.5 Hz, 4 H, Ar-H), 7.53 (s, 2 H, Ar-H), 7.44 (m, 3 H, Ar-H), 7.35(s, 2 H, SO2NH2 exchangeable with D2O), 7.22 (s, 1H, Ar-H), 2.37 (s, 3 H, CH3). 13C NMR (101 MHz, DMSO-d6): δ 157.95 (C1), 153.89 (C2), 147.14 (C3), 145.25 (C4), 144.12 (C5), 141.96 (C6), 137.51 (C7), 132.20 (C8), 131.83 (C9), 130.65 (C10), 129.62 (C11), 129.35 (C12), 129.24 (C13), 128.94 (C14), 127.25 (C15), 126.28 (C16), 109.39 (C17), 14.24 (C18). MS (EI): m/z 537 (M+). Anal. Calcd. For C24H20BrN5O3S: C, 53.54; H, 3.74; N, 13.01; Found: C, 53.55; H, 3.73; N, 13.00.
4-(5-phenyl-3-(2-(1-(p-tolyl)ethylidene)hydrazine-1-carbonyl)−1H-pyrazol-1-yl)benzenesulfonamide (AS-7): General procedure A, as yellowish white solid (79%); m.p.189℃. IR (cm− 1): 3332 (NH), 2954 (CH aliphatic), 1692 (CO), 1312, 1170 (SO2NH2). 1H NMR (400 MHz, DMSO- d6): δ 10.52 (s, 1H, NH exchangeable with D2O), 7.90 (d, J = 8.5 Hz, 2 H, Ar-H), 7.77 (s, 2 H, Ar-H), 7.61 (s, 2 H, Ar-H), 7.52 (s, 2 H, Ar-H), 7.44 (m, 3 H, Ar-H), 7.35 (s, 2 H, SO2NH2 exchangeable with D2O), 7.27 (d, J = 7.8 Hz, 2 H, Ar-H), 7.22 (s, 1H, Ar-H), 2.36 (s, 6 H, CH3). 13C NMR (101 MHz, DMSO- d6): δ 157.78 (C1), 155.31 (C2), 147.31 (C3), 145.20 (C4), 144.11 (C5), 141.98 (C6), 139.74 (C7), 135.55 (C8), 129.68 (C9), 129.61 (C10), 129.46 (C11), 129.35 (C12), 129.25 (C13), 127.23 (C14), 126.89 (C15), 126.27 (C16), 109.32 (C17), 21.33 (C18), 14.37 (C19). Anal. Calcd. for C25H23N5O3S: C, 63.41; H, 4.90; N, 14.79. Found: C, 63.42; H, 4.89; N, 14.80.
General procedure B for synthesis of compounds (AS-8: AS-9)
A mixture of hydrazide intermediate 4 (0.357 g, 1 mmol) and appropriate isothiocyanate derivatives (ethyl and tolyl isothiocyanate) (1 mmol) in absolute ethanol (30 mL) was heated under reflux for 12 h. Filter the formed solid, dried and recrystallized from absolute ethanol.
2-(5-phenyl-1-(4-sulfamoylphenyl)−1H-pyrazole-3-carbonyl)-N-(p-tolyl)hydrazine-1-carboxamide (AS-8): General procedure B, as white solid (79%); m.p. 209℃. IR (cm− 1): 3256 (NH), 3112 (CH aromatic), 2989 (CH aliphatic), 1684, 1667 (CO), 1322, 1107 (SO2NH2). 1H NMR (400 MHz, DMSO- d6): δ 10.95 (s, 1H, NH exchangeable with D2O), 10.45 (s, 1H, NH exchangeable with D2O), 9.73 (s, 1H, NH exchangeable with D2O), 7.89 (d, J = 8.6 Hz, 2 H, Ar-H), 7.55 (d, J = 8.6 Hz, 2 H, Ar-H), 7.52 (m, 2 H, Ar-H), 7.43 (m, 3 H, Ar-H), 7.35 (s, 2 H, SO2NH2 exchangeable with D2O), 7.30 (m, 1H, Ar-H), 7.17 (s, 1H, Ar-H), 7.13 (d, J = 8.2 Hz, 1H, Ar-H), 2.29 (s, 1H, CH3). 13C NMR (101 MHz, DMSO-d6): δ 161.02 (C1), 146.72 (C2), 144.82 (C3), 144.03 (C4), 141.98 (C5), 137.16 (C6), 129.62 (C7), 129.41 (C8), 129.33 (C9), 129.24 (C10), 129.17 (C11), 128.94 (C12), 127.22 (C13), 126.23 (C14), 126.06 (C15), 109.47 (C16), 108.63 (C17), 21.00 (C18). Anal. Calcd. For C24H22N6O4S: C, 58.76; H, 4.52; N, 17.13. Found: C, 58.77; H, 4.50; N, 17.14.
N-(4-ethylphenyl)−2-(5-phenyl-1-(4-sulfamoylphenyl)−1H-pyrazole-3-carbonyl)hydrazine-1-carboxamide (AS-9): General procedure B, as yellow crystals (89%); m.p. 199℃. 1H NMR (400 MHz, DMSO-d6): δ 9.31 (s, 1H, NH exchangeable with D2O), 8.05 (s, 1H, NH exchangeable with D2O), 7.89 (d, J = 8.6 Hz, 2 H, Ar-H), 7.52 (d, J = 8.6 Hz, 2 H, Ar-H), 7.50 (m, 2 H, Ar-H), 7.43 (m, 3 H, Ar-H), 7.32 (s, 2 H, SO2NH2 exchangeable with D2O), 7.14 (s, 1H, Ar-H), 3.48 (m, 3 H, 2 H of CH2CH3 and 1H of NH), 1.07 (t, J = 7.1 Hz, 3 H, CH2CH3). 13C NMR (101 MHz, DMSO-d6): δ 160.95 (C1), 146.69 (C2), 144.80 (C3), 144.01 (C4), 141.97 (C5), 129.61 (C6), 129.44 (C7), 129.41 (C8), 129.16 (C9), 127.21 (C10), 126.03 (C11), 119.13 (C12), 109.45 (C13), 19.00 (C14), 14.92 (C15). MS (EI): m/z 444 (M+). Anal. Calcd. For C19H20N6O4S: C, 53.26; H, 4.71; N, 19.61. Found: C, 53.25; H, 4.73; N, 19.59.
General procedure C for synthesis of compounds (AS-10 and AS-11)
A mixture of the hydrazide intermediate 4 (0.357 g, 1 mmol) and carbonyl compounds (phthalic anhydride/succinic anhydride) (1 mmol) in ethanol with a few drops of glacial acetic acid was heated near the boiling point for 8 h, then left to cool to room temperature. The solid formed was filtered off, followed by crystallization from ethanol to afford the desired products.
N-(1,3-dioxoisoindolin-2-yl)−5-phenyl-1-(4-sulfamoylphenyl)−1H-pyrazole-3-carboxamide (AS-10): General procedure C, as buff solid (80%); m.p. 179 ℃. IR (cm− 1): 3357 (NH), 3154 (CH aromatic), 2887 (CH aliphatic), 1686 (CO), 1349, 1163 (SO2NH2).1H NMR (400 MHz, DMSO- d6): δ 11.24 (s, 1H, NH exchangeable with D2O), 8.02 (m, 2 H, Ar-H), 7.88 (m, 2 H, Ar-H), 7.90 (d, J = 8.6 Hz, 2 H, Ar-H), 7.61 (d, J = 8.6 Hz, 2 H, Ar-H), 7.53 (m, 2 H, Ar-H), 7.44 (m, 3 H, Ar-H), 7.36 (s, 2 H, SO2NH2 exchangeable with D2O), 7.22 (s, 1H, Ar-H). 13C NMR (101 MHz, DMSO- d6): δ 165.65 (C1), 160.53 (C2), 145.56 (C3), 145.15 (C4), 144.36 (C5), 141.89 (C6), 135.95 (C7), 129.86 (C8), 129.72 (C9), 129.34 (C10), 129.23 (C11), 129.14 (C12), 127.22 (C13), 126.57 (C14), 124.37 (C15), 109.43 (C16). MS (EI): m/z 487 (M+). Anal. Calcd. For C24H17N5O5S: C, 59.13; H, 3.52; N, 14.37. Found: C, 59.15; H, 3.51; N, 14.38.
N-(2,5-dioxopyrrolidin-1-yl)−5-phenyl-1-(4-sulfamoylphenyl)−1H-pyrazole-3-carboxamide (AS-11): General procedure C, as white solid (66%); m.p. 199 ℃. IR (cm− 1): 3260 (NH), 3028 (CH aromatic), 2989 (CH aliphatic), 1686 (CO), 1336, 1153 (SO2NH2).1H NMR (400 MHz, DMSO- d6): δ 9.94 (s, 1H, NH exchangeable with D2O), 7.92 (m, 2 H, Ar-H), 7.60 (m, 2 H, Ar-H), 7.52 (m, 2 H, Ar-H), 7.43 (m, 3 H, Ar-H), 7.33 (s, 2 H, SO2NH2 exchangeable with D2O), 7.17 (s, 1H, Ar-H), 2.86 (s, 4 H, CH2). 13C NMR (101 MHz, DMSO- d6): 174.71 (C1), 160.64 (C2), 145.46 (C3), 145.31 (C4), 144.26 (C5), 141.89 (C6), 129.34 (C7), 129.29 (C8), 129.22 (C9), 127.20 (C10), 126.50 (C11), 126.22 (C12), 109.07 (C13), 26.78 (C14). Anal. Calcd. For C20H17N5O5S: C, 54.66; H, 3.90; N, 15.94. Found: C, 54.67; H, 3.89; N, 15.95.
4-(3-(5-mercapto-1,3,4-oxadiazol-2-yl)−5-phenyl-1H-pyrazol-1-yl)benzenesulfonamide (AS-12): To hydrazide intermediate 4 (0.357 g, 1 mmol) dissolved in 50 mL absolute ethanol, (0.056 g, 1 mmol) of potassium hydroxide was added, and (0.19 g, 2.5 mmol) of carbon disulfide was slowly added dropwise with stirring. The mixture was subjected to reflux for 3 h followed by distillation of ethanol under reduced pressure. The residue was dissolved in water, filtered, and the filtrate was acidified with dilute 5% hydrochloric acid to pH 3 − 4. Filter the formed precipitated solid, washed, and dried to give a white solid, 89% yield and m.p.207℃. IR (cm− 1): 3361 (NH), 3157 (CH aromatic), 2925 (CH aliphatic), 1687 (CO), 1363, 1153 (SO2NH2), 1245 (C = S). 1H NMR (400 MHz, DMSO- d6): δ 7.91 (m, 2 H, Ar-H), 7.58 (m, 1H, Ar-H), 7.54 (m, 3 H, Ar-H), 7.44 (m, 3 H, Ar-H), 7.35 (s, 2 H, SO2NH2 exchangeable with D2O), 7.27 (s, 1H, Ar-H). 13C NMR (101 MHz, DMSO- d6): δ 177.80 (C1), 156.36 (C2), 145.71 (C3), 144.27 (C4), 141.68 (C5), 137.67 (C6), 129.86 (C7), 129.35 (C8), 129.33 (C9), 128.79 (C10), 127.28 (C11), 126.20 (C12), 108.39 (C13). MS (EI): m/z 399 (M+). Anal. Calcd. For C17H13N5O3S2: C, 51.12; H, 3.28; N, 17.53. Found: C, 51.11; H, 3.29; N, 17.55.
4-(3-(5-(4-bromophenyl)−1H-pyrazole-1-carbonyl)−5-phenyl-1H-pyrazol-1-yl) benzenesulfonamide (AS-13): Firstly, the enamineone of 4-bromoacetophenone was prepared as reported by mixing a mixture of 4-bromoacetophenone (0.995 g, 5 mmol) and DMF-DMA (2.7 mL, 2.38 g, 20 mmol) in DMF (2 mL) and heating at 150 ℃ for 20 h. The reaction mixture was evaporated; the crude residue was suspended with diethyl ether, and the suspension was stirred for 15 min. The solid was filtered and used without further purification for the next reaction as reported77. Subsequently, the hydrazide intermediate 4 (0.357 g, 1 mmol) and enamineone of 4-bromoacetophenone (0.253 g, 1 mmol) in glacial acetic acid (20 ml) were heated under reflux for 5–6 h. The formed solid precipitate, was filtered off, recrystallized from a mixture of ethanol and dioxane, and dried to give a brown solid, 87% yield and m.p. 277-9 ℃. IR (cm− 1): 3367 (NH), 3216 (CH aromatic), 2924 (CH aliphatic), 1629 (CO), 1304, 1177 (SO2NH2). 1H NMR (400 MHz, DMSO-d6): δ 9.08 (s, 1H, Ar-H), 8.53 (m, 1H, Ar-H), 8.22 (m, 1H, Ar-H), 7.89 (m, 3 H, Ar-H), 7.86–7.81 (m, 2 H, Ar-H), 7.78 (s, 3 H, Ar-H), 7.54–7.41 (m, 3 H, Ar-H), 7.41–7.34 (m, 2 H, SO2NH2 exchangeable with D2O), 7.26 (m, 1H, Ar-H), 6.76 (s, 1H, Ar-H). 13C NMR (101 MHz, DMSO-d6): δ 165.63 (C1), 143.61 (C2), 143.26 (C3), 142.47 (C4), 134.89 (C5), 132.53 (C6), 132.47 (C7), 132.09 (C8), 131.75 (C9), 131.62 (C10), 130.12 (C11), 129.46 (C12), 129.27 (C13), 129.07 (C14), 128.78 (C15), 127.05 (C16), 125.62 (C17), 125.02 (C18), 109.44 (C19). Anal. Calcd. For C25H18BrN5O3S: C, 54.75; H, 3.31; N, 12.77. Found: C, 54.76; H, 3.30; N, 12.77.
General procedure D for synthesis of compounds (AS-14 and AS-15)
A mixture of the hydrazide intermediate 4 (0.357 g, 1 mmol), aromatic acids (0.01 mol) was heated under reflux for 24 h in the presence of phosphorous oxychloride (0.76 g, 5 mmol). Cool the reaction mixture; pour it onto the crushed ice and let it sit overnight. The collected solid was filtered, dried, and recrystallized from ethanol.
4-(3-(5-(4-methoxyphenyl)−1,3,4-oxadiazol-2-yl)−5-phenyl-1H-pyrazol-1-yl)benzenesulfonamide (AS-14): General procedure D, as white solid (79%); m.p. 209℃. IR (cm− 1): 3429 (NH), 3115 (CH aromatic), 2926 (CH aliphatic), 1615 (CO), 1317, 1127 (SO2NH2). 1H NMR (400 MHz, DMSO-d6): δ 8.08 (m, 2 H, Ar-H), 7.60–7.78 (m, 3 H, Ar-H), 7.44 (m, 4 H, Ar-H), 7.37 (m, 6 H, 4 H of Ar-H and 2 H of SO2NH2), 7.18 (s, 2 H, Ar-H), 3.87 (s, 3 H, OCH3). 13C NMR (101 MHz, DMSO-d6): δ 164.30 (C1), 162.67 (C2), 159.53 (C3), 148.75 (C4), 145.40 (C5), 139.42 (C6), 138.09 (C7), 129.61 (C8), 129.28 (C9), 129.19 (C10), 129.09 (C11), 127.02 (C12), 125.46 (C13), 125.40 (C14), 115.95 (C15), 115.44 (C16), 108.34 (C17), 56.04 (C18). MS (EI): m/z 473.1 (M+); 90.89 (base peak). Anal. Calcd. For C24H19N5O4S: C, 60.88; H, 4.04; N, 14.79. Found: C, 60.90; H, 4.05; N, 14.80.
4-(5-phenyl-3-(5-phenyl-1,3,4-oxadiazol-2-yl)−1H-pyrazol-1-yl)benzenesulfonamide (AS-15): General procedure D, as white crystals (75%); m.p. 109 ℃. IR (cm− 1): 3388 (NH), 3116 (CH aromatic), 2992 (CH aliphatic), 1696 (CO), 1364, 1180 (SO2NH2). 1H NMR (400 MHz, DMSO-d6): δ 8.08 (m, 2 H, Ar-H), 7.64–7.78 (m, 3 H, Ar-H), 7.49–7.53 (m, 2 H, Ar-H), 7.42 (m, 5 H, Ar-H), 7.33 (s, 2 H, SO2NH2 exchangeable with D2O), 7.17 (s, 1H, Ar-H). 13C NMR (101 MHz, DMSO-d6): δ 160.96 (C1), 145.51 (C2), 139.48 (C3), 130.00 (C4), 129.31 (C5), 129.27 (C6), 129.14 (C7), 127.22 (C8), 127.03 (C9), 126.97 (C10), 126.88 (C11), 125.57 (C12), 125.52 (C13), 125.39 (C15), 123.95 (C16), 108.34 (C17). MS (EI): m/z 443.5 (M+). Anal. Calcd. For C23H17N5O3S: C, 62.29; H, 3.86; N, 15.79. Found: C, 62.27; H, 3.85; N, 15.80.
4-(3-(5-(chloromethyl)−1,3,4-oxadiazol-2-yl)−5-phenyl-1H-pyrazol-1-yl)benzenesulfonamide (AS-16): A mixture of the hydrazide intermediate 4 (0.357 g, 1 mmol), chloroacetic acid (0.0945 g, 1 mmol) in xylene was heated under reflux for 10 h in the presence of phosphorous oxychloride (0.15 g, 1 mmol). Cooling the reaction mixture and pouring it onto the crushed ice were carried out until producing a solid mass. Recrystallization was carried out from ethanol to afford a white solid (87%); m.p. 217℃. IR (cm− 1): 3265 (NH), 3156 (CH aromatic), 2963 (CH aliphatic), 1663 (CO), 1330, 1160 (SO2NH2). 1H NMR (400 MHz, DMSO- d6): δ 7.90 (m, 1H, Ar-H), 7.67–7.79 (m, 2 H, Ar-H),7.54–7.60 (m,1H, Ar-H) 7.40–7.42 (m, 4 H, Ar-H), 7.30–7.36 (m, 4 H, 2 H of Ar-H and 2 H of SO2NH2), 7.18 (s, 2 H, Ar-H), 3.87 (s, 3 H, OCH3). 13C NMR (101 MHz, DMSO- d6): δ 165.81 (C1), 163.24 (C2), 160.92 (C3), 144.85 (C4), 129.39 (C5), 129.30 (C6), 129.26 (C7), 129.22 (C8), 129.12 (C9), 127.03 (C10), 126.88 (C11), 125.43 (C12), 108.63 (C13), 33.60 (C14). MS (EI): m/z 415.1 (M+). Anal. Calcd. For C18H14ClN5O3S: C, 51.99; H, 3.39; N, 16.84. Found: C, 51.97; H, 3.41; N, 16.85.
Biological evaluation
Anti-inflammatory activity studies
In vitro COX-1 and COX-2 Inhibition assay
The IC50 assay for synthesized pyrazole derivatives AS-1–16 was performed in triplicate utilizing the inhibitory COX assay kit according to the manufacturer instructions (Cayman Chemical Company). The assay was performed depending on the direct measure prostaglandin 2α resulting from cyclooxygenase. The positive control for selective COX-2 inhibition activity was celecoxib (MW = 381.37), while diclofenac was resembled as a non-selective COX-1/COX-2 NSAID. Reaction buffer (Tris–HCl 0.1 M, pH 8.0, containing 5 mM EDTA and 2 mM phenol) with a volume of 20 µl that contained 10 µl from tested compounds (AS-1–16) was pre-incubated for 15 min at 37 °C with the enzyme before arachidonic acid addition. Afterword, a volume of 10 µl from arachidonic acid 10 mM was then added for 2 min at 37 °C to start the reaction. To stop the reaction, the addition of 1 N HCl 50 µl and saturated stannous chloride was added. Investigation was achieved by ovine 100 U of COX-1 and recombinant human COX-2. The resulting prostanoid was measured by the spectrophotometric method. The selectivity indices (SI, COX-1 IC50/COX-2 IC50) were relative to celecoxib, the standard COX-2 selective inhibitor [78,79,80].
Antiproliferative activity
Human cancer cell lines were used involving A-549 (lung cancer cells), PaCa-2 (pancreatic adenocarcinoma), PC-3 (prostate cancer cells), MCF-7 (breast cancer cells), and HT-29 (colorectal cancer cells) and were purchased from the American Type Culture Collection (ATCC, VA, USA). Dulbecco’s Modified Eagle Medium (DMEM) or RPMI-1640 culture media were prepared with fetal bovine serum (FBS) 10% and 1% penicillin-streptomycin, and the incubation of the cells was at 37 °C in 5% CO₂80. In 96-well microplates, cells were seeded to reach a density of 5 × 10⁴ cells/mL for 24 h. Afterward, cell types were stimulated with new synthesized pyrazole derivatives, AS-1: AS-16 which were dissolved in DMSO for 48 h. After 48-hr incubation, the used media were removed and the cells were washed two times with phosphate-buffered saline (PBS). A volume of 100 µL of 50 µg/mL in PBS Propidium Iodide (PI) staining solution was used and incubated at RT for 15 min in the dark. The dead cells were stained with PI by intercalating into DNA81. The fluorescence intensities were determined using a fluorescence plate reader at 535/617 nm excitation/emission. Data resulted in quantification as the % of viable cells relative to the untreated cells. The IC₅₀ was calculated using regression analysis (non-linear) in GraphPad Prism software (version 9.0). The process was performed in triplicate samples, and results were denoted as mean ± standard error of means (SEM)82.
Enzyme Inhibition assays
Tubulin polymerization Inhibition assay
The tubulin polymerization inhibition activity of the synthesized compounds AS-1, AS-2, and AS-14 was evaluated in vitro according to the method described by Abdelhameid et al.83. In brief, the purified tubulin proteins from bovine brain dissolved in G-PEM buffer (1 mM MgCl₂, 80 mM PIPES, 1 mM EGTA, and pH 6.9) supplemented with 1 mM GTP. The tested compounds at different concentrations (1–100 µM) were incubated at 37 °C in a 96-well plate with the tubulin solution, and the polymerization step was initiated by gradual heating, and the increase in absorbance at 1 min intervals for 60 min at 340 nm was recorded spectrophotometrically. To compare depolymerizing and polymerizing effects, vincristine, a tubulin destabilizer, and docetaxel, a tubulin stabilizer, were used, as reference standard84. In comparison to vehicle control (DMSO), the extent of β-tubulin polymerization inhibition was assessed by calculating the area under the curve (AUC) of the polymerization profile. Active inhibitors of β-tubulin polymerization, are considered for the compounds that significantly reduced the AUC in a concentration-dependent manner. The results were expressed as mean ± standard errors of means (SEM). Using non-linear regression analysis, the inhibitory concentration (IC₅₀) values for tubulin polymerization were determined85.
FAK inhibitory assay
The FAK inhibitory activity of the newly synthesized compounds was evaluated using kinase assay in vitro based on the quantification of phosphorylated FAK at Tyr397. Recombinant human FAK enzyme and a synthetic peptide substrate specific to FAK activity were used according to the protocol described by86. In brief, the tested compounds at different concentrations (typically 0.01–100 µM) were incubated at 30 °C for 60 min with the FAK enzyme in a kinase reaction buffer containing MgCl₂, ATP, and substrate peptide in 96-well microplates. After adding stop solution, the level of phosphorylated substrate was estimated by the phospho-Tyr397-specific ELISA kit method. The % inhibition was calculated in relation to vehicle control, and the IC₅₀ values were determined using nonlinear regression analysis. All experiments were performed in triplicate, and the resulted data were represented as mean ± standard error of means (SEM)87.
EGFR (epidermal growth factor receptor) inhibitory assay
The EGFR inhibitory activity of the synthesized compounds was evaluated based on the quantification of EGFR autophosphorylation using a cell-free in vitro kinase assay. The assay was carried out according to protocol of88. Different concentrations (typically 0.01–100 µM) from the compounds were incubated at 30 °C for 60 min in 96-well microplates with the EGFR enzyme. The termination of the reaction was done by the stop solution, and measuring of the phosphorylated substrate level was by a phospho-EGFR (Tyr1068) ELISA kit. Inhibitory activity was estimated in relation to vehicle-controls (0.5% DMSO), and the values of IC₅₀ were calculated using non-linear regression analysis with GraphPad Prism software. Experiment was performed as triplicate, and data were represented as mean ± standard error of means (SEM)89,90.
BRAF inhibitory activity
The in vitro inhibitory activity of BRAF for the newly synthesized compounds is based on the inhibitory effect of the compounds on the recombinant human BRAF V600E kinase. A commercially available BRAF kinase assay kit (e.g., Promega or Thermo Fisher) was used according to the manufacturer’s instructions. In brief, the recombinant BRAF enzyme (10–20 ng) was incubated with its specific substrate (a peptide containing the MEK phosphorylation motif) in kinase reaction buffer (MgCl₂, ATP, and DTT) with or without the tested compound in different concentrations (from 0.01 to 100 µM) for 60 min at 30 °C. The degree of phosphorylation of the substrate was estimated by fluorescence recognition. The BRAF % inhibition was analyzed in relation to negative control. The determination of the IC₅₀ values was calculated using version 9 of the GraphPad Prism91,92.
Cell cycle analysis
Cell cycle arrest and distribution were performed using the propidium Iodide (PI) Flow Cytometry Kit (ab139418, Abcam). This kit is designed for quantitative DNA content analysis in tissue culture cells. 5 × 105 cells were cultured in the absence and presence of the tested compound (AS-14). The adherent cells were fixed in 66% ethanol at + 4 °C for 2 h washed with 100 mL PBS, and incubated with propidium iodide (PI) and 200 µL RNase staining solution for 30 min in the dark at room temperature. Finally, propidium iodide fluorescence intensity was collected on FL2 of a flow cytometer and 488 nm laser excitation93.
Annexin V-FITC apoptosis analysis
Apoptosis analysis was carried out using a flow cytometer with an Annexin V-FITC/PI double staining apoptosis detection kit (K101, Biovision) to study apoptosis and cell cycle distribution of MCF-7 cells treated with compound AS-14. 5 × 105 cells were incubated with Annexin V-FITC and apoptosis was induced. Cells were collected and resuspended in 500 µL of 1X Binding buffer.5 µl of Annexin V-FITC, and 5 µL of propidium iodide (PI) were added, and cells were incubated for 5 min at room temperature in the dark. Annexin V-FITC binding by flow cytometry was analyzed using the FITC signal detector (FL1) and PI staining by the phycoerythrin emission signal detector (FL2)94,95.
Apoptosis regulatory genes
Evaluation of P53, Caspase7, BAX and bcl2 gene expression
Gene expression of P53 was evaluated by real-time quantitative PCR (qPCR) to evaluate the effect of the newly synthesized compounds. The total extracted RNA contents from the control and compound-treated MCF-7 cell line were performed using a commercial isolation kit for RNA (Qiagen RNeasy Mini Kit) according to the manufacturer’s directions. The extracted samples in terms of purity and concentration were estimated using a NanoDrop spectrophotometer. An amount of 1 µg from samples was transcribed reversely to form cDNA using (Thermo Scientific RevertAid Kit). The Green Master Mix, SYBR (Applied Biosystems) was estimated using (ABI 7500 Fast Real-Time PCR System). Specific primers used as follow; for P53: F 5’- CCTCAGCATCTTATCCGAGTGG − 3’ R 5’- TGGATGGTGGTACAGTCAGAGC − 3’, the primer sequence for caspase 7: F 5’- CGGAACAGACAAAGATGCCGAG − 3’ R 5’- AGGCGGCATTTGTATGGTCCTC-3’, for BAX: F 5’- TCAGGATGCGTCCACCAAGAAG-3’ R 5’- TGTGTCCACGGCGGCAATCATC-3’, the primer sequence for bcl2: F 5’- ATCGCCCTGTGGATGACTGAGT-3’ R 5’- GCCAGGAGAAATCAAACAGAGGC-3’ and for the housekeeping gene GAPDH: F 5’- GTCTCCTCTGACTTCAACAGCG-3’ R 5’- ACCACCCTGTTGCTGTAGCCAA-3’. The qPCR reactions were used in triplicate as the following cycle: denaturation for 10 min at 95 °C, denaturation of 40 cycles at 95 °C for 15 s and annealing/extension for 1 min at 60 °C. The relative gene expression levels (treated to the untreated cells) were calculated using the 2^−ΔΔCt method. Where; Ct (Cycle threshold), ΔCt = Ct (target gene) – Ct (reference gene) and ΔΔCt = ΔCt (treated sample) – ΔCt (control sample)96.
Evaluation of bcl-2 expression
The protein of interest in the samples and standards binds to the antibody coated on the plate. A biotin-conjugated antibody is added and binds to the protein captured by the first antibody. Streptavidin-HRP is added and binds to the biotin-conjugated antibody. The substrate solution is added to the wells to form the colored products. The reaction is then terminated by the addition of acid, and absorbance is measured at 450 nm. A standard curve is prepared to determine the protein concentration84.
Evaluation of Bax expression
The Human Bax ELISA kit is a complete kit for the quantitative determination of Bax in human cell lysates. The kit uses a monoclonal antibody to Bax immobilized on a microtiter plate to bind the Bax in the standards or sample. A recombinant human Bax-α standard is provided in the kit. After a short incubation the excess sample or standard is washed out and a biotinylated monoclonal antibody to Bax is added. This antibody binds to the Bax captured on the plate. After a short incubation the excess antibody is washed out, and streptavidin conjugated to Horseradish peroxidase is added, which binds to the biotinylated monoclonal Bax antibody. Excess conjugate is washed out, and substrate is added. After a short incubation, the enzyme reaction is stopped, and the color generated is read at 450 nm. The measured optical density is directly proportional to the concentration of Bax in either standards or samples84.
Caspase-7 activation assay
The human caspase-7 ELISA Kit is a solid phase sandwich an enzyme-linked immunosorbent assay for the measurement of human caspase. Adsorbed An anti-human caspases coating antibody onto microwells. This human caspase present in the sample or standard binds to antibodies adsorbed to the microwells. The polyclonal detection antibody (rabbit) binds to human caspase captured by the first antibody. Following incubation, unbound detection antibody is removed during a wash step. Anti-rabbit-IgG-HRP is added and binds to the detection antibody. Following incubation unbound anti-rabbit-IgG-HRP is removed during a wash step, and substrate solution reactive with HRP is added to the wells. A colored product is formed in proportion to the amount of human caspase present in the sample or standard. Acid is added, and absorbance is measured at 450 nm. A standard curve is prepared from seven human caspase standard dilutions, and human caspase concentration is determined97,98.
Computational study
Molecular Docking
Different crystal structures of COX-2 enzyme (PDB IDs: 1CX2, 2NT1, and 5IKR), tubulin protein (PDB IDs: 4O2B, 5LYJ, and 1JFF), Focal adhesion kinase (PDB IDs: 2JKK, 3BZ3, and 1K04), and EGFR kinase (PDB ID:1M17) were obtained from the Protein Data Bank (PDB) (https://www.rcsb.org, accessed on August 2, 2023, September 12, 2024, and December 5, 2025). The docking method was validated by redocking co-crystallized ligands with AutoDock 1.5.6 and Vina, with RMSD values ranging from 0.4174 to 1.413 Å (Supplementary Figure S1). ChemDraw software was used to create a 2D diagram of the ligand structures, which was then saved in mol format. Avogadro ver1.2.0 was used to convert the 2D structures to 3D, then to optimize them using the MMFF94 force field. A web-based software called CB-Dock2 was utilized for docking accessed on September 12–14, 2024, and December 6, 2025 (http://clab.labshare.cn/cb-dock/php/)99. The Discovery Studio program ver24.1.0.23298 created interaction and visualization profiles for the docked complexes.
Drug-likeness and Pharmacokinetic investigation
To determine the absorption, distribution, metabolism, elimination, and toxicity (ADMET) properties of the examined ligand, the open-source tool ADMETLab 2.0 from the Computational Biology & Drug Design Group (https://admetmesh.scbdd.com/, accessed on September 14, 2024) was utilized100.
Molecular dynamic simulation (MD)
MD simulations were performed using the Desmond package (Schrödinger LLC, NY, USA). Each system was simulated for 200 nanoseconds in the NPT ensemble at 310 K and 1 bar, following a brief 1‑picosecond relaxation step. The OPLS_2005 force field was applied, and long‑range electrostatics were handled with the particle‑mesh Ewald method using a 9.0 Å cutoff. Water molecules were represented with the simple point charge (SPC) model. To maintain system conditions, pressure was controlled with the Martyna–Tuckerman–Klein scheme, while temperature was regulated using the Nosé–Hoover thermostat. Non‑bonded forces were calculated with the r‑RESPA integrator, and trajectories were saved every 4.8 ps. For ligand preparation, the AMBER 14 package was used with the ff99 force field to perform energy minimization, add counterions, and perform MD simulations in explicit TIP4P water. Ligand geometries were further optimized using density functional theory (DFT) with a 6‑31G basis set. To ensure thorough conformational sampling, we ran ten independent simulations, each lasting 10 ns with a 0.001 ps time step. Finally, system stability was verified, and trajectory analysis was performed using cpptraj from AMBER Tools (Kumar et al., 2022)101.
Discussion
Chemistry
In the current study, the novel targeted derivatives were synthesized as signified in Scheme 1, 2 and 3. Our starting acyl hydrazide intermediate 4 was prepared according to the previously reported methods including Claisen Condensation and Knorr pyrazole synthesis method102,103,104,105,106.

The synthesis of compound 4. Reagents and reaction conditions: (a) Diethyl oxalate, sodium ethoxide, EtOH, rt, overnight; (b) (i) NaNO2/HCl, 0–5 ℃, (ii) SnCl2/HCl, overnight, refrigerator; c) EtOH, reflux, 12 h; d) NH2NH2.H2O, EtOH, reflux at 80℃, 6 h (65%).
The acyl hydrazide intermediate was allowed to be condensed with appropriate aldehydes or ketones in ethanol, producing the corresponding Schiff`s bases AS-2, AS-3, AS-6, and AS-7 in good yield. Confirmation of the Schiff`s bases structures was carried out using NMR spectra. 1H NMR spectra of aldimine compounds AS-2, AS-3, and AS-5 revealed the presence of a singlet peak in the range of δH: 8.47–8.74 ppm, representing the imine proton. However, of methyl group of ketamine compounds AS-6 and AS-7 was confirmed by appearance of a singlet peak at δH 2.37 and 2.36, respectively. Moreover, the 13C NMR spectrum of ketamine compounds AS-6 and AS-7 showed a signal at δC 14.24 and 14.37 ppm, respectively, representing the methyl group.

The synthesis of compound AS-1:AS-7. Reagents and reaction conditions: a) appropriate aldehyde/acetophenones, ethanol, drops of glacial acetic acid, reflux, 6–12 h.
Meanwhile, thiosemicarbazide derivatives AS-8 and AS-9 were prepared by reacting the acyl hydrazide intermediate 4 with the appropriate isothiocyanates in ethanol. 1H NMR spectra of compound AS-9 revealed triplet signal at δH 1.07ppm and quartet signal at 3.48ppm representing the ethyl group. The 13C NMR spectra was presented an Additional confirmation as a result of appearance of the signals in the range of δC 160–162 ppm ascribed to C═S. Additionally, N-Phthalimido-protected hydrazide AS-10 was obtained upon refluxing of the acyl hydrazide intermediate 4 with carbonyl compounds (phthalic anhydride/succinic anhydride) in ethanol with few drops of glacial acetic acid phthalic anhydride in acetic acid. NH group of compound AS-10 was confirmed through IR and H NMR spectrums. In IR spectra, NH group appeared at λ 3357 cm− 1, while, in 1H NMR spectra, it was shown at δ = 11.24ppm. Furthermore, C13 NMR spectra proved compound AS-10’s structure by appearance of carbonyl carbon of the phthalimido group at δ 165.38 ppm.

The synthesis of compound AS-8:AS-16. Reagents and reaction conditions: (a) isothiocyanate derivatives, ethanol, reflux, 6 h; (b) succinic anhydride or phthalic anhydrtide, ethanol, glacial acetic acid, reflux, 10 h; (c) CS2, ethanol, KOH, 12 h; (d) enaminone of 4-bromoacetophenone, glacial acetic acid, reflux,6 h; (e) Aromatic acid derivatives, POCl3, reflux, 12–24 h; (f) ClCH2COOH, POCl3, reflux, 12 h.
The oxadiazolthione derivative AS-12 was prepared from the reaction of the acyl hydrazide intermediate 4 with carbon disulfide in ethanol in the presence of potassium hydroxide. Its IR spectra shown the disappearance of bands corresponding to C═O group of the hydrazide intermediate 4 and the appearance of another band related to C═S group at 1245 cm − 1. Moreover, the pyrazole derivative AS-13 was obtained by reaction of the hydrazide intermediate 4 with enamineone of 4-bromoacetophenone in in glacial acetic acid under reflux. Furthermore, treatment of the hydrazide intermediate 4 with substituted benzoic acids in the presence of POCl3 produced the oxadiazole derivative AS-14 and AS-15. 1H NMR spectra of compound AS-14 revealed a singlet peak at δH: 3.87 ppm, signifying OCH3 group of P-anisidic acid. On the other hand, upon treatment, the hydrazide intermediate 4 with chloroacetic acid in xylene in the presence of POCl3 afforded the 2-chloromethyl-oxadiazole derivative AS-16. The 1H NMR spectral analyses confirmed the structure of AS-16 by the appearance of a singlet peak at δH: 5.16 ppm demonstrating the CH2 of chloromethyl group.
Biological evaluation
Anti-inflammatory activity studies
In vitro COX-1 and COX-2 Inhibition assay
The synthesized pyrazole derivatives AS-1: AS-16 were subjected to in vitro inhibition of ovine COX-1/COX-2 using colorimetric enzyme immunoassay (EIA) kits to screen the isozyme-specific inhibition. The minimum dose of the tested compounds that makes 50% inhibition (IC50) of both enzymes was detected to evaluate the effectiveness of the synthesized compounds. Furthermore, the COX-2 selectivity index (S.I.) values were calculated as [IC50 (COX-1)/IC50 (COX-2)] and compared to the standard drugs: Diclofenac as a non-selective NSAID and Celecoxib as a selective COX-2 inhibitor to predict the gastrointestinal safety profile of the newly synthesized compounds.
The results demonstrated that all the tested compounds displayed lower potency to the COX-1 isoenzyme (IC50 = 2.95–9.78 µM) compared to the non-selective standard drug, Diclofenac (IC50 = 3.71 µM). Accordingly, all tested compounds displayed weak inhibition activity to the COX-1 enzyme and exhibited moderate to potent COX-2 inhibitory activity. (Table 1& Fig. 5.)
In vitro 5-Lipoxygenase (5-LOX) inhibition assay
All the estimated derivatives suppressed 5-LOX enzyme with an IC50 range equal to 4.65–17.29 µM. Compound AS-14, which bears an oxadiazole moiety, was the most potent 5-LOX suppressor, with (IC50 = 4.65 ± 1.63 µM) revealing comparable inhibitory activity to that exerted by reference drug Quercetin (IC50 = 3.52 ± 0.05 µM). In addition, pyrazole hydrazones AS-6 (IC50 = 4.7 µM), AS-7 (IC50 = 5.68 µM), and AS-1 (IC50 = 5.89 µM) displayed good 5-LOX inhibitory activity (Table 1 & Fig. 5.).

Inhibitory effects of target compounds AS-1: AS-16 and reference drugs celecoxib, Diclofenac and Quercetin on the COX-1, COX-2 and 5-LOX enzymes.
IL-1β, IL-6 and TNF-α suppression assay
The outcome results of this assay were recorded in Table 2; Fig. 6. which revealed that all the tested derivatives suppressed IL-1β, IL-6 and TNF-α with various percentages. They suppressed IL-1β release with a percentage inhibition range of 26–75%, IL-6 releases with a percentage inhibition range of 15–61% and they suppressed TNF-α release with a percentage inhibition range of 36–91 to LPS control. The most suppressor effect was achieved by compound, AS-11, it was able to inhibit IL-1β (percentage inhibition = 75%), IL-6 (percentage inhibition = 61%), TNF-α (percentage inhibition = 91%). The least inhibitory result was assigned by the pyrazole-oxadiazole derivative AS-14 towards IL-1β, IL-6, and TNF-α secretion with inhibitory percentages of 26%, 15%, and 36%, respectively. (Table 2& Fig. 6.)

Effects of tested compounds AS-1: AS-16 on pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α).
Anti-proliferative activity
In this study, propidium iodide (PI) fluorescence assay was performed to evaluate the antiproliferative effect of new synthesized pyrazole derivatives AS-1: AS-16 on five different human cancer cell lines namely; PaCa-2 (pancreatic carcinoma cells), A-549 (epithelial cancer cells), MCF-7 (breast cancer cells), PC-3 (human prostate cancer cells) and HT-29 (colon cancer cells).The anticancer activity results are presented as growth inhibitory concentration of compounds to produce 50% cell growth inhibition (IC50 values). (Table 3 & Fig. 7.)
The MCF-10 A cells were treated with the synthesized compounds for 4 days and a 3-(4,5-dimethylthiazol-2-yl)−2,5- diphenyltetrazolium bromide (MTT) assay was used to.
measure the viability of cells. All compounds were discovered to be nontoxic with most of them exhibiting more than 90% cell viability.

Inhibitory effects of target compounds AS-1: AS-16 on the growth of human cancer cells.
It is obvious from the results, that the synthesized compounds demonstrated promising anticancer activity almost against all the cell lines tested to a reasonable extent. As illustrated in Table 3, the three most active compounds: hydrazone derivatives AS-1 and AS-2, and oxadiazol-yl pyrazole derivative AS-14, exhibited potent inhibition of cancer cell growth with IC50 ranging from 6.92 to 20.03 µM. All other compounds showed moderate effects against all five cancer cell lines. Pyrazole oxadiazole hybrid AS-14 was found to be the best antiproliferative agent against all cancer cell with the values of IC50 in the range of 6.92 ± 1.98 µM to 16.02 ± 1.41µM (Table 3). The three most potent compounds were selected for anticancer mechanistic experiments, including their effect on tubulin polymerization, FAK, EGFR, and BRAF.
Enzyme Inhibition assays
Tubulin polymerization Inhibition assay
Tubulin polymerase is a natural target for chemotherapeutic agents to promote apoptosis via suppression of microtubules growth and inhibition of cell division83. Compounds AS-1, AS-2, and AS-14 that showed potent cytotoxic activity were evaluated for their β-tubulin polymerization inhibitory activity using DPBS, Vincristine, and Docetaxel as reference drugs. Results showed that the three compounds exhibited weak activity compared to Vincristine. (Table 4)
The effect of all three selected synthetic compounds on tubulin polymerization has been summarized in Table 4. All three compounds were not active and showed the arbitrary units value close to Dulbecco’s phosphate-buffered saline (DPBS) that was used as a negative control and were discovered to be inactive, suggesting a mechanism of action other than tubulin inhibition for the observed cytotoxicity.
In comparison to model drug docetaxel, no compound exhibited promising microtubules stabilizing action. No synthetic compounds exhibited microtubule-stabilizing action comparable to docetaxel.
FAK inhibitory assay
Through effects on the stromal cells of the tumor microenvironment and on cancer cells, FAK stimulates metastasis and tumor progression. Cancer stem cell self-renewal, gene expression, survival, invasion, and cell movement are regulated by FAK’s kinase-dependent and independent functions. Small-molecule inhibitors of FAK have been discovered to decrease metastasis and tumor growth in numerous preclinical models with lesser adverse effects. The FAK inhibitory potency of compounds was investigated and the findings revealed all investigated, compounds were found to be less active FAK inhibitors (IC50 12.2 ± 0.8 to 17.1 ± 0.9 µM) than the reference drug erlotinib (IC50 7.5 ± 0.8 µM). (Table 4)
EGFR (epidermal growth factor receptor) inhibitory assay
EGFR assay was performed to assess the EGFR inhibitory potency of novel compounds, and the results are included in Table 4. The investigated compounds, AS-1, AS-2, and AS-14 exhibited inhibition of EGFR with IC50 ranging from 2.5 ± 0.4 to 5.9 ± 1.7 µM. According to data presented, the strongest EGFR inhibitory activity was shown by compound AS-14 selected from oxadiazolyl-pyrazole hybrid series with (IC50 2.5 ± 0.4 µM).
BRAF inhibitory activity
An in vitro assay was performed to investigate the BRAF inhibitory potential of most active synthesized compounds. According to data shown in Table 4, all investigated compounds showed IC50 in the range of 7.4 ± 1.6 to 18.5 ± 1.9 µM. The pyrazole derivative AS-14 bearing an oxadiazole moiety was discovered to be a strong BRAF inhibitor with an IC50 of 7.4 ± 1.6.
Cell cycle analysis
The Propidium Iodide (PI) Flow Cytometry Kit assay was applied to explore the cell distribution in varying phases of the cell cycle (G0-G1, S, and G2/M) and to detect the ability of the most potent compound AS-14, to induce apoptosis. MCF-7 cells were treated with the IC50 inhibitory dose (10.27 µM) of compound AS-14 and DNA contents were then analyzed. The percentage of MCF-7 cells in each phase and cell cycle distribution histogram of the stained DNA of treated MCF-7 and control untreated cells is illustrated in Fig. 8.

(A) Percentage of MCF-7 cells in G0/G1, S and G2/M phases after treatment with compound AS-14 compared to control cells. (B) DNA content distribution of cell cycle phases in PI stained MCF-7 cells treated with AS-14.
The results showed the that percentage of G0/G1 apoptosis induced by compound AS-14 on MCF-7 cells was 55.15% followed by a great decrease in S phase (18.66%) and a high percent of cell accumulation was observed at G2/M phase (26.19%) when compared to the control untreated cells (10.60%). Thus, it may be concluded that the increased propensity for cell cycle arrest by the investigated compound led to an arrest pattern at the G2/M phase. (Table 5)
Annexin V-FITC apoptosis analysis
Apoptosis, or programmed cell death, is regarded as a defense mechanism against cancer spreading since it is crucial in the elimination of neoplastic cells. In this work, Annexin V-FITC/PI double staining flow cytometry assay was applied to evaluate apoptosis induced by compound AS-14 on treated MCF-7 cells. The results showed a remarkable increase of cell population in the total, early, and late cellular apoptosis from 2.84%, 0.59%, and 0.18% (DMSO control) to 26.99%, 15.14%, and 8.59%, respectively. These results indicate that the tested compound significantly promotes apoptosis of cancer cells. Also, 1.57-fold increase in percentage of necrotic cells was observed. (Fig. 9.)

(A) Flow cytometry analysis of annexin V/PI stained MCF-7 cells treated with AS-14 compared to control cells; (B) Percentage of apoptosis of MCF-7 cells treated with AS-14 compared to control cells.
Apoptosis regulatory genes
After demonstrating that apoptosis was the underlying mechanism of AS-14 cytotoxicity, we investigated its effect on expression levels of various apoptosis regulatory genes. Real-time PCR quantified the gene expression levels of p53, bax, bcl-2, and caspase-7. Overexpression of p53, bax, and caspase-7 (5.57 folds, 4.13 folds, and 4.80 folds, respectively) was noted. On the other hand, the expression of anti-apoptotic bcl-2 gene was inhibited (0.28 folds) (Fig. 10.). These findings illustrate the role of AS-14 in triggering the apoptotic pathway and shed the light on a novel therapeutic drug in breast cancer.
Likely the apoptotic activity of AS-14 in this cell line is triggered by pathways involving up-regulation of the proapoptotic proteins p53, Bax, and caspase-7 and down-regulation of the anti-apoptotic protein, Bcl-2 (Fig. 10).

The effect of compound AS-14 on expression of proapoptotic proteins p53, bax, and caspase-7 and antiapoptotic protein Bcl-2 compared to untreated cancer cell line.
Computational studies
Molecular docking study
Compared to control drugs and based on the outcomes of enzyme inhibitory assays, the strength of the interaction between the ligands such as AS-14, AS-16, AS-3, AS-2, and AS-1 which exhibited potent, mediate, and low activity against COX-2, tubulin, FAK, and EGFR enzyme receptors or proteins, described as more negative binding-free energy (Fig. 11.). The ligands exhibited superior docking scores against COX-2 enzymes, especially 5IKR with − 11.3 and − 9.9 kcal/mol for AS-14 and AS-16, compared to the control, Celecoxib (−9.6 kcal/mol). Notably, higher docking scores from − 8.3 to −9.9 kcal/mol, were detected with tubulin proteins (PDB IDs: 4O2B, 5LYJ, and 1JFF), where AS-2 was the most potent, in agreed with Table 4 (Fig. 11.). The binding free energies calculated between ligands and the FAK and EGFR enzymes (PDB IDs: 2JKK, 3BZ3, 1K04, and 1M17) were comparable ranging from − 7.5 to −10.1 kcal/mol. AS-14 showed higher activity against 2JKK and 1K04, and was close to AS-2 in the interaction with 1M17.
Similar results were reported by Shaker et al. (2023) for novel pyridine-pyrimidine hybrids as selective COX-2 suppressors, using the same PDB enzymes, with docking scores ranging from − 9.6 to −11.2 kcal/mol. On the other hand, the ligands in the current study showed comparable results to the aryl pyridines examined against 5LYJ (−8.3 to −8.8 kcal/mol) as reported by He et al. (2020)107 and higher activities compared to cyclic lipopeptides (−6.3 to −7.0 kcal/mol) or novel tubulin polymerization inhibitors designed through 3d-QSAR with −7.0 and − 7.6 kcal/mol as the highest binding score against 1JFF and 4O2B108(Zhao et al., 2022). Along the same lines, pyrimidine-based FAK inhibitors (2JKK), in addition to novel drug derivatives such as defactinib and others, showed lower binding affinities for 3BZ3 and 1K04 than the ligands of the present study109,110,111. Finally, in agreement with our results, Soltan et al. (2025) showed a similar docking score (−11.4 kcal/mol) for the interaction of 1,5-diarylpyrazole carboxamide derivative with dual inhibition of EGFR target (1M17).
Molecular docking of phytochemicals with enzymes can reveal potential interactions. Still, discrepancies with in vitro and in vivo results are common, as shown in the current study, because docking doesn’t account for factors such as absorption, metabolism, and environmental conditions (Elshibani et al., 2025)112,113.

Binding free energy of the ligands with COX-2, tubulin, FAK, and EGFR enzymes compared to controls.
The interactions with higher docking scores of the AS-14 with 5IKR, 3BZ3, and 1M17 (−11.3, −9.5, and − 9.6 kcal/mol), and AS-2 with 1JFF (−9.9 kcal/mol), were illustrated in Fig. 11. The conventional hydrogen bond had the highest interaction strength, followed by the carbon-H bond, π-π bond, π-alkyl, and n-alkyl interactions114. Consequently, the higher binding affinity of the ligand-proteins is attributed to the conventional hydrogen bonding between amino or hydroxyl groups of ligand or moieties such as ARG A:469 and THR A:82, and carbonyl groups of GLU A:465, THR A:80, and ASN A:551 or sulfoxide group of the ligand (Fig. 12.). De Freitas and Schapira (2017)115 found that N–H.O interactions were more frequent than O–H.O interactions in molecular docking studies, where both were found in the present study results112. Tyrosine is one of the main protein residues that interact via π-stacking, as shown for TYR A:130 and 224115. In line with the present study, Brandl et al. (2001) found that half of Tyr-rings are acceptors in C-H.π interactions, while Ile and Leu represent the most donors, as shown in Fig. 12116.
In addition, π-sulfur (ligand-CYS A:36,47), Alkyl (ligand-PRO A:156), and π-alkyl (ligand-PRO A:153, 156, VAL A:46, 78, 484, LEU A:152, ALA A:452, MET A:499, LEU A:553, 567) hydrophobic interactions were also observed with (Fig. 12.). Cation– π shown in Fig. 12 between amino groups of LYS A:137 and ARG A:426 and the π-orbitals of the ligand are non-covalent electrostatic interactions between ions and aromatic systems117,118. Similarly, the interaction between an electron-deficient aromatic moiety and an anion has been accepted as a non-covalent contact and reported as anion–π as shown in Fig. 12C and D.

Interactions of the AS-14 with 5IKR (A), 3BZ3 (B), 1M17 (C), and AS-2 with 1JFF (D).
Computed Pharmacokinetic and toxicity properties of AS-14 (ADMET)
The drug-likeness of compounds, encompassing absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, is pivotal in drug discovery. The current study utilized the ADMETlab 2.0 tool to assess the ADMET properties of AS-14. As per the data in Table 6, AS-14 demonstrates the essential physicochemical properties required for a drug. It has met the criteria for Lipinski, Pfizer, and Golden Triangle rules but not the GSK rule due to unfavorable QED values based on various drug-likeness-related properties. According to ADMETlab 2.0, compounds complying with the Golden Triangle rule may exhibit a more advantageous ADMET profile.
AS-14 is optimal for Caco-2 permeability but inhibits P-glycoprotein (P-gp) efflux pumps, actively removing drugs from cells and reducing their intracellular concentrations. These pumps are crucial for secreting xenobiotics and protecting the central nervous system119. Additionally, the ligand exhibits an excellent human intestinal absorption (HIA) value, an alternative indicator for oral bioavailability (Table 6). AS-14’s high plasma protein binding (PPB) rate indicated a low therapeutic index. Also, the low percentage of fraction unbound in plasma (Fu) suggests a lower probability of efficiently crossing the cellular membranes and reaching their target sites. However, it has optimal volumes of distribution (VD), suggesting it can efficiently cross cellular membranes to reach its target sites. Similarly, AS-14 has a low blood-brain barrier (BBB) penetration capacity, which avoids CNS side effects.
AS-14 shows a high probability of interacting negatively with major Cytochrome P450 (CYP), which play a crucial role in drug metabolism in the liver and intestine. Inhibiting these enzymes can lead to serious drug interactions, while being substrates to these enzymes can lead to drug deactivation or the creation of more active metabolites120. Therefore, the performance of In vitro hepatocyte assays and CYP inhibition panels should confirm computational predictions. The hERG gene encodes a potassium channel that regulates the heart’s action potential and resting potential. Blocking hERG can lead to sudden death121. However, AS-14 is generally safe and does not block the hERG K + channel. On the other hand, AS-14 has moderate carcinogenicity and high rat oral acute toxicity (Table 6). Further, toxicological studies are necessary to ensure its safety due to adverse toxicity properties revealed by ADMET.
Molecular dynamics simulation
The molecular dynamics simulation was carried out for 200 ns under NPT conditions at 310 K, focusing on a protein-ligand complex in explicit solvent. The system included a protein with 551 residues (chain A, + 3 charge), a ligand composed of 81 atoms (C₂₄H₄₇N₅O₄S) with nine rotatable bonds, and approximately 15,766 water molecules together with neutralizing Na⁺ and Cl⁻ ions. Over the course of the simulation, the protein’s RMSD stabilized between 1.5 and 2.0 Å after equilibration, confirming that the structure remained stable. The ligand RMSD stayed within 2–3 Å, showing that it remained bound to the protein while retaining some flexibility inside the binding pocket (Fig. 13A).
Analysis of flexibility revealed RMSF peaks at the N- and C-termini and in loop regions, consistent with the expected dynamic behavior, while secondary structure elements, such as helices and strands, remained stable. The protein maintained ~ 31% helical and ~ 4% strand content, totaling ~ 35% secondary structure, with helices dominating throughout the trajectory (Fig. 13B).
Binding interactions were observed with residues including HIS39, CYS41, GLN42, ARG44, TYR55, PRO153/154, GLU67, GLN461, SER462, GLU465, LYS468, and ARG469. These interactions involved stable hydrophobic contacts (e.g., PRO153/154, TYR55), polar and hydrogen bonds (HIS39, GLN42, ARG44), and intermittent water bridges. Several of these contacts persisted for more than 30% of the simulation, underscoring the consistency of binding. Overall, hydrogen bonds (≈ 40–60% occupancy) and hydrophobic contacts (≈ 50–70%) dominated the interface, with ionic interactions (≈ 20–40%) and water bridges (≈ 10–30%) also contributing. This balance of polar and nonpolar forces created a robust, well-solvated environment that maintained the ligand’s binding throughout the 200 ns run. The absence of ligand dissociation or large-scale conformational changes confirmed the complex’s stability, while residues such as ARG44, ASN34, and LEU152 emerged as key binding hotspots. These results highlight opportunities for ligand optimization, particularly by reducing torsional strain and reinforcing polar contacts to improve affinity (Fig. 13C).
The ligand itself displayed diverse torsion profiles due to its nine rotatable bonds, with some torsions showing strain that may contribute entropically to binding. Its compactness was reflected in a radius of gyration of ~ 4.8–5.4 Å, consistent with its bulky structure, while solvent-accessible surface area (SASA) fluctuated between 60 and 180 Ų, suggesting partial burial within the pocket (Fig. 13D). Occasional intramolecular hydrogen bonds were observed, helping pre-organize the ligand’s conformation. Taken together, the protein remained structurally stable over the 200 ns trajectory, while the ligand demonstrated moderate stability supported by persistent hydrophobic and polar contacts.

RMSD (A), RMSF (B), Interaction profile (C), rGyr, intraHB, MolSA, SASA, and PSA (D) of the protein and ligand during the MD process.
Conclusion
Different pathological mechanisms of cancer made it one of the most multifaceted and serious medical condition. So, there is continuous need to develop multi-target directed ligand affording anticancer activity. Here, we focused on synthesis of the pyrazoles series AS-1:AS-16 as selective Cox-2 inhibition fragment. Integration other known anticancer fragments is an attempt to perform multi-target anticancer agent like hydrazone, thiosemicarbazone, phthalic derivatives and oxadiazole derivatives. Pyrazoles compounds were investigated for their COX inhibitory activities and antiproliferative action; most of the tested compounds showed moderate to high action. Notably, AS-1, AS-2 and AS-14 displayed pronounced activities and were subjected to extra testing to detect the preferred target among tubulin polymerase, FAK, EGFR and BRAF enzymes. Fortunately, all investigated compounds showed excellent IC50 in the range of 7.4 ± 1.6 to 18.5 ± 1.9 µM, especially compound AS-14 bearing oxadiazole moiety was discovered to be strong BRAF inhibitors with IC50 7.4 ± 1.6. Subsequently, further analyses were performed to confirm AS-14’s mechanism of action through cell cycle analysis and apoptosis marker analysis. AS-14 showed arrest pattern at the G2/M phase, in addition to its ability to up-regulate the proapoptotic proteins p53, bax, and caspase-7 and down-regulate the anti-apoptotic protein, bcl-2. All these results were consistent with the molecular docking and molecular dynamic simulation outcomes confirming validity of our rational. Concerning the toxicity and pharmacokinetics of compound AS-14, it showed optimal volumes of distribution (VD), suggesting it can efficiently cross cellular membranes to reach its target sites. Similarly, the ligand has a low blood-brain barrier (BBB) penetration capacity, which avoids CNS side effects. AS-14 is generally safe and does not block the hERG K + channel. The toxicological studies of AS-14 are necessary to ensure its safety due to adverse toxicity properties revealed by ADMET, especially against CYP enzymes. Finally, we can conclude that AS-14 can be considered as an excellent promising multi-target anticancer agent.
Data availability
The datasets used during the current study available from the corresponding author on reasonable request.
References
Deo, S. V. S., Sharma, J. & Kumar, S. GLOBOCAN 2020 report on global cancer burden: challenges and opportunities for surgical oncologists. Ann. Surg. Oncol. 29, 6497–6500 (2022).
Tang, L. et al. Genetic association between HER2 and ESR2 polymorphisms and ovarian cancer: A meta-analysis. Onco Targets Ther. 11, 1055–1066 (2018).
Cai, F. F. et al. Epigenetic therapy for breast cancer. Int. J. Mol. Sci. 12, 4465–4476 (2011).
DeVita, V. T. & Chu, E. A history of cancer chemotherapy. Cancer Res. 68, 8643–8653 (2008).
Kim, W. Y. Therapeutic targeting of lipid synthesis metabolism for selective elimination of cancer stem cells. Arch. Pharm. Res. 42, 25–39 (2019).
Zanardi, E., Bregni, G., De Braud, F. & Di Cosimo, S. Better together: targeted combination therapies in breast cancer. Semin Oncol. 42, 887–895 (2015).
Augusto, T. V., Georgina, C. S., Rodrigues, C. M. P., Teixeira, N. & Amaral, C. Acquired resistance to aromatase inhibitors: where we stand! Endocr. Relat. Cancer. 25, R283–R301 (2018).
Li, X. et al. The multi-molecular mechanisms of tumor-targeted drug resistance in precision medicine. Biomed. Pharmacotherapy. 150, 113064 (2022).
Mokhtari, R. B. et al. Combination therapy in combating cancer. Oncotarget 8, 38022–38043 (2017).
Jordan, M. A. & Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer. 4, 253–265 (2004).
Kumar, A., Sharma, P. R. & Mondhe, D. M. Potential anticancer role of colchicine-based derivatives: an overview. Anticancer Drugs. 28, 250–262 (2016).
Pecoraro, C., Carbone, D., Cascioferro, S. M., Parrino, B. & Diana, P. Multi or Single-Kinase inhibitors to counteract drug resistance in cancer: what is new? Curr. Med. Chem. 30, 776–782 (2022).
Lechertier, T. & Hodivala-Dilke, K. Focal adhesion kinase and tumour angiogenesis. J. Pathol. 226, 404–412 (2012).
Yoon, H., Dehart, J. P., Murphy, J. M. & Lim, S. T. S. Understanding the roles of FAK in cancer: Inhibitors, genetic Models, and new insights. J. Histochem. Cytochem. 63, 114–128 (2015).
Li Petri, G. et al. New imidazo[2,1-b][1,3,4]thiadiazole aerivatives anhibit FAK phosphorylation and potentiate the antiproliferative effects of gemcitabine through modulation of the human equilibrative nucleoside transporter-1 in peritoneal mesothelioma. Anticancer Res. 40, 4913–4919 (2020).
Lee, B. Y., Timpson, P., Horvath, L. G. & Daly, R. J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 146, 132–149 (2015).
Wang, X. et al. Targeting focal adhesion kinase (FAK) in cancer therapy: A recent update on inhibitors and PROTAC degraders. Eur J. Med. Chem 276, 116678 (2024).
Ott, G. R. et al. Discovery of clinical candidate CEP-37440, a selective inhibitor of focal adhesion kinase (FAK) and anaplastic lymphoma kinase (ALK). J. Med. Chem. 59, 7478–7496 (2016).
Shanthi, E. et al. Focal adhesion kinase inhibitors in the treatment of metastatic cancer: A patent review. Expert Opin. Ther. Pat. 24, 1077–1100 (2014).
Jones, S. F. et al. A phase i study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest. New. Drugs. 33, 1100–1107 (2015).
Mendelsohn, J. & Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 33, 369–385 (2006).
Greig, S. L. & Osimertinib First Global Approval Drugs 76, 263–273 (2016).
Shi, Y. et al. Clinical Activity, and pharmacokinetics of alflutinib (AST2818) in patients with advanced NSCLC with EGFR T790M mutation. J. Thorac. Oncol. 15, 1015–1026 (2020). Safety.
Tran, P. N. & Klempner, S. J. Profile of rociletinib and its potential in the treatment of non-small-cell lung cancer. Lung Cancer: Targets Therapy. 7, 91–97 (2016).
Ho, C. C. et al. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J. Thorac. Oncol. 12, 567–572 (2017).
Bearz, A., De Carlo, E., Doliana, R., Schiappacassi, M. & Acquired BRAF V600E mutation as resistant mechanism after treatment with Third-Generation EGFR tyrosine kinase inhibitor. J. Thorac. Oncol. 12, e181–e182 (2017).
Dankner, M., Rose, A. A. N., Rajkumar, S., Siegel, P. M. & Watson, I. R. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene 37, 3183–3199 (2018).
Greten, F. R. & Grivennikov, S. I. Inflammation and cancer: Triggers, Mechanisms, and consequences. Immunity 51, 27–41 (2019).
Elinav, E. et al. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer. 13, 759–771 (2013).
Wang, X. et al. EGFR signaling promotes inflammation and cancer stem-like activity in inflammatory breast cancer. Oncotarget 8, 67904–67917 (2017).
Rayburn, E. R., Ezell, S. J. & Zhang, R. Anti-inflammatory agents for cancer therapy. Mol. Cell. Pharmacol. 1, 29–43 (2009).
Chavey, C. et al. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 9, 1–11 (2007).
Zappavigna, S. et al. Anti-inflammatory drugs as anticancer agents. Int. J. Mol. Sci. 21, 1–29 (2020).
Zamarron, B. F. & Chen, W. Dual roles of immune cells and their factors in cancer development and progression. Int. J. Biol. Sci. 7, 651–658 (2011).
Yan, L., Anderson, G. M., DeWitte, M. & Nakada, M. T. Therapeutic potential of cytokine and chemokine antagonists in cancer therapy. Eur. J. Cancer. 42, 793–802 (2006).
Kraus, S. & Arber, N. Cyclooxygenase-2 expression and recurrence of colorectal adenomas: effect of aspirin chemoprevention. Curr. Colorectal Cancer Rep. 7, 5–7 (2011).
Denkova, A. G., Liu, H., Men, Y. & Eelkema, R. Enhanced cancer therapy by combining radiation and chemical effects mediated by nanocarriers. Adv. Ther. (Weinh). 3, 1–17 (2020).
Mittal, A., Dixit, S. & Rana, S. Anticancer effects of chemotherapy and nature products. J. Med. Discovery. 2, 1–8 (2017).
Bailey, J., Oliveri, A. & Levin, E. Regulation of inflammation in cancer by eicosanoids Emily. Prostaglandins Other Lipid Mediat. 2396, 27–36 (2011).
Sharma, V. et al. Recent advancement in the discovery and development of COX-2 inhibitors: Insight into biological activities and SAR studies (2008–2019) Vrinda. Bioorg Chem, 103007, (2019).
Ruzov, M., Rimon, G., Pikovsky, O. & Stepensky, D. Celecoxib interferes to a limited extent with aspirin-mediated Inhibition of platelets aggregation. Br. J. Clin. Pharmacol. 81, 316–326 (2016).
Yin, D. et al. Celecoxib reverses the glioblastoma chemo-resistance to Temozolomide through mitochondrial metabolism. Aging 13, 21268–21282 (2021).
Vital-Reyes, V. et al. Celecoxib inhibits cellular Growth, decreases Ki-67 expression and modifies apoptosis in ovarian cancer cell lines. Arch. Med. Res. 37, 689–695 (2006).
Park, W., Oh, Y. T., Han, J. H. & Pyo, H. Antitumor enhancement of celecoxib, a selective Cyclooxygenase-2 inhibitor, in a Lewis lung carcinoma expressing Cyclooxygenase-2. J. Experimental Clin. Cancer Res. 27, 1–9 (2008).
Dai, Z. J. et al. Antitumor activity of the selective cyclooxygenase-2 inhibitor, celecoxib, on breast cancer in vitro and in vivo. Cancer Cell. Int. 12, 1–8 (2012).
Shyamsunder, S., Lu, Z., Takiar, V. & Waltz, S. E. Challenges and resistance mechanisms to EGFR targeted therapies in head and neck cancers and breast cancer: insights into RTK dependent and independent mechanisms. Oncotarget 16, 508–530 (2025).
Notarangelo, T., Sisinni, L., Condelli, V. & Landriscina, M. Dual EGFR and BRAF Blockade overcomes resistance to Vemurafenib in BRAF mutated thyroid carcinoma cells. Cancer Cell. Int. 17, 1–9 (2017).
Wang, Y. X., Gao, J. X., Wang, X. Y., Zhang, L. & Liu, C. M. Antiproliferative effects of selective cyclooxygenase-2 inhibitor modulated by nimotuzumab in estrogen-dependent breast cancer cells. Tumour Biol. 33, 957–966 (2012).
Kang, J. H. et al. Involvement of Cox-2 in the metastatic potential of chemotherapy-resistant breast cancer cells. BMC Cancer 11, 334 (2011).
Rothschild, S. I. & Gautschi, O. Crizotinib in the treatment of non-small-cell lung cancer. Clin. Lung Cancer. 14, 473–480 (2013).
Akamine, T., Toyokawa, G., Tagawa, T. & Seto, T. Spotlight on lorlatinib and its potential in the treatment of NSCLC: the evidence to date. Onco Targets Ther. 11, 5093–5101 (2018).
Santos, N. E., Carreira, A. R. F., Silva, V. L. M. & Braga, S. S. Natural and biomimetic antitumor pyrazoles, a perspective. Molecules 25(6), 1364 (2020).
Roubal, K., Myint, Z. W., Kolesar, J. M. & Erdafitinib A novel therapy for FGFR-mutated urothelial cancer. Am. J. Health-System Pharm. 77, 346–351 (2020).
Harras, M. F. & Sabour, R. Design, synthesis and biological evaluation of novel 1,3,4-trisubstituted pyrazole derivatives as potential chemotherapeutic agents for hepatocellular carcinoma. Bioorg. Chem. 78, 149–157 (2018).
Fizazi, K. et al. Re: darolutamide in nonmetastatic, castration-resistant prostate cancer. J. Urol. 202, 660–661 (2019).
Li, L. Y. et al. Potent hydrazone derivatives targeting esophageal cancer cells. Eur. J. Med. Chem. 148, 359–371 (2018).
Patil, S., Kuman, M. M., Palvai, S., Sengupta, P. & Basu, S. Impairing powerhouse in colon cancer cells by Hydrazide-Hydrazone-Based small molecule. ACS Omega. 3, 1470–1481 (2018).
Interactions, C. M. et al. Synthesis of new Naphthyl aceto Hydrazone-Based metal complexes: micellar Interactions, DNA Binding, Antimicrobial, and cancer Inhibition studies. Molecules 26, 1–18 (2021).
Fan, C., Zhao, J., Zhao, B., Zhang, S. & Miao, J. Novel complex of copper and a salicylaldehyde pyrazole hydrazone derivative induces apoptosis through up-regulating integrin β4 in vascular endothelial cells. Chem. Res. Toxicol. 22, 1517–1525 (2009).
Boström, J., Hogner, A., Llinàs, A., Wellner, E. & Plowright, A. T. Oxadiazoles in medicinal chemistry. J. Med. Chem. 55, 1817–1830 (2012).
Helft, P. R., Slaven, J. E., Cottingham, A. H., Ma, M. A. R. & Torke, A. M. Design, synthesis, Docking and QSAR study of substituted benzimidazole linked oxadiazole as cytotoxic agents, EGFR and erbB2 receptor inhibitors Md. Eur. J. Med. Chem. 126, 853–869 (2016).
Liu, K., Lu, X., Zhang, H. J., Sun, J. & Zhu, H. L. Synthesis, molecular modeling and biological evaluation of 2-(benzylthio)-5-aryloxadiazole derivatives as anti-tumor agents. Eur. J. Med. Chem. 47, 473–478 (2012).
Ruel, R. et al. Discovery and preclinical studies of 5-isopropyl-6-(5-methyl-1,3,4-oxadiazol-2-yl)-N-(2-methyl-1H-pyrrolo[2,3-b]pyridin-5-yl)pyrrolo[2,1-f][1,2,4]triazin-4-amine (BMS-645737), an in vivo active potent VEGFR-2 inhibitor. Bioorg. Med. Chem. Lett. 18, 2985–2989 (2008).
Rajak, H. et al. 2,5-Disubstituted-1,3,4-oxadiazoles/thiadiazole as surface recognition moiety: design and synthesis of novel hydroxamic acid-based histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 21, 5735–5738 (2011).
Valente, S. et al. 1,3,4-Oxadiazole-containing histone deacetylase inhibitors: anticancer activities in cancer cells. J. Med. Chem. 57, 6259–6265 (2014).
Ihmaid, S. K. et al. Design of molecular hybrids of phthalimide-triazole agents with potent selective MCF-7/HepG2 cytotoxicity: Synthesis, EGFR inhibitory effect, and metabolic stability. Bioorg Chem 111, 104835 (2021).
Kunos, C. A. & Ivy, S. P. Triapine radiochemotherapy in advanced stage cervical cancer. Front. Oncol. 8, 1–7 (2018).
Fan, C. D. et al. A novel copper complex of salicylaldehyde pyrazole hydrazone induces apoptosis through up-regulating integrin β4 in H322 lung carcinoma cells. Eur. J. Med. Chem. 45, 1438–1446 (2010).
Kamal, A. et al. Combretastatin linked 1,3,4-oxadiazole conjugates as a potent tubulin polymerization inhibitor. Bioorg. Chem. 65, 126–136 (2016).
Rejmund, M. et al. Piperazinyl fragment improves anticancer activity of triapine. PLoS One 13(4), e0188767 (2018).
Pavase, L. S., Mane, D. V. & Baheti, K. G. Anti-inflammatory exploration of sulfonamide containing diaryl pyrazoles with promising COX-2 selectivity and enhanced gastric safety profile. J. Heterocycl. Chem. 55, 913–922 (2018).
Abdelrahman, K. S. et al. Development and Assessment of 1,5–Diarylpyrazole/Oxime Hybrids Targeting EGFR and JNK–2 as Antiproliferative Agents: A Comprehensive Study through Synthesis, Molecular Docking, and Evaluation. Molecules, 28, (2023).
Shen, H. C. & Hammock, B. D. Discovery of inhibitors of soluble epoxide hydrolase: A target with multiple potential therapeutic indications. J. Med. Chem. 53, 1789–1808 (2011).
Abdelazeem, A. H. et al. Discovery of novel urea-diarylpyrazole hybrids as dual COX-2/sEH inhibitors with improved anti-inflammatory activity and highly reduced cardiovascular risks. Eur. J. Med. Chem. 205, 112662 (2020).
López-Maldonado, E. A. et al. Synthesis, characterization and biological activity of schiff bases based on Chitosan and acetyl acetophenone and 2-phenyl-2-acetophenone derivatives: experimental and theoretical approach. J Mol. Struct 1326, 141079 (2025).
Inci, G. H., Cem, Y., Synthesis of potential anticancer & effective compounds containing pyrazole – benzensulfonamide moieties, enlightenment of their structures and their cytotoxic activities. WO2019/17857, 2019, A2 (2019).
Dou, G. et al. Clean and efficient synthesis of isoxazole derivatives in aqueous media. Molecules 18, 13645–13653 (2013).
Mohamed, M. S., Mansour, Y. E., Amin, H. K. & El-Araby, M. E. Molecular modelling insights into a physiologically favourable approach to eicosanoid biosynthesis Inhibition through novel thieno[2,3-b]pyridine derivatives. J. Enzyme Inhib. Med. Chem. 33, 755–767 (2018).
Lu, X. Y. et al. Design, synthesis and evaluation of benzenesulfonamide-substituted 1,5-diarylpyrazoles containing phenylacetohydrazide derivatives as COX-1/COX-2 agents against solid tumors. RSC Adv. 6, 22917–22935 (2016).
Animal Cell Culture Guide. Atcc (2014).
Riccardi, C. & Nicoletti, I. Analysis of apoptosis by Propidium iodide staining and flow cytometry. Nat. Protoc. 1, 1458–1461 (2006).
Sebaugh, J. L. Ã. Guidelines for accurate EC50 / IC50 Estimation. Pharm. Stat. 10, 128–134 (2011).
AbdElhameid, M. K., Labib, M. B., Negmeldin, A. T., Al-Shorbagy, M. & Mohammed, M. R. Design, synthesis, and screening of ortho-amino thiophene Carboxamide derivatives on hepatocellular carcinomaas VEGFR-2Inhibitors. J. Enzyme Inhib. Med. Chem. 33, 1472–1493 (2018).
Abdelazeem, A. H. et al. Novel Diphenylthiazole derivatives with multi-target mechanism: Synthesis, Docking study, anticancer and anti-inflammatory activities. Bioorg. Chem. 75, 127–138 (2017).
El-Sherief, H. A. M. et al. Synthesis, anticancer activity and molecular modeling studies of 1,2,4-triazole derivatives as EGFR inhibitors. Eur. J. Med. Chem. 156, 774–789 (2018).
Slack-davis, J. K. et al. Cellular characterization of a novel focal adhesion. J. Biol. Chem. 282, 14845–14852 (2007).
Long, Z. et al. Design, synthesis and biological evaluation of 4-arylamino-pyrimidine derivatives as focal adhesion kinase inhibitors. Bioorg Chem 140, 106792 (2023).
Hynes, N. E., Lane, H. A. & Miescher, F. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer. 5, 341–354 (2005).
Abou-Zied, H. A., Youssif, B. G. M., Mohamed, M. F. A., Hayallah, A. M. & Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and Docking studies of novel Xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg Chem 89, 102997 (2019).
Hisham, M., Youssif, B. G. M., Osman, E. E. A., Hayallah, A. M. & Abdel-Aziz, M. Synthesis and biological evaluation of novel Xanthine derivatives as potential apoptotic antitumor agents. Eur. J. Med. Chem. 176, 117–128 (2019).
Arora, R. et al. Design, synthesis and characterisation of a novel type II B-RAF paradox breaker inhibitor. Eur J. Med. Chem 250, (2023).
Al-Wahaibi, L. H. et al. Design and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg Chem 104, 104260(2020).
Cui, Z. et al. Design, synthesis and evaluation of azaacridine derivatives as dual-target EGFR and Src kinase inhibitors for antitumor treatment. Eur. J. Med. Chem. 136, 372–381 (2017).
El-Sherief, H. A. M., Youssif, B. G. M., Bukhari, S. N. A., Abdel-Aziz, M. & Abdel-Rahman, H. M. Novel 1,2,4-triazole derivatives as potential anticancer agents: Design, synthesis, molecular Docking and mechanistic studies. Bioorg. Chem. 76, 314–325 (2018).
Ramadan, M. et al. Design and synthesis of new pyranoquinolinone heteroannulated to Triazolopyrimidine of potential apoptotic antiproliferative activity. Bioorg Chem 105, 104392 (2020).
Karagül, M. İ., Aktaş, S., Yetkin, D., Bayrak, G. & Çelikcan, D. P53, Bcl2 and Bax Expression and Apoptosis in Perifosine and Vitamin D-Treated Endometrial Cancer Cell Line (HEC1A). in 1564MDPI AG, (2018). https://doi.org/10.3390/proceedings2251564
Smith, G. et al. Design, synthesis, and biological characterization of a caspase 3/7 selective Isatin labeled with 2-[18f]fluoroethylazide. J. Med. Chem. 51, 8057–8067 (2008).
Abdelall, E. K. A., Elshemy, A. H., Labib, H. & Mohamed, E. A. M. B. F. Characterization of novel heterocyclic compounds based on 4-aryl-4H-chromene scaffold as anticancer agents: Design, synthesis, antiprofilerative activity against resistant cancer cells, dual β-tubulin/c-Src inhibition, cell cycle arrest and apoptosis induction. Bioorg Chem 120, 105591 (2022).
Liu, Y. et al. CB-Dock2: improved protein-ligand blind Docking by integrating cavity detection, Docking and homologous template fitting. Nucleic Acids Res. 50, W159–W164 (2022).
Xiong, G. et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 49, W5–W14 (2021).
Kumar, B. K. et al. Pharmacophore based virtual screening, molecular docking, molecular dynamics and MM-GBSA approach for identification of prospective SARS-CoV-2 inhibitor from natural product databases. J. Biomol. Struct. Dyn. 40 (3), 1363–1386 (2022).
Sidique, S. et al. Design and synthesis of pyrazole derivatives as potent and selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). Bioorg. Med. Chem. Lett. 19, 222–225 (2009).
Bechmann, N. et al. Novel (pyrazolyl)benzenesulfonamides with a nitric oxide-releasing moiety as selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett. 25, 3295–3300 (2015).
Yoon, J. Y., Lee, S. & Shin, H. Recent advances in the regioselective synthesis of pyrazoles. Curr. Org. Chem. 15, 657–674 (2011).
Ibrahim, H. S. et al. Isatin-pyrazole benzenesulfonamide hybrids potently inhibit tumor-associated carbonic anhydrase isoforms IX and XII. Eur. J. Med. Chem. 103, 583–593 (2015).
Abdelazeem, A. H. et al. Discovery of novel urea-diarylpyrazole hybrids as dual COX-2/sEH inhibitors with improved anti-inflammatory activity and highly reduced cardiovascular risks. Eur J. Med. Chem 205, 112662 (2020).
He, J. et al. Synthesis, biological Evaluation, and molecular Docking of arylpyridines as antiproliferative agent targeting tubulin. ACS Med. Chem. Lett. 11, 1611–1619 (2020).
Cob-Calan, N. N. et al. Molecular Docking and dynamics simulation of protein β-tubulin and antifungal Cyclic lipopeptides. Molecules 24, 1–10 (2019).
Wang, F. et al. Molecular description of pyrimidine-based inhibitors with activity against FAK combining 3D-QSAR analysis, molecular Docking and molecular dynamics. Arab. J. Chem. 14, 103144 (2021).
Shi, M. et al. Molecular Docking, molecular dynamics Simulations, and free energy calculation insights into the binding mechanism between VS-4718 and focal adhesion kinase. ACS Omega. 7, 32442–32456 (2022).
Yang, H. et al. Design, synthesis, biological evaluation and molecular docking of novel F-18-labeled focal adhesion kinase inhibitors as potential tumor radiotracers. Molecules, 29, (2024).
Osama, M. et al. Hiroyuki Konno, Design, synthesis, and Biological Evaluation of Novel 1,5-diarylpyrazole Carboxamides with Dual Inhibition of EGFR and COX-2 for the Treatment of Cancer and Inflammatory Diseases, Bioorg Med Chem Lett., 130, 130416 (2025).
Elshibani, F. A. et al. GC-MS and HPLC chemical profile, antioxidant, anti-acetylcholinesterase, and anti-diabetic activities of Libyan Salvia lanigera herb extract and essential oil. Sci Rep. 15(1), 31853 (2025).
Ritika, A. et al. In Silico prediction, characterization and molecular Docking studies on Glutathione-S-transferase as a molecular sieve for toxic agrochemicals explored in survey of North Indian farmers. Heliyon 7, e07875 (2021).
De Ferreira, R. & Schapira, M. A systematic analysis of atomic protein-ligand interactions in the PDB. Medchemcomm 8, 1970–1981 (2017).
Brandl, M., Weiss, M. S., Jabs, A., Sühnel, J. & Hilgenfeld, R. C-H···π-interactions in proteins. J. Mol. Biol. 307, 357–377 (2001).
Mahadevi, A. S. & Sastry, G. N. Cation-π interaction: its role and relevance in chemistry, biology, and material science. Chem. Rev. 113, 2100–2138 (2013).
Mohamed, H. S. H. & Ahmed, S. A. Reviewing of synthesis and computational studies of Pyrazolo pyrimidine derivatives. J. Chem. Reviews. 1, 183–232 (2019).
Obakiro, S. B. et al. Phytochemical, Cytotoxicity, and Antimycobacterial Activity Evaluation of Extracts and Compounds from the Stem Bark of Albizia coriaria Welw ex. Oliver. Evidence-based Complementary and Alternative Medicine, 2022. (2022).
Durán-Iturbide, N. A., Díaz-Eufracio, B. I. & Medina-Franco, J. L. In Silico ADME/Tox profiling of natural products: A focus on BIOFACQUIM. ACS Omega. 5, 16076–16084 (2020).
Adem, Ş., Yırtıcı, Ü., Aydın, M., Rawat, R. & Eyüpoğlu, V. Natural flavonoids as promising 6-phosphogluconate dehydrogenase inhibitor candidates: in Silico and in vitro assessments. Arch. Pharm. (Weinheim). 1–14. https://doi.org/10.1002/ardp.202300326 (2023).
Acknowledgements
This work was funded by the Deanship of Graduate Studies and Scientific Research at Jouf University under grant No. (DGSSR-2025-FC-01022).
Funding
The Deanship of Graduate Studies and Scientific Research at Jouf University under grant No. (DGSSR-2025-FC-01022).
Author information
Authors and Affiliations
Contributions
M. A. A., Funding acquisition and writing - review & editing; S. N. A. B., validation and methodology; M. H. E. format analysis and investigation; M. E., Investigation, and format analysis; E. M. M., resources and data curation; A. M., visualization; W. A., methodology; M. S. A., methodology and writing original draft; A. F., methodology, writing original draft and software, H. A. H. E. methodology, writing original draft and administration; A. G. S. E., methodology, conceptualization and writing original draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Abdelgawad, M.A., Bukhari, S.N.A., Elkomy, M.H. et al. In-silico studies, synthesis, and pharmacological screening of novel multitarget diphenylpyrazole scaffold as EGFR/BRAF and cyclooxygenase-2 inhibitors. Sci Rep 16, 1973 (2026). https://doi.org/10.1038/s41598-025-33006-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-33006-6
