
Abstract
Fibrotic diseases are involved in 45% of deaths in the United States. In particular, fibrosis of the kidney and lung are major public health concerns due to their high prevalence and lack of existing treatment options. Here, we harness the pathophysiological features of fibrotic diseases, namely leaky vasculature and aberrant extracellular matrix (ECM) protein deposition (i.e. collagen), to target an anti-fibrotic biologic and a small molecule drug to disease sites of fibrosis, thus improving the therapeutic potential of both the biologic and small molecule in mouse models of both lung and kidney fibrosis. First, we identify and validate two collagen-targeting drug delivery systems that preferentially accumulate in fibrotic organs: von Willebrand Factor’s A3 domain (VWF-A3) and decorin-derived collagen-binding peptide-conjugated micelles (CBP-micelles). We then engineer and recombinantly express novel candidate biologic therapies based on the anti-inflammatory cytokine IL-10: A3-IL-10 and A3-Serum Albumin-IL-10 (A3-SA-IL-10). Simultaneously, we stably encapsulate the potential anti-fibrotic water-insoluble drug, rapamycin, in CBP-micelles. We show that these novel formulations of therapeutics bind to collagen in vitro and that their efficacy in mouse models of lung and kidney fibrosis is improved, compared to free, untargeted drugs. Our results demonstrate that collagen-targeted anti-fibrotic drugs may be next generation therapies of high clinical potential.
Similar content being viewed by others


Introduction
Fibrosing diseases—including pulmonary fibrosis, congestive heart failure, liver cirrhosis, and end-stage kidney disease—are involved in 45% of deaths in the United States1,2. Of these diseases, kidney and lung fibrosis, caused by the excessive production and deposition of extracellular matrix (ECM) proteins in the kidney and lung interstitium, respectively, are among the most challenging to treat and eventually lead to end-stage kidney disease (ESKD) and lung failure. The two FDA approved treatments for fibrosis, pirfenidone and nintedanib2,3, slow, but fail to reverse, the progression of fibrosis via poorly understood mechanisms of action4,5. Currently, the only intervention capable of restoring kidney and lung function is organ transplantation6. However, the number of patients awaiting a transplant far exceeds the available supply of donor organs7. Hence, there remains an unmet need for new therapies that can better target and treat the fibrotic organs.
Phenotypically, fibrosis occurs when tissue injury activates macrophages to secrete growth factors, enzymes, and cytokines that stimulate fibroblast migration and proliferation8. Recruited fibroblasts, in turn, produce large amounts of extracellular matrix (ECM) proteins including collagens type I and III and fibronectin, resulting in tissue that is rich in collagen and characterized by leaky vasculature9,10,11. Vascular leakiness also contributes to the pathogenesis of lung fibrosis11,12 and kidney fibrosis13,14.
Kidney and lung fibrosis progression can be mirrored in mice through several injury models, including the unilateral ureteral obstruction (UUO) model, in which one of the kidneys is blocked via ligation of the ureter15,16,17, and the bleomycin-induced pulmonary fibrosis model, in which the interstitium of the lungs is compromised by the agent bleomycin, respectively18. Collagen 1 is upregulated in UUO-induced fibrotic kidneys19,20, and is also upregulated in bleomycin-induced lung fibrosis21.
A number of protein and small molecule-based therapies are currently being explored for the treatment of fibrosing diseases. Interleukin-10 (IL-10) is an immunosuppressive cytokine that plays a critical role in preventing inflammatory and autoimmune diseases22. When IL-10 binds to the heterodimeric IL-10 receptor, the JAK/STAT signaling pathway is activated, suppressing the production of pro-inflammatory mediators including TNF-α and IL-1β22. IL-10 treatment also reportedly reduces antigen presentation and phagocytosis by antigen presenting cells and inhibits pathogenic T helper cell proliferation, while expanding immunosuppressive regulatory T cells23. Due to its established role in suppressing macrophage and myofibroblast-mediated inflammation, IL-10 is a promising candidate for cytokine therapy in fibrosis24. IL-10 gene and recombinant protein delivery has been reported to suppress bleomycin-induced pulmonary fibrosis by inhibiting the production of TGF-β1 by alveolar macrophages, lung fibroblasts, and myofibroblasts25,26. IL-10 reduces collagen deposition in UUO-induced kidney fibrosis27 and bleomycin-induced lung fibrosis28. Treatment with recombinant IL-10 has also been reported to reduce liver fibrosis in chronic hepatitis C patients who did not respond to interferon-based therapy29. Despite these promising results, the therapeutic utility of recombinant IL-10 has been hindered by the inability to reach sufficiently high local concentrations within the fibrotic microenvironment.
Rapamycin (sirolimus) is a natural macrocyclic lactone used clinically as an orally-administered immune suppressant that operates through inhibition of the mammalian target-of-rapamycin (mTOR) receptor30,31. Rapamycin is an anti-inflammatory and autophagy-inducing molecule, and has been reported to inhibit the progression of both kidney32 and lung33 fibrosis via increased expression of TGF-α and epidermal growth factor receptor (EGFR) signaling. Rapamycin also reduces collagen deposition in UUO-induced kidney fibrosis34 and bleomycin-induced lung fibrosis35. However, the therapeutic use of intravenously infused rapamycin is hindered by its low solubility in water and the difficulty of its delivery, resulting in unwanted systemic side effects31.
These therapies are particularly promising because IL-10 (Supplementary Fig. 1) is downregulated in idiopathic pulmonary fibrosis (IPF, data from IPF cell atlas36). Both IL10 receptor (IL10R, Supplementary Fig. 2) and mTOR (Supplementary Fig. 3) are variously down- and up-regulated in cell types found in IPF. This indicates that increasing the local concentration of IL-10 might have an immediate anti-fibrotic effect. While the amount of mTOR is not increased in fibrosis (based on the IPF cell atlas or other datasets37), mTOR activation is correlated with kidney fibrosis38, and inhibition of mTOR ameliorates kidney fibrosis in mice32.
Here, we engineer delivery systems that bind to exposed collagen I and III prevalent in kidney and lung fibrosis. To target the cytokines to the leaky vasculature of fibrotic tissues, we used protein engineering to recombinantly fuse the von Willebrand Factor A3 domain (A3-VWF), a collagen-binding domain, to the cytokine IL-10. We also engineered collagen binding peptide-conjugated micelles for the encapsulation of the small molecule rapamycin. We further compared the collagen-targeting strategy with a separate strategy of binding to fibronectin Extra Domain A (FN-EDA), a splice variant of fibronectin (FN) prevalent in fibrosis. We then evaluated the delivery ability of these drug delivery systems and demonstrated superior efficacy to unmodified counterparts in mouse models of fibrosis (diagrammatic sketch in supplementary Fig. 4). These findings support the use of innovative collagen-binding approaches to improve the clinical potential of anti-fibrotic drugs through accumulation at sites of fibrosis.
Results
The leakiness of the fibrotic tissue vasculature exposes ECM proteins normally unavailable in blood circulation11,12,13,14. In this work, we harnessed this phenomenon by engineering therapies to actively bind collagen at sites of fibrosis. We sought to accumulate IL-10, an anti-inflammatory cytokine, or rapamycin, a small molecule anti-fibrotic. Our laboratory has previously identified several peptide and protein-based ligands that bind to exposed ECM and accumulate in tissues: (1) von Willebrand Factor A3 domain (VWF-A3, ~ 20 kDa), which binds to collagens I and III39; (2) a fibronectin-extra-domain-A (FN-EDA) antigen-binding fragment (α-FN-EDA Fab, ~ 50 kDa) with affinity for FN-EDA40,41; and (3) decorin-derived collagen-binding peptide (CBP, ~ 2 kDa) that binds to collagens I and III42. While the protein IL-10 (~ 19 kDa) can be fused to VWF-A3 or α-FN-EDA Fab targeting domains via recombinant protein engineering, the small molecule rapamycin is water insoluble and requires additional encapsulation. For this purpose, we developed a CBP-conjugated micelle for the stable encapsulation of rapamycin.
Rapamycin-CBP-micelle development and characterization
Rapamycin is a well-validated anti-inflammatory small-molecule antibiotic that is being explored as an anti-fibrotic drug due to its ability to inhibit fibroblast proliferation43,44. However, its low water solubility makes drug formulation difficult and its low bioavailability results in the systemic side effects31. As a result, the use of rapamycin as an intravenously-administered drug is limited. We developed a targeted drug delivery system engineered to both efficiently solubilize rapamycin and target it to the fibrotic, collagen-rich tissues. To encapsulate rapamycin, we use a block copolymer of poly(ethylene glycol) (pEG) and poly(phenylalanine) (pPhe) which forms nanometer-scale micelles that have been reported as good carriers of hydrophobic small molecule drugs45. The pEG114-pPhe20 polymer of about 8 kDa was synthesized as previously described45 (Supplementary Fig. 5A), and its block lengths confirmed by 1HNMR (Supplementary Fig. 5B) and MALDI-TOF (Supplementary Fig. 5C). The polymer forms micelles when dispersed in water at the concentration above their critical micellar concentration (CMC) of 0.01mM (Supplementary Fig. 6A). Hydrophobic compounds encapsulated in pEG114-pPhe20 micelles are released through passive diffusion following Korsmeyer-Peppas drug release kinetic, as demonstrated by in vitro release kinetic study using model small molecule dye rhodamine B (Supplementary Fig. 6B).
In our approach, we additionally linked the micelles with a decorin-derived collagen binding peptide (CBP, CNNNLHLERL) via a small-molecule heterobifunctional linker (Supplementary Fig. 7). First, peptide was reacted with the linker to arm it with azide-reactive dibenzocyclooctine (DBCO) functional group, yielding peptide-DBCO. Next, azide-modified micelles were reacted with a peptide-DBCO, resulting in peptide-micelle conjugate. As a non-binding control, a scrambled-sequence peptide (SCR, CLLNEHLNNR) was conjugated to the micelles in place of CBP. Peptide-micelle conjugates were first characterized by Dynamic Light Scattering (DLS) to determine nanoparticle size (Supplementary Fig. 8A). We observed an increase in micelle diameter from 18.0 nm to 24.6 nm upon CBP and SCR peptide conjugation (Supplementary Fig. 8A). In addition, cryogenic electron microscopy (cryo-EM) images of the conjugates confirmed their size of about 15–20 nm and demonstrated their semi-spherical shape (Supplementary Fig. 8B). There were no obvious differences observed between cryo-EM images of conjugated and unmodified micelles (Supplementary Fig. 8B). The presence of CBP peptide conjugated to nanoparticles was evidenced by performing a reactive site-competition reaction experiment. First, azide-modified micelles were either reacted with peptide-DBCO or left unreacted. Subsequently, azide-reactive fluorescent dye (AF647-DBCO) was added to both peptide-reacted and unreacted micelles; reaction could happen only if free azide groups were available. Based on size exclusion chromatography (SEC) elution profiles (Supplementary Fig. 9A) we observed that AF647-DBCO reacted only with unreacted micelles (fractions 15–25), providing a proof of a successful conjugation of the peptide. Next, we examined if the CBP micelles could bind to collagen in vitro to enable their accumulation in collagen-rich environment (Supplementary Fig. 9B). To demonstrate this, we incubated fluorescently labeled CBP-micelles or SCR-micelles (non-binding control) with collagen type I sponge hydrogels (Helistat, cut-out to ca. 0.2 cm in diameter). After an hour of incubation at 37 °C, the gels were taken out, washed with PBS and imaged using IVIS imaging. Collagen gels incubated with CBP-micelles had significantly higher fluorescence than gels incubated with the SCR-micelles. Taken together, data shown in Supplementary Fig. 9A-B demonstrated the presence of the peptide on the micelles and the preserved affinity to collagen type I after the conjugation.
VWF-A3 and CBP-micelles accumulate to both fibrotic kidney and lungs
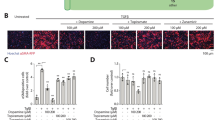
Next, we tested the ability of these novel peptide and antibody targeting agents to accumulate either the fibrotic kidney, induced by unilateral ureteral obstruction (UUO), or to accumulate in the fibrotic lung, induced by bleomycin instillation (Fig. 1). For this purpose, we insulted mouse kidneys or mouse lungs in two separate studies and allowed fibrosis to develop in these organs for 1 week. At 1 week, UUO-insulted kidneys and bleomycin-insulted lungs have increased collagen I deposition19,21,34. We then intravenously injected fluorescently labeled (1) α-collagen II Fab, (2) α-FN-EDA Fab, (3) VWF-A3, (4) CBP-micelles and (5) scrambled CBP-micelles at equimolar concentrations and compared the fluorescence of the harvested organs after 24–48 h for kidney fibrosis or lung fibrosis respectively via IVIS (Fig. 1, Supplementary Figs. 10, 11).The α-collagen II Fab served as a control for the α-FN-EDA Fab, because fibrotic tissues tend not to express collagen II, and thus we wouldn’t expect to see antigen-specific binding of the α-collagen II Fab. The α-collagen II Fab also serves as control for non-specific accumulation of similarly sized proteins in fibrotic tissue due to leaky vasculature.

Fluorescently labeled VWF-A3 and CBP-micelles target to both fibrotic kidney and lungs. The left kidneys of mice were insulted with UUO ligation, and the lungs of other mice were insulted with intranasally instilled bleomycin (75 µg). These mice were allowed to become fibrotic for 1 week and were then injected i.v. with fluorescently labeled (1) α-collagen II Fab, (2) α-FN-EDA Fab, (3) VWF-A3, (4) CBP-micelles or (5) Scrambled CBP-micelles at equimolar concentrations. Heart, lung, spleen, kidneys, and liver were harvested 24 h after injection for fibrotic kidney experiments and 48 h after injection for fibrotic lung experiments. Fluorescence intensity was measured via IVIS (images in Supplemental Fig. 9) and the normalized fluorescence units as % of total fluorescence was calculated for healthy and fibrotic kidney and lungs. N of 3 (PBS) or 4 (other conditions). * = statistical significance of P < 0.05 between fluorescence of fibrotic organs vs. fluorescence of healthy organs, Student’s t-test. Error bars are SEM.
In this fibrosis accumulation study, we observed unexpectedly that α-FN-EDA Fab accumulated in fibrotic kidneys, but not to fibrotic lungs (Fig. 1). This result may be due to differences in the amount fibronectin-EDA present in the kidney and lung fibrosis models used in the study. Further investigation outside of the scope of this study is needed to explore this hypothesis. While α-FN-EDA appears not to be suitable for accumulating bioactive macromolecules to fibrotic lungs, it may be used in the future as a drug carrier in kidney fibrosis. However, in the same accumulation study, VWF-A3 displayed greater accumulation in both fibrotic kidneys and lungs compared to the healthy organs (Fig. 1). This likely occurs due to high levels of exposed collagen I and III present in both disease models46,47. We also evaluated whether CBP-micelles could bind exposed collagen in fibrotic kidneys and lungs (Fig. 1). We observed that CBP-micelles preferentially accumulated in fibrotic kidneys with total fluorescence units exceeding 20% in both mouse models (Fig. 1). Scrambled CBP-micelles also accumulated in the fibrotic tissues, but to a lesser extent than the actively targeted CBP-micelles (Fig. 1). The observed accumulation of scrambled CBP-micelles, especially in the lung fibrosis model, is likely due to the physical effect of the passive accumulation of nanoparticles to tissues with increased vascular permeability48,49. Because VWF-A3 and CBP-micelles successfully accumulated in both fibrotic kidneys and lungs, we selected these delivery systems as carriers of IL-10 and rapamycin, respectively.
Collagen-targeting IL-10 treatment suppresses kidney and lung fibrosis
We first recombinantly fused the VWF-A3 to IL-10 (A3-IL-10) and produced this fusion protein in human embryonic kidney cells (Supplementary Fig. 11)39,42,50,51. To prolong the half-life of the collagen-targeted IL-10, we incorporated mouse serum albumin (A3-SA-IL-10, ~ 66 kDa) (Supplementary Fig. 11) as described previously50. Following affinity and size exclusion chromatography, the purity of A3-IL-10 and A3-SA-IL-10 variants was confirmed by SDS-PAGE (Supplementary Fig. 12A). Affinity measurements were then performed to confirm the ability of IL-10 variants (A3-IL-10 and A3-SA-IL-10) to bind to IL-10 receptor alpha (Supplementary Fig. 12B) and collagen I and III (Supplementary Fig. 13A, B).
To determine if collagen-binding A3-IL-10 and A3-SA-IL-10 could treat kidney fibrosis more effectively than unmodified IL-10, we administered these therapies in a mouse model of kidney fibrosis. In this model, fibrosis was induced in the left kidney via UUO ligation and 7 days post UUO, mice were intravenously injected with 20 μg (or molar equivalent) of IL-10, A3-IL-10, and A3-SA-IL-10. Mice were then sacrificed 14 days post UUO, and both injured and fibrotic kidneys were resected, fixed, sectioned, and stained for collagen I using immunohistochemistry. The extent of fibrosis in uninjured (Fig. 2A), untreated fibrotic (Fig. 2B), and IL-10 treated fibrotic kidneys (Fig. 2C-E) was then assessed by quantifying the ratio of fibrotic (collagen I high) to healthy (collagen I low) tissue area (Fig. 2F). While unmodified IL-10 treatment slightly reduced extent of kidney fibrosis, only collagen-binding A3-IL-10 and A3-SA-IL-10 significantly reduced the disease area ratio. Furthermore, the reduction of fibrotic to healthy tissue area ratio in mice treated with A3-SA-IL-10 was significantly lower than mice treated with unmodified IL-10.

A single injection of 20 µg of A3-IL-10 or A3-SA-IL-10 rescues the fibrotic damage from UUO insult to mouse kidneys. The descending ureter of the left kidney was surgically ligated, and 20 µg (or molar equivalent) IL-10, A3-IL-10, or A3-MSA-IL-10 was injected i.v. after 7 days. Kidneys were resected 14 days post UUO insult (7 days post injection), mounted, and assessed via immunohistochemistry for collagen I. The amount of positive IHC staining was compared to the overall amount of kidney tissue, per image. Representative images for (A) uninjured kidneys, (B) fibrotic kidneys, and fibrotic kidneys treated with (C) IL-10, (D) A3-IL-10, and (E) A3-SA-IL-10. (F) Area ratio (x100) of fibrotic to healthy tissue calculated based on histology images. N = 6. * = statistical significance of P < 0.05, one-way ANOVA vs. fibrosis control, Welch’s correction. Comparison between IL-10 and A3-SA-IL-10 is Student’s t-test. Error bars are SEM.
We next evaluated the efficacy of collagen-binding A3-IL-10 and A3-SA-IL-10 in a mouse model of lung fibrosis. For this purpose, we insulted the lungs with bleomycin and treated the mice by intravenously injecting 10 µg (or molar equivalent) of IL-10, A3-IL-10 and A3-SA-IL-10 at 7, 10, 14 and 17 days post-bleomycin insult (Fig. 3A-H). Because bleomycin injury is associated with weight loss in mice47,52, we weighed the mice, but noted no significant changes in weight between the experimental groups (Fig. 3I). The mice were then euthanized 21 days post insult. A hydroxyproline assay was performed on the right lung lobe to quantify the amount of collagen, which is positively correlated with ongoing fibrotic disease (Fig. 3J). From this assay, we determined that A3-IL-10 significantly reduced the amount of collagen in the right lobe of the fibrotic lungs, whereas unmodified IL-10 had no significant impact on this metric (Fig. 3J). The left lung lobes were also resected, fixed, sectioned, and stained for Masson’s trichrome, a dye used to visualize collagen present in fibrotic tissue sections. The stained histology images were blindly scored using the Ashcroft scale, an established method for quantifying the extent of lung fibrosis (Fig. 3A-H). This assessment confirmed the results of the hydroxyproline assay. A3-IL-10 treatment significantly reduced Ashcroft score compared fibrotic untreated and unmodified IL-10 treated mice (Fig. 3A-H). Taken together, these results suggest that collagen targeting via recombinant fusion of VWF-A3 to IL-10 improves the therapeutic efficacy of IL-10 in mouse models of kidney and lung fibrosis. In the kidney fibrosis model, the addition of half-life prolonging serum albumin to the A3-IL-10 construct further improved efficacy, whereas in the lung fibrosis model, this modification did not show any additional benefit over A3-IL-10. The differences in efficacy due to serum albumin fusion may have been influenced by the different dosing regimens in the two mouse models. In the lung fibrosis model, the mice were dosed more frequently with the IL-10 variants, which may not have taken advantage of the prolonged half-life of the serum albumin fused molecule. Future studies can be performed to further optimize the dose of collagen-targeted therapies.

A3-IL-10 rescues the fibrotic damage in mouse model of lung fibrosis. Lungs insulted with 75 µg bleomycin; 1 week following instillation mice were left untreated (A, E) or injected i.v. with a total of 40 µg (or molar equivalent) in 4 injections of 10 µg on days 7, 10, 14 and 17 post bleomycin insult with either (B, F) IL-10, (C, G) A3-IL-10, (D, H) A3-SA-IL-10. Insets are pictured and the size bar is 2000 μm. (I) Mouse body weights after bleomycin insult and treatment. (J) Collagen content from the right, multi-lobed lung assessed by hydroxyproline assay. (K) Blinded Ashcroft scoring. N = 8. * = statistical significance of P < 0.05, significance vs. fibrotic lungs calculated by 2-way ANOVA (with Fisher’s LSD post-test). Comparison between IL-10 and A3-IL-10 is Student’s t-test. Error bars are SEM.
Rapamycin encapsulated collagen-targeting micelles suppress kidney and lung fibrosis
Having demonstrated that the recombinant fusion of collagen-targeting domains to the biologic drug IL-10 improves its therapeutic efficacy in mouse models of fibrosis, we next sought to evaluate whether the encapsulating the small molecule drug rapamycin in collagen-binding peptide-conjugated micelles could also improve efficacy in these models. For this purpose, we induced kidney fibrosis in the left kidney via UUO ligation. Then, we injected 3 mg/kg of rapamycin as a free molecule or a molar equivalent amount of rapamycin encapsulated in CBP-micelles (rapamycin-CBP-micelles) at 7-, 9-, 11- and 13-days post disease induction (Fig. 4). On day 14, mice were sacrificed, and tissues were processed and analyzed as described above. The extent of fibrosis in uninjured (Fig. 4A), untreated fibrotic (Fig. 4B), free rapamycin treated fibrotic (Fig. 4C) and rapamycin-CBP-micelles treated fibrotic (Fig. 4D) kidneys was then assessed by quantifying the ratio of fibrotic (collagen I high) to healthy (collagen I low) tissue area (Fig. 4E). While both rapamycin treatments significantly reduced kidney fibrosis, rapamycin-CBP-micelles treatment showed the greatest reduction in collagen I fibrotic-to-healthy disease area ratio. Because the induction of fibrosis has been reported to alter blood chemistry parameters53, we compared the levels of markers of liver damage and kidney damage (alanine aminotransferase (ALT), albumin, amylase, aspartate aminotransferase (AST), total bilirubin, blood urea nitrogen (BUN), calcium, creatinine, creatine kinase, total bilirubin, and uric acid), in the serum of uninjured, fibrotic and rapamycin-treated fibrotic mice. Of these, rapamycin treatment changed the levels of alanine aminotransferase (ALT) (Supplementary Fig. 14A), aspartate aminotransferase (AST) (Supplementary Fig. 14B) and total bilirubin (Supplementary Fig. 14C), In this study, we observed that kidney fibrosis induction reduces ALT, AST and total bilirubin levels in the serum (Supplementary Fig. 14A-C), which is consistent with published reports on the response of ALT, AST, and bilirubin to fibrosis53. While treatment of fibrotic mice with free rapamycin did not affect biochemistry markers, we observed that treatment with collagen-targeting rapamycin CBP-micelles restored ALT, AST and total bilirubin levels to those of uninjured mice (Supplementary Fig. 14A-C).

Rapamycin and rapamycin-CBP-micelle treatments rescue the fibrotic damage from UUO insult to mouse kidneys. The descending ureter of the left kidney was surgically ligated, and 3 mg/kg (or molar equivalent) rapamycin and rapamycin-CBP-micelles was injected i.v. at 7, 9, 11, and 13 days post UUO ligation. Kidneys were resected 14 post UUO insult, mounted, and assessed via immunohistochemistry for collagen I. Representative images for (A) uninjured kidneys, (B) fibrotic kidneys, (C) and fibrotic kidneys treated with (C) rapamycin, and (D) rapamycin-CBP-micelles. (E) The amount of positive IHC staining was compared to the overall amount of kidney tissue, per image. N = 6. * = statistical significance of P < 0.05, one-way ANOVA vs. fibrosis control, Welch’s correction. Comparison between rapamycin and rapamycin-CBP-micelles is Student’s t-test. Error bars are SEM.
After demonstrating the efficacy of rapamycin formulated in collagen targeting CBP-micelles in kidney fibrosis, we next sought to evaluate its efficacy in a mouse model of lung fibrosis. For this purpose, we induced lung fibrosis via bleomycin instillation into the lungs. Fibrotic mice were then treated with intravenous administration of 3 mg/kg of rapamycin as a free molecule or encapsulated in CBP-micelles (rapamycin-CBP-micelles) at 7, 9, 11, 13, 15, 17 and 19 days post disease induction (Fig. 5A-H). Throughout the experiment, we observed no significant changes in weight between the experimental groups (Fig. 5I). The mice were euthanized 21 days post insult and the lungs were processed and analyzed as described above. Based on the hydroxyproline assay, the administration of rapamycin CBP-micelles significantly reduced the amount of collagen in the right lobe of the fibrotic lungs, whereas free rapamycin treatment had no significant impact on this metric (Fig. 5J). Similarly, blinded analysis of Masson’s trichrome staining of lung histology sections (Fig. 5A-H) showed that rapamycin-CBP-micelles treatment reduced Ashcroft score of fibrotic left lungs to healthy lung levels and that the score was significantly lower compared to free rapamycin treated fibrotic mice (Fig. 5K). In this study, we also analyzed blood chemistry markers of uninjured, untreated fibrotic and rapamycin-treated fibrotic mice. For this purpose, we quantified the concentration of markers of lung, heart and liver damage (alanine aminotransferase (ALT), albumin, amylase, aspartate aminotransferase (AST), total bilirubin, blood urea nitrogen (BUN), calcium, creatinine, creatine kinase, CO2, total bilirubin, and uric acid) in the serum. While rapamycin-CBP micelles did not significantly improve CO2 levels in the bloodstream (Supplementary Fig. 15A), treatment with rapamycin-CBP-micelles restored creatine kinase (Supplementary Fig. 15B), resulting in a statistically significant reduction vs. both the untreated disease state and the injection of free rapamycin. Both free rapamycin and rapamycin-CBP micelles brought ALT (Supplementary Fig. 15C) and AST (Supplementary Fig. 15D) levels to those of uninjured mice. Taken together, these results suggest that collagen targeting of rapamycin via its encapsulation in CBP-micelles improves the therapeutic efficacy of rapamycin in mouse models of kidney and lung fibrosis.

Rapamycin and rapamycin-CBP-micelle treatments rescue the fibrotic damage in a mouse model of lung fibrosis. Lungs were insulted with 75 µg bleomycin and 1 week later mice were left untreated (B, F) or injected i.v. with 3 mg/kg of free rapamycin (C, G) or rapamycin-CBP-micelles (D, H) on days 7, 9, 11, 13, 15, 17 and 19. Healthy lungs (A, E) were imaged for reference. Insets are pictured and the size bar is 2000 microns. (I) Mouse weights after bleomycin insult and treatment. (J) Collagen content from the right, multi-lobed lung assessed by hydroxyproline assay. (K) Blinded Ashcroft scoring. N = 7 for untreated fibrosis, N = 4 for rapamycin-treated fibrosis, and N = 6 for rapamycin-CBP-micelles treated fibrosis. * = statistical significance of P < 0.05, significance vs. fibrotic lungs calculated by 2-way ANOVA (with Fisher’s LSD post-test). Comparison between rapamycin and rapamycin-CBP micelles is Student’s t-test. Error bars are SEM.
Therapeutic outcomes could be further improved by optimizing the dose of rapamycin administered and the treatment regimen. The dose of rapamycin used in this study (3 mg/kg) is lower than doses previously used in the treatment of kidney32 and lung33 fibrosis. The dose used (3 mg/kg), administered every other day, is also comparable to the lowest average daily dose found in the relevant literature (1.5 mg/kg)54. Our CBP-micelle technology can also be adapted to formulate other anti-fibrotic water-insoluble molecules currently in development to improve their solubility, pharmacokinetics, toxicity and efficacy.
Discussion
Fibrosing diseases are refractory to treatment, and currently available therapies only mildly reduce the symptoms of disease. A major goal of the ongoing fibrosis research is to identify new clinical treatments that are able to reverse damage and restore function of affected organs. In this work, we have demonstrated that novel collagen-binding protein-based and nanoparticle-based drug delivery systems can be harnessed to target systemically administered bioactive molecules to the leaky and collagen-rich fibrotic organs. We have engineered and validated formulations of two candidate drugs: IL-10, an anti-inflammatory biologic, and rapamycin, an anti-fibrotic small molecule water-insoluble drug. In concert, our studies demonstrated improved efficacy of A3-IL-10/A3-SA-IL-10 and rapamycin-CBP-micelles in mouse models of kidney and lung fibrosis.
We cannot be certain of the mechanism IL-10 and rapamycin used in our study to reduce the amount of collagen in fibrotic kidneys and lungs. However, both IL-10 and rapamycin are both anti-inflammatories that affect fibroblasts and macrophages, but have also been reported to reduce collagen in fibrosis of the lung and kidney27,28,34,35.
Our novel therapies outperformed the unmodified drugs in improving the phenotype of fibrosis-affected organs. The technologies presented in this work can be easily adapted in the future to deliver other biologics as well as water-insoluble medicines. Taken together, our findings indicate that collagen-targeted approaches can be used to develop versatile next generation anti-fibrotic therapies with high clinical potential.
Experimental section
Study design
This study was designed to screen for fibrosis-targeting agents from several collagen-targeting proteins, peptide-decorated micelles and antibodies. Group size was selected taking into account the reported variability in mouse kidney and pulmonary fibrosis models. To eliminate cage effects, mice were randomized into treatment groups within a cage. To ensure reproducibility, treatment was performed by multiple researchers over the course of this study. Kidneys and lungs were also resected by multiple researchers, and blinded scoring was performed on the lung fibrosis histology images.
Production of α-fibronectin-EDA fab
The sequence encoding the anti-fibronectin-EDA Fab (α-FN-EDA Fab) was synthesized and subcloned into the mammalian expression vector pSecTag A. Suspension-adapted HEK-293 F cells were routinely maintained in serum-free FreeStyle 293 Expression Medium (Gibco). On the day of transfection, cells were inoculated into fresh medium at a density of 1 × 106 cells/mL, and then 2 µg/mL plasmid DNA, 2 µg/ml linear 25 kDa polyethylenimine (Polysciences), and OptiPRO SFM medium (4% final concentration, Thermo Fisher) were sequentially added. The culture flask was agitated by orbital shaking at 135 rpm at 37 °C in the presence of 5% CO2. 7 days after transfection, the cell culture medium was collected by centrifugation and filtered through a 0.22 μm filter. Next, the cell culture medium was loaded onto a HiTrap MabSelect 5 mL column (GE Healthcare), using an ÄKTA Pure 25 (GE Healthcare). After washing the column with PBS, protein was eluted with 0.1 M sodium citrate (pH 3.0). Purification was carried out at 4 °C. The α-FN-EDA Fab was verified as > 90% pure by SDS-PAGE (performed on 4–20% gradient gels (Bio-Rad)).
Production and purification of recombinant A3-IL-10 and A3-SA-IL-10
Recombinant VWF-A3 cytokine fusion proteins were produced as previously described39,42,51. Briefly, the sequences encoding the A3 domain of VWF (A3), a (GGGS)2 flexible linker, mouse serum albumin (SA), a (GGGS)2 flexible linker and mouse IL-10 (A3-SA-IL-10) or A3, (GGGS)2 and mouse IL-10 (A3-IL-10) were synthetized and subcloned into the mammalian expression vector pcDNA3.1(+) by GenScript. To enable affinity-based purification, a sequence encoding (His)6 was added to the carboxy terminus of each protein. The amino acid sequences of the proteins are shown in Supplementary Fig. 11. Suspension-adapted HEK293F cells were maintained in serum-free Free Style 293 Expression Medium (Gibco). On the day of transfection, HEK cells were resuspended in fresh medium at a density of 106 cells mL−1. A concentration of 2 µg mL−1 plasmid DNA and 2 µg mL−1 plasmid DNA linear 25 kDa polyethyleneimine (Polysciences) dissolved in OptiPRO serum free medium (Thermo Fisher) at 4% final volume were added sequentially to the suspension cells. After 7 days of culture, cell supernatants were collected, centrifuged at 4,000 rpm for 10 min at 4 °C, and filtered through a 0.22 μm filter. Proteins were purified via affinity chromatography and size exclusion chromatography using an ÄKTA pure 25 (Cytiva) as described previously50. Protein purity was assessed by SDS-PAGE (Supplementary Fig. 12A)39,42,51. The purified proteins were then tested for endotoxin using the HEK-Blue TLR4 reporter cell line, and the endotoxin levels were confirmed to be below 0.01 EU mL−139,42,51. Protein concentration was determined via absorbance at 280 nm using a NanoDrop spectrophotometer (Thermo Scientific).
Detection of A3-IL-10 and A3-SA-IL-10 binding to collagens
Surface plasmon resonance (SPR) measurements were performed using the Biacore X100 SPR system (Cytiva) as described previously42. To measure the affinity of A3-IL-10 and A3-SA-IL-10 for collagen proteins, recombinant human type I or type III collagen (Millipore Sigma) were immobilized on a CM5 sensor chip (Cytiva) by standard amine coupling method for around 1500 resonance units (RUs) and blocked with ethanolamine according to the manufacturer’s instructions. A reference cell was also blocked with ethanolamine. Binding assays were carried out at room temperature and the Kd values of A3-IL-10 and A3-SA-IL-10 were determined by fitting the 1:1 Langmuir binding model to the data using BIAevaluation software (Cytiva) and are displayed in Supplementary Fig. 13A, B.
Detection of A3-IL-10 and A3-SA-IL-10 binding to IL-10 receptor
An ELISA was performed to determine the affinity of IL-10 variants for IL-10 receptor as described previously for A3-cytokine variants39. 96-well ELISA plates (Greiner Bio One) were coated with 4 µg ml−1 recombinant mouse IL-10 receptor alpha (R&D Systems) overnight at 4 °C. The wells were then washed in PBS with 0.05% Tween 20 (PBS-T) and blocked with 2% BSA in PBS-T for 1 h at room temperature. 0 to 4 µg ml−1A3-IL-10 or A3-SA-IL-10 (wild type IL-10 molar equivalent) were added to the wells and incubated for 2 h at room temperature. After three washes with PBS-T, wells were then incubated for 1 h at room temperature with biotinylated antibody against IL-10 (Invitrogen) and then incubated with HRP-conjugated streptavidin (Invitrogen) for 30 min at room temperature. After five washes, the bound A3-IL-10 and A3-SA-IL-10 were detected by tetramethylbenzidine substrate measurement of absorbance at 450 nm with subtraction of 570 nm as described previously39. The apparent KD values were obtained by nonlinear regression analysis in Prism software (v9, GraphPad Software assuming one-site specific binding) and are displayed in Supplementary Fig. 12B.
Synthesis of pEG114-pPhe20 copolymers
Both methoxy-pEG114-pPhe20 and N3-pEG114-pPhe20 polymers were prepared by ring-opening polymerization of L, D-Phenylalanine N-carboxyanhydride (Phe-NCA; made in house from L, D-Phenylalanine and triphosgene) using amino-terminated methoxy-pEG114-NH2 and N3-pEG114-NH2, respectively, as initiators45. Briefly, Phe-NCA (50 mg, 0.25 mmol) and respective pEG114-NH2 (50 mg, 0.01 mmol) were added to anhydrous dimethylformamide: tetrahydrofuran solution (DMF: THF 1:1 v/v, 2 mL) under a nitrogen atmosphere for further reaction time of 24 h at 35 °C (Supplementary Fig. 4A). The copolymers were purified by multiple precipitation in diethyl ether and hexanes, and dried under vacuum for further use. The yield of polymerization was 66% for methoxy-pEG114-pPhe20 and 71% for N3-pEG114-pPhe20. The polymers were characterized using 1H NMR and MALDI-TOF to confirm the composition and purity (Supplementary Fig. 4B, C).
Determination of critical micelle concentration (CMC)
The CMC of block copolymer pEG114-pPhe20was determined by fluorescence and using pyrene as a probe based on previously reported procedures55. Briefly, micellar dispersions of pEG114-pPhe20 were prepared at various concentrations (0.1–0.13 mM) with a fixed concentration of pyrene of 0.6 µM. The excitation spectra of pyrene from 300 to 360 nm were monitored as 390 nm for each dilution using Horiba Scientific Fluoromax 4 fluorimeter. CMC was determined by plotting the ratio of the intensities at 335 and 332 nm (I3/I1) versos concentration of pEG114-pPhe20.
In vitro release kinetic study
To study the kinetics of drug release from pEG114-pPhe20 micelles, model drug/dye Rhodamine B was first encapsulated in pEG114-pPhe20. The free dye was removed using Zeba desalting columns with MWCO of 7 kDa. The release kinetic was assessed in vitro by employing the dialysis method56. Equal amount of Rhodamine B, either as a free dye or formulated within pEG114-pPhe20 micelles, was placed in a tube capped with a dialysis membrane of MWCO of 10 kDa. The formulations were dialyzed against 10x volume of PBS buffer. The profiles were obtained by periodically withdrawing aliquots of the dialysis buffer to measure the amount of released Rhodamine B via fluorescence spectroscopy (ex/em 550/600) using Horiba Scientific Fluoromax 4 fluorimeter.
Synthesis of AF647-labeled pEG114-pPhe20 copolymers
Alexa Fluor™ 647 Succinimidyl ester (AF647-NHS, Invitrogen) was used to label pEG114-pPhe20 copolymers for fluorescence imaging biodistribution study evaluation. Briefly, AF647-NHS (1 mg) was added to pEG114-pPhe20 (25 mg) in anhydrous dichloromethane (1 mL) and stirred for 24 h. The pEG114-pPhe20-AF647 conjugates were purified by precipitation in diethyl ether.
Synthesis of peptide-linker azide-reactive conjugates
Both collagen binding peptide (CBP, sequence: CNNNLHLERL, Genscript) and scrambled sequence peptide (SCR, sequence: CLLNEHLNNR, Genscript) were modified with heterobifunctional Sulfo-DBCO Maleimide linker (Click Chemistry Tools) to arm the peptide with an azide-reactive group (dibenzocyclooctyne group, DBCO). Briefly, Sulfo-DBCO Maleimide linker (0.44 mg, 1.0 eq) was added to peptide (1 mg, 1.1 eq) solution in DMSO and allowed to react overnight (Supplementary Fig. 6). The peptide-linker was used crude without further modification.
Formulation and characterization of peptide-micelle conjugate nanoparticles
Copolymer micelles were formed by rehydration of the polymer film and subsequent sonication of resulted suspension (Supplementary Fig. 6). Briefly, methoxy-pEG114-pPhe20-AF647 (9.5 mg) and N3-pEG114-pPhe20-AF647 (0.5 mg) solution (to ensure 5%mass of azide-terminated polymer) in tetrahydrofuran was transferred to a 1.5 mL glass vial and the polymer film was formed under nitrogen gas flow. Next, phosphate buffered saline (PBS, 1 mL) and the solution was sonicated for 45 min until almost-clear uniform suspension was formed. The suspension was then filtered with 0.22 μm PVDF syringe filter (ThermoFisher) and peptide-linker solution (at 1.1:1 linker: N3 ratio) was added. The mixture was allowed to react at room temperature for 6 h (Supplementary Fig. 6) and then was dialyzed against PBS using Slyde-A-Lyzer™ Dialysis Cassette (7 K MWCO, 3mL, ThermoFisher) to remove unreacted peptide-linker and any remaining free fluorescent dye (AF647). Obtained micelle-peptide nanoparticle solutions: 5%N3-pEG114-pPhe20, CBP-pEG114-pPhe20, and SCR-pEG114-pPhe20 were characterized using Dynamic Light Scattering (DLS) (Supplementary Fig. 7A) and cryogenic electron microscopy (cryo-EM) (Supplementary Fig. 7B) to determine their size, zeta potential and morphology.
Formulation and characterization of rapamycin-loaded CBP-micelles
Rapamycin (MedChemExpress) (5 mg) in tetrahydrofuran (200 µL) was added in to methoxy-pEG114-pPhe20-AF647 (9.5 mg) and N3-pEG114-pPhe20-AF647 (0.5 mg) in tetrahydrofuran (300 µL) in 1.5 mL glass vial. The polymer-drug film was formed under nitrogen gas flow. Next, phosphate buffered saline (PBS, 1 mL) and the solution was sonicated for 45 min until almost-clear uniform suspension was formed. The suspension was then filtered with 0.22 μm PVDF syringe filter (ThermoFisher) and peptide-linker solution (at 1.1:1 linker: N3 ratio) was added. After reacting for 6 h, the formulation was ready for use. The concentration of rapamycin was determined by UV-VIS at 290 nm, using a standard curve method.
Formulation of rapamycin for intravenous injection
Free rapamycin saline solution was prepared by diluting a stock solution of rapamycin in DMSO: PEG300:Tween80:saline (5:40:5:50 v/v) to a desired concentration.
Production of fluorescently labeled antibodies
α-FN-EDA Fab, and VWF-A3 were fluorescently labeled with Cy7 using sulfo-Cy7 N-hydroxysuccinimide ester (Lumiprobe) according to the manufacturer’s instruction, as previously described47. Unreacted Cy7 was removed by dialysis against PBS.
In vivo biodistribution study
An in vivo biodistribution study was conducted as previously described47, with minor adjustments. 7 days following bleomycin insult or UUO surgery, 50 µg (or molar equivalent) of Cy7-α-FN-EDA Fab, Cy7-VWF-A3, and Cy7 were injected via tail vein. 24 h later or 48 h later (Fig. 1), heart, lungs, spleen, kidneys, and liver were resected and imaged via IVIS (Xenogen) under the following conditions: f/stop, 2; optical filter excitation, 710 nm; emission, 780 nm; exposure time, 5 s; small binning.
Bleomycin induced pulmonary fibrosis model
All the animal experiments performed in this work were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Chicago, and was reported in this manuscript using ARRIVE guidelines. All mouse experiments were performed under supervision with protocols approved by the University of Chicago IACUC, and the study was performed in accordance with the relevant guidelines and regulations. Male and female mice were acquired at 8 weeks of age (Jackson laboratories, Bar Harbor, ME). Mouse lungs were instilled with 0.075 units bleomycin (75 µg, Fresenius Kabi, Switzerland) suspended in endotoxin-free PBS, as previously described47. Briefly, mice were anesthetized via isoflurane inhalation (2%). Mice were then placed upright on an angled surface and a 200 µL narrow pipet was placed at the entrance of their throat. 50 µL of bleomycin/PBS was dispensed to the entrance of the throat, and mice were allowed to inhale. Mice were then weighed and placed on a heating pad to recuperate.
Following bleomycin insult, mice were injected via tail-vein with 10 µg (or molar equivalent) of IL-10, A3-IL-10, or A3-SA-IL-10 on 7, 10, 14, and 17 days after bleomycin insult. Another group of mice were injected with 3 mg/kg of rapamycin (or molar equivalent) or rapamycin-CBP-micelles via tail-vein injection 7, 9, 11, 13, 15, 17 and 19 days following bleomycin insult.
Mice were euthanized at 21 days post insult via injection of euthasol (Covetrus, Portland, ME) instead of CO2 inhalation to prevent the potential damage of the lungs.
Lung resection and fibrosis scoring
Lungs were harvested, and the right and left lobes were separated. The left lobe was fixed in 4% paraformaldehyde overnight, mounted in paraffin, sectioned into 5 μm slices, and stained using Masson’s trichrome. Stained lungs were scanned at high resolution using a CRi Panoramic SCAN 40x Whole Slide Scanner (Perkin-Elmer, Waltham, MA), and were read for fibrosis using a modified Ashcroft method, as previously described47. Lungs were read unlabeled by another researcher uninvolved with animal treatment.
The right lobe of the lung was frozen, and dehydrated using a tissue lyophilizer (Labconco, Kansas City, MO), weighed, and was assessed for collagen content by hydroxyproline assay57. Briefly, dried lungs were digested in 6 N HCl/PBS at 100 °C for 24 h. Supernatant from this digestion was added to 96 well plates and treated sequentially with chloramine-T solution and Ehrlich’s solution at 65 °C for 15 min to facilitate the color change reaction. Color was read at 561 nm. Quantification was provided by use of a hydroxyproline (Sigma) dilution series, which was transformed into a standard curve.
Kidney unilateral ureteral obstruction (UUO) fibrosis model
UUO surgery was performed as previously described47, with adjustments. Briefly, mice were anesthetized via 2% isoflurane inhalation, and injected with meloxicam (1 mg/kg), buprenorphine (0.1 mg/kg) in a saline solution, subcutaneously. Mice were laid on their right side and an abdominal incision used to visualize the left ureter. The left ureter was ligated in the middle section of the ureter with two ties (2 mm apart) using 7 − 0 silk sutures. Peritoneum was then closed with 5 − 0 vicryl sutures and skin closed with 5 − 0 nylon sutures.
Mice were injected via tail-vein with 20 µg (or molar equivalent) of IL-10, A3-IL-10, or A3-SA-IL-10 7 days post-UUO ligation. Another group of mice were injected with 3 mg/kg (or molar equivalent) of rapamycin or rapamycin-CBP-micelles via tail-vein injection 7, 9, 11, 13, 15, 17 and 19 days following UUO insult. Two weeks post UUO, mice were sacrificed via CO2 inhalation, and their kidneys harvested. At this point, the placement of UUO ligation was confirmed for each mouse.
Assessment of fibrosis in kidneys
Kidney fibrosis was assessed as previously described47. Briefly, right (healthy) and left (fibrotic) kidneys were placed in 4% PFA for 24 h, mounted in paraffin, sectioned into 5 mm full kidney slices, and stained using immunohistochemistry (IHC) for collagen I (1:4000, polyclonal rabbit, lifespan biosciences, Seattle WA) via a Bond-Max autostaining system (Leica biosystems, Lincolnshire, IL). Stained kidneys were scanned at high resolution using a CRi Panoramic SCAN 40x Whole Slide Scanner (Perkin-Elmer).
Images were equalized in size and converted to .tif files using CaseViewer. Images were then imported into ImageJ, scale set for conversion between microns and pixels, and deconvoluted with the “H DAB” deconvolution option. A threshold of the resulting blue image was set at 215 to see how many pixels were negative for collagen I, and a threshold of the brown (IHC positive) image set at 185 to see how many pixels were positive for collagen I. Use of the Bond-Max autostaining system produced kidney immunohistochemistry staining with high reproducibility, allowing comparison of staining intensity between slides.
Biochemistry analysis of serum for markers of kidney damage
At the time of euthanasia, blood was collected via submandibular bleed into protein low-bind tubes and allowed to coagulate for 2 h on ice. Coagulated blood was then centrifuged at 10,000xg for 10 min, and serum collected. Serum was then diluted 4x in MilliQ water before being placed on deck on an Alfa Wassermann VetAxcel Blood Chemistry Analyzer. All tests requiring calibration were calibrated on the day of analysis and quality controls were run before analyzing samples. Serum tests were run according to kit instructions, and creatinine kinase concentration was normalized to calcium ion concentrations to account for sample hemolysis.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software, and P < 0.05 was considered statistically significant. Either Student’s t-test, 2-way ANOVA (with Fisher’s LSD post test), or 1-way ANOVA with Welch’s correction was used to compare groups.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Wynn, T. A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214 (2), 199–210 (2008).
Wynn, T. A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 4 (8), 583–594 (2004).
Spagnolo, P. Novel treatments for idiopathic pulmonary fibrosis. Am. J. Med. 128 (5), 447 - 449 (2015).
Xaubet, A., Serrano-Mollar, A. & Ancochea, J. Pirfenidone for the treatment of idiopathic pulmonary fibrosis. Expert Opin. Pharmacother. 15 (2), 275–281 (2014).
Knuppel, L. et al. A novel antifibrotic mechanism of nintedanib and Pirfenidone. Inhibition of collagen fibril assembly. Am. J. Respir Cell. Mol. Biol. 57 (1), 77–90 (2017).
Hewitson, T. D. Fibrosis in the kidney: is a problem shared a problem halved? Fibrogenesis Tissue Repair. 5 (Suppl 1), S14 (2012).
OPTN. The Organ Procurement and Transplantation Network. Web-based statistical inquiry [online]. Accessed 2016 Aug 6 (2016).
Schultz, G. S. & Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair. Regen. 17 (2), 153–162 (2009).
Frantz, C., Stewart, K. M. & Weaver, V. M. The extracellular matrix at a glance. J. Cell. Sci. 123 (Pt 24), 4195–4200 (2010).
Zuk, A. & Bonventre, J. V. Acute kidney injury. Annu. Rev. Med. 67, 293–307 (2016).
Probst, C. K. et al. Vascular permeability in the fibrotic lung. Eur. Respir J. 56(1), p.1900100 (2020).
Tager, A. M. et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 14 (1), 45–54 (2008).
Cai, A., Chatziantoniou, C. & Calmont, A. Vascular permeability: regulation pathways and role in kidney diseases. Nephron 145 (3), 297–310 (2021).
Goligorsky, M. S. Permissive role of vascular endothelium in fibrosis: focus on the kidney. Am. J. Physiol. Cell. Physiol. 326 (3), C712–C723 (2024).
Bander, S. J. et al. Long-term effects of 24-hr unilateral ureteral obstruction on renal function in the rat. Kidney Int. 28 (4), 614–620 (1985).
Chevalier, R. L., Forbes, M. S. & Thornhill, B. A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 75 (11), 1145–1152 (2009).
Ucero, A. C. et al. Unilateral ureteral obstruction: beyond obstruction. Int. Urol. Nephrol. 46 (4), 765–776 (2014).
Walters, D. M. & Kleeberger, S. R. Mouse models of bleomycin-induced pulmonary fibrosis. Curr Protoc Pharmacol, Chapter 5: p. Unit 5 46. (2008).
Oxburgh, L., et al., (Re)Building a Kidney. J Am Soc Nephrol, 2017. 28(5): p. 1370-1378.
Atkinson, J. et al. Refining the unilateral ureteral obstruction mouse model: no Sham, no shame. Lab. Anim. 55 (1), 21–29 (2021).
Gul, A. et al. Pulmonary fibrosis model of mice induced by different administration methods of bleomycin. BMC Pulm Med. 23 (1), 91 (2023).
Iyer, S. S. & Cheng, G. Role of Interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 32 (1), 23–63 (2012).
Rachmawati, H. et al. Pharmacokinetic and biodistribution profile of Recombinant human interleukin-10 following intravenous administration in rats with extensive liver fibrosis. Pharm. Res. 21 (11), 2072–2078 (2004).
Steen, E. H. et al. The role of the Anti-Inflammatory cytokine Interleukin-10 in tissue fibrosis. Adv. Wound Care (New Rochelle). 9 (4), 184–198 (2020).
Nakagome, K. et al. In vivo IL-10 gene delivery attenuates bleomycin induced pulmonary fibrosis by inhibiting the production and activation of TGF-beta in the lung. Thorax 61 (10), 886–894 (2006).
Shamskhou, E. A. et al. Hydrogel-based delivery of Il-10 improves treatment of bleomycin-induced lung fibrosis in mice. Biomaterials 203, 52–62 (2019).
Jung, K., et al., Interleukin-10 Protects against Ureteral Obstruction-Induced Kidney Fibrosis by Suppressing Endoplasmic Reticulum Stress and Apoptosis. Int J Mol Sci, 23(18), p.10702 2022.
Li, Y. et al. IL-10 deficiency aggravates cell senescence and accelerates BLM-induced pulmonary fibrosis in aged mice via PTEN/AKT/ERK pathway. BMC Pulm Med. 24 (1), 443 (2024).
Nelson, D. R. et al. Interleukin 10 treatment reduces fibrosis in patients with chronic hepatitis C: a pilot trial of interferon nonresponders. Gastroenterology 118 (4), 655–660 (2000).
Li, J., Kim, S. G. & Blenis, J. Rapamycin: one drug, many effects. Cell. Metab. 19 (3), 373–379 (2014).
Woo, H. N. et al. Preclinical evaluation of injectable sirolimus formulated with polymeric nanoparticle for cancer therapy. Int. J. Nanomed. 7, 2197–2208 (2012).
Chen, G. et al. Rapamycin ameliorates kidney fibrosis by inhibiting the activation of mTOR signaling in interstitial macrophages and myofibroblasts. PLoS One. 7 (3), e33626 (2012).
Korfhagen, T. R. et al. Rapamycin prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am. J. Respir Cell. Mol. Biol. 41 (5), 562–572 (2009).
Wu, M. J. et al. Rapamycin attenuates unilateral ureteral obstruction-induced renal fibrosis. Kidney Int. 69 (11), 2029–2036 (2006).
Han, Q. et al. Inhibition of mTOR ameliorates bleomycin-induced pulmonary fibrosis by regulating epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 500 (4), 839–845 (2018).
Neumark, N. et al. The idiopathic pulmonary fibrosis cell atlas. Am. J. Physiol. Lung Cell. Mol. Physiol. 319 (6), L887–L893 (2020).
Cao, Y. et al. Decreased expression of urinary mammalian target of Rapamycin mRNA is related to chronic renal fibrosis in IgAN. Dis. Markers. 2019, 2424751 (2019).
Marques-Ramos, A. & Cervantes, R. Expression of mTOR in normal and pathological conditions. Mol. Cancer. 22 (1), 112 (2023).
Ishihara, J. et al. Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci. Transl Med. 11(487), p. eaau3259 (2019).
Villa, A. et al. A high-affinity human monoclonal antibody specific to the alternatively spliced EDA domain of fibronectin efficiently targets tumor neo-vasculature in vivo. Int. J. Cancer. 122 (11), 2405–2413 (2008).
Rybak, J. N. et al. The extra-domain A of fibronectin is a vascular marker of solid tumors and metastases. Cancer Res. 67 (22), 10948–10957 (2007).
Mansurov, A. et al. Collagen-binding IL-12 enhances tumour inflammation and drives the complete remission of established immunologically cold mouse tumours. Nat. Biomed. Eng. 4 (5), 531–543 (2020).
Molina-Molina, M. et al. Anti-fibrotic effects of Pirfenidone and Rapamycin in primary IPF fibroblasts and human alveolar epithelial cells. BMC Pulm Med. 18 (1), 63 (2018).
Plate, M., Guillotin, D. & Chambers, R. C. The promise of mTOR as a therapeutic target pathway in idiopathic pulmonary fibrosis. Eur. Respir Rev. 29(157), p. 200269 (2020).
Wen, H. et al. Redox-mediated dissociation of PEG-polypeptide-based micelles for on-demand release of anticancer drugs. J. Mater. Chem. B. 4 (48), 7859–7869 (2016).
Katsumata, K. et al. Targeting inflammatory sites through collagen affinity enhances the therapeutic efficacy of anti-inflammatory antibodies. Sci. Adv. 5 (11), peaay1971 (2019).
White, M.J.V., et al., Blocking antibodies against integrin-α3, -αM, and -αMβ2 de-differentiate myofibroblasts, and improve lung fibrosis and kidney fibrosis. Scientific Reports. 14(1), p. 21623 2024.
Wei, Y. & Zhao, L. Passive lung-targeted drug delivery systems via intravenous administration. Pharm. Dev. Technol. 19 (2), 129–136 (2014).
Yao, Y. et al. Nanoparticle-Based drug delivery in cancer therapy and its role in overcoming drug resistance. Front. Mol. Biosci. 7, 193 (2020).
Yuba, E. et al. Suppression of rheumatoid arthritis by enhanced lymph node trafficking of engineered Interleukin-10 in murine models. Arthritis Rheumatol. 73 (5), 769–778 (2021).
Ishihara, A. et al. Prolonged residence of an albumin-IL-4 fusion protein in secondary lymphoid organs ameliorates experimental autoimmune encephalomyelitis. Nat. Biomed. Eng. 5 (5), 387–398 (2021).
Michael, J. V. et al. Myofibroblast differentiation is governed by adhesion mechanics, and inhibition of the stress sensor Talin2 reverses lung fibrosis. 2021. bioRxiv, DOI: https://doi.org/10.1101/2021.06.07.447403.
Ray, L. et al. A comparative study of serum aminotransferases in chronic kidney disease with and without end-stage renal disease: need for new reference ranges. Int. J. Appl. Basic. Med. Res. 5 (1), 31–35 (2015).
Yoshizaki, A. et al. Treatment with Rapamycin prevents fibrosis in tight-skin and bleomycin-induced mouse models of systemic sclerosis. Arthritis Rheum. 62 (8), 2476–2487 (2010).
Li, H. et al. Influence factors on the critical micelle concentration determination using pyrene as a probe and a simple method of Preparing samples. R Soc. Open. Sci. 7 (3), 192092 (2020).
Sethi, M. et al. Effect of drug release kinetics on nanoparticle therapeutic efficacy and toxicity. Nanoscale 6 (4), 2321–2327 (2014).
Bergheim, I. et al. Critical role of plasminogen activator inhibitor-1 in cholestatic liver injury and fibrosis. J. Pharmacol. Exp. Ther. 316 (2), 592–600 (2006).
Acknowledgements
We thank the Human Tissue Resource Center of the University of Chicago for histology analysis. We thank the Integrated Light Microscopy Core of the University of Chicago for Imaging. We would also like to acknowledge guidance on fibrosis models from Dr. Anne I. Sperling. We thank the CTC Microsurgery Core at Northwestern University for performing the initial surgical procedures and University of Chicago Animal Research Center for help and assistance. We acknowledge Tera Lavoie from the Advanced Electron Microscopy Facility at the University of Chicago for technical assistance. Supplemental Figure 4 was created in BioRender. White, M. (2025) https://BioRender.com/3237odt
Funding
This work was supported in part by the University of Chicago (to JAH) and the Rebuilding the Kidney consortium (RBK, to JAH).
Author information
Authors and Affiliations
Contributions
Conceptualization: MJVW, MMR, EB, JAH. Methodology: MJVW, MMR, EB. Investigation: MJVW, MMR, EB, EY, AS, HS, ZJZ, LTG, SC, ATA. Visualization: MJVW, MMR, EB. Funding acquisition: JAH. Project administration: MJVW, JAH Supervision: MJVW, JAH. Writing – original draft: MJVW, MMR, EB, LTG. Writing – review & editing: MJVW, MMR, EB, JAH.
Corresponding author
Ethics declarations
Competing interests
JAH is founder and shareholder of HeioThera, Inc, which has licensed the variants of IL-10 described herein from the University of Chicago. The other authors declare that they have no competing interests.
Ethical approval
All the animal experiments performed in this work were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Chicago, and was reported in this manuscript using ARRIVE guidelines. All mouse experiments were performed under supervision with protocols approved by the University of Chicago IACUC, and the study was performed in accordance with the relevant guidelines and regulations.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
White, M.J.V., Raczy, M.M., Budina, E. et al. Engineering IL-10 and rapamycin to bind collagen leads to improved anti fibrotic efficacy in lung and kidney fibrosis. Sci Rep 15, 13279 (2025). https://doi.org/10.1038/s41598-025-94073-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94073-3
