
Abstract
Intramammary dry-off treatment is widely considered an effective method for preventing and curing intramammary infection (IMI) in lactating cows; however, it is not commonly used in small ruminants like goats. Therefore, this study was designed to evaluate the effect of an approved cefazolin-based intramammary treatment on the milk microbiota of Alpine dairy goats during the dry and early lactation periods. Sixty goats were randomly selected based on bacteriological results and randomly allocated into the control group (CG) or the treatment group (TG). Cefazolin 250 mg (Cefovet A, Dopharma, Firenze, Italy) was administered to the TG group at dry-off, whereas the CG received no treatment. Pooled milk samples were collected at dry-off (T1; 52 samples), colostrum (T2; 46 samples), and 5–10 days in milk (T3; 55 samples) for bacteriological analysis, somatic cell count (SCC), and 16 S rRNA gene sequencing. SCC levels were initially high in both groups at T1 (TG: 1,588,000 cells/mL; CG: 1,629,000 cells/mL), which significantly decreased at T3 (TG: 148,000 cells/mL; CG: 153,000 cells/mL). Notably, the TG had fewer infected mammary glands than the CG at T3 (p = 0.0248), while no differences were found at T1 or T2. Despite the reduction in SCC and infection rates, cefazolin-based treatment did not significantly affect the alpha- and beta-diversity between the TG and CG. On the other hand, shifts in microbial composition, including fluctuations in Firmicutes, Proteobacteria, and Actinobacteria, were primarily due to the lactation stage rather than the treatment. Differential abundance analyses identified non-pathogenic genera, such as Acinetobacter, Bacteroides, and Paracoccus, that varied between groups at different timepoints. The study provided insights into the effects of cefazolin-based dry goat treatment on goat milk microbiota and its changes during the lactation cycle, demonstrating its potential to reduce SCC and mammary gland infections without significant alterations to the milk microbiota.
Similar content being viewed by others

Introduction
Goat milk production is of great importance in Europe1, where approximately 19.3 million goats, equal to 1.9% of the world’s goat population, provide 2.8 million tons of milk (15.1% of global production) and 201.3 thousand tons of cheese (35.1% of global production). Among the European goat population, 8.3% are reared in Italy, contributing 0.61 million tons of milk and 6.04 thousand tons of cheese2. Among the medium-sized breeds, the cosmopolitan Alpine goat from Switzerland is predominant in Piedmont and Lombardy3. The most common economic problem in the dairy industry, intramammary infection (IMI), results in lower milk yield and quality, higher somatic cell counts (SCCs), and veterinary treatment and management costs, leading to early animal replacement and culling4,5. In dairy cows, intramammary treatment has been regarded as the most effective cure and preventive measure during the dry-off period, eliminating existing infections and reducing new IMI risk factors for several decades6. On the other hand, mastitis in small ruminants, which is caused mostly by Staphylococcus spp. (including S. aureus) and Mannheimia hemolytica, has proven to be particularly difficult to treat, and persistent IMIs have become very common throughout lactation and dry periods7,8. In small ruminants, few molecules are officially approved for intramammary therapy, and few reports exist on their use at dry-off9. Dry-off at the end of lactation refers to the cessation of milking, occurring 6–8 weeks prior to kidding and enabling the goat to regenerate mammary tissue and prepare for the subsequent lactation. Intramammary antibiotic usage during dry period has been considered as the most effective measure to control IMI and reduce SCC in cows as well as in sheep and goats9,10. Intramammary dry therapy in cows can cure existing infections and prevent new IMIs over the dry period, leading to lower new IMI incidence rate at the beginning of next lactation11,12,13. It has been stated that 60–90% of existing IMI are cured14,15 and 50% of new IMI are decreased after dry treatment in cows9,14,15. Dry-goat therapy (DGT) can cure 66–100% of existing IMIs9,16 and reduce 1.9–12.7% of new IMIs9.
Cefazolin, a first-generation cephalosporin that shows in vitro activities against Gram-positive and Gram-negative bacteria, is the only drug approved in Italy for goats at dry-off, but scientific evidence regarding its efficacy and safety in goats is still under discussion. Cefazolin has minimal oral absorption and results in low plasma and tissue concentrations when administered via intramammary infusion. Lactating animals excrete it unchanged in urine, with only trace amounts found in milk. According to Sato et al., after intramammary administration in cows, serum levels of cefazolin peaked at 0.06 µg/ml one hour after administration, and no cefazolin was detected in milk 72 h later17. Although specific studies on cefazolin’s withdrawal period in goats are lacking, cefuroxime, a similar cephalosporin antibiotic, has a withdrawal period of approximately 95.3 h (± 17.23) in low-producing Saanen dairy goats18. Additionally, cefazolin-based intramammary treatments, including those used during dry-off, have been approved by EU regulatory authorities. The European Medicines Agency has confirmed the safety, efficacy, and residue profiles for cefazolin in milk19,20. Furthermore, antimicrobial treatment, together with species, breed, and animal health, can alter the milk microbiota and its protective role in the mammary gland21,22,23,24,25. Milk has been previously defined as an environment rich in microbial diversity26,27. This complex community of microorganisms is essential not only for ensuring the quality and safety of dairy products but also for improving animal health28. Thus, understanding the milk microbiota is crucial for developing strategies to increase animal health and production29,30,31. Among the essential culture-independent techniques for the assessment of genetic wealth, metagenomics represents a powerful tool that uses sequence-based and functional-based approaches32. In livestock, the milk microbiota has been previously studied in dairy cattle27,33 to explore the complex interactions between commensal organisms and pathogenic bacteria in both healthy and infected animals. Firmicutes, Proteobacteria, Bacteroidetes, and Actinobacteria have been found to be the main bacterial phyla shaping the structure of the bovine milk microbiota under healthy conditions, while a reduced abundance of Bacteroidetes has been demonstrated in mastitis patients34. Alpha-diversity denotes the diversity within a single sample, typically measured by the Chao1 index (richness) and the Shannon index (diversity), while beta-diversity refers to the variation in microbial community composition between samples or groups25. Factors determining the composition of goat milk microbiota encompass lactation stage, health state, udder infection, and season, which influence both alpha- and beta-diversity25,35. Previous results on cows treated with antibiotics revealed that the diversity and richness of the microbiota were not significantly different between animals with or without intramammary ceftiofur hydrochloride33, whereas another study36 reported increased alpha diversity in the milk microbiota of mastitis cases treated with third-generation cephalosporin. Few studies25,37,38,39 have been published on milk microbiota in small ruminants, highlighting the dominance of the phylum Actinobacteria, followed by the high presence of Firmicutes, Proteobacteria, and Bacteroidetes40. The phylum Firmicutes, an important group of bacteria in both goat milk and cow milk21, is predominantly represented by a variety of potentially beneficial bacterial genera and potential mastitis agents, such as Staphylococcus (S. arlettae, S. capitis, S. delphini, S. epidermidis, S. equorum, S. hominis, S. lentus, S. pasteuri, S. sciuri, S. simulans, S. schleiferi, S. vitulinus, S. warneri, and S. xylosus), Streptococcus, Lactococcus, Lactobacillus, Lacticaseibacillus, Enterococcus, and Veillonella40. As occurs in the intestinal microbiota of healthy goats41, the homeostatic balance of the milk microbiota of the mammary gland should be extremely beneficial to the host. Previous results on the structure and metabolism of the rumen microbiota indicated that antimicrobial use in goats affects both the rumen fluid and mucosal microbiota, reducing functional redundancy42. Similarly, mastitis therapy with intramuscular infusions containing enrofloxacin induced an alteration in the milk microbiota and caused a disturbance in abundance, with negative effects on the health and physiological functions of the host38. To our knowledge, this is the first study in which the milk microbiota at dry-off and in early lactation was investigated in healthy and infected Alpine goats with and without cefazolin intramammary treatment.
Results
Bacteriological culture and SCC
The culture results revealed that the dominant bacterial species in CG was S. caprae (29%) at T1, whereas the dominant bacterial species in T2 and T3 were S. equorum (33%) and Serratia marcescens (11%), respectively. In the TG, S. equorum was the dominant bacteria at T1 (15%) and T2 (22%), while Serratia marcescens at T3 (7%), as reported in Table 1.
At T1, the average SCC was 1,588,000 cells/mL in the TG and 1,629,000 cells/mL in the CG. At T3, the average SCC decreased to 148,000 cells/mL in the TG and 153,000 cells/mL in the CG. No significant differences were observed between the two groups (Fig. S1).
Figure 1 presents the proportions of healthy and infected goat mammary glands in the CG and TG at the three timepoints considered. No differences emerged at T1 (p value = 0.8867) or T2 (p value = 1.000), whereas the TG had a lower proportion of infected mammary glands than the CG did at T3 (p value = 0.0248).
Goat milk microbiome
Changes in the composition of the goat milk microbiota
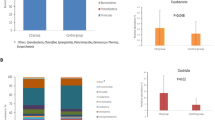
Figure 2 shows the relative abundances of bacterial phyla at T1, T2 and T3 in both the TG and CG. At T1, the three most abundant phyla were Firmicutes, Actinobacteria, and Proteobacteria, accounting for 41.6%, 29.7% and 23.1%, respectively. Firmicutes increased at T2 (47.3%) and decreased at T3 (36.2%), Actinobacteria remained stable over time (29.5% and 27.7%, at T2 and T3, respectively), whereas Proteobacteria decreased at T2 (18.6%) and increased at T3 (29.9%). Additional phyla with average relative abundances higher than 1% over time included Bacteroidetes (2.5–4.6%) and Verrucomicrobia (0.3–1.6%). In terms of normalized absolute counts, significant changes (p value < 0.05, from Eq. (2)) were observed for Chloroflexi, Deinococcus-Thermus, Euryarchaeota, Fibrobacteres, and Verrucomicrobia. The detailed composition with the relative proportions of each phylum in both groups and all timepoints is summarized in S1, and pairwise contrasts between phylum abundances along timepoints were shown in Fig. S2. Additionally, the core goat milk microbiota, consisting of genera present in more than 90% of the samples, is summarized in Table 2. Moving from the aggregated phylum to the unaggregated OTU level, according to Eq. (2), a total of 35 genera were differentially abundant between the CG and TG at different timepoints (8 genera at T1, 11 genera at T2 and 16 genera at T3). The abundances of the genera Acinetobacter, Atopostipes, Bacteroides, Iamia, Methylobacterium, Paracoccus, Ruminococcaceae UCG-005, Stenotrophomonas and Turicibacter differed significantly between the two experimental groups (Fig. 3 and S2). None of these genera appear to be related to any pathogenic groups; most of these groups were significant because they were present at only one timepoint.
Alpha- and beta-diversity
According to Eq. (1), neither the alpha diversity indices were significantly different between healthy and infected milk samples at T1 (p > > 0.05), nor the Bray-Curtis dissimilarities between the two groups (p = 0.437) (S3). This justified the use of the statistical models described below, where healthy and infected milk samples were analyzed jointly to investigate differences among timepoints and between experimental groups (CG and TG).
We first examined the overall composition of the goat milk microbiome, irrespective of the experimental group. For alpha- and beta-diversity, the effects of timepoint from Eq. (3) and from PERMANOVA were consistently significant (Figs. 4 and 5).
In this study, we estimated alpha diversity indices for species richness (ACE, Chao1, and observed number of OTUs) and diversity (Shannon, Simpson, Fisher and inverse Simpson indices), as shown in Fig. 4. From Eq. (2), we determined that the alpha diversity of the milk microbiota in dairy goats changed significantly over time, specifically at T2 and T3, with respect to T1, but we did not observe a significant effect of the treatment on the alpha diversity of the milk microbiota (CG vs. TG, S4).
On the basis of the milk microbiome, the Bray‒Curtis dissimilarity matrix was used to analyze the distances between CG and TG across timepoints (beta diversity). Figure 5 shows the multidimensional scaling plot (first two dimensions) of Bray‒Curtis distances between groups at T1 (Fig. 5A), T2 (Fig. 5B) and T3 (Fig. 5C) and among timepoints irrespective of treatment (Fig. 5D). PERMANOVA (S5) revealed that the timepoint effect was significant (p value = 0.00099) but not the treatment effect (p value = 0.758).
Analysis of the Firmicutes: Bacteroidetes ratio (F: B ratio)
The F: B ratio was greater at T2 (CG = 57.54, TG = 88.56) compared to T1 (CG = 36.35, TG = 27.62) and T3 (CG = 21.3, TG = 10.47), with large within-group variation within groups. The F: B ratios are not distributed normally but are skewed (skewness coefficient = 5.30) and typically have heavy asymmetrical tails (the ratio between the mean absolute deviation and standard deviation = 0.473). The results from the model indicated that there was no significant effect of treatment on the F: B ratio in the goat milk microbiome (p value = 0.67068), whereas the effect of timepoint was highly significant (p value = 0.000305).
Discussion
This study investigated the effect of an approved cefazolin-based intramammary treatment on goats at different timepoints. The results provide some understanding of the goat milk microbiome during these critical periods and highlight the negligible impact of antibiotic therapy on microbial diversity and richness.
Checking the validity of our baseline samples by the analysis of alpha diversity indices revealed no significant differences in microbial richness or evenness between CG and TG at T1 (dry-off), indicating that random milk samples from both healthy and infected goats, as representative of the herd goat population, presented similar bacterial compositions.
Although the treatment groups did not present differences in terms of the indices of alpha diversity, the bacteriological results revealed that antibiotic use during the dry-off period (T1) improved the cure rate and reduced the risk of new IMI during the early lactation period (T3). Contrasting and comparing the main pathogens in goats with and without cefazolin at T3, the lower proportion of infected mammary glands in the TG than in the CG indicated the efficiency of DGT in inhibiting bacterial proliferation and diminishing mastitis cases.
At T2 and T3, changes in microbial diversity were evident in both TG and CG, confirming that the milk microbiota varied with time and with the lactation stage, as previously demonstrated in dairy cattle43. Additionally, beta diversity analysis provided evidence that time was the primary factor influencing microbial composition and suggested that physiological dynamics are likely much greater than treatment effects for driving community structure. These findings from alpha- and beta-diversity analyses were further confirmed by the significant variations in the F: B ratio obtained within the same group, which were attributed mainly to timepoints rather than to cefazolin treatment; therefore, the physiological fluctuations associated with the dry-off and postpartum periods have a relatively greater influence on the microbial composition than does antibiotic treatment12,43. The F: B ratio, which is traditionally used to describe shifts in the human gut microbiome, has recently been applied to studies of the dairy milk microbiota as well12, whose observations agreed with our results suggesting the importance of physiological transitions in milk microbiota modulation12,44. Similar patterns in microbial stability and resilience in response to antibiotic treatment were observed in cows treated with cephalonium dehydrate and cloxacillin12,45, as well as in goats treated with enrofloxacin38. Notably, across the TG and CG, the core milk microbiota was composed predominantly of Firmicutes, Actinobacteria, and Proteobacteria, representing the three most abundant phyla, as previously reported46. Firmicutes increased at T2, followed by a decrease at T3, whereas Proteobacteria showed the opposite trend. In contrast, Actinobacteria remained stable over time, suggesting that some microbial taxa may be resilient to various physiological changes and antibiotic treatments43. Previous results on the temporal stability of milk microbiota after antibiotic treatment have also highlighted a reservoir of core bacteria that can be relatively rapidly restored to preperturbation levels12. At the genus level, the core goat milk microbiota was predominantly composed of Staphylococcus spp., Cutibacterium spp., and Streptococcus spp. that remained stable over time, highlighting the genus-level composition of the core microbiota in healthy caprine milk47. The persistence of specific genera, such as Staphylococcus and Streptococcus, in bacterial populations after antibiotic treatment is consistent with previous findings in cows treated with cefazolin, where the antibiotic reduced the pathogen load without significantly disrupting the core milk microbiota12,45, but it was found for the first time in goats, highlighting the novelty of our study.
Furthermore, the abundances of other genera, such as Acinetobacter, Atopostipes, Bacteroides, and Stenotrophomonas, significantly differed between the CG and TG at various timepoints and remained relatively unaffected by antibiotic treatment. Further investigations are needed to understand the actual impact of these significant changes observed at the genus level, since the alpha- and beta-diversity analyses revealed no significant differences between the two experimental groups. In small ruminants, particularly sheep, the effects of antibiotics on the milk microbiome are highly variable. A study on antibiotics such as oxytetracycline and penicillin‒streptomycin used at dry-off reported that both microbial shifts and resilience were influenced by factors such as dosage, timing, and animal health status38. The limited impact of cefazolin on the milk microbiota observed in our study suggests that Alpine goats may exhibit a degree of microbial stability in response to cefazolin.
Based on the bacteriological results, administering cefazolin during dry-off (T1) significantly impacted the milk bacteriology. The administration of cefazolin during T1 had a long-lasting effect in diminishing the presence of certain microorganisms. This indicates that cefazolin-based dry-off treatment considerably decreases the occurrence of these microorganisms in the udder. Some commensal bacteria may become opportunistic, leading to IMI in dairy animals48.
Conclusion
This study highlights the efficacy of cefazolin-based dry goat therapy for better udder health management in small ruminants, especially goats. The treatment significantly reduced SCC and IMI incidence rates by early lactation, showing its efficacy in preventing new infections. While cefazolin effectively inhibited mastitis causative agents, its overall impact on the milk microbiota was minimal, suggesting that it can be used without significantly disrupting microbial communities in milk. Observed microbiota shifts were primarily linked to lactation stages rather than antibiotic intervention, with stable key phyla such as Firmicutes, Proteobacteria, and Actinobacteria. These findings validate the use of cefazolin as targeted antibiotic therapy for managing mastitis without disrupting microbial balance. Future work could further investigate its long-term effects, resistance profiles, and applicability across breeds and conditions to optimize animal and udder health, and milk quality.
Materials and methods
Ethics statements
The current longitudinal study was carried out at a commercial Alpine goat farm in Lombardy, Italy. The experimental design and procedures performed on the goats were reviewed and approved by the Ethics Committee for Animal Welfare of the University of Milan, Italy (documentation no. OPBA 118–2023) and followed the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. All methods were performed in accordance with the relevant guidelines and regulations.
Animal selection and antibiotic treatments
The study was conducted from November 2022 to April 2023. The herd consisted of 150 Alpine goats with an average milk production of 747 L per lactation and an average bulk tank SCC of 2,238,000 cells/mL of milk. The bulk tank composition included 3.8% fat, 3.5% protein, 4.6% lactose, and 2.6% casein, with a daily milk production of 2.5 L/goat. The goats were housed in a free stall housing system with straw-deep bedding. A total of 106 dairy goats in their first (63) and second (43) lactation periods were recruited 7 days before drying on the basis of the following criteria: (i) good health conditions without signs of clinical mastitis and (ii) neither systemic nor intramammary antibiotic treatment or anti-inflammatory medication within 30 days prior to drying. Milk samples were collected from each udder half and cultured for bacteriological analysis following the NMC recommendations49 at the Laboratorio di Malattie Infettive degli Animali (MiLab, Università degli Studi di Milano, Lodi, Italy). For each positive sample, microbiological identification of bacterial colonies was performed with MALDI-TOF MS with the MALDI Biotyper System (Bruker Daltonik GmbH, Bremen, Germany) via the direct transfer method as previously described50,51. On the basis of the bacterial culture results, 60 of these 106 goats were selected for this study: 30 healthy goats with both udder-half milk samples analyzed as culture negative and 30 infected goats with at least one udder-half milk sample analyzed as culture positive for one or two pathogens. Health status at enrollment was adopted only as a blocking factor when the two experimental groups were balanced. Information about age, number of lactations, days in milk (DIM), expected date of kidding, history of clinical mastitis, use of antibiotics, and milk production was recorded for all selected goats before drying. At dry-off, the 60 goats were randomly assigned to two experimental groups: a control group (CG: 30 goats; 15 healthy and 15 infected) without antibiotics and a treatment group (TG: 30 goats; 15 healthy and 15 infected) with antibiotics. Each half udder of a TG goat was treated with an intramammary suspension of cefazolin 250 mg (Cefovet A, Dopharma, Firenze, Italy). Among the 60 selected goats, 5 (CG: 2; TG: 3) were excluded for the use of antibiotics (n = 3) and culling (n = 2) during the study period. The remaining 55 (CG: 28; TG: 27) goats were analyzed.
Sampling time, microbiological analysis and somatic cell count
A total of 165 pooled milk samples were collected from each goat at three different timepoints: dry-off (T1), colostrum (T2) and 5–10 DIM (T3), following the guidelines of the National Mastitis Council49. This resulted in 55 samples at each timepoint, with 28 from the CG and 27 from the TG. The milk samples were immediately transferred to MiLab for microbiological analysis as previously described, and SCC was performed with a Bentley Somacount 150 (Bentley Instrument, Chaska, MN, USA). As 12 milk samples were lost during the process, a total of 153 samples were retained for further analysis, as shown in Fig. 6.
DNA extraction
Genomic DNA extraction from the 153 pooled milk samples was carried out following a previously developed protocol52. The quality and quantity of the extracted DNA were evaluated with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA), and the extracted DNA was stored at -20 °C until sequencing.
16 S rRNA-gene sequencing and bioinformatics processing
The amplification of bacterial DNA was accomplished with the help of primers that have been reported in the scientific literature44,53. These primers focus on the V3-V4 hypervariable regions of the 16 S rRNA gene. All of the samples were subjected to a PCR amplification of 25 µL. A total of 2 µL of genomic DNA (5 ng/L) was mixed with 12.5 µL of Phusion High-Fidelity Master Mix 2X (Thermo Fisher Scientific, Walthem, MA, USA) and 0.2 µL of each primer (100 M). In addition, blank controls were used, meaning that no DNA template was added to the process. Applied Biosystem 2700 thermal cycler (Thermo Fisher Scientific, Waltham, MA, USA) was used to carry out the initial step of the amplification process. Following a denaturing step at 98 °C for 30 s, the samples were subjected to 25 cycles consisting of annealing at 56 °C for 1 min, extension at 72 °C for 1 min, and a final extension at 72 °C for 7 min. Following the 16 S Metagenomic Sequencing Library Preparation Protocol (Illumina, San Diego, California, USA), amplicons were cleaned with Agencourt AMPure XP (Beckman Coulter Inc., Brea, California, USA), and libraries were created in accordance with the protocol. The generated libraries were subjected to real-time PCR via KAPA Library Quantification Kits (Kapa Biosystems, Inc., Massachusetts, USA). After that, they were pooled in an equimolar proportion and sequenced via a single MiSeq (Illumina) run with paired-end reads that were 2 × 250 bases in length.
The quality of the demultiplexed paired-end reads obtained from the sequencing of the 16S rRNA gene was initially evaluated via FastQC54. After that, the reads were cleaned by eliminating primers and adapters via the Python utility Cutadapt55 and then trimmed for quality via the C + + tool Sickle56, with a Phred threshold > 20 (that is, the end portion of the reads was eliminated if it was of low quality). Following the completion of the cleaning process, the forward and reverse paired-end reads were combined via the Python pipeline known as Microbial Community Analysis (MICCA)57. More specifically, the function known as “mergepairs” was utilized with default values, which included a minimum overlap length of 32 and a maximum number of mismatches in the overlap region of 8. After the readings were assembled, they were checked for quality and reads that were missing or uncalled bases or had an estimated error rate greater than 1% (1 error in 100 bases) were discarded. Using the denoising method that was implemented in the MICCA function ‘otu’ (method ‘denovo_unoise’), all of the remaining reads were utilized to identify operational taxonomic units (OTUs)58. In the end, the OTUs that were identified were classified via the MICCA function known as “classify” to assign taxa that were annotated in the SILVA132 reference database59. The parameters that were used for this classification were as follows: for each OTU, the maximum number of hits that can be considered for taxonomic classification is 3; a taxon is assigned if it is present in at least half of the hits; and an OTU is rejected if the proportion of alignment to the reference sequence is less than 0.75. The obtained OTU table was filtered by removing the least represented OTUs with < 15 counts in fewer than 3 samples. On the basis of the filtered OTUs, the following alpha diversity indices were calculated: the number of observed OTUs, ACE, Chao1, Fisher, Simpson, Shannon, and inverse Simpson indices, among others. To calculate the alpha diversity indices, the function estimate_richness from the phyloseq R package was used60,61. The cumulative-sum scaling method was then utilized to normalize the filtered OTU counts to account for the unequal sequencing depth62. The Bray‒Curtis distances between samples were determined by using the filtered and normalized OTU counts as the basis63. The nonparametric evaluation of between-group (CG/TG) and pairwise Bray‒Curtis dissimilarities among timepoints was carried out via permutational analysis of variance (999 permutations)64.
Statistical analysis
Both experimental groups, TG and CG, included healthy goats and those affected by IMI. At T1, the milk microbiota data were analyzed to assess any differences between samples from healthy goats and those with IMI.
where yij represents the alpha-diversity index values for sample i, udder_statusj represents the presence or absence of IMI, and eij represents the model residuals. Similarly, pairwise Bray–Curtis dissimilarities at T1 between healthy and IMI milk samples were evaluated via the permutational analysis of variance approach described above.
The association between mammary health status (healthy/infected) and antibiotic treatment (CG/TG) was evaluated via a chi-square test for independence, which is based on the difference between observed and expected frequencies and on one degree of freedom for the resulting χ2 random variable.
The differential OTU abundance between groups (CG/TG) was evaluated at each timepoint with the following linear model:
where yij represents the CSS-normalized counts for sample i, treatmentj represents the treatment effect (antibiotic-TG or control-CG), and eij represents the model residuals.
Alpha diversity indices were analyzed via a linear model with both timepoint and treatment effects:
where all the elements are as in Eq. (1), with the addition of the timepoint effect in classes (T1, T2, T3).
The Firmicutes: Bacteroidetes (F: B) ratio was calculated for all samples (both groups, all timepoints) from normalized OTU counts. The F: B ratios tend to be nonnormally distributed, with pronounced skewness and fat tails. Therefore, we opted for a regression model approach with Cauchy-distributed errors, as the Cauchy distribution accommodates fat (heavy) tails better than the Gaussian distribution does65,66. The same model as in Eq. (2) was used, where yijk was the F: B ratio for sample i belonging to treatment j at timepoint k, and eijk ~ Cauchy (0,1). To fit this Cauchy regression model, the R package heavy was used67.
Software
Reads from 16 S rRNA gene sequencing were analyzed via the QIIME pipeline version 1.9 68, which was also employed to calculate the majority of diversity indices. The ACE index and sample-based rarefaction were calculated via our own scripts for Python (scripts available at https://github.com/filippob/Rare-OTUs-ACE.git) and R statistical programming language tools (scripts available at https://github.com/filippob/sampleBasedRarefaction). Plots were created utilizing the ggplot2 R package [F3]69. Further data manipulation and analysis were conducted via the R environment for statistical computation70.

Proportion of healthy and infected udders in the TG (treatment group) and CG (control group) at T1 (dry-off), T2 (colostrum), and T3 (5–10 DIM).

Relative proportions of bacterial phyla at each timepoint (T1, T2, and T3) in the milk microbiomes of all the goats (all) and the CG and TG separately.

A bar plot demonstrating the significant differences (p value < 0.05) in the abundance of OTUs/ASVs between the CG and TG at three timepoints. The figure displays a color gradient where darker shades represent lower p values, indicating higher degrees of statistical significance. The blue and orange colors represent the differences in the normalized microbial population between CG and TG, with blue indicating positive differences and orange indicating negative differences.

Significance of alpha diversity differences between the CG and TG at timepoints T2 and T3 with respect to T1.

Multidimensional scaling plots of the Bray‒Curtis distances between the milk microbiomes of the goat samples. A, B, and C refer to the distances between the CG and TG at T1, T2, and T3, respectively. D shows the distances between timepoints irrespective of the treatment.

Overview of the study design. A total of 60 goats were selected based on bacteriological culture results and randomly treated with the antibiotic cefazolin at dry-off (TG) or dried without receiving any treatment (CG). A total of 153 pooled milk samples were analyzed for SCC, bacteriology, and 16 S rRNA gene sequencing. The figure was created with BioRender.com.
Data availability
The DNA sequences obtained and analysed in this study are accessible in the NCBI SRA reposi-tory under BioProject ID SUB14836770. Furthermore, additional datasets included in this study are available upon request from the first author and the corresponding author.
Abbreviations
- ARRIVE:
-
Animal Research Reporting of In Vivo Experiments
- CG:
-
Control group
- DIM:
-
Days in milk
- DGT:
-
Dry-goat therapy
- IMI:
-
Intramammary infection
- MICCA:
-
Microbial Community Analysis
- OTUs:
-
Operational taxonomic units
- S. aureus :
-
Staphylococcus aureus
- SCC:
-
Somatic cell count
- T1:
-
Timepoint 1
- T2:
-
Timepoint 2
- T3:
-
Timepoint 3
- TG:
-
Treatment group
References
Albenzio, M. et al. Somatic cell count in sheep milk. Small Ruminant Res. 176, 24–30 (2019).
Morales, F.D.A.R., Genís, J.M.C. and Guerrero, Y.M. Current status, challenges and the way forward for dairy goat production in Europe. Asian-Australas J. Anim. Sci. 32, 1256–1265 (2019).
Agradi, S. et al. Comparison of female verzaschese and camosciata delle alpi goats’ hematological parameters in the context of adaptation to local environmental conditions in semi-extensive systems in Italy. Animals 12, 1703 (2022).
Contreras, A. et al. Mastitis in small ruminants. Small Ruminant Res. 68, 145–153 (2007).
Dejyong, T. et al. An economic analysis of high milk somatic cell counts in dairy cattle in Chiang Mai, Thailand. Front. Vet. Sci. 9, 958163 (2022).
Krömker, V. & Leimbach, S. Mastitis treatment—reduction in antibiotic usage in dairy cows. Reprod. Domest. Anim. 52, 21–29 (2017).
Plummer, P. J. & Plummer, C. Diseases of the Mammary. Sheep & Goat Medicine-E-Book: Sheep & Goat Medicine-E-Book 442 (2011).
Giguère, S., Prescott, J. F. & Dowling, P. Antimicrobial Therapy in Veterinary Medicine (Wiley, 2013).
McDougall, S. & Anniss, F. Efficacy of antibiotic treatment at drying-off in curing existing infections and preventing new infections in dairy goats. Mastitis Dairy. Production: Curr. Knowl. Future Solutions Wageningen Acad. Publishers 523–528 (2005).
Baştan, A. et al. The effects of dry-off therapy on milk somatic cell count in Saanen goats. Turk. J. Vet. Anim. Sci. 39, 550–555 (2015).
Harmon, R. J., Crist, W. L., Hemken, R. W. & Langlois, B. E. Prevalence of minor Udder pathogens after intramammary dry treatment1. J. Dairy. Sci. 69, 843–849 (1986).
Biscarini, F. et al. A randomized controlled trial of teat-sealant and antibiotic dry-cow treatments for mastitis prevention shows similar effect on the healthy milk microbiome. Front. Vet. Sci. 7, (2020).
Petzer, I. M., Karzis, J., Donkin, E. F., Webb, E. C. & Etter, E. Somatic cell count thresholds in composite and quarter milk samples as indicator of bovine intramammary infection status. Onderstepoort J. Vet. Res. 84, 1–10 (2017).
Browning, J. W., Mein, G. A., Barton, M., Nicholls, T. J. & Brightling, P. Effects of antibiotic therapy at drying off on mastitis in the dry period and early lactation. Aust Vet. J. 67, 440–442 (1990).
Williamson, J. H., Woolford, M. W. & Day, A. M. The prophylactic effect of a dry-cow antibiotic against Streptococcus uberis. N Z. Vet. J. 43, 228–234 (1995).
Poutrel, B., de Crémoux, R., Ducelliez, M. & Verneau, D. Control of intramammary infections in goats: impact on somatic cell counts3. J. Anim. Sci. 75, 566–570 (1997).
Sato, K. et al. Effects of temporary cessation of milking following intramammary cefazolin sodium infusion on residual antibiotic in lactating dairy cows. J. Vet. Med. Sci. 83, 814–818 (2021).
Karzis, J., Donkin, E. F. & Petzer, I. M. Intramammary antibiotics in dairy goats: withdrawal periods of three intramammary antibiotics compared to recommended withdrawal periods for cows. Onderstepoort J. Vet. Res. 74, 217–222 (2007).
McDougall, S. & Anniss, F. Efficacy of antibiotic treatment at drying-off in curing existing infections and preventing new infections in dairy goats. in Mastitis in dairy production 523–528Wageningen Academic, (2005).
Virto, M., Santamarina-García, G., Amores, G. & Hernández, I. Antibiotics in dairy production: Where is the problem? Dairy 3, 541–564 (2022).
Oikonomou, G. et al. Microbiota of cow’s milk; Distinguishing Healthy, sub-clinically and clinically diseased quarters. PLoS One. 9, e85904 (2014).
Bhatt, V. D. et al. Milk microbiome signatures of subclinical mastitis-affected cattle analysed by shotgun sequencing. J. Appl. Microbiol. 112, 639–650 (2012).
Li, N. et al. Variation in raw milk microbiota throughout 12 months and the impact of weather conditions. Sci. Rep. 8, 2371 (2018).
Hoque, M. N. et al. Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Sci. Rep. 9, 13536 (2019).
Polveiro, R. C. et al. Distinguishing the milk microbiota of healthy goats and goats diagnosed with subclinical mastitis, clinical mastitis, and gangrenous mastitis. Front. Microbiol. 13, 918706 (2022).
Ruegg, P. L. The bovine milk microbiome – an evolving science. Domest. Anim. Endocrinol. 79, 106708 (2022).
Addis, M. F. et al. The bovine milk microbiota: insights and perspectives from-omics studies. Mol. Biosyst. 12, 2359–2372 (2016).
Little, C. L. et al. Microbiological quality of retail cheeses made from raw, thermized or pasteurized milk in the UK. Food Microbiol. 25, 304–312 (2008).
Zhang, R., Huo, W., Zhu, W. & Mao, S. Characterization of bacterial community of raw milk from dairy cows during subacute ruminal acidosis challenge by high-throughput sequencing. J. Sci. Food Agric. 95, 1072–1079 (2015).
Chen, X., Xu, J., Ren, E., Su, Y. & Zhu, W. Co-occurrence of early gut colonization in neonatal piglets with microbiota in the maternal and surrounding delivery environments. Anaerobe 49, 30–40 (2018).
Salman, M. M., Nawaz, M., Yaqub, T. & Mushtaq, M. H. Investigation of milk microbiota of healthy and mastitic Sahiwal cattle. BMC Microbiol. 23, 304 (2023).
Dos Santos, D. F. K., Istvan, P., Quirino, B. F. & Kruger, R. H. Functional metagenomics as a tool for identification of new antibiotic resistance genes from natural environments. Microb. Ecol. 73, 479–491 (2017).
Bonsaglia, E. C. R. et al. Milk microbiome and bacterial load following dry cow therapy without antibiotics in dairy cows with healthy mammary gland. Sci. Rep. 7, 8067 (2017).
Gryaznova, M. V. et al. Microbiota of cow’s milk with udder pathologies. Microorganisms 9, 1974 (2021).
Tarrah, A. et al. New insights into the raw milk microbiota diversity from animals with a different genetic predisposition for feed efficiency and resilience to mastitis. Sci. Rep. 12, 13498 (2022).
Ganda, E. K. et al. Longitudinal metagenomic profiling of bovine milk to assess the impact of intramammary treatment using a third-generation cephalosporin. Sci. Rep. 6, 37565 (2016).
McInnis, E. A., Kalanetra, K. M., Mills, D. A. & Maga, E. A. Analysis of raw goat milk microbiota: Impact of stage of lactation and lysozyme on microbial diversity. Food Microbiol. 46, 121–131 (2015).
Polveiro, R. C. et al. Effects of enrofloxacin treatment on the bacterial microbiota of milk from goats with persistent mastitis. Sci. Rep. 10, 4421 (2020).
Zhang FuXin, Z. F. et al. Bacterial diversity in goat milk from the Guanzhong area of China. (2017).
Lauková, A. et al. Microbiome associated with slovak raw goat milk, trace minerals, and vitamin E Content. Int J Food Sci 4595473 (2022). (2022).
DeGruttola, A. K., Low, D., Mizoguchi, A. & Mizoguchi, E. Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel Dis. 22, 1137–1150 (2016).
Shen, H., Lu, Z., Xu, Z. & Shen, Z. Antibiotic pretreatment minimizes dietary effects on reconstructure of rumen fluid and mucosal microbiota in goats. Microbiologyopen 7, e00537 (2018).
Derakhshani, H., Plaizier, J. C., De Buck, J., Barkema, H. W. & Khafipour, E. Composition of the teat canal and intramammary microbiota of dairy cows subjected to antimicrobial dry cow therapy and internal teat sealant. J. Dairy. Sci. 101, 10191–10205 (2018).
Filippone Pavesi, L. et al. Effect of the selective dry cow therapy on Udder Health and milk microbiota. Antibiotics 12, 1259 (2023).
Patangia, D. V., Grimaud, G., Linehan, K., Ross, R. P. & Stanton, C. Microbiota and resistome analysis of colostrum and milk from dairy cows treated with and without dry cow therapies. Antibiotics 12, 1315 (2023).
Li, Z., Wright, A. D. G., Yang, Y., Si, H. & Li, G. Unique bacteria community composition and co-occurrence in the milk of different ruminants. Sci. Rep. 7, 40950 (2017).
Toquet, M., Gómez-Martín, Á. & Bataller, E. Review of the bacterial composition of healthy milk, mastitis milk and colostrum in small ruminants. Res. Vet. Sci. 140, 1–5 (2021).
Piessens, V. et al. Distribution of coagulase-negative Staphylococcus species from milk and environment of dairy cows differs between herds. J. Dairy. Sci. 94, 2933–2944 (2011).
Adkins, P. R. F. & Middleton, J. R. Laboratory Handbook on Bovine Mastitis (National Mastitis Council, 2017).
Monistero, V. et al. Genotyping and antimicrobial susceptibility profiling of Streptococcus uberis isolated from a clinical bovine mastitis outbreak in a dairy farm. Antibiotics 10, 644 (2021).
Rosa, N. M., Penati, M., Fusar-Poli, S., Addis, M. F. & Tola, S. Species identification by MALDI-TOF MS and gap PCR–RFLP of non-aureus Staphylococcus, Mammaliicoccus, and Streptococcus spp. associated with sheep and goat mastitis. Vet. Res. 53, 84 (2022).
Cremonesi, P. et al. Technical note: Improved Method for Rapid DNA extraction of Mastitis pathogens directly from milk. J. Dairy. Sci. 89, 163–169 (2006).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences 108, 4516–4522 (2011).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. Preprint at (2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Joshi, N. A. & Sickle, F. J. N. A sliding-window, adaptive, quality-based trimming tool for FastQ files 2011. Computer program (2021). https://github.com/najoshi/sickle
Albanese, D., Fontana, P., De Filippo, C., Cavalieri, D. & Donati, C. MICCA: a complete and accurate software for taxonomic profiling of metagenomic data. Sci. Rep. 5, 9743 (2015).
Rosen, M. J., Callahan, B. J., Fisher, D. S. & Holmes, S. P. Denoising PCR-amplified metagenome data. BMC Bioinform. 13, 1–16 (2012).
Glöckner, F. O. et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 261, 169–176 (2017).
McMurdie, P. J. & Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 8, e61217 (2013).
Biscarini, F. et al. Rumen microbiome in dairy calves fed copper and grape-pomace dietary supplementations: composition and predicted functional profile. PLoS One. 13, e0205670 (2018).
Paulson, J. N., Stine, O. C., Bravo, H. C. & Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods. 10, 1200–1202 (2013).
Bray, J. R. & Curtis, J. T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 326–349 (1957).
Anderson, M. J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46 (2001).
Feller, W. An Introduction to probability theory and its applications, Volume 2 vol. 81 (Wiley, 1991).
Taleb, N. N. Statistical consequences of fat tails: Real world preasymptotics, epistemology, and applications. arXiv preprint arXiv:10488 (2020).
Osorio, F. & Package’heavy’ Robust Estimation Using Heavy-Tailed Distributions. R package version 0.38. 196. Preprint at (2019). https://doi.org/https://cran.r-project.org/package=heavy
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7, 335–336 (2010).
Wickham, H. ggplot2: Elegant Graphics for data Analysis New York (NY: Springer, 2009).
R Core Team. A language and environment for statistical computing. (no Title) (2021).
Acknowledgements
The authors wish to acknowledge Rachele Mezzetti, Marinella Buzzini, Massimo Buzzini, and Maria Paola Rossi for allowing this experiment to be performed on their farm and Mario Villa, DVM, for the valuable contribution and supervision during the trial. Additionally, the authors acknowledge the support of the APC central fund of the University of Milan (Università degli Studi di Milano), Italy.
Funding
The research was supported by internal funding of Paolo Moroni.
Author information
Authors and Affiliations
Contributions
Conceptualization: P.M.; Methodology: D.H., A.J.M., V.M., F.B., S.F.P., A.M., V.B., and P.C.; Investigation: D.H. and P.C.; Software: F.B.; Data validation: D.H. and P.M.; Data curation: F.B., D.H., and P.M.; Formal analysis: F.B., and V.B.; Visualization: D.H. and F.B.; Resources: D.H., F.B., and P.M.; Writing-original draft preparation: D.H., F.B., V.M., P.C., and B.C.; Writing-review and editing: P.M., M.F.A., F.B., V.B., M.M., B.C., P.C., and E.T.; Supervision: P.M.; Project administration: P.M.; All authors contributed to the manuscript and approved the submitted version of the manuscript for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All experiments were approved by the Ethics Committee for Animal Welfare of the University of Milan, Italy (documentation no. OPBA 118–2023).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hossain, D., Biscarini, F., Monistero, V. et al. Use of approved cefazolin based intramammary dry goat treatment and its effects on the milk microbiota. Sci Rep 15, 2250 (2025). https://doi.org/10.1038/s41598-025-85120-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85120-0
