
Abstract
Expression of the PD-1 protein by tumor cells is relatively common and has been shown to exert proliferation-inhibitory effects across various tumor types, including T-cell malignancies, non-small cell lung cancer, and colon cancer. However, harnessing this tumor suppressor pathway is challenging because PD-1 activation by PD-L1 also suppresses normal T-cell function. We hypothesized that cancer antigen-specific TCR-T cells engineered to express PD-L1 could selectively activate the PD-1 pathway in tumor cells while simultaneously preventing self-inhibition by knocking out intrinsic PD-1 expression in TCR-T cells. To test this hypothesis, we co-expressed a MAGE-C2-specific recombinant TCR and the PD-L1-encoding CD274 gene in normal human T cells in which the PDCD1 gene was knocked out. These engineered TCR-T cells targeted MAGE-C2-expressing malignant cells, activating PD-1 signaling to suppress tumor proliferation while maintaining suppressed PD-1 signaling in the TCR-T cells themselves. To evaluate the tumor-suppressive potential of this approach, we compared the efficacy of PDL1-MC2-TCR-TPD1⁻ cells against subtypes lacking PD-L1 expression, PD-1 knockout, or both. Our findings demonstrated that this TCR-T model exhibited significantly enhanced cytotoxic efficacy compared to other subtypes in vitro, ex vivo, and in vivo. These results suggest that the targeted activation of intrinsic PD-1 signaling in T-cell malignancies inhibits tumor proliferation and, when combined with PD-1 inhibition in TCR-T cells, synergistically enhances their cancer-suppressing efficacy. This study provides a foundation for novel cancer treatment strategies.
Similar content being viewed by others


Introduction
Despite breakthroughs and advances, cancer treatment remains challenging. Among current therapies with encouraging effects, adoptive immunotherapy and immune checkpoint inhibitor (ICI) therapy have shown remarkable efficacy in some types of cancer. However, adverse effects still occur and the underlying mechanisms are not fully understood. New therapeutic strategies and improvements to existing treatments are in constant demand.
In adoptive immunotherapy, engineered T cell receptor T (TCR-T) cells have properties that are superior to those of CAR-T cells in recognizing both the cell surface and intracellular cancer-specific antigens presented by MHC molecules. The latter is characteristic of a wide range of solid tumors. Furthermore, TCR-T cells have a high activity for target cells because TCRs require a low amount of the pMHC complex for T-cell activation. Current research and clinical trials on TCR-T cell therapy targeting various antigens are ongoing. The FDA recently approved afami-cel, the first TCR-T cell therapy targeting MAGE-A41[,2. Members of the melanoma antigen gene (MAGE) family, consisting of cancer-testis antigens that can be divided into type I and type II MAGE subfamilies, are among the most frequently used candidates for selecting cancer-specific antigens for TCR-T cells because of their natural characteristics of cancer-specific and intracellular expression. Among MAGE members, MAGE-C2 (MC2) is a promising antigen associated with various types of cancers, including lung cancer, breast cancer, prostate cancer, liver cancer, and multiple myeloma3,4. Typically, high-efficacy TCR-T clones are identified using tumor samples or patient blood. Several MC2-specific TCR clones have been screened from a melanoma patient who responded to immunotherapy, and their sequences were analyzed5,6,7. These clinically proven, high-efficacy TCR clones are convenient for further research. Additionally, the combination of TCR-T cell therapy with ICI therapy has been shown to enhance the antitumor efficacy of TCR-T cell therapy, which may help overcome some hurdles such as T cell exhaustion-related resistance caused by immune checkpoint signaling in T cells.
ICI therapies aim to promote the activation of immune cells, especially CD8 + cytotoxic T lymphocytes; however, current methods of immune checkpoint inhibition do not target specific cell types(s). Several lines of research suggest that the cell-intrinsic PD-1/PD-L1 axis has antitumor effects on a wide range of cancer types, including T-cell lymphoma (TCL) and lung, colon, pancreatic, breast, and bone cancers8,9,10,11,12,13,14. PD-1 inhibition may be a “double-edged” sword, in that it activates normal immune cells and promotes malignancy. Inhibition of PD-1 in cancer cells may facilitate cancer development by promoting cancer-promoting PI3K/AKT signaling or overactivating TCR signaling in T cell malignancies8,9,10,11,12,13,14. Targeted PD-1 inhibition in normal T cells should be considered as an optimized therapy. In addition, the effects of purposefully activating PD-1 in malignant cells and the combined effects of PD-1 inhibition in normal T cells are important topics of investigation.
In this study, we engineered PD-L1-expressing/PD-1-null/MC2-specific TCR-T cells that function as targeted PD-1 activators in cancer cells, while maintaining their own PD-1 suppression, in addition to their direct cytotoxicity to MC2-pMHC-expressing cancer cells. The general antitumor effect of this TCR-T model was synergistically greater than that of its subtype models, which lacked PD-1 or PD-L1 gene modulation in vitro, ex vivo, and in vivo.
Materials and methods
Human ethics statement
All procedures involving human participants were approved by the Medical Ethics Committee of Inner Mongolia Medical University (No. 202301150). The research was conducted in accordance with the ethical principles of the Declaration of Helsinki. Written informed consent was obtained from all patients and healthy volunteers prior to their participation. Participant privacy and confidentiality were strictly maintained.
Animal ethics statement
All animal experiments were approved by the Animal Ethics Committee of Inner Mongolia Medical University (ethics review number: ZMU21-2301-034). Animal procedures were performed in compliance with the European Parliament Directive 2010/63/EU on the protection of animals used for scientific purposes and the ARRIVE guidelines 2.0 for laboratory animal care and use.
Cell lines and reagents
K562, T2, Jurkat, HH, Hut-78, and 293T cell lines were obtained from ATCC and cultured in RPMI-1640 medium supplemented with 10% FBS. Primary malignant cells from patients with TCL were cultured in RPMI-1640 medium supplemented with 20% FBS. Retrovirus-packaged 293T cells were cultured in DMEM supplemented with 10% FBS. Human PBMCs and CD8 + T cells (HYcells, Shanghai, China) were cultured in RPMI-1640 medium supplemented with 20% FBS and stimulated with 1% activator T Cell TansAct (Miltenyi Biotec, Charlestown, USA) and 600 U/mL IL-2 (PeproTech, Cranbury, USA). All cell lines were authenticated using short tandem repeat (STR) markers and tested for mycobacterial contamination via Mycoplasma agar culture. Nivolumab, 5-azacitidine, PMA, and ionomycin were purchased from Sigma‒Aldrich (St. Louis, USA). Brefeldin A (BFA) and carboxyfluorescein diacetate succinimidyl ester (CFSE) were obtained from Thermo Fisher (Waltham, USA). D-luciferin potassium salt was purchased from Yeasen (Shanghai, China). The lentiviral vector pHR_LV-NS1 (modified from pLVX-Puro by replacing CMV with an SFFV promoter) and the packaging vectors psPAX2 and pMD2.G were obtained from Infinity Biotechnology (Xi’an, China) and Addgene, respectively. Cas9-mRNA and PDCD1 sgRNA were synthesized using GenScript (Nanjing, China). Lipid nanoparticles (LNPs) used for RNA encapsulation were obtained from Infinity Biotechnology (Xi’an, China). Anti-CD8a-APC, anti-HLA-A2-PE, anti-IFN-γ-FITC, anti-PD-1, and anti-IFN-γ-FITC antibodies were obtained from BioLegend (San Diego, USA). Recombinant PD-L1 protein was obtained from ProSci (Poway, USA). The MC2ALKDVEERV (336−344)-HLA-A*02:01 PE-conjugated tetramer was synthesized by MBL Life Sciences (Tokyo, Japan).
Human healthy donors and patient information
In vitro experiments were conducted using T cells from two independent healthy donors to ensure biological replicates and experimental reproducibility. Ex vivo experiments utilized malignant T cells from CTL patient donors, screened into PD-1-positive and negative groups, respectively.
Vector construction, cell transduction, and peptide pulsing
Recombinant cloning vectors bearing the target genes were synthesized by Tsingke Biotech (Beijing, China), and the target genes were cloned, inserted into the lentiviral vector pHR_LV-NS1, and confirmed by sequencing. Packaging was performed by co-culturing the recombinant vector pHR_LV-NS and the packaging plasmids psPAX2 and pMD2.0, with 293T packaging cells. For lentiviral transduction, T cells were activated with 600 U/mL IL-2 and transducted with different recombinant TCR gene-bearing lentiviruses or empty lentiviral vectors. K562 cells were transducted with MC2ALKDVEERV-HLA-A*02:01 or PDCD1 gene-bearing recombinant lentiviruses to construct the target cells. For peptide pulsing, T2 cells were incubated with 10 µM control peptide or MAGE-C2ALKDVEERV peptide synthesized by GenScript (Nanjing, China) for 2 h at 37 °C to generate target T2 cells.
Coculture and ELISA
The indicated effector TCR-T cells and target cells were co-cultured at effector/target ratios of 1:1 or 2:1 for 24, 48, or 72 h. For each effector/target cell co-culture assay, the number of TCR-T cells added was determined based on the proportion of tetramer-positive cells in each sample to ensure that all groups contained an equal number of effector cells. The cells were processed for the flow cytometry detection of cytokines or markers using related staining agents or fluorescently labeled antibodies. For some experiments, the killing or inhibition efficiency of each group was analyzed. For ELISA, engineered TCR-T cells were co-cultured with target cells for 24 h, and the secreted TNF-α in the medium was measured using an ELISA kit following the manufacturer’s instructions.
Flow cytometry
In general, cells were washed and labeled with fluorophore-labeled antibodies or MC2336−344-HLA-A*02:01 tetramers at the manufacturer’s recommended concentrations, incubated at room temperature for 30 min, resuspended in FACS buffer as single-cell suspensions, and assessed using a BD flow cytometer. For the detection of live cells, cells were stained with 1 µM CFSE for 20 min at 37 °C in the dark, co-cultured with TCR-T cells, and subjected to flow cytometry before and after co-culture for the indicated times. Intracellular cytokine staining (ICS) was used to detect IFN-γ production by the T cells. Briefly, cells were stimulated with the indicated target cells for 24 h, after which the protein transport inhibitor, BFA, was added to retain cytokines within the cells. An anti-CD8a-APC antibody was added and the cells were incubated for 30 min at 4 °C. The cells were then fixed, permeabilized, and incubated with an anti-IFN-γ-FITC antibody at room temperature for 15 min before detection by flow cytometry. All flow data were analyzed using the FlowJo software.
Gene knockout
Cas9-mRNA and PD-1-sgRNA were encapsulated in the LNPs using an iFLYTEK microfluidic mixer (Anhui, China). The cells were transfected with LNP-mRNA (Cas9-mRNA/PD-1-sgRNA; 500 ng/well) in a 24-well plate. After transfection, the cells were cultured for 24 h, and gene knockout was assessed by flow cytometry using anti-PD-1 antibodies.
In vivo studies
NCG immunodeficient mice were obtained from GemPharmatech (Nanjing, China). Mice were subcutaneously injected with 1 × 106 luciferase-labelled MC2-A02-K562PD1+ cells (MC2-A02-K562PD1+/Luc). Six days after injection, mice bearing xenograft tumors were treated with control or two types of TCR-T cells (PDL1-MC2-TCR-TPD1− or MC2-TCR-TPD1−) via IV injection at low or high doses of 1x or 5 × 106 cells/100µl of saline, respectively. The mice were then observed for 25 days and scanned for luciferase signals every 5 days using an IVIS Perkin Elmer Lumina LT Imager (Shelton, USA). D-luciferin potassium salt was intraperitoneally (IP) injected intraperitoneally before each scan. The mice were euthanized on the day of the 6th scan, tumors were collected via surgical resection, and the tumor volume was measured.
Euthanasia Method: At the end of the experiment, animals were humanely euthanized to minimize suffering. Euthanasia was performed using carbon dioxide (CO₂) inhalation in a gradually filled chamber, followed by cervical dislocation to ensure death. The method was selected based on the guidelines of the American Veterinary Medical Association (AVMA) for the humane euthanasia of laboratory animals. All procedures were carried out under the supervision of trained personnel to ensure animal welfare.
Statistical information
Statistical differences between groups were analyzed using one-way analysis of variance (ANOVA). The experiments were conducted in triplicate. Data are presented as mean ± SD, with statistical significance defined at P < 0.05.
Results
Design of effector TCR-T and target cell models
To generate TCR-T cell models, we engineered three key cellular modifications through recombinant gene engineering: (1) MC2-targeting TCR expression, (2) exogenous PD-L1 expression, and (3) PDCD1 gene knockout (PD1−). For experimental purposes, we introduced these modifications individually or in combination to construct four distinct TCR-T cell models:
-
1.
MC2-targeting TCR-T cells (MC2-TCR-T cells).
-
2.
MC2-targeting and PD-1-knockout TCR-T cells (MC2-TCR-TPD1− cells).
-
3.
PD-L1-expressing and MC2-targeting TCR-T cells (PDL1-MC2-TCR-T cells).
-
4.
PD-L1-expressing, MC2-targeting, and PD-1-knockout TCR-T cells (PDL1-MC2-TCR-TPD1− cells).
Additionally, we constructed two engineered MC2-targeting TCR-Jurkat models, with or without PD-L1 expression, as parallel TCR-T models:
-
1.
MC2-TCR-expressing Jurkat cells (MC2-TCR-J cells).
-
2.
PD-L1-expressing MC2-TCR-Jurkat cells (PDL1-MC2-TCR-J cells).
A summary of the six TCR-T models and their respective functions is provided in Table 1.
For the target cell models, we utilized the K562 and T2 cell lines to generate three engineered MC2-expressing models, with or without PD-1 expression. Additionally, we selected the wild-type MC2+/PD1+ (dual-positive) TCL line HH, the MC2+/PD1− Hut78 line, and MC2+ primary cultures derived from multiple patients with T-cell lymphoma, which exhibited either PD1+ or PD1− phenotypes, as target cells (Table 2).
Engineering of MC2-targeting TCR-T and target cell lines
The gene sequence of a rearranged TCR clone targeting the HLA-A02:01-restricted MC2 peptide ALKDVEERV336–344was originally identified and sequenced in a melanoma patient with a clinical history of tumor regression following immunotherapy5 (TCR-Vα3 GenBank#: EU427374.1; TCR-Vβ28 GenBank#: EU427375.1). To generate MC2-TCRH-T cells, we first inserted the full sequence of this human TCR clone into a lentiviral vector and integrated it into the genome of human T cells. Previous studies have reported that engineered human TCRs often exhibit instability in binding to the peptide–MHC (pMHC) complex, whereas murine–human hybrid TCRs demonstrate improved TCR pairing, enhanced TCR–CD3 binding, and increased antitumor activity15,16,17. To enhance avidity and functional activation, we replaced the constant (C) regions of the α and β chains in the recombinant MC2-TCR coding sequences with their murine homologs, thereby generating hybrid MC2-TCRHM-T cells (Fig. 1A). The sequences of the TCR subunits (V, C, and J) used for TCR construction are provided in the supplementary data (Tables S1 and S2). All in vitro experiments were conducted with T cells from two different donors, and results were consistent across biological replicates.
K562 is an MC2-negative CML cell line that lacks HLA class I and II expression and has been widely used as an in vitro target for cytotoxic T cells18. K562 cells do not express PD-119. To construct target cells, we transducted the recombinant MC2336−344-HLA-A*02:01 gene alone or together with the PDCD1 gene into K562 cells via lentiviral vectors to establish two MC2-positive K562 cell models that were PD-1 negative (MC2-A02-K562) and positive (MC2-A02-K562PD1+), as validated by flow cytometry (Fig. 1B). T2 is a human lymphoma-derived TAP (transporter associated with antigen processing) -deficient cell line that expresses empty HLA-A*02:01. It does not present peptides derived from cytosolic antigens, but can instead present pulse-loaded peptides. Therefore, we pulse-loaded the MC2336−344 peptide onto T2 cells as another MC2-positive target cell model.
MC2-TCR-T cell optimization and validation
The effectiveness of TCR-T cells largely depends on their affinity for the specific peptide-MHC present on the surface of cancer cells. To determine the binding ability of our constructed TCR-T cells to the MC2-pMHC complex, we custom synthesized an MC2ALKDVEERV-HLA-A*02:01 tetramer conjugated with a PE fluorophore. We first confirmed that the murine sequence-modified TCRHM had superior tetramer-binding and T-cell activation capabilities compared to its original form, TCRH. As shown in Fig. 1C, compared with transduction with TCRH, transduction with TCRHM increased the percentage of cells bound to the tetramer from 7.92 to 23.5% (≈ 3-fold) for CD8 + T cells and 14–41% (≈ 3-fold) for all PBMCs. To compare the cytotoxic effects of MC2-TCRH-T and MC2-TCRHM-T cells on target cells, two types of cells were co-cultured with MC2-A02-K562 and MC2-T2 cells for 48 h. The activation of TCR-T cells was measured using ICS staining for IFN-γ production and ELISA detection of TNF-α secretion. As shown in Fig. 1D and E, both types of cells were activated, but comparisons revealed that MC2-TCRHM-T cells presented greater avidity, activation ability, and cytotoxicity to both types of target cells than MC2-TCRH-T cells. Based on these results, we applied TCRHM only to the TCR-T constructs in subsequent experiments (hereafter referred to as TCR-T cells; i.e., the HM suffix was omitted).

Construction and optimization of MC2-TCR-T cells. A Schematic map showing the modified MC2-TCR design, in which the constant (C) regions of the α and β chains were replaced with mouse homologous sequences. B Engineered MC2-A02-K562 and MC2-A02-K562PD1+ target cells were assessed for exogenous MC2-HLA-A02 (upper) and PD-1 protein expression (lower) using the anti-HLA-A02 and anti-PD-1 antibodies, respectively. The results are shown as flow dot plots; empty vector-transducted K562 and MC2-A02-K562 cells served as controls. C TCR-T cells with human (MC2-TCRH-T) or modified human-murine hybrid (MC2-TCRHM-T) TCRs were examined for specific binding to the MC2-pMHC complex by an MC2336−344-HLA-A*02:01-PE tetramer via flow cytometry. D, E Histogram showing IFN-γ production (D) and TNF-α secretion (E) of TCR-T cells bearing the indicated human or human-murine hybrid TCRs, as measured by intracellular cytokine staining (ICS) or ELISA, when cocultured with MC2-T2 or MC2-A02-K562 target cells, respectively. Control peptide (HPV16-E6)-loaded T2 cells (C-T2) and empty HLA-A*02:01-transducted K562 cells (A02-K562) served as controls. The results are from three individual replicates. One-way ANOVA was used for statistical analysis. Different lowercase letters (a, b, c…) above the bars designate statistically significant differences (P < 0.05).
PD-L1 exogenous expression in MC2-TCR-T cells inhibited their function
Recent studies have shown that PD-1 is a tumor suppressor14, and its inhibition may trigger carcinogenesis8,9,10. Therefore, we next explored whether the targeted activation of PD1 signaling in malignant T cells by exogenous PD-L1 delivered by MC2-TCR-T cells can enhance the tumor-suppressive effect of MC2-TCR-T cells. We transducted the PD-L1 coding gene CD274, along with the engineered coding sequence of the MC2-TCR recombinant protein, into PBMCs or human primary CD8 + T cells using a lentiviral vector to establish a PDL1-MC2-TCR-T cell model. Exogenous PD-L1 expression was assessed and the cells were sorted for PD-L1 expression using flow cytometry (Fig. 2A). We initially assessed the ability of PDL1-MC2-TCR-T cells to specifically bind to the MC2-pMHC complex using the MC2336−344-HLA-A*02:01 tetramer. Since the K562 cell line does not express PD1, we constructed PD1 and MC2 co-expressing MC2-A02-K562PD1+ target cells and examined the cytotoxic effects of PDL1-MC2-TCR-T cells on MC2-T2 or MC2-A02-K562PD1+ cells by co-culturing for 72 h. These findings revealed that increasing PD-L1 expression in MC2-TCR-T cells resulted in a significant decrease in TCR expression and binding ability to the MC2-pMHC complex (Fig. 2B). Similarly, when co-cultured with MC2-A02-K562PD1+ or MC2-T2 cells, PDL1-MC2-TCR-T cells presented significantly lower activation levels and killing capacities, as indicated by IFN-γ and TNF-α production (Fig. 2C–G), than MC2-TCR-T cells, which do not express exogenous PD-L1.

Exogenous expression of PD-L1 in MC2-TCR-T cells inhibited TCR expression and its functions. A Flow dot plot showing PD-L1 on T cells after CD274 gene transduction. B PDL1-MC2-TCR-T cells and MC2-TCR-T cells were compared for their affinity for the MC2336−344-HLA-A*02:01 tetramer, as detected by flow cytometry. C, D Representative flow dot plots and histogram showing IFN-γ production in PDL1-MC2-TCR-T and MC2-TCR-T cells stimulated with MC2-T2 cells via ICS staining. E, F Representative flow dot plots and histogram showing IFN-γ production in PDL1-MC2-TCR-T and MC2-TCR-T cells stimulated with MC2-A02-K562PD1+ cells via ICS staining. G TNF-α secretion by PDL1-MC2-TCR-T and MC2-TCR-T cells after stimulation by coculture with MC2-T2 (left) and MC2-A02-K562PD1+ (right) cells, respectively. Control peptide (HPV16-E6)-loaded T2 cells (C-T2) and empty HLA-A*02:01-transducted K562 cells (A02-K562) served as controls. The results are from three individual replicates. One-way ANOVA was used for statistical analysis. Different lowercase letters (a, b, c…) above the bars designate statistically significant differences (P < 0.05).
Function evaluation of the MC2-TCR-T cells with exogenous PDL1 expression and endogenous PD-1 knockout
To prevent self-inhibition by the exogenously expressed PD-L1 protein, as observed in the co-culture assay (Fig. 2), we knocked out the endogenous PDCD1 (PD-1) gene in these TCR-T cells using the CRISPR/Cas9 system. PD-1 knockout efficiency was assessed by flow cytometry. PD-1 expression has been reported at varying percentages in human CD8+ T lymphocytes, with a mean frequency of 28%20. In our study, PD-1 gene knockout reduced the proportion of PD-1+ cells from 35.5 to 9%, demonstrating high efficiency (Fig. 3A, B). This resulted in the establishment of the PDL1-MC2-TCR-TPD1− model.
Efficient expression of MC2-TCR, its affinity for the MC2-MHC complex, and PD-L1 expression were confirmed through flow cytometry analysis. As shown in Fig. 3, detection of MC2-TCR expression using a specific tetramer revealed that MC2-TCR was expressed in approximately 50% of the cells across the three TCR-T models (Fig. 3C, D left), indicating exogenous PDL1 expression does not affect MC2-TCR binding to the MC2-pMHC complex. As expected, PD-L1 expression was observed only in the PD-L1-MC2-TCR-TPD1− model, demonstrating the efficiency of CD274 gene transduction and confirming the absence of endogenous PD-L1 expression in other TCR-T cells (Fig. 3C). Notably, PD-L1 expression was present in 100% of the MC2-TCR-expressing cells (Fig. 3D right), suggesting that MC2-TCR could serve as a marker for PD-L1 expression in PD-L1-MC2-TCR-TPD1− cells. Therefore, in subsequent assays involving the PDL1-MC2-TCR-TPD1− model, the number of cells was determined based on tetramer-binding detection.
Next we evaluated the function of these TCR-T cells by measuring cytokine release upon activation by PD-1-positive target cell model MC2-A02-K562PD1+ (Table 2). The IFN-γ production and TNF-α secretion were measured by ICS staining/flow cytometry and ELISA, respectively. Notably, the PDL1-MC2-TCR-TPD1− cells exhibited significantly higher levels of IFN-γ release and TNF-α secretion upon activation compared to all other groups, highlighting the marked enhancement of TCR-T cell function with this design (Fig. 3E–G). Additionally, a comparison between MC2-TCR-T and MC2-TCR-TPD1− cells revealed that knocking out endogenous PD-1 in TCR-T cells enhanced their capacity for TNF-α secretion (Fig. 3G).

Exogenous PD-L1 expression and endogenous PD-1 knockout enhanced MC2-TCR-T cytokine secretion. A Representative flow cytometry dot plots illustrating PD-1 expression in two TCR-T models before and after PD-1 knockout, detected using an anti-PD-1 antibody. The PD-1+ population is highlighted within the squared regions. B Histogram quantifying the percentage of PD-1+ cells from panel A. C Representative flow cytometry dot plots showing dual detection of MC2-TCR and PD-L1 expression in different TCR-T models using the MC2336−344-HLA-A*02:01 tetramer and an anti-PD-L1 antibody. D Histograms depicting the percentages of tetramer-bound cells post-transduction (left) and the proportion of PD-L1-expressing cells within the tetramer-bound population (right). E IFN-γ production in various TCR-T cells stimulated with MC2-A02-K562PD1+, measured via intracellular cytokine staining (ICS). F Histogram showing the quantitative analysis of ICS-positive cells. G TNF-α secretion, assessed using an ELISA kit, in various TCR-T cells stimulated with MC2-A02-K562PD1+. All results represent data from three independent replicates. Statistical analysis was performed using one-way ANOVA. Different lowercase letters ( a, b, c…) above the bars designate statistically significant differences (P < 0.05).
PDCD1 gene knockout in PDL1-MC2-TCR-T cells significantly increased cell cytotoxicity to target cells both in vitro and ex vivo
Cytotoxicity, the most critical function of TCR-T cells, was assessed through co-culture experiments with target cells. This study comprehensively compared PD-L1-MC2-TCR-TPD1−cells with other TCR-T subtypes. The cytotoxicity analysis focused on four TCR-T cell models: MC2-TCR-T, MC2-TCR-TPD1−, PD-L1-MC2-TCR-T, and PD-L1-MC2-TCR-TPD1− cells. Additionally, a sample from the MC2-TCR-T model pretreated with the PD-1 blocker nivolumab (MC2-TCR-T + Nivo) was included to compare the effects of PDCD1 gene knockout with those of clinically used PD-1 inhibitors in T cells.
Various types of MC2-positive cells were used as target cells in the co-culture experiments, categorized into PD-1-positive and PD-1-negative groups. As shown in Table 2, the PD-1-positive group included the engineered K562 cell line MC2-A02-K562PD1+, the HH cell line, and primary malignant T cells from TCL patients P1 and P2. The PD-1-negative group included the engineered K562 cell lines MC2-A02-K562 and A02-K562, MC2 peptide-loaded T2 cells line (MC2-T2), the Hut78 cell line, and primary malignant T cells from TCL patients P3 and P4. Since DNA demethylating agents upregulate MC2 expression in cancer cell lines, we pretreated the HH and Hut78 cell lines with 5-azacitidine to enhance MC2 expression6 (Figure S3A). All cell lines and primary malignant T cells from TCL patients were analyzed by PCR using specific primers (Table S3) to identify MC2-positive cells, as well as to distinguish PD-1-positive (HH, P1, and P2) from PD-1-negative (Hut78, P3, and P4) T cells (Figure S3B).
As shown in Fig. 4, co-culture experiments demonstrated that among all TCR-T models, PDL1-MC2-TCR-TPD1− cells exhibited the highest killing efficacy against MC2-A02-K562PD1+ cells, significantly surpassing all other models. The ranking of TCR-T cell killing efficiency, from highest to lowest, was as follows: PDL1-MC2-TCR-TPD1− cells, MC2-TCR-TPD1− cells, MC2-TCR-T + Nivo cells, MC2-TCR-T cells, TPD1− cells, normal CD8+ T cells, PDL1-MC2-TCR-T cells, C-TCR-T cells (HIV-targeting TCR-T cells, used as a non-relevant control), and PDL1-T cells (T cells with exogenously expressed PD-L1) (Fig. 4A and C). We also evaluated different TCR-T/target cell ratios and determined that the optimal condition was a 2:1 TCR-T/target cell ratio at 72 h, with cytotoxicity displaying a dose-dependent response (Fig. 4B). Additionally, we observed a trend of increasing cytotoxicity from 24 to 72 h (Figure S1).
To determine whether PD-L1 delivered by TCR-T cells specifically enhanced the killing of PD-1-positive target cells, we performed co-culture experiments using both PD-1-positive and PD-1-negative target cells. As shown in Fig. 4D, PDL1-MC2-TCR-TPD1− cells demonstrated greater cytotoxic efficacy than MC2-TCR-TPD1− cells in the PD-1-positive group. However, in the PD-1-negative group, PDL1-MC2-TCR-TPD1− cells exhibited similar killing efficacy to MC2-TCR-TPD1− cells (Fig. 4E). Notably, both PDL1-MC2-TCR-TPD1− and MC2-TCR-TPD1− cells outperformed all other TCR-T cell models in cytotoxicity. Treatment of control empty A02-K562 cells did not result in a killing effect, indicating that the engineered TCR-T cells had high specificity.
To confirm the role of PD-L1 in increasing the cytotoxicity of TCR-T cells, we treated PD-1-positive target cells, that is, MC2-A02-K562PD1+ cells, PD-1-positive T lymphoma HH cells, and PD-1-positive T lymphoma primary cells, with 20 µg/mL nivolumab for 24 h and co-cultured them with PDL-1-MC2-TCR-TPD1− cells for 72 h. The results showed that the PD-1 blockade completely reversed the increased killing caused by PD-L1 (Fig. 4F).
To further investigate the suppressive effect on target cells mediated by the PD-L1-PD-1 interaction, we utilized non-cytotoxic Jurkat cells as a parallel TCR-T model. Jurkat cells do not express high levels of perforin or granzyme, and therefore, remain non-cytotoxic even when expressing a TCR. Additionally, unless activated, Jurkat cells do not express PD-121,22, making them a natural PD-1-negative cell model. When Jurkat cells were engineered to co-express PD-L1 and MC2-TCR, they did not undergo intrinsic PD-1 activation-related TCR inhibition, allowing us to specifically analyze the effects of PD-L1-PD-1 signaling in the absence of cytotoxic mechanisms.
We constructed two engineered TCR-Jurkat cell models, PDL1-MC2-TCR-J and MC2-TCR-J, which express MC2-TCR with and without PD-L1, respectively. We performed parallel coculture assays to compare their efficacy against PD-1-expressing MC2-A02-K562PD1+ cells. Similar to PDL1-MC2-TCR-TPD1− cells, PDL1-MC2-TCR-J cells had greater cytotoxic functions and killing efficacy than MC2-TCR-Jurkat cells when co-cultured with MC2-A02-K562PD1+ cells. Assessment of viable cells via CFSE staining/flow cytometry at 24 and 48 h revealed an average increase in the killing percentage of 15.5% at 24 h and 25% at 48 h (Fig. 4G and H).

PDL1-MC2-TCR-T1 cells exhibited enhanced tumor-suppressing efficacy in vitro and ex vivo. A Representative flow cytometry plots showing live MC2-A02-K562PD1+ cells cocultured with MC2-TCR-TPD1−, PDL1-MC2-TCR-T, and PDL1-MC2-TCR-TPD1− cells and stained with CFSE. B Comparison of the efficacy of coincubation with PDL1-MC2-TCR-TPD1− and MC2-TCR-TPD1− cells at effector/target ratios of 1:1 and 2:1 for 72 h. C Histogram showing the percentages of MC2-A02-K562PD1+ cells killed when cocultured with different TCR-T cells for 72 h. T: normal CD8 + T cells; PDL1-T: normal CD8 + T cells engineered with exogenously expressed PD-L1; C-TCR-T: engineered HIV-targeting TCR-T cells as nonrelevant control TCR-T cells; TPD1− cells: normal CD8 + T cells with PDCD1 gene knockout; and MC2-TCR-T + Nivo: MC2-TCR-T cells pretreated with 20 µg/mL nivolumab for 72 h. D, E Histograms showing the inhibitory effects of C-TCR-T, MC2-TCR-T, MC2-TCR-TPD1−, PDL1-MC2-TCR-T and PDL1-MC2-TCR-TPD1− cells on engineered or natural cell lines and TCL primary malignant cell cultures that were MC2 and PD1 dual positive (D) or MC2 positive but PD1 negative (E). Empty HLA-A*02:01-transducted K562 cells were used as a control. F Comparison of the killing efficacy of MC2-TCR-TPD1− and PDL1-MC2-TCR-TPD1− cells on the indicated PD1-positive lines and PDL1-MC2-TCR-TPD1− cells on the same target lines and cells pretreated with 20 µg/mL nivolumab. G Representative flow cytometry dot plots showing the inhibitory effects of MC2-TCR-J and PDL1-MC2-TCR-J cells on MC2-A02-K562PD1+ cells at the 0, 24 and 48 h time points. H Histogram showing the quantitative and statistical analysis of the inhibitory effects in G. All the inhibition or killing percentages were determined by CFSE staining. All results represent data from three independent replicates. Statistical analysis was performed using one-way ANOVA. Different lowercase letters (a, b, c…) above the bars designate statistically significant differences (P < 0.05).
Antitumor effects of PDL1-MC2-TCR-TPD1− cells in vivo
By manipulating PD-1 and/or PD-L1 expression in TCR-T cells, we established four MC2-TCR-T cell models: MC2-TCR-T cells, MC2-TCR-TPD1− cells, PDL1-MC2-TCR-T cells, and PDL1-MC2-TCR-TPD1− cells. PDL1-MC2-TCR-TPD1− and MC2-TCR-TPD1− cells showed greater efficacy than the other cell models in the in vitro and ex vivo assays. Among them, PDL1-MC2-TCR-TPD1− cells showed greater efficacy than MC2-TCR-TPD1− cells, a finding that is supported by the mechanisms of PD-1 activation in malignant cells. To further test whether PDL1-MC2-TCR-TPD1− cells had a greater antitumor effect than MC2-TCR-TPD1− cells in vivo, we used immunodeficient NCG mice to establish an MC2-A02-K562PD1+ xenograft model to compare the effects of these two types of engineered TCR-T cells on tumors. The NCG (NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt) mouse is a variant of the NSG mouse, in which the Prkdc (protein kinase, DNA activated, catalytic polypeptide) and Il2rg (common gamma chain receptor) genes are completely knocked out and are capable of hosting xenograft cells. We subcutaneously injected mice with MC2-A02-K562PD1+ cells genetically labeled with luciferase (MC2-A02-K562PD1+/Luc) to establish a tumor-bearing NCG mouse model. We injected MC2-TCR-TPD1− and PDL1-MC2-TCR-TPD1− cells at both low (1 × 106) and high (5 × 106) doses, together with a high dose of control TCR-T cells, into tumor-bearing mice (Fig. 5A). The five groups of mice were scanned for luciferase signals every five days, and tumor volume was measured at the end of the observation period. As shown in Fig. 5B, C, D, and E, both the tumor luciferase signals and the tumor tissue volume revealed that, compared with MC2-TCR-TPD1−cells, PDL1-MC2-TCR-TPD1− cells were more effective at suppressing tumor growth. In addition, the effects of high doses of MC2-TCR-TPD1− and PDL1-MC2-TCR-TPD1− cells were greater than those of low doses. In general, the in vivo results were consistent with the in vitro and ex vivo results, that is, PDL1-MC2-TCR-TPD1− cells had a significantly greater killing effect on target cancer cells than PD-L1-lacking variant MC2-TCR-TPD1− cells. We also assessed the safety of TCR-T cell therapy, and none of the animals in the treatment groups exhibited signs of systemic off-target toxicity.

PDL1-MC2-TCR-TPD1− cells had greater tumor-suppressing efficacy than MC2-TCR-TPD1− cells did in vivo. A Schematic diagram of the animal experimental design. Five groups of mice were inoculated with MC2-A02-K562PD1+ cells and treated with two doses (1 × 106 or 5 × 106) of MC2-TCR-TPD1− or PDL1-MC2-TCR-TPD1− cells, as well as one dose of control TCR-T cells (5 × 106). The tumors were scanned for luminescence signals at 0 d, 5 d, 10 d, 15 d, 20 d, and 25 d. The mice were sacrificed at the end of the observation period (25 d), and tumors were surgically dissected. IVISI BLI (in vivo imaging system, bioluminescence imaging). B Pictures of the five groups of mice scanned for luminescence signals at the end of the observation period (note: In some animals, strong signals were saturated and could only be measured by the scanner). C Tumor growth curves for the five groups of mice, plotted at 0 d, 5 d, 10 d, 15 d, 20 d and 25 d, using quantitative luminescence signals. D Images of tumors surgically dissected from five groups of mice treated with the indicated TCR-T cells. E The histogram shows the comparison of the average tumor volumes (cm3) for the five groups of mice. The data are presented as means ± SDs. Statistical analysis was performed using one-way ANOVA. Different lowercase letters ( a, b, c…) above the bars designate statistically significant differences (P < 0.05).
Discussion
The use of TCR-T cell therapy is increasing, but it is still in the early stages of clinical application. In addition, ICI therapy is promising but not fully satisfactory. These fields of basic and preclinical research have garnered substantial attention23,24,25,26,27. Using genetic approaches, we successfully integrated PD-L1 delivery into malignant cells and PD-1 knockout TCR-T cells into an engineered MC2-targeting TCR-T cell model, and achieved enhanced tumor suppression efficacy in vitro, ex vivo and in vivo.
In the cancer microenvironment, the effects of ICIs can be more complicated than those of simply activating T cells because of their influence on immune cells, other normal cells, and malignant cells. Recent evidence indicated that PD-1 is a tumor suppressor in T-cell lymphoma and other types of cancer cells8,9,10,11,12,13,14. The significance of PD-1 as a tumor suppressor leads to two insights into cancer treatment: (1) Therapeutic activation of PD-1 signaling in malignant cells may induce tumor-suppressing effects, and (2) Systemic PD-1 inhibition, such as the current clinical use of ICIs, may may have consequences that counteract the tumor-suppressing effect or, in some conditions, increase cancer growth by inhibiting PD-1 in cancer cells. In clinical practice, a small fraction of patients experience hyperprogressive disease (HPD), which is characterized by the acceleration of cancer growth, during PD-1 inhibition therapy28. Therefore, targeted PD-1 activation or inhibition in specific cell types or populations could be treatment strategies worthy of exploration; however, no related in vitro or in vivo studies have been reported. For the first time, our study revealed that the combined manipulation of PD-1 signaling significantly enhanced cancer-specific TCR-T cell cytotoxicity.
We explored targeted PD-1 activation in malignant cells in the context of cancer antigen TCR-T cell therapy. The activation of PD-1 signaling may depend on the existence and binding of PD-L1. We utilized PD-1-negative and non-cytotoxic Jurkat cells21,22 as a parallel TCR-T model to evaluate the effect of exogenous PD-L1 on target cell inhibition. The engineered MC2-TCR-J and PDL1-MC2-TCR-J subtypes were used as effector cells in co-culture with K562 target cells, both of which exhibited inhibitory effects on target cell growth. However, the inhibitory effect of PDL1-MC2-TCR-J cells was significantly greater than that of MC2-TCR-J cells (Fig. 4H), indicating the role of the PD-1/PD-L1 axis in suppressing target cell proliferation. Since Jurkat cells lack direct cytotoxic activity, we speculate that the inhibition of target cells by MC2-TCR-J may result from indirect cytotoxic mechanisms, such as cytokine secretion.
In TCL and other malignancies, PD-L1 levels are consistently low, whereas PD-1 levels are high29,30. A lack of or inefficient PD-L1 in the proximity of cancer cells and subsequent intrinsic PD-1 inactivation could be contributing factors to cancer development or progression. Thus, the use of PD-L1 could be a strategy to assist in treatment. However, similar to systemic PD-1 inhibition, which causes off-target effects, systemic administration of PD-L1 triggers the inhibition of normal T cells or TCR-T cells in the context of TCR-T cell therapy. In addition, infusion or injection of recombinant PD-L1 protein may cause intolerable general toxicity. Therefore, seeking a method for limited and targeted PD-L1 delivery specifically to cancer cells is a key step in increasing treatment efficacy.
Methods involving the co-expression of a protein with a specific recombinant targeting structure, such as the co-expression of a CD8a with TCR in TCR-T cells (next-generation of afami-cel), have been studied in adaptive immunology31. In this study, we successfully adopted MC2-specific TCR-T cells as the delivery vehicle via a co-expression system of MC2-TCRs and PD-L1 for this purpose. The results of this TCR-T model revealed, for the first time, that targeting PD-1 activation in cancer cells with exogenous PD-L1 enhanced the tumor-suppressing efficacy of TCR-T cells in vitro and ex vivo. The advantage of using MC2-TCR-T cells as vehicles is that TCR-T cells exhibit cancer-specific cytotoxicity and are a powerful therapy. Our results demonstrate that cancer antigen-specific TCR-T cells fit the purpose of cancer cell-specific PD-L1 delivery (Figs. 3 and 4). Although our study focused on MC2-specific TCR-T cells, the model of exogenous PD-L1 expression in cancer-specific TCR-T cells has general significance for applications to other cancer antigen-specific TCR-T cells.
In engineered TCR construction, we confirmed the importance of using murine homology, which has been clinically applied in afami-cel32. Please note that the stronger activation signals and increased cytokine production (IFN-γ/TNF-α) observed in MC2-T2 co-cultures compared to MC2-A02-K562 cells (Figs. 1 and 2) can be attributed to differences in antigen presentation efficiency. T2 cells, due to their TAP deficiency, rely on exogenous peptide loading, which results in a higher density of peptide–MHC complexes on the cell surface, leading to stronger TCR engagement33. In contrast, MC2-A02-K562 cells rely on viral vector-mediated expression of the MC2 peptide–HLA-A02:01 complex, which may result in variable antigen processing and presentation efficiency34. Additionally, differences in co-stimulatory molecule expression and peptide processing pathways between the two cell types could further contribute to the observed differences in TCR-T cell activation.
We also applied targeted PD-1 inhibition in T cells to TCR-T cells. ICIs have been used in various cancer therapies including adoptive immunotherapy35. The benefits of PD-1 inhibition are mainly attributed to the activation and expansion of normal immune cells, such as CD8 + and CD4 + T cells36,37. As stated above, the current methods of ICI administration are not tissue-specific and may influence many cell types. Targeting PD-1 inhibition specifically for immune cells should be considered to avoid the promotion of malignancy caused by PD-1 inhibition29. The mechanism of HPD remains unclear, but it may be related to the impact of ICIs on cancer cells.
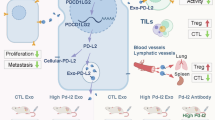
Additionally, the influence of PD-1 inhibition on various normal cell types other than immune cells may involve the risk of triggering carcinogenesis due to the overactivation of TCR/PI3K/mTOR signaling8,9,10,11,12,13,14. Clinically, ICI therapies are associated with the occurrence of secondary T-cell lymphomas or other cancers in patients with non-T-cell primary malignancies38,39,40. PD-1 silencing using gene editing technology has been shown to effectively increase the potency of adoptive cytotoxic T cells41,42. In this study, we used the CRISPR/Cas9 gene editing method to knockout PDCD1 in engineered TCR-T cells to generate PD-1-null MC2-TCR-TPD1− and PDL1-MC2-TCR-TPD1− cells (Fig. 3). The application of gene editing technology has two advantages over ICIs: first, it completely avoids the influence of off-target PD-1 inactivation on other cell types, especially malignant cells; second, it can be limited to a specific engineered T cell subpopulation to avoid the overactivation of the whole T cell population, which may cause complications such as a “cytokine storm”. The proposed mechanisms of this engineered PDL1-MC2-TCR-TPD1− cells are summarized in Fig. 6.

Interactions between engineered T cells and target cancer cells. The PDL1-MC2-TCR-TPD1− cells are engineered to co-express a MAGE-C2-specific recombinant TCR and PD-L1 proteins in normal human CD8 + T cells, with the PD-1-encoding PDCD1 gene knocked out. The exogenously expressed PD-L1 activates the PD-1 receptor on the surface of cancer cells while maintaining PD-1 suppression in the T cells. Activation of PD-1 inhibits PI3K-mediated carcinogenic signaling and subsequent cell proliferation. Binding of MC2-TCR with MC2 pMHC induces T cell activation and directs cytotoxicity toward target cells through the secretion of cytokines TNF-α and IFN-γ. Silencing of PD-1 in T cells enhances T cell activation and expansion. Red lines denote inhibitory effects, and “X” indicates knockdown of PDCD1 genes.
The important benefit of PD-1 inhibition in TCR-T cell treatment is that it helps overcome engineered T cell exhaustion, which is usually a major hurdle in TCR-T cell treatment and clinically manifests as resistance to treatment43,44. In addition, PD-1 inhibition is necessary to maintain the function of engineered PD-L1-secreting TCR-T cells. When we engineered a co-expression system of bioactive MC2-specific TCR and PD-L1 proteins to establish a PDL1-MC2-TCR-T model, we found that PD-L1 expression led to strong self-inhibition that almost completely eliminated the function of TCR-T cells. A comparison of PDL1-MC2-TCR-T cells and MC2-TCR-T cells co-cultured with target MC2-A02-K562 cells revealed that extra PD-L1 expression did not increase, but rather hindered the cytotoxicity of TCR-T cells to the target cells (Fig. 2). Since engineered PD-L1 expression by TCR-T cells inevitably leads to PD-1 activation in TCR-T cells, we hypothesized that the inhibition of PD-1 in T cells would restore the functions of TCR-T cells. Indeed, the PD-1 knockout variant, PDL1-MC2-TCR-TPD1− cells, showed enhanced cytotoxicity (Fig. 3E–G). Therefore, PD-1 inhibition in cells is a critical component of PD-L1 expression-related design to maintain the functions of PDL1-MC2-TCR-T cells.
The results generated from PDL1-MC2-TCR-TPD1−cells revealed that the combination of three functions, cancer antigen-specific TCR-T cells, PD-1 inhibition in T cells, and PD-1 activation in malignant cells, was more effective than any single or two combined functions of TCR-T cells in vitro and ex vivo (Figs. 4 and 5). Pretreatment of target cells with the PD-1 inhibitor nivolumab completely blocked the increased cytotoxicity of PDL1-MC2-TCR-TPD1− on PD-1 + cells, indicating that the extra killing capability of PDL1-MC2-TCR-TPD1− cells is dependent on the PD-1-PD-L1 interaction on effector-target cells (Fig. 4F).
Our in vivo assays revealed that PDL1-MC2-TCR-TPD1− cells had significantly greater tumor-suppressing efficacy than non-PD-L1 variant MC2-TCR-TPD1− cells in a dose-dependent manner in mice bearing MC2 and PD-1 dual-positive K562 xenograft tumors (Fig. 5). Although our in vivo experiments were performed in immunodeficient mice, PDCD1 gene knockout mimics the actions of ICIs on effector TCR-T cells in the immunocompetent human body. Therefore, targeted PD-1 activation in malignant T cells and PD-1 inhibition in normal T cells in our in vivo assay, to a certain extent, reflect the actual condition of the human body and have clinical significance. The data showed for the first time that PD-1 activation in cancer cells via exogenous PD-L1 delivered by TCR-T cells synergistically increased the tumor-suppressing efficacy of TCR-T cells in vivo. The safety and specificity were assessed using in vivo assays. The cross-reaction of the MAGE member MAGE-A3 with brain tissue, which can lead to severe life-threatening consequences, has been reported45. We did not find any safety issues, such as exaggerated poor conditions or quick death of the animals, which may be related to major off-target toxicity to normal tissues. All treated tumor-bearing mice survived until the end of the observation period (25 days). The in vivo results, along with the in vitro and ex vivo findings, provide evidence that PDL1-MC2-TCR-TPD1− cells acquired enhanced tumor-suppressing capabilities and warrant further investigation for their potential clinical use.
The mechanisms by which PD-L1 enhances TCR T-cell killing efficacy in cancer cells are not completely clear. Treatment of PD-1 + T-lymphoma cells with recombinant PD-L1 protein in an in vitro experiment inhibited proliferation, but did not result in a direct killing effect (Figure S2). The mechanisms of PD-1 activation/inhibition in benign and malignant cells can be complicated and tissue/cancer stage specific. Current data concerning PD-L1 expression in tumor tissues are controversial because reports of both high PD-L1 expression and low PD-L1 expression in tumor tissues exist46,47,48,49. Despite being a factor that activates tumor suppression, PD-L1 expression is frequently reported to be upregulated in cancer cells upon immune cell infiltration to activate the PD-1 receptor on T cells. We do not know if and how cancer cells escape the suppression caused by self-activation of the PD-1/PD-L1 axis. The underlying mechanisms and clinical significance of the PD-1/PD-L1-mediated effects on the TCR-T cell model in our study need to be further investigated. Furthermore, despite the absence of major off-target toxicity, the cross-reaction of these TCR-T cells with normal tissues still needs to be ruled out in future studies.
In conclusion, our study establishes that cancer-specific TCR-T cells can successfully deliver ligands to cancer antigen-expressing malignant cells and induce target receptor activation for the purpose of cancer treatment. Our new strategy exploits TCR-T cell cancer-specific cytotoxicity, PD-1 inhibition in T cells, and PD-1 activation in cancer cells, thereby increasing the efficacy of cancer-specific TCR-T cell therapy for inhibiting cancer. Therefore, this work has translational and clinical importance for designing new or optimized TCR-T cell and ICI-based cancer therapies.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding authors upon reasonable request.
References
Keam, S. J. Afamitresgene autoleucel: first approval. Mol. Diagn. Ther. 28, 861–866. https://doi.org/10.1007/s40291-024-00749-3 (2024).
Sanderson, J. P. et al. Preclinical evaluation of an affinity-enhanced MAGE-A4-specific T-cell receptor for adoptive T-cell therapy. Oncoimmunology 9, 1682381. https://doi.org/10.1080/2162402X.2019.1682381 (2020).
Weon, J. L. & Potts, P. R. The MAGE protein family and cancer. Curr. Opin. Cell. Biol. 37, 1–8. https://doi.org/10.1016/j.ceb.2015.08.002 (2015).
Schooten, E., Di Maggio, A., van Bergen En Henegouwen, P. M. P. & Kijanka, M. M. MAGE-A antigens as targets for cancer immunotherapy. Cancer Treat. Rev. 67, 54–62. https://doi.org/10.1016/j.ctrv.2018.04.009 (2018).
Ma, W. et al. Two new tumor-specific antigenic peptides encoded by gene MAGE-C2 and presented to cytolytic T lymphocytes by HLA-A2. Int. J. Cancer. 109, 698–702. https://doi.org/10.1002/ijc.20038 (2004).
Kunert, A. et al. MAGE-C2-specific TCRs combined with epigenetic drug-enhanced antigenicity yield robust and tumor-selective T cell responses. J. Immunol. 197, 2541–2552. https://doi.org/10.4049/jimmunol.1502024 (2016).
Lurquin, C. et al. Contrasting frequencies of antitumor and anti-vaccine T cells in metastases of a melanoma patient vaccinated with a MAGE tumor antigen. J. Exp. Med. 201, 249–257. https://doi.org/10.1084/jem.20041378 (2005).
Wartewig, T. & Ruland, J. PD-1 tumor suppressor signaling in T cell lymphomas. Trends Immunol. 40, 403–414. https://doi.org/10.1016/j.it.2019.03.005 (2019).
Wang, X. et al. Tumor cell-intrinsic PD-1 receptor is a tumor suppressor and mediates resistance to PD-1 blockade therapy. Proc. Natl. Acad. Sci. U S A. 117, 6640–6650. https://doi.org/10.1073/pnas.1921445117 (2020).
Wartewig, T. et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 552, 121–125. https://doi.org/10.1038/nature24649 (2017).
Du, S. et al. Blockade of tumor-expressed PD-1 promotes lung cancer growth. Oncoimmunology 7, e1408747. https://doi.org/10.1080/2162402X.2017.1408747 (2018).
Zha, H. et al. Non-canonical PD-1 signaling in cancer and its potential implications in clinic. J. Immunother Cancer. https://doi.org/10.1136/jitc-2020-001230 (2021).
Ludin, A. & Zon, L. I. Cancer immunotherapy: the dark side of PD-1 receptor Inhibition. Nature 552, 41–42. https://doi.org/10.1038/nature24759 (2017).
Lin, X. et al. Regulatory mechanisms of PD-1/PD-L1 in cancers. Mol. Cancer. 23, 108. https://doi.org/10.1186/s12943-024-02023-w (2024).
Cohen, C. J., Zhao, Y., Zheng, Z., Rosenberg, S. A. & Morgan, R. A. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 66, 8878–8886. https://doi.org/10.1158/0008-5472.CAN-06-1450 (2006).
Yu, Z. et al. Poor immunogenicity of a self/tumor antigen derives from peptide-MHC-I instability and is independent of tolerance. J. Clin. Invest. 114, 551–559. https://doi.org/10.1172/JCI21695 (2004).
Yao, Z. et al. Focusing on CD8(+) T-cell phenotypes: improving solid tumor therapy. J. Exp. Clin. Cancer Res. 43, 266. https://doi.org/10.1186/s13046-024-03195-5 (2024).
Britten, C. M. et al. The use of HLA-A*0201-transfected K562 as standard antigen-presenting cells for CD8(+) T lymphocytes in IFN-gamma ELISPOT assays. J. Immunol. Methods. 259, 95–110. https://doi.org/10.1016/s0022-1759(01)00499-9 (2002).
Eichholz, K. et al. Anti-PD-1 chimeric antigen receptor T cells efficiently target SIV-infected CD4 + T cells in germinal centers. J. Clin. Invest. https://doi.org/10.1172/JCI169309 (2024).
Saito, H. et al. PD-1 expression on Circulating CD8(+) T-Cells as a prognostic marker for patients with gastric cancer. Anticancer Res. 39, 443–448. https://doi.org/10.21873/anticanres.13132 (2019).
Li, Z. et al. The immunostimulative effect and mechanisms of a novel mouse Anti-Human PD-1 monoclonal antibody on Jurkat lymphocytic cells cocultured with hepatoma cells. Onco Targets Ther. 13, 12225–12241. https://doi.org/10.2147/OTT.S281397 (2020).
Ding, H., Wu, X. & Gao, W. PD-L1 is expressed by human renal tubular epithelial cells and suppresses T cell cytokine synthesis. Clin. Immunol. 115, 184–191. https://doi.org/10.1016/j.clim.2005.01.005 (2005).
Baulu, E., Gardet, C., Chuvin, N. & Depil, S. TCR-engineered T cell therapy in solid tumors: state of the art and perspectives. Sci. Adv. 9, eadf3700. https://doi.org/10.1126/sciadv.adf3700 (2023).
Zhang, J. & Wang, L. The emerging world of TCR-T cell trials against cancer: A systematic review. Technol. Cancer Res. Treat. 18, 1533033819831068. https://doi.org/10.1177/1533033819831068 (2019).
Ikeda, H. T-cell adoptive immunotherapy using tumor-infiltrating T cells and genetically engineered TCR-T cells. Int. Immunol. 28, 349–353. https://doi.org/10.1093/intimm/dxw022 (2016).
Shafer, P., Kelly, L. M. & Hoyos, V. Cancer therapy with TCR-engineered T cells: current strategies, challenges, and prospects. Front. Immunol. 13, 835762. https://doi.org/10.3389/fimmu.2022.835762 (2022).
Zhao, Q. et al. Engineered TCR-T cell immunotherapy in anticancer precision medicine: pros and cons. Front. Immunol. 12, 658753. https://doi.org/10.3389/fimmu.2021.658753 (2021).
Toki, M. I., Syrigos, N. & Syrigos, K. Hyperprogressive disease: A distinct pattern of progression to immune checkpoint inhibitors. Int. J. Cancer. 149, 277–286. https://doi.org/10.1002/ijc.33429 (2021).
Saulite, I. et al. Blockade of programmed cell death protein 1 (PD-1) in Sezary syndrome reduces Th2 phenotype of non-tumoral T lymphocytes but may enhance tumor proliferation. Oncoimmunology 9, 1738797. https://doi.org/10.1080/2162402X.2020.1738797 (2020).
Gatalica, Z. et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomarkers Prev. 23, 2965–2970. https://doi.org/10.1158/1055-9965.EPI-14-0654 (2014).
Anderson, V. E. et al. Enhancing efficacy of TCR-engineered CD4 + T cells via coexpression of CD8alpha. J. Immunother. 46, 132–144. https://doi.org/10.1097/CJI.0000000000000456 (2023).
Klebanoff, C. A., Chandran, S. S., Baker, B. M., Quezada, S. A. & Ribas A. T cell receptor therapeutics: immunological targeting of the intracellular cancer proteome. Nat. Rev. Drug Discov. 22, 996–1017. https://doi.org/10.1038/s41573-023-00809-z (2023).
Ito, K. et al. Preparation of antibodies to guinea pig IgE and its use for enzyme-linked immunosorbent assay of IgE antibodies. Int. Arch. Allergy Appl. Immunol. 77, 438–444. https://doi.org/10.1159/000233823 (1985).
Groth, C. G. et al. Use of rabbit anti-human thymocyte Globulin in cadaver kidney transplantation. Preliminary experiences. Scand. J. Urol. Nephrol. Suppl. 64, 200–204 (1981).
Deng, R. et al. PD-1 Blockade potentially enhances adoptive cytotoxic T cell potency in a human acute myeloid leukaemia animal model. Hematology 23, 740–746. https://doi.org/10.1080/10245332.2018.1486357 (2018).
Zuazo, M. et al. Systemic CD4 immunity as a key contributor to PD-L1/PD-1 blockade immunotherapy efficacy. Front. Immunol. 11, 586907. https://doi.org/10.3389/fimmu.2020.586907 (2020).
Carter, L. et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur. J. Immunol. 32, 634–643. https://doi.org/10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9 (2002).
Anand, K. et al. T-cell lymphoma secondary to checkpoint inhibitor therapy. J. Immunother Cancer. https://doi.org/10.1136/jitc-2019-000104 (2020).
Marks, J. A., Parker, D. C., Garrot, L. C. & Lechowicz, M. J. Nivolumab-associated cutaneous T-cell lymphoma. JAAD Case Rep. 9, 39–41. https://doi.org/10.1016/j.jdcr.2020.12.033 (2021).
Miao, K., Yu, S., Ni, J., Zhang, X. & Zhang, L. Second primary tumor after immune checkpoint inhibitor therapy: a case report. Thorac. Cancer. 13, 1076–1078. https://doi.org/10.1111/1759-7714.14327 (2022).
Menger, L. et al. TALEN-mediated inactivation of PD-1 in tumor-reactive lymphocytes promotes intratumoral T-cell persistence and rejection of established tumors. Cancer Res. 76, 2087–2093. https://doi.org/10.1158/0008-5472.CAN-15-3352 (2016).
Zhao, Z. et al. CRISPR knock out of programmed cell death protein 1 enhances anti-tumor activity of cytotoxic T lymphocytes. Oncotarget 9, 5208–5215. https://doi.org/10.18632/oncotarget.23730 (2018).
Kim, S. P. et al. Adoptive cellular therapy with autologous tumor-infiltrating lymphocytes and T-cell receptor-engineered T cells targeting common p53 neoantigens in human solid tumors. Cancer Immunol. Res. 10, 932–946. https://doi.org/10.1158/2326-6066.CIR-22-0040 (2022).
Chapuis, A. G. et al. T-cell therapy using interleukin-21-primed cytotoxic T-cell lymphocytes combined with cytotoxic T-cell lymphocyte antigen-4 blockade results in long-term cell persistence and durable tumor regression. J. Clin. Oncol. 34, 3787–3795. https://doi.org/10.1200/JCO.2015.65.5142 (2016).
Morgan, R. A. et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 36, 133–151. https://doi.org/10.1097/CJI.0b013e3182829903 (2013).
Xu, D. et al. PD-L1 is a tumor suppressor in aggressive endometrial cancer cells and its expression is regulated by miR-216a and LncRNA MEG3. Front. Cell. Dev. Biol. 8, 598205. https://doi.org/10.3389/fcell.2020.598205 (2020).
Patel, S. P. & Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 14, 847–856. https://doi.org/10.1158/1535-7163.MCT-14-0983 (2015).
Ai, L., Xu, A. & Xu, J. Roles of PD-1/PD-L1 pathway: signaling, cancer, and beyond. Adv. Exp. Med. Biol. 1248, 33–59. https://doi.org/10.1007/978-981-15-3266-5_3 (2020).
Dong, P., Xiong, Y., Yue, J., Hanley, S. J. B. & Watari, H. Tumor-intrinsic PD-L1 signaling in cancer initiation, development and treatment: beyond immune evasion. Front. Oncol. 8, 386. https://doi.org/10.3389/fonc.2018.00386 (2018).
Acknowledgements
This work was supported by the Inner Mongolia Science & Technology Project Plan (2019GG037), the Natural Science Research Funds of Inner Mongolia (2022MS08009), and the Inner Mongolia High Education Institutes Young Science and Technology Talent Project (NYJT22013).
Author information
Authors and Affiliations
Contributions
FXZ contributed to the design and execution of all experiments and analyzed the data. XZ and YT contributed to the analysis of the data and execution of the in vitro, ex vivo and in vivo assays. HXY, HTP, RWA and WGYMR performed cell culture and flow cytometry. CY and WL performed the ELISA and PCR experiments under the guidance of FW. JQW conceived, designed, analyzed, and supervised the study. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Consent for publication
All authors agreed on the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, F., Zhang, X., Tang, Y. et al. Engineered PD-L1 co-expression in PD-1 knockout and MAGE-C2-targeting TCR-T cells augments the cytotoxic efficacy toward target cancer cells. Sci Rep 15, 11894 (2025). https://doi.org/10.1038/s41598-025-92209-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-92209-z
