
Abstract
Atrial fibrillation (AF) is a predominant cardiac arrhythmia with unclear etiology. This study used bioinformatics and machine learning to explore the relationship between mitochondrial energy metabolism-related genes (MEMRGs) and immune infiltration in AF. The datasets GSE31821, GSE41177, and GSE79768 were retrieved from the Gene Expression Omnibus (GEO) database, and differential expression analysis identified 59 mitochondrial energy metabolism-related differentially expressed genes (MEMRDEGs) associated with AF. Key MEMRDEGs were selected using the least absolute shrinkage and selection operator (LASSO) and support vector machine (SVM) methods, and a diagnostic model was developed. Immune infiltration was assessed using single-sample gene set enrichment analysis (ssGSEA) and the microenvironment cell population counter (MCPcounter). The diagnostic model, based on the key genes ACAT1, ALDH1L2, HTT, OGDH, and SLC25A3, achieved an area under the curve (AUC) of 0.903. Significant differences in immune cell composition were observed between the AF and control groups. ALDH1L2 was positively correlated with most immune cells, while SLC25A3 showed a negatively correlated with the monocytic lineage. The findings indicate that MEMRGs interact with immune responses in AF, offering insights into the potential molecular mechanisms and therapeutic targets for AF.
Similar content being viewed by others


Introduction
Atrial fibrillation (AF) is the predominant sustained arrhythmia and results from the sudden onset of aberrant electrical signals in the atria, which override the heart’s natural pacemaker and disrupt its ability to regulate heart rhythm1. The frequency and occurrence of AF have been progressively increasing as individuals age and chronic condition management has improved. By 2050, the expected incidence of AF is projected to be 6 to 16 million in the United States, 14 million in Europe, and at least 72 million in Asia2,3. According to the Seventh National Population Census in China, the number of AF patients in 2020 was estimated to exceed 12 million. Considering that approximately one-third of these individuals are unaware of their condition, leading to undiagnosed cases of paroxysmal AF, it is probable that the actual number of AF patients exceeds the estimated figure mentioned above4. AF, a frequently occurring arrhythmia, substantially elevates the risk of severe consequences, including stroke, embolism, and heart failure, thus contributing to increased mortality3. Current primary treatments for AF include heart rate control, catheter ablation, and anticoagulation therapy. Despite extensive research, outcomes remain suboptimal, with AF remaining a significant contributor to mortality and increasing healthcare costs1. Moreover, the underlying mechanisms of AF remain unclear. Therefore, developing a model for risk prediction and elucidating possible biomarkers and treatment targets for AF is crucial for enhancing diagnostic accuracy, intervention efficiency, and overall prognosis for patients with AF.
Mitochondrial energy metabolism refers to the series of biochemical processes within mitochondria, the organelles responsible for generating adenosine triphosphate (ATP) through oxidative phosphorylation5. ATP is the primary energy currency of cells, and its production is vital for numerous cellular functions. In energy-demanding tissues such as the heart, mitochondrial energy metabolism plays a critical role in maintaining cellular function6. During AF, the repetitive activation of the atrial myocardium increases the ATP demand, potentially leading to an ATP shortage and the accumulation of reactive oxygen species (ROS), which can disrupt normal mitochondrial function and further compromise energy metabolism7,8. Extensive research has been conducted on the correlation between mitochondrial dysfunction and AF. Malfunction of mitochondria may result in inadequate ATP and excessive ROS generation, damaging calcium ion homeostasis in myocardial cells and the excitability of membranes, ultimately leading to AF9,10. Moreover, there is a correlation between mitochondrial malfunction and reduced ATP levels, as well as between the deterioration of the mitochondrial membrane potential and mitochondrial network fragmentation. These factors ultimately contribute to impaired contractile function and the advancement of AF in both experimental and clinical settings7. The current findings indicate that mitochondrial dysfunction is a crucial factor in the pathological processes that cause AF. Nevertheless, the connection between mitochondrial energy metabolism and AF remains uncertain.
Recent studies have increasingly focused on the roles of mitochondrial dysfunction and immune cell infiltration (ICI) in the pathophysiology of AF11,12. Mitochondrial DNA mutations and oxidative stress disrupt cellular energetics, contributing to the arrhythmogenic substrate13. Immune infiltration, particularly in the left atrial appendage, correlates with structural remodeling and the perpetuation of AF14. While mitochondrial dysfunction and immune responses have been studied separately, their synergistic interactions in AF remain poorly understood. Most existing AF diagnostic models primarily rely on clinical parameters or single-omics features, leaving a gap in integrating mitochondrial energy metabolism-related genes (MEMRGs) with immune infiltration dynamics15. This study aims to fill this gap by combining bioinformatics and machine learning approaches to investigate the interplay between MEMRGs and immune infiltration in AF pathogenesis, providing valuable insights into AF development and potential therapeutic targets.
With rapid advancements in bioinformatics and the accumulation of large amounts of disease data, big data-driven approaches have emerged as significant avenues for understanding the biological characteristics of diseases. Against this backdrop, advanced machine learning algorithms, including nonnegative matrix factorization (NMF)16, least absolute shrinkage and selection operator (LASSO) regression17, and support vector machines (SVMs)18,] have gained widespread application. These algorithms have demonstrated efficacy in screening, diagnosing, prognosticating, and identifying molecular targets for multiple diseases19.
Herein, a thorough analysis of many Gene Expression Omnibus (GEO) gene expression datasets was conducted to investigate MEMRGs in AF. Bioinformatics techniques and machine learning approaches were integrated to identify the main genes linked to mitochondrial function that may serve as biomarkers for AF risk prediction and therapeutic targets. An AF diagnostic model was created and verified, and key genes were identified. In addition, we developed interaction networks for messenger RNA‒RNA-binding protein (mRNA-RBP) and messenger RNA-transcription factor (mRNA-TF) with the ENCORI, CHIPBase, and hTFtarget databases20,21,22. Furthermore, an examination was implemented to ascertain the connection between the number of immune cells and these genes. These findings provide a deeper understanding of AF pathogenesis, highlighting the potential roles of mitochondrial dysfunction, immune system dysregulation, and their interactions in AF development and progression.
Materials and methods
Data download
The gene expression data for cardiac tissues from AF patients were obtained from the GEO database23 (https://www.ncbi.nlm.nih.gov/geo/) via the GEOquery R package24. The specific datasets retrieved included GSE31821, GSE4117725, and GSE79768 26.
The GSE31821 dataset contains auricular tissues from 4 AF patients and 2 normal controls. GSE41177 consisted of 38 atrial tissues from 32 AF patients and 6 normal controls. GSE79768 contains 26 heart tissue samples, including 14 AF atrial and 12 normal heart tissue samples. All three datasets mentioned were obtained from Homo sapiens via the GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array chip platform. The data analysis was performed using these samples. Comprehensive information on each dataset is provided in Table 1.
The sva R package27 was implemented to debatch the datasets GSE31821, GSE41177, and GSE79768, resulting in combined GEO datasets (combined datasets). These combined dataset datasets comprised 50 AF and 20 normal samples. The combined datasets were subsequently normalized with the R package limma28, and the annotation probes were subsequently standardized. Principal component analysis (PCA)29 was then executed on the expression matrices prior to and following batch effect elimination to verify its effectiveness. The PCA method can decrease data dimensionality and visualize features in 2D or 3D graphs.
Using the GeneCards database (https://www.genecards.org/)30 and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database31 (https://www.kegg.jp/kegg/pathway.html) database is a collection of MEMRGs. Within the GeneCards database, a search was conducted using the term “mitochondrial energy metabolism.” Only the entries classified as “Protein Coding” and MEMRGs with a relevance score exceeding 1 were retained. Overall, 43 MEMRGs were identified. After merging with the 147 MEMRGs obtained from the KEGG pathway database to remove duplicates, 185 MEMRGs were identified, and the particular data are presented in Table S1.
AF-related MEMRDEGs
The samples in the merged GEO datasets were classified into AF and normal groups on the basis of their characteristics. Research on differential gene expression was executed with limma (R package). Differentially expressed genes (DEGs) were identified on the basis that |logFC| had to be greater than 0 and the p value had to be less than 0.05 32. Genes with a p value less than 0.05 were categorized into elevated (|logFC| > 0) and downregulated (|logFC| > 0) genes.
To identify MEMRDEGs associated with AF, DEGs with |logFC| > 0 and p value < 0.05 were intersected with the genes involved in mitochondrial energy metabolism (MEMRGs). The intersection was visualized with a Venn diagram. Additionally, volcano and differential sorting plots were created with the ggplot2 R package to plot the differential expression study outcomes.
Gene ontology (GO) functional and KEGG pathway enrichment analyses
GO33 is a widely implemented approach for extensive functional enrichment investigations involving biological process (BP), cell component (CC), and molecular function (MF) terms. KEGG34 is an extensively applied database that contains comprehensive data on genomes, disorders, biological pathways, medications, and different linked subjects. The clusterProfiler (R package)35 was used to execute GO functional and KEGG pathway enrichment studies of the MEMRDEGs. The enrichment outcomes were ascertained with the criteria of a p value < 0.05 and an FDR value (q value) < 0.05.
Gene set enrichment analysis (GSEA) and gene set variation analysis (GSVA)
GSEA36 is implemented to ascertain the gene arrangement in a predetermined gene set inside a gene table, rated using their link with the phenotype. This strategy enables researchers to ascertain its impact on the phenotype. The genes from the merged datasets were first rated according to their logFC values. Afterward, the R program clusterProfiler was used to perform GSEA on all the genes in the merged datasets. GSEA was conducted with several parameters: the number of seeds was set to 2020, 1000 computations were performed, any gene set had to have a minimum of 10 genes, and fewer than 500 genes were permitted in any gene set. The Molecular Signatures Database (MSigDB)37 was accessed via https://www.gsea-msigdb.org/to acquire c2 gene sets (Cp. All. V2022.1. Hs. Symbols.). GSEA was conducted via the GMT file, specifically including all canonical pathways, which consisted of 3050 gene sets. The GSEA data were filtered on the basis of a significance threshold of a p value < 0.05 and a false discovery rate (FDR) of q value < 0.25.
GSVA38 is a nonparametric and unsupervised analytical method. This approach examines gene set enrichment in nuclear transcriptome data obtained from microarray analysis. This is accomplished by transforming the gene expression matrix, which compares the gene expression levels across several samples, into a matrix that examines the expression levels inside each individual sample. Using this approach, we can determine the pathways enriched in different samples. The “h.all.v7.4.symbols.gmt” reference gene set was obtained with MSigDB to determine changes in gene function in various groups of the GEO dataset (combined datasets). Significant enrichment was determined at a p value < 0.05. The expression matrix was subjected to GSVA, and the enriched pathways were discovered in the findings of the GSVA enrichment analysis.
Construction of the AF diagnostic model
Logistic regression (LR) research was executed on the MEMRDEGs to construct AF diagnosis models utilizing the combined GEO datasets. LR was implemented to investigate the connection between the independent and binary dependent variables, which consisted of the AF and normal groups. A significance criterion of P < 0.05 was implemented to examine the MEMRDEGs. Afterwards, an LR model was constructed. An LR model was constructed to investigate the correlation between these variables and AF more thoroughly.
Following the identification of the MEMRDEGs via an LR model, we employed an SVM algorithm (45) to construct an SVM classification model. We then optimized the model by selecting a subset of MEMRDEGs that yielded the greatest accuracy and the smallest error rate. The LASSO regression method was subsequently executed with glmnet (R package)39, with the settings specified as set. seed (500) and family="binomial.” The analysis incorporated the MEMRDEGs in the LR model. The LASSO regression method expands upon linear regression, addresses the issue of model overfitting, and enhances generalization by including a penalty component, which is the product of lambda and the slope absolute value. The LASSO regression study findings are shown with a diagnostic model and variable trajectory graphs. The AF diagnostic model was created on the basis of the outcomes of the LASSO regression study, with the detected MEMRDEGs being referred to as model genes.
Finally, the LASSO risk score was estimated via the LASSO regression analysis risk coefficients. The MEMRDEGs identified by the LASSO and SVM methods were overlapped to identify crucial genes (mRNAs) for further investigation. The risk score was estimated with the following formula:
Validation of the AF diagnostic model
Afterward, a nomogram for the MEMRDEGs was generated with the rms package in R. In a rectangular coordinate system, a nomogram is a visual depiction that uses distinct line segments and clusters to highlight the functional link between numerous separate elements. This relationship is generated via multivariate regression studies. A scale was constructed to characterize each element in the model throughout the multivariate regression analysis. A cumulative score was subsequently computed to forecast the probability of an event occurring.
Calibration plots are often implemented to ascertain the model’s predictive performance by graphically comparing the actual and expected probabilities across various situations. It is mostly used to perform fitting analyses on models generated via the LR method in real-world situations.
Decision curve analysis (DCA) is a valuable approach for determining the practicality of prediction models, diagnostic tests, and genetic indicators in a clinical setting. To estimate the effectiveness of our LR model, we implemented the ggDCA tool in R to produce a DCA plot. This figure graphically represents the model’s precision and its capacity to differentiate between positive and negative instances.
Moreover, the pROC (R package) was used to produce receiver operating characteristic (ROC) curves in the GEO datasets and compute the area under the curve (AUC). To ascertain the diagnostic precision of the linear predictors in the LR model for forecasting AF occurrence. The AUC values of the ROC curve varied between 0.5 and 1. The diagnostic performance is deemed superior when the AUC value approaches 1.
mRNA-RBP and mRNA-TF interaction networks
The ENCORI database20 (version 3.0) (https://starbase.sysu.edu.cn/) integrates miRNA-ncRNA, miRNA‒mRNA, ncRNA-RNA, RNA‒RNA, RBP-ncRNA, and RBP-mRNA interactions derived from CLIP-seq and DSB-seq data (for plants). This application offers a user-friendly interface for investigating possible microRNA targets. We applied the ENCORI database to identify RBPs that potentially interact with our pivotal genes (mRNAs). On the basis of the database results, we then built a network that depicts these mRNA–RBP interactions.
The CHIPBase database21 (version 3.0) (https://rna.sysu.edu.cn/chipbase/) contains ChIP-seq data for DNA-binding proteins. It is utilized to identify numerous binding sites and base sequence matrices and anticipate millions of transcription factors (TFs) and interactions with gene transcription control. The hTFtarget database, which can be accessed at http://bioinfo.life.hust.edu.cn/hTFtarget, is an extensive repository of information concerning the regulation of human TFs and their target genes. We searched for TFs with binding affinity for crucial genes (mRNAs) via the CHIPBase (version 3.0) and hTFtarget databases.
Differential expression analysis of key genes
To elucidate potential mechanisms, biological features, and pathways underlying DEGs in AF, we executed a differential expression analysis with the Wilcoxon rank-sum test. The critical gene expression levels between the AF and normal sample groups in the merged GEO datasets in this test were evaluated. The ggplot2 package in R was subsequently employed to visualize the DEGs.
On the basis of the differential expression analysis, we selected crucial genes and further plotted ROC curves in the GEO dataset (combined datasets) for further analysis and visualization. The ROC curve40 is a visual tool that ascertains the most effective model, eliminates less effective models, or establishes appropriate thresholds within a given model. The ROC curve visually depicts the sensitivity and specificity of the continuous variables. The AUC of the ROC curve varies from 0.5 to 1, with larger AUC values suggesting superior diagnostic ability. AUC values between 0.5 and 0.7 suggest poor accuracy, whereas values between 0.7 and 0.9 suggest good accuracy. Values over 0.9 imply an elevated accuracy level. The proc package in R was used to visualize the ROC curves of the filtered key genes in the GEO dataset (combined datasets) and calculate the area under the curve (AUC) to evaluate the diagnostic effectiveness of the expression of the MEMRDEGs for patients with AF.
Immune infiltration analysis (ssGSEA)
The ssGSEA41 approach was implemented to ascertain the proportionate abundance of each immune cell. First, many types of ICIs have been identified, including activated CD8 T cells, activated dendritic cells, gamma-delta T cells, natural killer (NK) cells, regulatory T cells, and different human immune cell subtypes. Afterward, ssGSEA enrichment scores were applied to determine the relative abundance of each immune cell in each sample. Patients with a p value less than 0.05 were excluded from the ICI matrix. The group comparison plots show the changes in the amount of ICI used between the AF and normal groups. In addition, the pheatmap R package was used to create correlation heatmaps depicting the outcomes of the correlation study conducted on immune cells and MEMRDEGs in AF.
Immune infiltration assay (MCPcounter)
MCPcounter42 was used to ascertain the ICI and different stromal cells from the expression data. MCPcounter is capable of calculating abundance estimates for eight different immune cell groups, incorporating CD4 + T, CD8 + T, and NK cells; B lymphocytes; and monocytes, dendritic cells, neutrophils, and cytotoxic lymphocytes (which include CD8 + T and cytotoxic innate lymphocyte NK cells). Furthermore, this approach was applied to compute the prevalence of two nonimmune stromal cell groups, namely, fibroblasts and endothelial cells, that lack immunity. We assessed the correlation between the abundance estimates generated by MCPcounter and the antigen gene expression levels with Spearman’s rank correlation coefficient p value less than 0.05 was considered significant.
Functional similarity analysis
GO annotation semantic comparison is used to quantify the similarity of genes and genomes, serving as a crucial basis for several bioinformatics analytic techniques. We used the GOSemSim (version 50) program to compute the semantic similarity of the GO concepts linked to our primary genes. This analysis considered all three aspects of GO: BP, CC, and MF. To obtain each gene pair’s single similarity score, we computed the geometric mean of the similarity values across these three GO aspects. Finally, we implemented the ggplot2 package in R to create visualizations that effectively communicate the outcomes of our functional similarity analysis.
Statistical analysis
The data were examined and processed with R software (version 4.2.2). To assess the significance of normally distributed continuous variables between two groups, an independent Student’s t-test was applied unless otherwise specified. The Wilcoxon rank sum test was employed to detect discrepancies among normally distributed variables. The Kruskal‒Wallis test was used to compare three or more groups. Spearman correlation analysis was applied to estimate the correlation coefficient among distinct compounds. All p values were two-sided unless otherwise stated, and a p value less than 0.05 was deemed significant.
Results
Technology roadmap (Fig. 1).

Technology roadmap. PCA Principal Component Analysis, GSEA Gene Set Enrichment Analysis, GSVA Gene Set Variation Analysis, DEGs Differentially Expressed Genes, MEMRGs Mitochondrial Energy Metabolism-Related Genes, MEMRDEGs Mitochondrial Energy Metabolism-Related Differentially Expressed Genes, GO Gene Ontology, KEGG Kyoto Encyclopedia of Genes and Genomes, LASSO Least Absolute Shrinkage and Selection Operator, SVM Support Vector Machine, ROC Receiver Operating Characteristic, RBP RNA-binding protein, TF Transcription Factor.
Data collection and correction
The sva R tool was implemented to alleviate batch effects in the AF datasets GSE31821/41,177/79,768, involving the generation of merged GEO datasets. Gene expression distribution boxplots were employed to compare datasets before and after batch effect removal (Fig. 2A-B). PCA plots were subsequently utilized to assess the distribution of low-dimensional features prior to and following batch effect removal (Fig. 2C-D). The presence of batch effects in each sample from the AF dataset was significantly mitigated after using batch elimination techniques, as shown by the distribution box and PCA plots.

Debatching of the dataset. (A, B) Boxplots plot of GEO Datasets (Combined Datasets) before to (A) and after (B) normalization. (C, D) PCA plots of GEO datasets (Combined Datasets) before (C) and after (D) batch effect elimination. PCA: Principal Component Analysis. Blue represents the GSE31821 dataset, red represents the GSE41177 dataset, and purple indicates the GSE79768 dataset.
AF-related MEMRDEGs
Data from integrated GEO datasets were allocated into AF and normal groups. The differential analysis of the integrated GEO datasets was executed with the limma R package, which compared the gene expression levels of the AF and normal groups to identify DEGs between the two groups. There were 7618 DEGs in the combined GEO datasets that matched the criterion of having a logFC over 0 and a p value below 0.05. According to the volcano plots, a total of 3636 genes were upregulated, matching the requirements of logFC > 0 and p value < 0.05. Additionally, the expression of 3982 genes decreased, matching the criteria of logFC < 0 and p value < 0.05 (Fig. 3A).

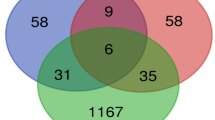
Differential gene expression analysis. (A) Volcano plot of DEGs analysis comparing the AF and normal groups in GEO Datasets (Combined Datasets). (B) Venn diagram of DEGs and MEMRGs in GEO Datasets (Combined Datasets). (C) Heatmap of differential expression of MEMRDEGs in GEO datasets (Combined Datasets). DEGs Differentially Expressed Genes, MEMRGs Mitochondrial Energy Metabolism-Related Genes, MEMRDEGs Mitochondrial Energy Metabolism-Related Differentially Expressed Genes.
MEMRDEGs were identified based on the criteria of |logFC| > 0 and a p value < 0.05. The intersection of DEGs and MEMRGs was then determined with a Venn diagram (Fig. 3B). A total of 59 MEMRDEGs were subsequently identified (Table S2). The changes in MEMRDEG expression between the AF and normal groups in the integrated GEO datasets were further analyzed. The pheatmap R package was used to plot a heatmap of the emerging differential sequences (Fig. 3C).
GO and KEGG enrichment studies
GO and KEGG enrichment studies were applied to further explore the relationships between BPs, CCs, MFs, and biological pathways of the 59 MEMRDEGs and AF (Table 2). We found that the 59 MEMRDEGs were enriched mostly in BPs associated with AF, including the production of precursor metabolites and energy, small molecule catabolic processes, ATP metabolic processes, and energy derivation via organic molecule oxidation. In terms of CCs, the MEMRDEGs were significantly enriched in many CCs, including the mitochondrial matrix, mitochondrial inner membrane, oxidoreductase complex, and mitochondrial protein-containing complex.
In the case of MFs, the MEMRDEGs were enriched in oxidoreductase activity, namely, reactions involving aldehyde or oxo group donors and nicotinamide adenine dinucleotide or nicotinamide adenine dinucleotide phosphate as acceptors. They were also shown to have active transmembrane transporter activity and electron transfer activity. A KEGG enrichment study revealed that the MEMRDEGs were enriched in oxidative phosphorylation, diabetic cardiomyopathy, Huntington’s disease, chemical carcinogenesis-reactive oxygen species, Parkinson’s disease, amyotrophic lateral sclerosis, pathways of neurodegeneration-multiple diseases, carbon metabolism and different biological pathways.
The outcomes of the GO and KEGG pathway enrichment studies are displayed with bubble plots (Fig. 4A-B). Simultaneously, network plots for BPs, CCs, MFs, and biological pathways were generated with the GO and KEGG enrichment studies (Fig. 4C-D). The connecting lines depict the links between molecules and their accompanying annotations, whereas larger nodes indicate a greater number of molecules in the relevant entry.

GO and KEGG enrichment analysis for MEMRDEGs. (A, B) GO and pathway (KEGG) enrichment analysis results of the MEMRDEGs illustrated through bubble plots: BP, CC, MF, and biological pathways (KEGG). The GO and KEGG terms are visualized on the abscissa. In the bubble plot, the bubble size reflects the gene count, whereas the color signifies the p value: red denotes a smaller p value, indicating greater significance, whereas blue indicates greater p values, suggesting lower significance. (C, D) GO and pathway (KEGG) enrichment analysis findings network diagram of the MEMRDEGs: BP, CC, MF (C), and KEGG (D). The red nodes correspond to objects, the blue nodes reflect molecules, and the lines depict the links between items and molecules. MEMRDEGs Mitochondrial Energy Metabolism-Related Differentially Expressed Genes, GO Gene Ontology, KEGG Kyoto Encyclopedia of Genes and Genomes, BP Biological Process, CC Cellular Component, MF Molecular Function. The screening criteria for GO and pathway (KEGG) enrichment analysis were a p value < 0.05 and an FDR value (q value) < 0.05.
Gene set enrichment analysis (GSEA)
Next, GSEA was applied to ascertain all gene expression implications in merged GEO datasets on AF and the BPs contributing. The link between the CCs and MFs affected is depicted in Fig. 5A; Table 3.

GSEA for combined datasets. (A) GSEA 5 biological function mountain maps of the GEO datasets (combined datasets). (B–F) GSEA showing that the MEMRDEGs were significantly enriched in the Wnt (B), Hedgehog (C), JAK-STAT (D), and WNT (B) pathways. TGF-beta (E), and PI3KCI pathways (F). GSEA gene set enrichment analysis. The selection standards of GSEA were a p value < 0.05 and an FDR value (q value) < 0.25.
We concluded that the genes present in the integrated GEO datasets were significantly enriched in various pathways, including the Wnt, Hedgehog, Janus kinase (JAK)-signal transducer and activator of transcription (STAT), transforming growth factor-beta (TGF-β), and phosphatidylinositol 3-kinase complex I (PI3KCI) pathways (Fig. 5b-F). Additionally, these genes were linked to different biologically related functions and signaling pathways.
Creation of an AF diagnostic model
First, a univariate LR was performed to estimate the diagnostic significance of the 59 MEMRDEGs in AF. The results revealed that 52 MEMRDEGs were significant in the LR model (P < 0.05) (Table S3).
The 52 MEMRDEGs were subsequently utilized to construct a LASSO regression model through LASSO regression analysis, which served as the diagnostic model for AF. The LASSO regression model and the LASSO variable trajectory diagrams (Fig. 6A-B) were applied to display the LASSO regression model. Sixteen MEMRDEGs, referred to as model genes, were incorporated into the LASSO regression model, including ACAT1, ALDH18A1, ALDH1A1, ALDH1L2, ALDH4A1, CYP2U1, HADHA, HTT, IDH3G, NDUFB7, NDUFS3, NDUFS6, OGDH, SLC25A3, TANGO2, and TP53.

Creation of a diagnostic model for AF. (A) Diagnostic model plot of the MEMRDEGs of the LASSO regression in the merged datasets. (B) Visualization of the LASSO diagnostic model’s variable trajectories. (C) The SVM method identified the genes with the lowest error rate. (D) The gene number with the greatest accuracy was acquired via the SVM method. (E) Venn diagram illustrating the overlap between the LASSO and SVM methods. LASSO Least absolute shrinkage and selection operator, SVM Support Vector Machine, MEMRDEGs Mitochondrial energy metabolism-related differentially expressed genes.
Finally, we built an SVM model using the 52 MEMRDEGs and the SVM technique. We identified the genes that had the smallest mistake rate (Fig. 6C) and the greatest accuracy (Fig. 6D). The outcomes indicated that the SVM model manifested the maximum accuracy level when the number of genes was 8. The eight MEMRDEGs included SLC25A3, OGDH, HTT, ACAT1, ACSBG2, ALDH1L2, GPI, and NFATC4.
The intersection of the MEMRDEGs in the LASSO regression and the SVM models was utilized to identify key genes, yielding five key genes (ACAT1, ALDH1L2, HTT, OGDH, and SLC25A3) that were used for subsequent analysis. A Venn diagram was generated for visualization (Fig. 6E).
Confirmation of the AF diagnostic model
To further confirm the significance of the AF diagnosis model, a nomogram was developed according to model genes with the integrated GEO datasets to highlight their interrelationships (Fig. 7A). The outcomes revealed that the OGDH expression level was significantly involved in the utility of the AF diagnostic model compared with the other model genes.

Diagnostic and validation analysis of AF. (A) Nomograms of model genes in combined GEO datasets for atrial fibrillation (AF) diagnostic models. (B, C) Calibration curve (B) and DCA plot (C) of model genes in integrated GEO datasets (combined datasets) for the AF diagnostic model. (D) ROC analysis of linear predictors of LR models in GEO datasets (combined datasets). (E) Heatmap of the GSVA outcomes between the high/low groups of linear predictors in the LR model. GSVA, gene set variation analysis. The selection standards of GSVA were a p value < 0.05 and an FDR value (q value) < 0.25. Red and blue colors reflect the high/low groups of linear predictors of the LR model, respectively. The blue and red colors represent mitigated and elevated values, respectively, in the heatmaps. The vertical axis of the calibration curve reflects the net benefit, whereas the horizontal axis reflects the threshold or threshold risk. DCA, decision curve analysis; ROC, receiver operating characteristic; AUC > 0.9 manifested elevated accuracy. AUC, area under the curve; logistic regression, LR.
A calibration study was subsequently conducted to ascertain the accuracy and discriminative ability of the AF diagnostic model. This incorporated construction of a calibration curve (Fig. 7B). Although the calibration curve slightly deviates from the ideal model’s diagonal line, it demonstrates a good fit overall, indicating the reliable predictive performance of the model.
Moreover, DCA was applied to ascertain the clinical usefulness of the AF diagnostic model using the integrated GEO datasets. The results revealed that the model net benefit was greater within a certain range and remained consistently superior to the “all positive” and “all negative” scenarios (Fig. 7C), suggesting its promising clinical application potential.
Additionally, ROC curves were generated for the linear predictors of the LR model in the AF and normal groups on the basis of integrated GEO datasets (Fig. 7D). ROC curve analysis revealed that the diagnostic performance of the LR model on the basis of the GEO dataset was satisfactory.
GSVA was applied to all the genes in the merged GEO datasets to explore the h.all.v7.4.symbols.gmt gene set differential expression between high and low (high/Low) groups of linear predictors in the LR model, and the outcomes are shown in Table 4. The GSVA results revealed significant distinctions between the high/low groups of linear predictors in the LR model in gene sets encompassing the following hallmark p53 pathway: tumor necrosis factor-alpha (TNF-α) signaling/nuclear factor-kappa B (NF-κB), epithelial‒mesenchymal transition, early estrogen response, inflammatory response, oxidative phosphorylation, interleukin 2 (IL2)/STAT5 signaling, and late estrogen response (P < 0.05).
Finally, the differential expression patterns between the high/low groups of linear predictors in the LR model were analyzed on the basis of the GSVA results and visualized via a heatmap (Fig. 7E).
Creation of mRNA–RBP and mRNA–TF interaction networks
mRNA-RBP data from the ENCORI database were utilized to anticipate the interaction between RBPs and the five key genes (ACAT1, ALDH1L2, HTT, OGDH, and SLC25A3). The Cytoscape program was subsequently implemented to create a graphical depiction of the mRNA‒RBP interaction network (Fig. 8A), revealing that the five key genes interact with 26 RBPs, forming 65 pairs of mRNA‒RBP interaction connections (Table S4).

mRNA-RBP, mRNA-TF interaction network. (A) Key gene-RBP interaction network. (B) Key gene-transcription factor interaction network. Yellow oval genes are mRNAs. The blue ovals represent RBP, and the purple ovals represent TF. RBP: RNA-binding protein. TF: transcription factor.
Furthermore, TFs that interact with the five essential genes were identified by searching the CHIPBase and hTFtarget databases. The mRNA‒TF interaction network was subsequently created with Cytoscape software (Fig. 8B). Ultimately, three key genes (ALDH1L2, OGDH, and SLC25A3) interact with 15 TFs, forming 21 pairs of mRNA‒TF interaction connections (Table S5).
Differential expression analysis of key genes between the AF and normal groups in integrated GEO datasets
Violin plots ((Fig. 9A) were generated to illustrate the differential expression of five key genes (ACAT1, ALDH1L2, HTT, OGDH, and SLC25A3) between the AF and normal groups in the merged GEO datasets. The results revealed that the expression levels of the five key genes exhibited highly significant differences between the two groups.

Differential expression analysis of key genes between AF and normal groups in integrated GEO datasets. (A) Analysis of the main genes between the AF and normal groups in merged GEO datasets. (B) The results of correlation heatmaps for essential genes in integrated GEO datasets. (C) Functional similarity analysis of important genes. (D–H) ROC curve analysis of the crucial genes SLC25A3 (D), OGDH (E), HTT (F), ACAT1 (G), and ALDH1L2 (H) in the integrated GEO datasets. ***P < 0.001 denotes highly statistically significant; *P < 0.05 reflects statistical significance. Diagnostic performance improves as AUC approaches 1. An AUC value greater than 0.9 suggests a high accuracy level, whereas an AUC value between 0.7 and 0.9 suggests a moderate accuracy level. Red represents and signifies the AF and normal groups, respectively. ROC receiver operating characteristic, AUC area under the curve, AF atrial fibrillation.
Moreover, a correlation analysis was executed with the full expression matrix of the five essential genes in the integrated GEO datasets. This analysis resulted in the generation of a correlation heatmap (Fig. 9B). OGDH was shown to be positively correlated with ACAT1, SLC25A3, and HTT, whereas SLC25A3 was negatively correlated with ALDH1L2, OGDH, and HTT.
Afterward, a functional similarity analysis was performed on the five critical genes. GOSemSim, an R program, was employed to determine the semantic similarity of gene products, gene clusters, sets, and terms of GO. The obtained findings were then shown graphically via boxplots (Fig. 9C). OGDH was determined to have the greatest functional similarity with the other important genes.
Finally, ROC curves for the five key genes (ACAT1, ALDH1L2, HTT, OGDH, SLC25A3) in the integrated GEO datasets were plotted (Fig. 9D-H). SLC25A3 had high accuracy between the two groups (AUC > 0.9), whereas OGDH, HTT, ACAT1, and ALDH1L2 exhibited moderate accuracy (0.7 < AUC < 0.9).
ICI treatment via SsGSEA and MCPcounter
The ssGSEA method was employed to ascertain 28 ICI levels between the AF and normal groups on the basis of integrated GEO datasets. The abundances of 28 ICIs in the AF and normal groups were compared via a boxplot (Fig. 10A). The results revealed that 21 immune cells, including myeloid-derived suppressor T cells, effector memory CD8 T cells, neutrophils, mast cells, monocytes, central memory CD8 T cells, regulatory T cells, immature B cells, plasmacytoid dendritic cells, NK cells, central memory CD4 T cells, activated dendritic cells, activated CD8 T cells, immature dendritic cells, eosinophils, memory B cells, CD56bright NK cells, gamma delta T cells, type 1 T helper cells, activated CD4 T cells, and macrophages, were significantly different between the two groups (P < 0.05).

Immune infiltration analysis with ssGSEA and MCPcounter. (A) ssGSEA of 28 immune cells between the AF and normal groups according to integrated GEO datasets. (B) Heatmap for the connection between the abundance of ICIs and essential genes of the ssGSEA algorithm (P < 0.05). (C) Heatmap showing the connections between key genes and ICI abundance calculated using the MCPcounter algorithm. The red circles indicate a positive association between important genes and the amount of ICI, with a larger circle indicating a greater correlation. The blue circles indicate a negative correlation between genes and the amount of ICI, with a larger circle indicating a greater correlation. ns indicates not significant (P > 0.05); *P < 0.05 indicates significance; **P < 0.01 indicates great significance; ***P < 0.001 indicates statistical significance. ssGSEA single-sample gene set enrichment analysis, MCPcounter microenvironment cell population counter.
The correlation heatmap revealed that five key genes (ACAT1, ALDH1L2, HTT, OGDH, and SLC25A3) were correlated with the abundance of 21 significant (P < 0.05) ICIs (Fig. 10B). ALDH1L2 was positively correlated with most immune cells, whereas SLC25A3 was inversely correlated with most immune cells.
The MCPcounter method was implemented to compute the connection between the five pivotal genes and the amount of ICI in both the AF and normal groups via integrated GEO datasets (Fig. 10C). The results revealed a link between the five essential genes and ten distinct kinds of immune cells, namely, B lineage, CD8 T, cytotoxic lymphocyte, endothelial, fibroblast, monocytic lineage, myeloid dendritic, neutrophil, NK, and T cells. HTT and OGDH were associated with favorable connections between CD8 + T cells and myeloid dendritic cells. SLC25A3 and HTT were negatively correlated with the monocyte lineage.
Discussion
AF is a complicated cardiac arrhythmia with poorly understood molecular mechanisms. Recent research has indicated that inflammation and oxidative stress are significant factors in structural and electrical changes in atrial tissues, which are involved in the development of AF43,44. Inflammation induced by the immune response can lead to tissue damage and fibrosis, disrupting normal electrical conduction in the atria. Oxidative stress arises from an imbalance between ROS generation and the protective antioxidant defense system, further exacerbating cellular damage. As vital organelles for cellular energy production, mitochondria play critical roles in myocardial energy metabolism and the redox state45,46. During energy metabolism, mitochondria generate ROS, excessive levels of which might result in cellular damage and the onset of arrhythmias47. Therefore, a better understanding of the connection between mitochondrial energy metabolism and AF might offer novel insights and therapeutic concepts from an energy metabolism perspective.
To further explore the pathogenesis of AF, we conducted a comprehensive analysis using GEO datasets and a mitochondrial energy metabolism-related gene set, yielding 59 MEMRDEGs. GO and KEGG enrichment studies revealed that these genes were enriched primarily in various mitochondrial components and participated in processes related to energy metabolism and ATP synthesis. These abnormalities might lead to the occurrence of AF, indicating their potentially significant roles in regulating energy metabolism processes during AF onset and progression. These findings emphasize the importance of mitochondria in AF development. In addition, a GSVA enrichment study was performed to further elucidate the molecular pathways implicated in AF. These genes were significantly enriched in Wnt, Hedgehog, JAK-STAT, TGF-beta, and PI3KCI in AF, suggesting their pivotal roles in cardiovascular system homeostasis and pathophysiology. These outcomes offer more evidence that AF etiology involves numerous molecular pathways beyond mitochondrial energy metabolism alone. Notably, the PI3KCI pathway regulates critical BPs, including cell proliferation, survival, and metabolism48, with its enrichment in AF potentially indicating the significance of dysregulated cell proliferation and metabolism in disease progression.
The 59 MEMRDEGs were analyzed via a single-factor LR model to ascertain their diagnostic value in AF and construct a corresponding diagnostic model. Fifty-two genes exhibited statistical significance in the LR model. Subsequently, LASSO algorithms and SVM methods were employed to screen the 52 genes further, ultimately identifying five key genes: ACAT1, ALDH1L2, HTT, OGDH, and SLC25A3. To integrate the results from both models, we selected genes that were identified by both the LASSO regression and SVM algorithms, ensuring that the selected genes had robust and consistent support from multiple models. These genes likely play pivotal roles in the pathogenesis of AF.
ACAT1, expressed in macrophages, promotes the accumulation of intracellular cholesterol esters, playing a critical role in early atherosclerotic lesions49. Although direct evidence linking ACAT1 to AF is lacking, its heightened expression in the AF group suggests its potential involvement in disease pathogenesis, possibly through its association with the proinflammatory state in AF. This conclusion is consistent with that of Li et al.50. HTT, well-known for its association with Huntington’s disease, is involved in cellular stress responses and mitochondrial dynamics51,52. Mounting evidence suggests that HTT not only plays critical roles in embryonic development and neural maturation but also may be important in cardiovascular diseases53,54,55. While no reports currently link HTT to AF, the current investigation revealed significant hindrance in HTT expression in the AF group compared with the normal group, suggesting a potential association between HTT deficiency or mutation and AF occurrence. OGDH, an key enzyme in the tricarboxylic acid cycle, plays a critical role in cellular energy metabolism56. Its dysregulation may disrupt the tricarboxylic acid cycle, affecting the energy supply and metabolic balance in cardiac cells. The upregulation of OGDH not only activates the mitogen-activated protein kinase signaling pathway but also upregulates the expression of proinflammatory cytokines, crucial risk factors in AF57,58. Our data also revealed that OGDH was highly expressed in the AF group and was positively correlated with myeloid dendritic cells, suggesting its significant role in metabolic‒immune interactions in AF. Finally, SLC25A3, a mitochondrial phosphate transporter, is essential for ATP synthesis and mitochondrial function59. Dysregulation of SLC25A3 may impair ATP production, thereby contributing to AF development.
Further correlation analysis of the five key genes revealed that OGDH was positively correlated with ACAT1, SLC25A3, and HTT. SLC25A3 was negatively correlated with ALDH1L2, OGDH, and HTT, suggesting potential shared regulatory mechanisms or interactions among them during the process of AF. Additionally, functional similarity analysis indicated that, compared with other important genes, OGDH presented greater functional similarity. The construction of an interaction network based on ALDH1L2, OGDH, and SLC25A3 revealed complex interactions with RBPs and TFs, providing clues for further understanding their regulatory roles in AF pathogenesis.
Furthermore, GSVA revealed several biological pathways crucial for AF pathogenesis, including the p53, TNF-α/NF-κB, inflammatory response, oxidative phosphorylation, and the IL-2/STAT5 signaling pathways. Inflammation is an AF risk factor, and the immunological response associated with inflammation contributes to AF initiation and progression58,60. Investigations have shown that macrophages act primarily through cytokine release, with TNF-α and IL-1β being the most important60. Neutrophils serve as major sources of ROS and myeloperoxidase (MPO), which are involved in the promotion of fibrosis in the pathogenesis of AF61. Mast cells might be involved in AF occurrence through platelet-derived growth factor subunit A (PDGF-A) mediation11. The roles of T and B cells in AF remain insufficiently studied; T cells are involved mainly in modulating innate immune responses, and B cells potentially exert their effects through self-antibody secretion60.
The ssGSEA methodology revealed significant disparities in 21 distinct immune cell types between patients with AF and control patients. The AF patients manifested a significant increase in the presence of effector and central memory CD8 + T cells, neutrophils, mast cells, and monocytes, which contrasted with the findings of the normal group. Conversely, AF patients manifested a significant hindrance in the quantity of regulatory T, immature B, NK, and central memory CD4 T cells. These results indicate that some types of immune cells may exhibit greater activity levels in AF patients, possibly contributing to the development and progression of the illness. Elevated levels of neutrophils and macrophages exacerbate AF by promoting fibrosis and inflammation, whereas decreased regulatory T cells may fail to control abnormal immune responses, furthering disease development. Additionally, reduced levels of NK cells and plasmacytoid dendritic cells may indicate weakened antiviral and antitumor immune functions in AF patients. Overall, these variations revealed the crucial involvement of the immune response in the pathological mechanisms of AF.
Afterward, the connections between the five essential genes and these immune cells were examined. The results revealed a positive correlation between ALDH1L2 and most immune cells, but SLC25A3 exhibited a negative correlation with most immune cells. Further validation via the MCPcounter algorithm revealed positive correlations between HTT and CD8 T cells and OGDH and myeloid dendritic cells and negative correlations between SLC25A3 and the monocytic lineage, underscoring the interplay between MEMRGs and the immunological response in AF. AF is a complex arrhythmia with multiple contributing factors. While mitochondrial dysfunction and immune responses have been studied separately, this research uniquely integrates both aspects by analyzing MEMRGs and immune cell dynamics in AF60,62. This integrated approach provides a comprehensive understanding of the multifactorial mechanisms underlying AF pathogenesis, revealing how mitochondrial energy metabolism disruption, coupled with immune cell infiltration, amplifies AF onset and progression. To enhance the predictive accuracy of our AF progression and risk model, we incorporated immune cell dynamics alongside mitochondrial gene expression profiles. By examining correlations between immune cells and key mitochondrial genes, we refined our model to better capture the complex interactions between immune responses and mitochondrial dysfunction. This significantly increased the model’s accuracy, as immune infiltration is key to AF pathogenesis. Moreover, the integration of immune cell changes allowed for more personalized risk assessments, offering insights into potential therapeutic targets. However, the precise molecular mechanisms underlying this interaction warrant further investigation.
In addition, the value of the AF diagnostic model was verified, and its effectiveness was evaluated. Calibration curve analysis revealed that predicted probabilities closely matched actual outcomes. DCA showed significant net benefits across various risk thresholds, suggesting the model can enhance clinical decision-making by accurately identifying at-risk patients. ROC analysis yielded an AUC of 0.903 (95% confidence interval: 0.823–0.983), indicating excellent discriminatory power. These results suggest the model improves prediction accuracy over traditional methods, enabling earlier, more precise identification of potential AF patients, especially those without clinical symptoms. It highlights immune responses’ critical role in AF pathogenesis, offering new strategies for personalized therapy. While the model showed excellent performance in the current datasets, data heterogeneity and distinct feature distributions across datasets may affect performance. Future studies will incorporate independent datasets for external validation to assess the model’s robustness and generalizability. Additionally, exploring different analysis methods will ensure the model maintains high diagnostic performance across various conditions.
Despite the strong potential of this study, several limitations exist, which may act as barriers to the clinical translation of the model. Data integration from three platforms may introduce batch effects, potentially affecting findings’ robustness. The limited sample size of the GEO datasets necessitates further power analysis and sample size expansion for validation. The lack of appropriate GEO datasets prevents distinction of AF clinical subtypes, which should be addressed in future research. The absence of experimental validation, such as reverse transcription PCR or Western blotting, limits the clinical confirmation. Although the model shows strong potential, further experimental studies and clinical validation are required to confirm its clinical applicability and performance.
Conclusions
This study integrated MEMRDEGs with machine learning to construct an AF prediction model, identifying five genes (ACAT1, ALDH1L2, HTT, OGDH, and SLC25A3) strongly associated with AF risk. Immune infiltration analysis linked these genes to immunological responses in AF, offering new perspectives on AF development and therapeutic targets. By using LASSO and SVM algorithms, our model is the first to integrate mitochondrial function and immune response into the AF diagnostic framework. Quantifying immune cell dynamics optimized the model’s predictive power, providing valuable perspectives for personalized risk assessment and treatment strategies. Future research is essential to confirm the clinical applicability and generalizability of these findings, particularly for therapeutic interventions based on these discoveries.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.The datasets supporting the results of this study are publicly available and can be accessed as follows: Gene expression data: The data used in this study are available from the Gene Expression Omnibus (GEO) repository, accessible at the following links: GSE31821: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE31821 GSE41177: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE41177 GSE79768: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi? acc=GSE79768 KEGG pathway gene sets: The gene sets hsa00190, hsa05415, hsa05016, hsa05208, and hsa05012 were sourced from KEGG (Kyoto Encyclopedia of Genes and Genomes), and permission to use these materials has been obtained from KEGG. The KEGG database can be accessed at https://www.kegg.jp.
References
Brundel, B. J. J. M. et al. Atrial fibrillation. Nat. Rev. Dis. Primers. 8, 21 (2022).
Tsao, C. W. et al. Heart disease and stroke Statistics-2023 update: A report from the American heart association. Circulation 147, e93–e621 (2023).
Kornej, J., Börschel, C. S., Benjamin, E. J. & Schnabel, R. B. Epidemiology of atrial fibrillation in the 21st century: novel methods and new insights. Circ. Res. 127, 4–20 (2020).
Du, X. et al. Atrial fibrillation prevalence, awareness and management in a nationwide survey of adults in China. Heart 107, 535–541 (2021).
Lopaschuk, G. D., Karwi, Q. G., Tian, R., Wende, A. R. & Abel, E. D. Cardiac energy metabolism in heart failure. Circ. Res. 128, 1487–1513 (2021).
Li, T. et al. Defective Branched-Chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to Ischemia-Reperfusion injury. Cell. Metab. 25, 374–385 (2017).
Wiersma, M. et al. Mitochondrial dysfunction underlies cardiomyocyte remodeling in experimental and clinical atrial fibrillation. Cells 8, 1202 (2019).
Emelyanova, L. et al. Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am. J. Physiol. Heart Circ. Physiol. 311, H54–63 (2016).
Moslehi, J., DePinho, R. A. & Sahin, E. Telomeres and mitochondria in the aging heart. Circ. Res. 110, 1226–1237 (2012).
Clark, A. & Mach, N. The crosstalk between the gut microbiota and mitochondria during exercise. Front. Physiol. 8, 319 (2017).
Liao, C. et al. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J. Clin. Invest. 120, 242–253 (2010).
Zhang, Y. L. et al. Chemokine receptor CXCR-2 initiates atrial fibrillation by triggering monocyte mobilization in mice. Hypertension 76, 381–392 (2020).
Parahuleva, M. S. et al. Pro-Inflammatory and calcium handling remodeling underlies spontaneous onset of paroxysmal atrial fibrillation in JDP2-Overexpressing mice. Int. J. Mol. Sci. 21, 9095 (2020). Structural.
Hohmann et al. _2020_Inflammatory cell infiltration in left atrial appendageal tissues of patients.pdf.
Zhang, Y. et al. A machine learning-based model for predicting paroxysmal and persistent atrial fibrillation based on EHR. BMC Med. Inf. Decis. Mak. 25, 51 (2025).
Gaujoux, R. & Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 11, 367 (2010).
Tibshirani, R. The Lasso method for variable selection in the Cox model. Stat. Med. 16, 385–395 (1997).
Ma, S. et al. Predicting the ecotoxicity of ionic liquids towards Vibrio fischeri using genetic function approximation and least squares support vector machine. J. Hazard. Mater. 283, 591–598 (2015).
Rau, C. S. et al. Machine learning models of survival prediction in trauma patients. J. Clin. Med. 8, 799 (2019).
Wang, W., Zhang, J., Wang, Y., Xu, Y. & Zhang, S. Identifies microtubule-binding protein CSPP1 as a novel cancer biomarker associated with ferroptosis and tumor microenvironment. Comput. Struct. Biotechnol. J. 20, 3322–3335 (2022).
Zhou, K. R. et al. ChIPBase v2.0: decoding transcriptional regulatory networks of non-coding RNAs and protein-coding genes from ChIP-seq data. Nucleic Acids Res. 45, D43–D50 (2017).
Zhang, Q. et al. hTFtarget: A comprehensive database for regulations of human transcription factors and their targets. Genomics Proteom. Bioinf. 18, 120–128 (2020).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 41, D991–995 (2013).
Davis, S. & Meltzer, P. S. GEOquery: a Bridge between the gene expression omnibus (GEO) and bioconductor. Bioinformatics 23, 1846–1847 (2007).
Yeh, Y. H. et al. Region-specific gene expression profiles in the left atria of patients with valvular atrial fibrillation. Heart Rhythm. 10, 383–391 (2013).
Tsai, F. C. et al. Differential left-to-right atria gene expression ratio in human sinus rhythm and atrial fibrillation: implications for arrhythmogenesis and thrombogenesis. Int. J. Cardiol. 222, 104–112 (2016).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The Sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Ben Salem, K. Ben Abdelaziz, A. Principal component analysis (PCA). Tunis Med. 99, 383–389 (2021).
Safran, M. et al. GeneCards Version 3: the human gene integrator. Database (Oxford) baq020 (2010). (2010).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Tien, F. M. et al. Dysregulated immune and metabolic pathways are associated with poor survival in adult acute myeloid leukemia with CEBPA bZIP in-frame mutations. Blood Cancer J. 14, 15 (2024).
Mi, H., Muruganujan, A., Ebert, D., Huang, X. & Thomas, P. D. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 102, 15545–15550 (2005).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Engebretsen, S. & Bohlin, J. Statistical predictions with Glmnet. Clin. Epigenetics. 11, 123 (2019).
Mandrekar, J. N. Receiver operating characteristic curve in diagnostic test assessment. J. Thorac. Oncol. 5, 1315–1316 (2010).
Xiao, B. et al. Identification and verification of Immune-Related gene prognostic signature based on SsGSEA for osteosarcoma. Front. Oncol. 10, 607622 (2020).
Zheng, H., Liu, H., Ge, Y. & Wang, X. Integrated single-cell and bulk RNA sequencing analysis identifies a cancer associated fibroblast-related signature for predicting prognosis and therapeutic responses in colorectal cancer. Cancer Cell. Int. 21, 552 (2021).
Dobrev, D., Heijman, J., Hiram, R., Li, N. & Nattel, S. Inflammatory signalling in atrial cardiomyocytes: a novel unifying principle in atrial fibrillation pathophysiology. Nat. Rev. Cardiol. 20, 145–167 (2023).
Balan, A. I., Halațiu, V. B. & Scridon, A. Oxidative stress, inflammation, and mitochondrial dysfunction: A link between obesity and atrial fibrillation. Antioxid. (Basel). 13, 117 (2024).
Xie, Y. et al. The coupling of Mitoproteolysis and oxidative phosphorylation enables tracking of an active mitochondrial state through mitotimer fluorescence. Redox Biol. 56, 102447 (2022).
Wang, Q. et al. Reduced immunity regulator MAVS contributes to Non-Hypertrophic cardiac dysfunction by disturbing energy metabolism and mitochondrial homeostasis. Front. Immunol. 13, 919038 (2022).
Tascanov, M. B. et al. Relationships between paroxysmal atrial fibrillation, total oxidant status, and DNA damage. Rev. Port Cardiol. (Engl Ed). 40, 5–10 (2021).
Yu, J. S. L. & Cui, W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 143, 3050–3060 (2016).
Liang, Y. et al. Homocysteine-mediated cholesterol efflux via ABCA1 and ACAT1 DNA methylation in THP-1 monocyte-derived foam cells. Acta Biochim. Biophys. Sin (Shanghai). 45, 220–228 (2013).
Li, W., Wang, L., Wu, Y., Yuan, Z. & Zhou, J. Weighted gene co–expression network analysis to identify key modules and hub genes associated with atrial fibrillation. Int. J. Mol. Med. 45, 401–416 (2020).
Zhu, Y. et al. Progressive cardiac arrhythmias and ECG abnormalities in the Huntington’s disease BACHD mouse model. Hum. Mol. Genet. 29, 369–381 (2020).
Ismailoglu, I. et al. Huntingtin protein is essential for mitochondrial metabolism, bioenergetics and structure in murine embryonic stem cells. Dev. Biol. 391, 230–240 (2014).
Nickel, A., Löffler, J. & Maack, C. Myocardial energetics in heart failure. Basic. Res. Cardiol. 108, 358 (2013).
Schroeder, A. M. et al. Cardiac dysfunction in the BACHD mouse model of Huntington’s disease. PLoS One. 11, e0147269 (2016).
Child, D. D. et al. Cardiac mTORC1 dysregulation impacts stress adaptation and survival in Huntington’s disease. Cell. Rep. 23, 1020–1033 (2018).
Liu, Y. et al. N 6-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science 365, 1171–1176 (2019).
Hao, Y., Wang, Z., Wang, X., Zhan, W. & Wu, D. OGDH is involved in sepsis induced acute lung injury through the MAPK pathway. J. Thorac. Dis. 13, 5042–5054 (2021).
Hu, Y. F., Chen, Y. J., Lin, Y. J. & Chen, S. A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 12, 230–243 (2015).
Peoples, J. N. et al. Loss of the mitochondrial phosphate carrier SLC25A3 induces remodeling of the cardiac mitochondrial protein acylome. Am. J. Physiol. Cell. Physiol. 321, C519–C534 (2021).
Liu, Y., Shi, Q., Ma, Y. & Liu, Q. The role of immune cells in atrial fibrillation. J. Mol. Cell. Cardiol. 123, 198–208 (2018).
Rudolph, V. et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat. Med. 16, 470–474 (2010).
Pool, L., Wijdeveld, L. F. J. M., de Groot, N. M. S. & Brundel, B. J. J. M. The role of mitochondrial dysfunction in atrial fibrillation: translation to druggable target and biomarker discovery. Int. J. Mol. Sci. 22, 8463 (2021).
Funding
Supported by Fujian Provincial Natural Science Foundation of China (No. 2020J011293).
Author information
Authors and Affiliations
Contributions
CYL, WL and GAZ designed the study. XJY and ZSC analyzed the experimental data and drafted the manuscript. All the authors have read and approved the final manuscript. XJY, CYZ, and ZCY were responsible for confirming the authenticity of the original data.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
GEO belong to public databases. The patients involved in the database have obtained ethical approval. Users can download relevant data for free for research and publish relevant articles. Our study is based on open source data, so there are no ethical issues and other conflicts of interest.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, X., Lan, W., Lin, C. et al. Atrial fibrillation risk model based on LASSO and SVM algorithms and immune infiltration of key mitochondrial energy metabolism genes. Sci Rep 15, 6681 (2025). https://doi.org/10.1038/s41598-025-91047-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91047-3
