
Abstract
Preeclampsia (PE) is a major pregnancy-specific cardiovascular complication posing latent life-threatening risks to mothers and neonates. The contribution of immune dysregulation to PE is not fully understood, highlighting the need to explore molecular markers and their relationship with immune infiltration to potentially inform therapeutic strategies. We used bioinformatics tools to analyze gene expression data from the Gene Expression Omnibus (GEO) database using the GEOquery package in R. Differential expression analysis was performed using the DESeq2 and limma packages, followed by analysis of variance to identify immune-related differentially expressed genes (DEGs). Several machine learning algorithms, including least absolute shrinkage and selection operator (LASSO), bagged trees, and random forest (RF), were used to select immune-related signaling genes closely associated with the occurrence of PE. Our analysis identified 34 immune source–related DEGs. Using the identified PE- and immune source–related genes, we constructed a diagnostic forecasting model employing several ML algorithms. We identified six types of statistically significant immune cells in patients with PE and discovered a strong relationship between biomarkers and immune cells. Moreover, the immune-derived hub genes for PE exhibited strong binding capabilities with drugs, such as alitretinoin, tretinoin, and acitretin. This study presents a robust prediction model for PE that integrates multiple machine learning–derived immune-related biomarkers. Our results indicate that these biomarkers may outperform previously reported molecular signatures in predicting PE and provide insights into the mechanisms underlying immune dysregulation in PE. Further validation in larger cohorts could lead to their clinical application in PE prediction and treatment.
Similar content being viewed by others

Introduction
Preeclampsia (PE) is a severe pregnancy-specific cardiovascular disorder. The American College of Obstetricians and Gynecologists defines PE as a condition in which pregnant women with previously healthy blood pressure develop high blood pressure, proteinuria, or multiple organ damage after 20 weeks of pregnancy1. Systemic perfusion and multiorgan damage—including renal and liver dysfunction, hematological and neurological complications, pulmonary edema, and convulsions—pose life-threatening risks to mothers and neonates2. PE can lead to maternal death, severe complications, or the need for intensive care as well as an increased likelihood of cesarean delivery. The effects of PE on the fetus include conditions such as intrauterine growth restriction, oligohydramnios, bronchopulmonary dysplasia, and premature birth. Globally, PE is responsible for the deaths of approximately 70,000 pregnant women and 500,000 fetuses each year3.
Despite its significant health burden, the etiology and pathogenesis of PE remain poorly understood. PE is a heterogeneous disorder likely involving multiple factors, mechanisms, and signaling pathways. The most widely accepted cause of PE is shallow placental trophoblast implantation, which impedes the remodeling of the uterine spiral artery, leading to placental ischemia and hypoxia and subsequently resulting in PE. Other causes and pathogenetic mechanisms include an overactive inflammatory immune response, vascular endometrial cell damage, genetic factors, and nutritional status3. No effective medication has been developed to delay or treat PE, and the current treatment methods have various limitations. The only effective treatment for PE is terminating the pregnancy (i.e., placental delivery)4. Therefore, new approaches for treating PE will have a crucial effect on the lives of pregnant women with PE and their fetuses.
In contemporary biomedical research, PE has garnered significant attention for its pathological mechanisms. However, although existing studies have elucidated its fundamental characteristics and impacts during pregnancy, detailed insights into its pathogenesis and contributing factors are lacking. Multiple studies have focused on immune cell infiltration and PE using bioinformatics5,6,7,8. Single-cell transcriptomics (scRNA-seq) and machine learning (ML) methods have also been used to assess the risk of PE9. However, compared with previous studies, the strength of our study lies in screening immune-related potential molecular markers and constructing a PE prediction model, both based on multiple ML algorithms. We downloaded the immune-related differentially expressed genes (IRDEGs) and gene expression profiles from the Gene Expression Omnibus (GEO) datasets to explore the changes and biological mechanisms of IRDEGs at the maternal–fetal interface in patients with PE. Subsequently, we investigated the potential relationship between immune cells and PE. We used ML algorithms to construct PE prediction, training, and validation models based on PE-associated immune-derived genes. Finally, we explored PE-related small-molecule compounds and ligand receptors to identify potential treatment targets for PE. This study aimed to address this gap by conducting an in-depth analysis of IRDEGs and elucidating the specific mechanisms by which immune responses contribute to PE development. The findings enhance our understanding of the pathophysiological processes underlying PE and offer potential novel insights and therapeutic targets for hypertensive disorders of pregnancy.
Results
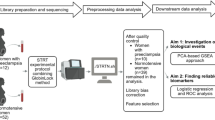
Flowchart for the Comprehensive analysis of preeclampsia -related immune source associated genes (Fig. 1)
IRDEG and potential immune source–related molecular marker screening

Flowchart for the Comprehensive Analysis of Preeclampsia -related Immune Source–Associated Genes (PE.ISGs).DEGs, differentially expressed genes; IRGs, immune-related genes; IRDEGs, immune-related differentially expressed genes; RF, random forest; LASSO, least absolute shrinkage and selection operator; LVQ, learning vector quantization; PE.ISG, Preeclampsia-related immune source–associated gene; NB, naive Bayes; SVM, support vector machine; LogitBoost, boosted logistic regressions; AdaBoost, AdaBoost classification trees; KNN, K-nearest neighbors; PPI, protein–protein interaction.
According to the grouping of the GSE60438 dataset, we divided the samples into the PE and control groups. To analyze the differences in gene expression values between the two groups, we used the R package limma for differential analysis to identify differentially expressed genes (DEGs). The results revealed that in the GSE60438 dataset, 358 DEGs met the threshold of |logFC| > 0.25 and adjusted P < 0.05. Of these, 191 were upregulated genes (logFC > 0.25 and adj. p < 0.05) and 167 were downregulated genes (logFC < − 0.25 and adj. p < 0.05), as indicated by the dataset variance analysis and represented in a volcano plot (Fig. 2A). To obtain IRDEGs, we took the intersection of DEGs and immune-related genes (IRGs) and created a Venn diagram (Fig. 2B). We identified a total of 34 IRDEGs (Table S1).

Differential Gene Expression and PE.ISG Analysis using Machine Learning Algorithms. (A) Volcano plot of differentially expressed genes (DEGs) between PE and control groups in dataset GSE60438. (B) Venn diagram of DEGs and IRGs. (C) Frequency distribution of LASSO algorithm results over 1,000 repeated experiments. The x-axis represents different gene sets, whereas the y-axis represents frequency. Bar chart values indicate the frequency of results for each gene set. (D) Bagged trees algorithm value distribution for different variables and model precision. The x-axis represents the source of preeclampsia immune source–related genes (PE.ISGs), whereas the y-axis represents the precision model’s optimal value. (E) Precision value distribution of the RF algorithm for different numbers of variables. The x-axis represents the source of PE.ISG, and the y-axis represents the precision model’s optimal value. (F) Bayesian algorithm precision value distribution with different numbers of variables. The x-axis represents the source of PE.ISG, and the y-axis represents the precision model’s optimal value. (G) Wrapper feature selection (Boruta) algorithm. The x-axis represents the name of each variable, and the y-axis shows the Z value of each variable. The box plot displays the Z-scores calculated during the model analysis: the green box represents important variables, the yellow box represents tentative attributes, and genes in the green and yellow boxes were selected for inclusion in the model. (H) Importance values of key genes identified using the learning vector quantization (LVQ) algorithm.PE, preeclampsia; IRGs, immune-related genes; IRDEGs, immune-related differentially expressed genes; DEGs, differentially expressed genes; PE.ISG, preeclampsia immune source–related gene; LASSO, least absolute shrinkage and selection operator; RF, random forest; LVQ, learning vector quantization; ML, machine learning.
To evaluate the diagnostic value of the 34 identified IRDEGs in PE, we performed a logistic regression analysis. The results demonstrated that all 34 IRDEGs were statistically significant (p < 0.05). We further screened the logistic regression analysis results of the IRDEGs and explored their characteristics and relationship with PE. In this study, we used bagged trees, Bayesian analysis, learning vector quantization (LVQ), the wrapper (Boruta) method, and six ML algorithms (including least absolute shrinkage and selection operator [LASSO] and random forest [RF]) to analyze the relationship between the IRDEGs from our logistic regression analysis and PE event occurrence in the training set GSE60438. We screened PE-related immune source–associated genes (PE.ISGs).
Using the LASSO algorithm (Fig. 2C), we identified 10 PE.ISGs closely related to PE event occurrence: ADIPOR2, CYLD, DDX17, LCN6, NEDD4, PF4V1, PSMC4, RXRG, UBR1, and VEGFA. Using the bagged trees algorithm (Fig. 2D), we identified 16 PE.ISGs closely connected to PE event occurrence: RXRG, NEDD4, NR1D1, CD72, ADIPOR2, NR2C1, S100A11, TMSB4X, LCN6, FGF11, DDX17, IGF2R, UBR1, PF4V1, HSPA4, and VEGFA. Using the RF algorithm (Fig. 2E), we selected 7 PE sources closely associated with PE-related events: RXRG, NEDD4, NR1D1, TMSB4X, NR2C1, CD72, and FGF11. Applying the Bayesian algorithm (Fig. 2F), we identified two PE.ISGs (RXRG and NR1D1) that are closely related to PE event occurrence. Employing the wrapper (Boruta) algorithm (Fig. 2G), we screened 16 PE.ISGs closely connected to PE event occurrence: RXRG, NR1D1, TMSB4X, NR2C1, DDX17, ADIPOR2, NEDD4, CD72, VEGFA, LCN6, PSMD14, PSMC4, HSPA4, FGF11, FABP5, and IGF2R. Finally, using the LVQ algorithm (Fig. 2H), we identified all 34 PE.ISGs closely related to PE event occurrence.
Construction of PE prediction model
First, because different ML algorithms focus on different aspects, we synthesized the results obtained from multiple algorithms to establish a more stable prediction model. We retained the genes in at least four ML algorithm results as PE.ISGs, resulting in a total of 11 genes (Fig. 3A): ADIPOR2, CD72, DDX17, FGF11, LCN6, NEDD4, NR1D1, NR2C1, RXRG, TMSB4X, and VEGFA.

Diagnostic Model Analysis by Machine Learning Algorithms. (A) Visualization of the upset plot of six different ML algorithms. (B) Chromosomal mapping of six PE.ISGs. (C) Prediction model for PE incidence constructed using seven ML algorithms and validated on the GSE75010 dataset. (D) Visualization of the ROC curve of the RF algorithm for predicting PE events. (E) Performance graph showing the predictive efficacy of PE.ISGs and three groups of published PE-related molecular markers. (F) Heatmap of PE.ISG-related immune genes and three groups of published PE-related molecular markers in dataset GSE60438, along with the prediction of immune efficacy in the dataset GSE75010. The mean AUC represents the average AUC value.PE, preeclampsia; PE.ISG, preeclampsia immune source–related gene; ML, machine learning; RF, random forest; SVM, support vector machine; LogitBoost, boosted logistic regression; KNN, K-nearest neighbors; AdaBoost, AdaBoost classification trees; ROC, receiver operating characteristic; AUC, area under the curve. AUC values between 0.5 and 0.7 indicate low accuracy, whereas AUC values between 0.7 and 0.9 indicate moderate accuracy.
Next, through the analysis of the RCircos R package, we mapped the chromosomal positioning of the 11 PE.ISGs (Fig. 3B). The chromosomal location maps indicated that most PE.ISGs were associated with chromosomes 9, 12, and 17: LCN6 and CD72 on chromosome 9, NR2C1 and ADIPOR2 on chromosome 12, and NR1D1 and FGF11 on chromosome 17.
To investigate the relationship between PE.ISGs and PE event occurrence, we downloaded the PE-related datasets GSE60438 and GSE75010 from the GEO database. First, we used seven ML algorithms to construct models predicting PE events based on the training set GSE60438. The best prediction model was selected by comparing the area under the curve (AUC) of the different algorithms. The seven ML algorithms we used were as follows: naive Bayes (NB), RF, support vector machine (SVM), AdaBoost classification tree (AdaBoost), enhanced logistic regression (LogitBoost), k-nearest neighbors (k-NN), and Cancerclass. We repeated five cross-validations 10 times for each model to optimize the parameters. Subsequently, we evaluated and compared the AUC values of the different ML models using the validation set GSE75010 (Fig. 3C). Our results indicated that the RF algorithm constructed a prediction model with higher diagnostic ability than the other algorithms (AUC = 0.70) (Fig. 3D).
To verify the predictive capacity of PE.ISGs and their advantages, we compared the PE.ISG-based ML models with three groups of published PE-related molecular markers. Our results indicated that in the GSE60438 and GSE75010 datasets, PE.ISGs had a better predictive capacity for treatment efficacy (Fig. 3E). Finally, we used heatmaps to demonstrate the immune response prediction capacity of the PE.ISGs and the three groups of published PE markers in datasets GSE60438 and GSE75010. The results showed that PE.ISGs provided better predictions of immunotherapeutic responses than the published PE-related molecular markers in all datasets (Fig. 3F). Therefore, the PE.ISG-based model exhibited higher accuracy for PE prediction than single molecular markers.
CIBERSORT immune analysis
We assessed LM22 characteristics using CIBERSORT to assess the distribution states of immune cells and compared the distributions of different immune cells between the PE and control groups using Wilcoxon rank-sum tests (Fig. 4A). Our results revealed statistically significant (p < 0.05) expression levels in six immune cell types between the PE and control groups: CD4 + naive T cells, CD4 + resting memory T cells, activated natural killer (NK) cells, monocytes, M1 macrophages, and resting mast cells.

Immune Infiltration Analysis using the CIBERSORT Algorithm. (A) Comparative analysis of immune cell infiltration abundance in dataset GSE60438. (B) Correlation heatmap of immune cell infiltration. (C-F) Dot plots showing correlations between PE.ISGs and immune cells in dataset GSE60438.PE, preeclampsia; PE.ISG, preeclampsia immune source–related gene. * represents p value < 0.05 (statistically significant); * * * p value < 0.001, highly statistically significant. The absolute value of the correlation coefficient (r value) below 0.3 indicates weak or no correlation, values between 0.3 and 0.5 indicate weak correlation, and values between 0.5 and 0.8 indicate moderate correlation. In the group comparison diagram, pink represents the PE group and blue represents the control group. In the correlation heatmap, red represents a positive correlation and blue represents a negative correlation.
Subsequently, we performed a correlation analysis to investigate the relationship between PE.ISGs and various immune cell distributions in the GSE60438 dataset (Fig. 4B). Our results demonstrated that PE.ISGs were strongly correlated with the abundance of CD4 + naive and resting memory T cells as well as infiltrating monocytes. Specifically, we observed significant negative correlations between NR2C1 and CD4 + naive T cells (r = − 0.534, p = 9.18e-05) and between LCN6 and monocytes (r = − 0.484, p = 0.000497). Additionally, we detected significant positive correlations between LCN6 and CD4 + resting memory T cells (r = 0.627, p = 1.86e − 06) and between CD72 and monocytes (r = 0.628, p = 1.78e − 06) (Fig. 4C-F).
Protein–protein interaction (PPI) and regulatory network construction
First, we performed a PPI analysis and constructed a PPI network of 11 PE.ISGs using the STRING database (Fig. 5A). The results indicated that four PE-related immune source–associated hub genes (hub PE.ISGs) were identified: ADIPOR2, NR1D1, NR2C1, and RXRG.

PPI Network and Regulatory Network Analysis. (A) STRING database for computing the source of PE.ISG PPI network. (B) Hub PE.ISG mRNA-miRNA regulatory network (C) mRNA-TF regulatory network of hub PE.ISGs. (D) Hub PE.ISG mRNA-RBP regulatory network.
PE.ISG, preeclampsia immune source–related gene; PPI, protein–protein interaction; TF, transcription factor; RBP, RNA-binding protein.Red represents mRNA, light blue represents miRNA, dark blue represents TF, and pink represents RBP.
Second, using the StarBase database, we accessed the microRNAs (miRNAs) associated with the hub PE.ISGs and constructed an mRNA-miRNA regulatory network, visualized using Cytoscape software (Fig. 5B). Among these, we identified one hub PE.ISG and 30 associated miRNAs (Table S2).
Third, using the ChIPBase database, we identified transcription factors (TFs) that bind the hub PE.ISGs. Using Cytoscape software, we constructed and visualized the mRNA-TF regulatory network (Fig. 5C), which contains two sources of hub PE.ISGs and 23 TFs (Table S3).
Finally, based on the StarBase database to predict hub PE.ISGs related to RNA-binding proteins (RBPs), we constructed and visualized an mRNA-RBP regulatory network using Cytoscape software (Fig. 5D). The network revealed two hub PE.ISGs and 27 RBPs (Table S4).
Drug susceptibility prediction and molecular docking
We used the DGIdb and cMAP databases to explore the potential interactions between hub PE.ISGs and PE-related small-molecule compounds. The drug–gene interaction network indicated multiple potentially sensitive drugs or small-molecule compounds for different hub PE.ISGs (Fig. 6A). Subsequently, we searched for potential therapeutic drugs and reliable targets in the cMAP database based on the hub genes associated with the hub PE.ISGs and screened the ligand–receptor interaction pairs with a “cMAP Connectivity Score > 70,” yielding the following: TBK1 and BX-795 as well as EDNRB and IRL-2500.

Gene–Drug Interaction Network and Molecular Docking. (A) Interaction network of hub PE.ISGs and small-molecule compounds. Red represents the hub PE.ISGs, light blue represents small-molecule compounds in the cMAP database, and dark blue represents small-molecule compound contained in DGIdb database. (B-D) Visualization of the docking results. RXRG with alitretinoin (B), RXRG with tretinoin (C), and RXRG with acitretin (D). From left to right, the panels show the global docking map and the interaction forces map, respectively.PE.ISG, preeclampsia immune source–related gene. The color gradient on the protein surface changes from green to orange to red, indicating a transition from hydrophilic to hydrophobic amino acids. The blue dashed lines represent hydrogen bonds, light blue dashed lines represent weak hydrogen bonds, gray dashed lines represent hydrophobic forces, yellow dashed lines represent ionic bonds, and green dashed lines represent π-π conjugation.
Finally, we performed blind docking of hub PE.ISGs and small-molecule compounds using the CB-Dock2 website. The hub PE.ISG RXRG displayed strong binding affinity for alitretinoin, tretinoin, and acitretin (Vina Scores: −10.3, − 8.3, and − 7.5 kcal/mol, respectively) (Fig. 6B–D). The following amino acids were involved in the interaction with alitretinoin through hydrogen bonds, weak hydrogen bonds, hydrophobic forces, and ionic bonds: VAL266, ILE269, ALA272, ALA273, GLN276, ASN307, LEU310, ILE311, SER313, PHE314, ARG317, LEU326, LEU327, ALA328, VAL343, ILE346, PHE347, VAL350, CYS433, HIS436, LEU437, and PHE440. For tretinoin, the following amino acids were involved through hydrophobic interactions and ionic bonds: VAL266, ILE269, CYS270, ALA272, ALA273, GLN276, TRP306, ASN307, LEU310, ILE311, SER313, PHE314, ARG317, LEU327, ALA328, VAL343, ILE346, VAL350, CYS433, HIS436, LEU437, and PHE440. For acitretin, the interaction involved hydrogen bonds, weak hydrogen bonds, ionic bonds, and hydrophobic forces with the following amino acids: VAL266, ILE269, ALA272, ALA273, GLN276, LEU280 ASN307, LEU309 LEU310, ILE311, SER313, PHE314, ARG317, LEU327, ALA328, VAL343, ILE346, PHE347, ARG372, CYS433, HIS436, LEU437, and PHE440.
Discussion
PE is a hypertensive disorder during pregnancy. Depending on the onset before or after 34 weeks of gestation, the condition can be classified as early-onset or late-onset. However, the causes of PE and its optimal clinical management remain unclear10. Risk factors for PE include pre-pregnancy high blood pressure, kidney disease, and diabetes11. The pivotal effector mechanisms of PE include endothelial dysfunction, impaired angiogenesis, inadequate uterine spiral artery remodeling, and insufficient trophoblast invasion, all of which are associated with immune factors1213.
Immune cells residing at the interface between the placenta and uterus are considered vital during pregnancy. An in-depth understanding of the immunological aspects of the mother-to-fetus interface is necessary to gain a better overview of PE pathogenesis14. Various innate and adaptive immune cells and mediators, including regulatory T cells, macrophages, NK cells, neutrophils, B cells, inflammatory cytokines, and anti-angiotensin type II 1 receptor autoantibodies, are involved in PE development15. However, PE-related immune dysregulation remains unclear. Therefore, further exploration of the relationship between immune mechanisms and PE is crucial.
Although an increasing number of studies have focused on the relationship between immune infiltration and PE through data mining, most relevant studies have focused on screening IRGs and immune cells5,6,16,17,18,19,20. In this study, we screened DEGs and immune cells using various ML methods and analyzed the correlation between immune cells and genes. Additionally, we screened potential drugs, thereby providing new perspectives for novel PE treatment strategies.
We analyzed the GSE60438 dataset to identify DEGs between PE and normal samples and obtained the intersection of DEGs and IRGs. A total of 34 IRDEGs were identified, and logistic regression analysis was performed to determine their diagnostic value in PE. This study confirmed that all 34 identified IRDEGs were PE-related. We designated 11 genes in at least four ML algorithm results as PE-related immunogens.
Moreover, our study is the first to demonstrate that NR2C1, NEDD4, CD72, LCN6, VIPR1, OGFR, PSMC4, UBR1, FGF11, TNFRSF14, TGFBR3, TRIM5, VCAM1, FABP5, CYLD, A2M, PF4V1, IGF2R, PSMD6, CD244, IL16, and S100A11 are PE-related genes. Nevertheless, their specific interactions and underlying effector mechanisms require further investigation.
We used seven ML algorithms—NB, RF, SVM, AdaBoost, augmented logistic regression (LogitBoost), k-NN, and Cancerclass—to construct a PE event prediction model and selected the best model by comparing the AUCs derived from the algorithms. Our results confirmed that the RF-based predictive model had the highest diagnostic capacity (AUC = 0.70).
A previous study demonstrated, using WGCNA analysis and the Evolutionary Approach, that DDX17 is a key gene for PE diagnostic biomarkers and therapeutic intervention targets21. DDX17 can act as a transcription coregulator and perform essential cellular functions, including splice site selection, RNA splicing, and secondary RNA structural rearrangement22,23,24. Our results also highlight DDX17 as an important molecular marker for PE prediction. Therefore, we hypothesize that DDX17 is crucial in PE pathogenesis.
The PPI network analysis demonstrated that the PE-associated hubs included ADIPOR2, NR1D1, NR2C1, and RXRG, which may play key roles in environmental changes in the uterus and pregnancy complications, including PE. Notably, our report on NR2C1 and RXRG represents the first mention of these markers in the context of PE. This breakthrough provides promising diagnostic biomarkers that could significantly bolster early detection and facilitate timely interventions, potentially enhancing prognoses for both expecting mothers and their fetuses. RXRG is a retinoid receptor gene, and bifenox can cause implantation disorders during early pregnancy by inhibiting RXRG expression25.
During pregnancy, estrogens cause changes in enzymatic activity, increase Ca2+-activated K + channel function, reduce inflammatory cytokine release and oxidative stress, and improve placental perfusion function. Moreover, estrogen levels are reportedly decreased in patients with PE26. Estrogen can affect immune responses by interacting with various estrogen receptor (ER)-expressing immune cells, such as T cells, B cells, dendritic cells, macrophages, neutrophils, and NK cells27. Interestingly, the expressions of RXRG and ER are positively correlated28. In this study, we demonstrated that RXRG is an IRDEG. Overall, RXRG is a potential PE surrogate factor that promotes PE development via changes in ER expression.
Furthermore, drug sensitivity prediction and molecular docking analysis revealed a strong binding force between RXRG and alitretinoin (Vina score = − 10.3 kcal/mol). This binding strength score indicates that alitretinoin is a strong candidate for drug development and therapeutic interventions targeting RXRG, providing a scientific basis for new drug development.
Adiponectin is an insulin-sensitizing hormone that exerts both pro- and anti-inflammatory effects. The adiponectin signaling receptors AdipoR1 and AdipoR2 are expressed in epithelial, endothelial, and immune cells29. ADIPOR2 protein and mRNA levels were significantly higher in patients with severe PE than in those with mild and normal pregnancy30. The results of this study were consistent with our findings, although the specific underlying mechanisms need to be further explored. The disturbance of maternal immune tolerance to the embryo at the mother-to-fetus interface may be related to PE onset. Recent studies have identified disease-associated IGFBP1 + SPP1 + extracellular villous trophoblast (EVT) cells of the decidua in patients with PE accompanied by newly discovered immune cell dysfunction31. Some studies have demonstrated that SP1 plays a vital role in decidualization. SP1 dysfunction could impair decidualization and contribute to PE32.
The PE- and immune-derived genes better predicted immunotherapeutic responses than the published PE-related molecular markers. In addition, our model predicted PE with higher accuracy than a single molecular marker. CIBERSORT can be used to analyze the distribution status of immune cells in diseases33. Our study identified increased infiltration of immune cells, including CD4+ naive T cells, CD4+ resting memory T cells, activated NK cells, monocytes, M1 macrophages, and resting mast cells. The invasion of abnormal EVT cells and spiral artery remodeling were associated with PE. During maternal tissue invasion, EVT cells express HLA-C, HLA-G, HLA-E, and HLA-F and interact with various cells, including uterine natural killer (uNK) cells, via different receptors34. Circulating monocyte subgroups in patients with PE are imbalanced35. Moreover, the number of M1 macrophages is increased in the placenta of patients with PE36.
This finding highlights the intricate immune dysregulation underlying PE and provides a more nuanced understanding of its pathophysiology from an immunological perspective. Exploring the interactions between these immune cells and specific molecular markers may pave the way for innovative studies into targeted therapies. These treatments could modulate the immune response in patients with PE, potentially reducing the severity and complications of the disease.
Additionally, we observed strong correlations between PE.ISGs and the abundance of CD4+ infantile T cells, CD4+ resting memory T cells, and monocyte infiltration. NR2C1 was significantly negatively correlated with CD4+ naive T cells. LCN6 was significantly negatively correlated with monocytes and positively correlated with CD4+ resting memory T cells. Finally, CD72 was significantly positively correlated with monocytes. These relationships are described for the first time in this study.
Monocytes are short-lived cells that mature in the circulation and can invade tissues upon inflammatory stimuli, developing into macrophages. Macrophages are abundant in the endometrium and are crucial for implantation and placentation during normal pregnancy37. A previous study described that in patients with severe PE, CD4+ T cell numbers increased and CD8+ T cells decreased compared with those during normal pregnancy38. Consequently, we hypothesized that NR2C1 and LCN6 regulate CD4+ infantile T cells, influencing CD4+ T cell and T cell CD4 memory resting populations.
This finding indicates the complex immune dysregulation underlying PE, thereby suggesting a more nuanced understanding of its pathophysiology from an immunological perspective. Exploring the interactions between these immune cells and specific molecular markers may lead to new avenues for targeted therapies. These treatments may help modulate the immune response in patients with PE, potentially reducing disease severity and associated complications.
Moreover, our drug–gene interaction network analysis identified potential therapeutic targets and small-molecule compounds (including TBK1, BX-795, EDNRB, IRL-2500, alitretinoin, tretinoin, and acitretin) that show strong binding affinities to PE-linked immune-derived hub genes. This underscores new avenues for drug repurposing and the creation of targeted therapies, paving the way for more effective PE treatments. Identifying compounds that specifically target and modulate the genes and pathways involved in PE pathogenesis could revolutionize the clinical management of this condition.
Our study has some limitations. First, it lacks laboratory experiments to validate the bioinformatics findings, which would strengthen the conclusions derived from computational analyses. Second, the sample sizes of the GSE60438 and GSE75010 datasets were relatively small, limiting the generalizability of the predictive models and molecular signatures. Third, clinically validated analyses were not performed to confirm the utility of PE-related immune genes in real-world settings. Finally, the use of multiple datasets introduces potential batch effects that may bias the results. Future studies should address these limitations by including larger cohorts, conducting experimental validations, and using robust statistical methods to mitigate batch effects.
In conclusion, this study highlighted the critical need for precision medicine approaches to manage pregnancy complications such as PE. By leveraging bioinformatics and ML techniques to unravel the complex interactions among genes, immune cells, and potential drug targets, personalized treatment strategies can significantly improve the outcomes of pregnant women and their babies.
Although this study identified molecular markers and immune mechanisms associated with PE via bioinformatics analysis, the absence of laboratory validation weakens the reliability of the conclusions. Additionally, the lack of clinically validated analyses limits the practical applicability of the identified immune genes in gestational hypertension. Future studies should focus on increasing sample sizes, conducting experimental validations, and applying robust statistical methods to reduce batch effects and enhance the credibility and utility of the study.
Materials and methods
Data extraction and IRDEGS
To develop immune-based PE prediction models for clinical precision medicine in PE, we used the R package GEOquery39 from the GEO database (https://www.ncbi.nlm.nih.gov/geo) to extract the PE GSE60438 40 and GSE75010 41 datasets. The Homo sapiens GSE60438 dataset and the GPL6884 data platform contained a total of 48 samples, of which 25 and 23 were PE and control samples, respectively. The Homo sapiens GSE75010 dataset and GPL6244 data platform contained 157 samples, of which 80 and 77 were PE and control samples, respectively. All PE and control samples were included in this study. Finally, we used the R package DESeq2 42 for the GSE60438 datasets and GSE75010 for standardizing, annotation probe, standardization, and normalization processing.
We used 2,483 source IRGs in subsequent analysis from the ImmPort database43 (https://www.immport.org/home) (Table S5). Furthermore, we identified three groups of PE molecular markers from previous publications, which were used for prediction model comparisons (Table S6)7,4445.
According to the sample grouping of the GSE60438 dataset, we divided the samples into PE and control groups. We used the R package limma to analyze the differences between the genes in the PE and control groups. We set | logFC | > 0.25 and adj. p < 0.05 as the threshold for DEGs. Among them, the logFC > 0.25 and adj. p < 0.05 genes were considered upregulated. Genes with logFC < − 0.25 and adj. p < 0.05 were considered downregulated. We visualized the differential analysis results using a volcano plot generated by the R package ggplot2.
To identify PE-associated IRDEGs, we performed a variance analysis of the GSE60438 dataset. All | logFC | > 0.25 and adj. p < 0.05 DEGs associated with IRGs were intersected, and the IRDEGs were mapped using a Venn diagram.
Machine learning algorithm sources for further immune-related molecular marker screening and PE prediction model construction
First, we performed logistic regression analysis based on the IRDEGs and GSE60438 training set. When the dependent variable was binary, namely the PE and control groups, we applied logistic regression to analyze the association between independent variables and the dependent variable using p < 0.05 as the criterion to screen IRDEGs.
Next, based on the IRDEGs included in the logistic regression analysis and the GSE60438 training set, we used six common ML algorithms to screen the IRDEGs: bagged trees, Bayesian, LVQ, wrapper (Boruta), LASSO, and RF. Finally, we selected genes identified in the results of at least four ML algorithms as PE.ISGs.
Among the algorithms, bagged trees is an ensemble learning algorithm that trains multiple decision trees by randomly selecting bootstrap samples of the data (with put-back sampling). The predictions of individual decision trees or their voting results are averaged to improve model stability and accuracy. The bagged trees algorithm reduces overfitting, which is useful for handling high-variance models.
Bayesian algorithms use prior knowledge and observations to update the probability distribution of the model, resulting in more accurate predictions and inferences. These algorithms are statistical inference methods based on Bayes’ theorem and are designed to deal with uncertainty and probabilistic inference. Among ML algorithms, Bayesian methods can be used for parameter estimation, model selection, and prediction.
LVQ algorithms are used in neural networks to classify patterns by creating a learning vector set in the feature space. These vectors represent different classes, and data points are assigned to the class represented by the closest learning vector. Although LVQ algorithms perform well on smaller datasets, they are limited when applied to large-scale data.
The wrapper (Boruta) algorithm is a feature selection algorithm that identifies the most important features from a large set of features. The RF algorithm compares the importance of the original features by introducing a set of virtual “shadow features.” The wrapper (Boruta) algorithm uses statistical methods to determine whether features are significant, thereby helping to reduce dimensionality and improve model performance.
The LASSO algorithm is a linear model for feature selection and regression analysis that adds an L1 regularization term (absolute value penalty) to optimize linear regression and push some coefficients to zero. The LASSO algorithm can effectively process high-dimensional data and reduce overfitting.
We used seven ML algorithms to construct the PE.ISG- and GSE60438 training set-based training model: NB, RF, SVM, AdaBoost, Strengthen Logistic Regression (Boosted Logistic Regression, LogitBoost), k-NN, and Cancerclass algorithms. We applied different parameters for these ML algorithms and used 50% cross-validation for hyper-parameter adjustment to improve the model’s performance. To ensure the model’s robustness, we repeated the optimization process using a different random seed 10 times for each resampling. For the Cancerclass algorithm, which does not require parameter tuning, we trained the model directly on the entire training set.
Finally, based on the prediction model constructed by the abovementioned classifiers, we used the GSE75010 validation set for the analysis. The classifier with the best classification performance in the validation set was selected to construct the PE prediction model.
PE.ISG comparisons
To further evaluate IRGs within PE.ISGs that have predictive value, we compared PE.ISGs with the three groups of PE molecular markers identified from previous studies. To assess PE prediction performance, three sets of molecular markers were compared with PE.ISG in the GSE60438 and GSE75010 datasets.
CIBERSORT immune infiltration analysis
CIBERSORT46 is based on linear support vector regression and was used to deconvolute the transcriptome expression matrix to assess immune cell composition and abundance in a mixture of cells. The CIBERSORT algorithm, combined with the LM22 feature gene matrix and filtering out data with immune cell enrichment scores higher than zero, obtained specific results for the immune cell infiltration matrix in the GSE60438 dataset.
Subsequently, we used the R package ggplot2 to create group comparison maps and visualize the expression differences of LM22 immune cells between the PE and control groups in the GSE60438 dataset. Finally, we used the R package heatmap to create heatmaps to highlight the results of correlation analysis between immune cells and PE.ISGs in the GSE60438 dataset.
PPI and regulatory network construction
The PPI network consisted of proteins that interact with each other and participate in signaling, gene expression regulation, and various life processes, such as energy and substance metabolism or cell cycle regulation. Systematic analysis of protein interactions in biological systems is particularly important for understanding the functional principles of corresponding proteins, the reaction mechanisms of biological signals, and energy and substance metabolism under specific physiological conditions (e.g., diseases) as well as the functional relationships between proteins.
The STRING database47 (https://string-db.org/) facilitates the search for interactions between known and predicted proteins. In this study, we used the STRING database based on PE.ISGs with a minimum interaction score > 0.150 (minimum required interaction score: low confidence [0.150]) to construct the PPI network of the PE.ISGs. The PPI network included genes as hub PE.ISGs.
TFs control gene expression after transcription through hub genes connected to immune-related source of hub PE.ISG interactions. We used the ChIPBase database48 (http://rna.sysu.edu.cn/chipbase/) to retrieve and analyze hub PE.ISG-regulating TFs and Cytoscape software to visualize the TF control network (mRNA-TF regulatory network).
In addition, miRNAs are crucial regulators of biological development and evolution. They regulate various target genes, and the same target gene can be regulated by multiple miRNAs. We used the StarBase v3.0 database49 to analyze the relationships between hub PE.ISGs and miRNAs and used Cytoscape software to visualize mRNA-miRNA regulatory networks.
RBPs50 are pivotal for gene regulation and various biological processes, such as RNA synthesis, alternative splicing, modification, transport, and translation. We predicted hub PE.ISG target RBPs using the StarBase v3.0 database (https://starbase.sysu.edu.cn/) and visualized the mRNA-RBP regulatory network using Cytoscape software.
Drug target prediction and molecular docking
We combined the DGIdb51 (https://dgidb.org/) and the cMAP (Connectivity Map) database (https://clue.io/) to explore the source of PE.ISGs and the interactions of small-molecule compounds to identify potential PE drug targets and therapies. The DGIdb is a drug–gene interaction database that provides information on the associations of genes with known or potential drugs. DGIdb contains > 14,000 drug–gene interactions, involving 2,600 genes and 6,300 drugs targeting these genes, as well as 6,700 other genes that could be potential targets for future drugs.
The cMAP database, developed by researchers at Harvard University, the University of Cambridge, and the Massachusetts Institute of Technology, is a gene expression database that uses gene expression differences after treating human cells with various compounds (including small molecules) to establish a biological application database of the interactions between distractors, gene expression, and disease. By combining the two databases, we identified the details of the interaction between PE.ISGs and small-molecule compounds and identified possible drugs associated with PE that could be beneficial for its treatment.
To investigate the mechanism of drug–PE.ISG interactions, we used the gene–drug correlation results predicted by cMAP. We selected small molecules with a “cMAP Connectivity Score” >70 and matched them with the corresponding source of PE.ISG. Subsequently, we used the CB-Dock2 website (24) (https://cadd.labshare.cn/cb-dock2/php/index.php) for molecular docking analysis. The CB-Dock2 website is a modified version of the CB-Dock server for protein–ligand blind docking, integrating cavity detection, docking, and homologous template fitting. Using three-dimensional (3D) protein and ligand structures, we predicted binding sites and affinities for computer-aided drug discovery. First, we downloaded the 3D molecular structures of alitretinoin (CID449171), tretinoin (CID444795), and acitretin (CID5284513) from the PubChem database52 (https://pubchem.ncbi.nlm.nih.gov). Subsequently, we obtained the X-ray crystal structure of PE from the protein structure database Protein Data Bank (PDB): RXRG (PDB code: 7A79)53. Finally, we used the AutoDock Vina program54 on the CB-Dock2 website to perform blind docking and visualize the PE.ISG and corresponding small-molecule compounds. The docking score of AutoDock Vina (Vina Score) indicates the binding strength as follows: > −4 kcal/mol, extremely weak or no binding; −7 kcal/mol, < Vina Score < − 4 kcal/mol, medium binding strength; < −7 kcal/mol, firm binding strength.
Statistical analysis
All data processing and analysis in this study were performed using R software (Version 4.2.2). Unless stated otherwise, we used an independent Student’s t-test to determine significant differences when comparing two groups of continuous variables with a normal distribution and the Mann–Whitney U test (Wilcoxon rank-sum test) to determine differences between variables that were not normally distributed. We used the Kruskal–Wallis test to compare three or more groups. Spearman’s correlation analysis was used to calculate the correlation coefficient between different molecules. All P-values were two-sided if not specified, and a p value < 0.05 was considered statistically significant.
Data availability
The datasets generated and analyzed during the current study are available in the Gene Expression Omnibus (GEO)(https://www.ncbi.nlm.nih.gov/geo).
Abbreviations
- PE:
-
Preeclampsia
- DEGs:
-
Differentially expressed genes
- ML:
-
Machine learning
- IRDEGs:
-
Immune-related differentially expressed genes
- GEO:
-
Gene Expression Omnibus
- TF:
-
Transcription factor
- LVQ:
-
Learning vector quantization
- LASSO:
-
Least absolute shrinkage and selection operator
- RF:
-
Random forest
- PPI:
-
Protein–protein interaction
- AUC:
-
Area under the curve
- NB:
-
Naive Bayes
- SVM:
-
Support vector machine
- AdaBoost:
-
AdaBoost classification tree
- LogitBoost:
-
Enhanced logistic regression, k-NN, k-nearest neighbors
- RBPs:
-
RNA-binding proteins
References
ACOG Practice Bulletin No. Gestational hypertension and preeclampsia. Obstet. Gynecol. 133. 202, 1. https://doi.org/10.1097/aog.0000000000003018 (2019).
Bajpai, D. Preeclampsia for the nephrologist: current understanding in diagnosis, management, and long-term outcomes. Adv. Chronic Kidney Dis. 27, 540–550. https://doi.org/10.1053/j.ackd.2020.05.001 (2020).
Rana, S., Lemoine, E., Granger, J. P., Karumanchi, S. A. & Preeclampsia Pathophysiology, challenges, and perspectives. Circul. Res. 124, 1094–1112. https://doi.org/10.1161/circresaha.118.313276 (2019).
Bakrania, B. A., George, E. M. & Granger, J. P. Animal models of preeclampsia: investigating pathophysiology and therapeutic targets. Am. J. Obstet. Gynecol. 226, S973–s987. https://doi.org/10.1016/j.ajog.2020.10.025 (2022).
Yang, M. Y., Ji, M. H., Shen, T. & Lei, L. Integrated analysis identifies four genes as novel diagnostic biomarkers which correlate with immune infiltration in preeclampsia. J. Immunol. Res. 2022 (2373694). https://doi.org/10.1155/2022/2373694 (2022).
Meng, Y., Li, C. & Liu, C. X. Immune cell infiltration landscape and immune marker molecular typing in preeclampsia. Bioengineered 12, 540–554. https://doi.org/10.1080/21655979.2021.1875707 (2021).
Wang, Y., Li, B. & Zhao, Y. Inflammation in preeclampsia: genetic biomarkers, mechanisms, and therapeutic strategies. Front. Immunol. 13, 883404. https://doi.org/10.3389/fimmu.2022.883404 (2022).
He, L., Zhan, F., Lu, L., Zhang, X. & Wu, J. Role of necroptosis and immune infiltration in preeclampsia: novel insights from bioinformatics analyses. BMC Pregnancy Childbirth. 23, 495. https://doi.org/10.1186/s12884-023-05821-0 (2023).
Wang, H. et al. A cost-effective machine learning-based method for preeclampsia risk assessment and driver genes discovery. Cell. Bioscience. 13 https://doi.org/10.1186/s13578-023-00991-y (2023).
Magee, L. A. et al. The 2021 International Society for the Study of Hypertension in Pregnancy classification, diagnosis & management recommendations for international practice. Pregnancy Hypertens. 27, 148–169. https://doi.org/10.1016/j.preghy.2021.09.008 (2022).
Wisner, K. Gestational hypertension and preeclampsia. MCN Am. J. Matern. Child Nurs. 44, 170. https://doi.org/10.1097/nmc.0000000000000523 (2019).
Geldenhuys, J., Rossouw, T. M., Lombaard, H. A., Ehlers, M. M. & Kock, M. M. Disruption in the regulation of immune responses in the placental subtype of preeclampsia. Front. Immunol. 9, 1659. https://doi.org/10.3389/fimmu.2018.01659 (2018).
Burke, S. D. & Karumanchi, S. A. Spiral artery remodeling in preeclampsia revisited. Hypertens. (Dallas Tex. : 1979). 62, 1013–1014. https://doi.org/10.1161/hypertensionaha.113.02049 (2013).
Erlebacher, A. Immunology of the maternal-fetal interface. Annu. Rev. Immunol. 31, 387–411. https://doi.org/10.1146/annurev-immunol-032712-100003 (2013).
Deer, E., Herrock, O., Campbell, N. & Cornelius, D. The role of immune cells and mediators in preeclampsia. Nat. Rev. Nephrol. 19, 257–270. https://doi.org/10.1038/s41581-022-00670-0 (2023).
Bai, L. et al. Machine learning and bioinformatics framework integration reveal potential characteristic genes related to immune cell infiltration in preeclampsia. Front. Physiol. 14, 1078166. https://doi.org/10.3389/fphys.2023.1078166 (2023).
Peng, Y., Hong, H., Gao, N., Wan, A. & Ma, Y. Bioinformatics methods in biomarkers of preeclampsia and associated potential drug applications. BMC Genom. 23, 711. https://doi.org/10.1186/s12864-022-08937-3 (2022).
Liu, Y., Xu, B. & Fan, C. Single-cell RNA sequencing and microarray analysis reveal the role of lipid-metabolism-related genes and cellular immune infiltration in pre-eclampsia and identify novel biomarkers for pre-eclampsia. Biomedicines 11 https://doi.org/10.3390/biomedicines11082328 (2023).
Wang, Y. et al. Bioinformatics analysis combined with clinical sample screening reveals that leptin may be a biomarker of preeclampsia. Front. Physiol. 13, 1031950. https://doi.org/10.3389/fphys.2022.1031950 (2022).
Huang, S. & Cai, S. Prediction of differentially expressed genes and a diagnostic signature of preeclampsia via integrated bioinformatics analysis. 5782637, (2022). https://doi.org/10.1155/2022/5782637 (2022).
Kondoh, K., Akahori, H., Muto, Y. & Terada, T. Identification of key genes and pathways associated with preeclampsia by a WGCNA and an evolutionary approach. Genes 13 https://doi.org/10.3390/genes13112134 (2022).
Hönig, A., Auboeuf, D., Parker, M. M., O’Malley, B. W. & Berget, S. M. Regulation of alternative splicing by the ATP-dependent DEAD-box RNA helicase p72. Mol. Cell. Biol. 22, 5698–5707. https://doi.org/10.1128/mcb.22.16.5698-5707.2002 (2002).
Rössler, O. G., Straka, A. & Stahl, H. Rearrangement of structured RNA via branch migration structures catalysed by the highly related DEAD-box proteins p68 and p72. Nucleic Acids Res. 29, 2088–2096. https://doi.org/10.1093/nar/29.10.2088 (2001).
Ha, M. & Kim, V. N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509–524. https://doi.org/10.1038/nrm3838 (2014).
Park, W., Park, H., Park, S., Lim, W. & Song, G. Bifenox compromises porcine trophectoderm and luminal epithelial cells in early pregnancy by arresting cell cycle progression and impairing mitochondrial and calcium homeostasis. Pestic. Biochem. Physiol. 196, 105628. https://doi.org/10.1016/j.pestbp.2023.105628 (2023).
Shu, C. et al. Estrogen and preeclampsia: potential of estrogens as therapeutic agents in preeclampsia. Drug. Des. Devel. Ther. 15, 2543–2550. https://doi.org/10.2147/dddt.s304316 (2021).
Schurz, H. et al. The X chromosome and sex-specific effects in infectious disease susceptibility. Hum. Genomics. 13 (2). https://doi.org/10.1186/s40246-018-0185-z (2019).
Joseph, C. et al. Retinoid X receptor gamma (RXRG) is an independent prognostic biomarker in ER-positive invasive breast cancer. Br. J. Cancer. 121, 776–785. https://doi.org/10.1038/s41416-019-0589-0 (2019).
Lim, J. Y. & Templeton, S. P. Regulation of lung inflammation by adiponectin. Front. Immunol. 14 https://doi.org/10.3389/fimmu.2023.1244586 (2023).
Tie, W. et al. Expressions of adiponectin receptors in placenta and their correlation with preeclampsia. Reproductive Sci. (Thousand Oaks Calif). 16, 676–684. https://doi.org/10.1177/1933719109334258 (2009).
Luo, F. et al. Single-cell profiling reveals immune disturbances landscape and HLA-F-mediated immune tolerance at the maternal-fetal interface in preeclampsia. Front. Immunol. 14, 1234577. https://doi.org/10.3389/fimmu.2023.1234577 (2023).
Zhang, Y. et al. Downregulation of decidual SP1 and P300 is associated with severe preeclampsia. J. Mol. Endocrinol. 60, 133–143. https://doi.org/10.1530/jme-17-0180 (2018).
Newman, A. M., Liu, C. L. & Green, M. R. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12, 453–457. https://doi.org/10.1038/nmeth.3337 (2015).
Gan, X., Ning, F. & Lash, G. E. Methods for co-culture of primary human extravillous trophoblast cells and uterine natural killer cells. Methods Mol. Biol. 2728, 183–194. https://doi.org/10.1007/978-1-0716-3495-0_16 (2024).
Gong, F. et al. The imbalance of circulating monocyte subgroups with a higher proportion of the CD14 + CD16 + CD163 + phenotype in patients with preeclampsia. Immunol. Lett. 253, 1–7. https://doi.org/10.1016/j.imlet.2022.11.005 (2023).
Yao, Y., Xu, X. H. & Jin, L. Macrophage polarization in physiological and pathological pregnancy. Front. Immunol. 10, 792. https://doi.org/10.3389/fimmu.2019.00792 (2019).
Faas, M. M., Spaans, F. & De Vos, P. Monocytes and macrophages in pregnancy and pre-eclampsia. Front. Immunol. 5, 298. https://doi.org/10.3389/fimmu.2014.00298 (2014).
Karkour, T. A., Abdelfattah, E. A., Abdel-Dayem, T. M., Kholeif, D. A. & Elssaidi, M. M. Cluster of differentiation 4/cluster of differentiation 8 ratio of T-lymphocyte subsets in Egyptian patients with severe pre-eclampsia. Egypt. J. Immunol. 30, 93–100 (2023).
Davis, S. GEOquery: a bridge between the Gene expression Omnibus (GEO) and BioConductor. Bioinformatics 23, 1846–1847 (2007).
Yong, H. E. et al. Genome-wide transcriptome directed pathway analysis of maternal pre-eclampsia susceptibility genes. PLoS One. 10, e0128230. https://doi.org/10.1371/journal.pone.0128230 (2015).
Leavey, K. et al. Unsupervised placental gene expression profiling identifies clinically relevant subclasses of human preeclampsia. Hypertension 68, 137–147. https://doi.org/10.1161/hypertensionaha.116.07293 (2016).
Love, M. I. & Anders, H. W. Moderated estimation of Fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Bhattacharya, S. et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci. Data. 5, 180015. https://doi.org/10.1038/sdata.2018.15 (2018).
Lin, J., Meng, Y., Song, M. F. & Gu, W. Network-based analysis reveals novel biomarkers in Peripheral blood of patients with Preeclampsia. Front. Mol. Biosci. 9, 757203. https://doi.org/10.3389/fmolb.2022.757203 (2022).
Liu, K., Fu, Q., Liu, Y. & Wang, C. An integrative bioinformatics analysis of microarray data for identifying hub genes as diagnostic biomarkers of preeclampsia. Biosci. Rep. 39 https://doi.org/10.1042/bsr20190187 (2019).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12, 453–457. https://doi.org/10.1038/nmeth.3337 (2015).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–d613. https://doi.org/10.1093/nar/gky1131 (2019).
Zhou, K. R. et al. ChIPBase v2.0: Decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 45, D43–d50. https://doi.org/10.1093/nar/gkw965 (2017).
Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 42, D92–97. https://doi.org/10.1093/nar/gkt1248 (2014).
Singh, A. RNA-binding protein kinetics. Nat. Methods. 18, 335. https://doi.org/10.1038/s41592-021-01122-6 (2021).
Freshour, S. L. et al. Integration of the drug–gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 49, D1144–D1151. https://doi.org/10.1093/nar/gkaa1084 (2020).
Kim, S. et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 49, D1388–d1395. https://doi.org/10.1093/nar/gkaa971 (2021).
Burley, S. K. et al. Protein Data Bank (PDB): the single global Macromolecular structure archive. Methods Mol. Biol. 1607, 627–641. https://doi.org/10.1007/978-1-4939-7000-1_26 (2017).
Eberhardt, J., Santos-Martins, D., Tillack, A. F., Forli, S. & AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61, 3891–3898. https://doi.org/10.1021/acs.jcim.1c00203 (2021).
Acknowledgements
This study was supported by the specialized scientific research fund projects of the First Hospital of Jilin University in lequn district (B022) and the Program of Academic Youth Development Fund of the First Hospital of Jilin University (JDYY14202326).
Author information
Authors and Affiliations
Contributions
Conceptualization: HYC, ZCW; Methodology: ZCW, LC, HYC; Investigation: ZCW; Visualization: ZCW, LC, GHL; Funding acquisition: HYC; Project administration: HCY, ZCW; Supervision: ZCW, LC, GHL, HYC; Writing – original draft: HCY, ZCW; Writing – review & editing: ZCW, LC, GHL, HYC.All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Z., Cheng, L., Li, G. et al. Development of immune-derived molecular markers for preeclampsia based on multiple machine learning algorithms. Sci Rep 15, 1767 (2025). https://doi.org/10.1038/s41598-025-86442-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-86442-9
