
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infects host cells via its spike (S) protein, which binds to the angiotensin-converting enzyme 2 (ACE2) receptor. Glycans are thought to influence this interaction by modulating the binding affinity between the S protein and its receptor. In this study, we screened 300 carbohydrate species using a glycan array to identify potential ligands that interact with the S proteins of the Delta and Omicron variants. Among the identified candidates, two aminoglycoside antibiotics, tobramycin and sisomicin, exhibited notable binding to the S protein. Surface plasmon resonance (SPR), circular dichroism (CD), and in silico docking analyses confirmed direct interactions between these aminoglycosides and the S protein, revealing distinct binding characteristics. Nuclear magnetic resonance (NMR) analysis further localized the tobramycin-binding site within the receptor-binding domain (RBD) of the S protein. Tobramycin and sisomicin showed a tendency to inhibit SARS-CoV-2 replication in human induced pluripotent stem cell (hiPSC)-derived lung organoids, though the effect did not reach statistical significance. Docking simulations using the trimeric S model suggested that aminoglycosides bind at an inter-subunit interface. These findings demonstrate that aminoglycosides can directly interact with the SARS-CoV-2 S protein and may serve as scaffolds for developing host-independent antiviral agents against SARS-CoV-2 and its variants.
Similar content being viewed by others
Introduction
Coronavirus disease 2019 (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)1,2. Similar to SARS-CoV-1, which emerged in 2003, SARS-CoV-2 is a positive-sense, single-stranded RNA virus sharing approximately 80% genome sequence homology with SARS-CoV-13,4. These coronaviruses possess four structural proteins: envelope (E), membrane (M), nucleocapsid (N), and spike (S)5. The S protein, localized on the viral surface, mediates viral entry by binding to angiotensin-converting enzyme 2 (ACE2) on the host cell membrane6,7. It consists of two subunits, S1 and S2, with the receptor-binding domain (RBD) located in S1, which directly interacts with ACE28. The RBD of SARS-CoV-2 exhibits 10- to 20-fold higher affinity for ACE2 compared to that of SARS-CoV-19, which likely accounts for the high infectivity and global spread of SARS-CoV-2.
A notable feature of SARS-CoV-2 is its rapid mutation rate, enabling immune evasion through structural alterations of viral proteins10,11. Variants are classified primarily based on mutations within the S protein12,13. Among them, the Delta variant (L452R mutation), first identified in India, became globally dominant14,15. Subsequently, the Omicron variant, first reported in South Africa in 2021, was designated as a variant of concern (VOC)16,17. The Omicron variant carries 26–32 mutations in the S protein, which may enhance infectivity and confer resistance to neutralizing antibodies18,19.
Recent detailed mapping and structural analyses revealed that S proteins of the Delta and Omicron variants are highly glycosylated20,21. The S protein undergoes both N- and O-glycosylation mediated by host-cell enzymes22. Moreover, various techniques confirmed that the S protein can interact with sialic acid in an enzyme-independent manner. Li et al. confirmed the interaction between the S protein and N-acetylneuraminic acid (Neu5Ac), which is predominantly found in human cells, by surface plasmon resonance analysis and mass spectrometry23. They showed that sialic acid binds the cleft between the 2 receptor-binding domains of the S proteins23. Nuclear magnetic resonance analysis performed by Unione et al. confirmed that the sialic acid directly binds with the S protein24. They showed that sialic acid binding site is located in the N-terminal domain of the S protein24. Interaction of sialic acid with the N-terminal domain was also shown by single-molecule Förster resonance energy transfer imaging25. In addition, glycolipids containing sialic acid is suggested to facilitate infection of SARS-CoV-226.
Given that sialic acid possesses affinity with the S protein and its importance to viral infection, we investigated whether specific carbohydrates or glycans other than sialic acid could interact with the S protein. To this end, we screened 300 carbohydrates for their ability to bind to the S proteins of the Delta and Omicron variants using a commercial glycan array. We identified several binding candidates, notably two aminoglycoside antibiotics—tobramycin and sisomicin. We further characterized their molecular interactions with the S protein using surface plasmon resonance (SPR), circular dichroism (CD) titration, nuclear magnetic resonance (NMR), and in silico docking analyses. Finally, we assessed the antiviral effects of tobramycin and sisomicin using human induced pluripotent stem cell (hiPSC)-derived lung organoids. Our findings suggest that aminoglycoside antibiotics could serve as potential scaffolds for the development of novel antiviral agents capable of inducing conformational changes in the SARS-CoV-2 S protein.
Materials and methods
Glycan array
The commercially available biotinylated recombinant S-protein of the SARS-CoV-2 Delta variant (B.1.617.2; catalog No.: AVI10878, R&D Systems) and Omicron variant (B.1.1.529; catalog No.: AVI11061, R&D Systems) were dissolved in DDW to prepare a stock of 500 μg/mL. Glycan array was performed using the RayBio® Glycan Array 300 kit (RayBiotech), and the procedure was carried out with some modifications to the manual. This kit contains two array slides, and the S-protein of the Delta variant was applied to the first slide, and the that of the Omicron variant was applied to the other slide. Each slide contains four sub-arrays, and the amount of protein applied to each sub-array was 0 μg, 1 μg, 5 μg, and 10 μg, resulting in final concentrations of 0 μM, 0.17 μM, 0.83 μM, and 1.67 μM, respectively (Fig. S1a). After application, the slides were incubated overnight at 4°C with gentle shaking. The solution containing the recombinant protein was removed, and the slides were washed with wash buffer I (5 times for 5 min) and wash buffer II (5 times for 5 min). The solution containing Cy3 equivalent dye-streptavidin, contained in the kit, was applied to each sub-array, and the slides were incubated at room temperature for 1 h with gentle shaking while protected from light. After removal of the solution, the slides were washed with wash buffer I for 15 min, wash buffer II for 5 min, and distilled water for 5 min with gentle shaking. The slides were completely dried and sent to RayBiotech for array scanning and analysis services.
Surface plasmon resonance (SPR) analysis
The procedures used in this study were modified from the previous report and the online guidebook27,28. Surface plasmon resonance (SPR) analysis was performed using a Biacore 3000 instrument (GE Healthcare; Fig. S1b). Sensor Chip SA was used with 10 mM phosphate-buffered saline (PBS) containing 150 mM NaCl (pH 7.4) as the running buffer. Biotinylated S proteins (20 μM, 120 μL) from the Delta and Omicron variants were immobilized onto the chip surface via streptavidin–biotin interaction. Sisomicin and tobramycin were injected at various concentrations (0, 5, 10, 20, 40, and 80 μM) in PBS at a flow rate of 30 μL/min. A schematic of the experimental setup is shown in Fig. S1b. A reference flow cell (without immobilized protein) was used for background subtraction. Binding responses were analyzed using the BIAevaluation software (GE Healthcare) with the steady-state affinity model.
Titration experiment with circular dichroism (CD) spectrum
The CD spectra were recorded in a basal solution containing 20 mM phosphate (pH 7.0), 100 mM NaCl, 0.1 mg/mL of recombinant Delta and Omicron S protein, that were same as those used for glycan array. The basal solutions were titrated with tobramycin or sisomicin, to final concentrations of 0.083, 0.289, 1.11, 3.16, 11.3, 31.6, and 112 μM. At each titration point, the mixtures were incubated at 5 °C for 10 min to reach equilibrium. The CD spectra were recorded using a J-1100 CD spectrometer equipped with a Peltier-type temperature controller (JASCO corporation, Tokyo) . Each spectrum was recorded in the spectral range of 250–200 nm with a spectral resolution of 0.1 nm, and 16 scans were averaged per titration point. The baseline spectrum of the basal solution was also recorded under identical conditions. The obtained CD spectra were subsequently subjected to least-squares fitting.
Determination of dissociation constants by the least-squares fitting
The baseline spectrum was subtracted from each titration spectrum. For each ligand, CD intensities were extracted at 5 nm intervals within the 250–200 nm range and used for least-squares fitting. Dissociation constants (Kd) were calculated assuming 1:1 complexation between the S protein and the ligand. Under this assumption, the theoretical titration curve for a given wavelength can be expressed as:
where ΔCD is the difference in the theoretical CD intensity at each titration point from its initial value, CDini and CDfin are the initial and final CD intensities, [P]0 is the protein concentration, [S] is the ligand concentration at each titration point, and Kd is the dissociation constant29. The theoretical CD intensities were fitted to the experimental data by the least squares method using the Solver add-in in Microsoft Excel (Microsoft, Redmond, WA).
Binding analysis in solution by nuclear magnetic resonance (NMR)
Insert DNA encoding the RBD (residues 323–532) of Wuhan variant was amplified from pGBW-m4046887 and cloned into pET28a( +) vector (Novagen) with a hexahistidine tag and a TEV protease cleavage site at both the N- and C-terminal ends. Plasmid pGBW-m4046887 was a gift from Ginkgo Bioworks & Benjie Chen (Addgene plasmid #145,730; http://n2t.net/addgene:145730; RRID: Addgene_145730). Uniformly 15N-labeled RBD (residues 323–532) of the Wuhan variant was expressed in E. coli using in an H2O-based M9 medium containing 15NH4Cl (1 g/L) and Celtone® base powder [15N, 98% + , (CIL), 0.2 g/L] and prepared by refolding from inclusion bodies as previously reported30, followed by further purification. Uniformly 15N-labeled RBD at a concentration of 100 μM was titrated with tobramycin or sisomicin and monitored using 1H-15N HSQC spectra. All experiments were performed at 25 °C in buffer containing 20 mM MES-NaOH, pH 6.5, 50 mM NaCl, and 10% 2H2O. Data were collected using a Bruker AVANCE NEO 600 spectrometer equipped with a cryogenic probe, processed using Bruker TopSpin 4.1.4 software, and analyzed using Sparky (T. D. Goddard and D. G. Kneller, Sparky 3, University of California). Assignments of the RBD amide signals corresponding to 175 residues out of 198 (88%) were transferred from the previous studies using BMRB 5126830 or BMRB 5080931.
Molecular docking simulations
The structure of the SARS-CoV-2 S protein Delta variant (B.1.617.2) in the one RBD-up conformation (PDB ID: 7V7O) was obtained from the PDBj repository (https://www.rcsb.org). The PDB file was preprocessed using UCSF Chimera to generate a PDBQT file32. Three-dimensional structures of sisomicin and tobramycin were generated with MoleView (https://molview.org). Using Avogadro, an open-source molecular builder and visualization tool (Version 1.2.0; http://avogadro.cc/)33, the conformations of both structures were converted to chair conformations with substituents in stable equatorial positions and then subjected to conformational searches. The resulting stable conformers were then converted from MOL format to PDBQT format using UCSF Chimera. Preliminary docking simulations of sisomicin and tobramycin with the SARS-CoV-2 S protein were conducted using AutoDock Vina (version 1.2.7)34,35 to identify potential binding sites. The simulations were performed targeting the protruding region on the top of the S protein, which is known to be an important site for binding to ACE2. As a result, one promising binding site was found where both sisomicin and tobramycin bound with the highest binding score respectively. To achieve a more precise docking simulation, the grid domain was restricted to a smaller region encompassing 20 amino acid residues located within 3 Å of the ligand molecules (chain B: Arg401, Asp403, Glu404, Ile416, Tyr451, Tyr493, Tyr503, Arg406, Gln407, Thr413, Gly414, Lys415, chain C: Tyr367, Ser369, Ala370, Phe372, Ser373, Thr374, and Phe375), which were designated as flexible residues. Two PDBQT files were generated using AutoDockTools (available from https://ccsb.scripps.edu/mgltools/) to define rigid and flexible components of the protein. Flexible docking simulations were performed with AutoDock Vina under the following conditions: center_x = 210, center_y = 227, center_z = 260, size_x = 30, size_y = 30, size_z = 35, energy_range = 3, exhaustiveness = 30, and num_modes = 20. The docking results were visualized and analyzed using UCSF Chimera and PyMOL. The best binding poses yielded docking scores of –7.271 kcal/mol for sisomicin and –7.366 kcal/mol for tobramycin.
Ethics statement
Our study has been performed in accordance with ARRIVE guidelines. All protocols involving specimens from human subjects recruited at Kyoto University were reviewed and approved by the Institutional Review Boards of Kyoto University (approval ID: R2379-3). We used human induced pluripotent stem cells established from the individual who had provided informed consent. All methods involving human-derived materials were performed in accordance with the relevant ethical guidelines and regulations.
Cell line of human induced pluripotent stem cells (hiPSC)
The procedures used in this study were modified from the previous report36. The hiPSC line 1383D6 (provided by Dr. Masato Nakagawa, Kyoto University) was maintained on 0.5 μg/cm2 recombinant human laminin-511 E8 fragments (iMatrix-511; Nippi)-coated plates in StemFit AK02N medium (Ajinomoto Healthy Supply). Cell passaging was performed every six days. For passaging, cell colonies were treated with TrypLE Select Enzyme (Thermo Fisher Scientific) for 10 min at 37°C and seeded in StemFit AK02N medium (Ajinomoto Healthy Supply) containing 10 μM Y-27632 (FUJIFILM Wako Pure Chemical).
Preparing of lung organoids derived from hiPSC
The procedures used in this study were modified from the previous report37. To start the differentiation, hiPSC colonies were treated with TrypLE Select Enzyme (Thermo Fisher Scientific) for 10 min at 37°C. After centrifugation, cells were seeded onto Matrigel Growth Factor Reduced Basement Membrane (Corning, NY, USA)-coated cell culture plates (2.0 × 105 cells/4 cm2) and cultured for 2 days. The differentiation of the human lung organoids was performed in serum-free differentiation (SFD) medium, composed of DMEM/F12 (3:1) (FUJIFILM Wako Pure Chemical and Thermo Fisher Scientific) supplemented with N2 (FUJIFILM Wako Pure Chemical), B-27 Supplement Minus Vitamin A (Thermo Fisher Scientific), 50 μg/mL ascorbic acid (STEMCELL Technologies), 1 × GlutaMAX (Thermo Fisher Scientific), 1% monothioglycerol (FUJIFILM Wako Pure Chemical), 0.05% BSA (Sigma-Aldrich), and 100 U/mL penicillin, 100 μg/mL streptomycin (P/S; Thermo Fisher Scientific). During days 0–1 of differentiation, cells were cultured with SFD medium supplemented with 10 μM Y-27632 (FUJIFILM Wako Pure Chemical) and 100 ng/mL recombinant Activin A (R&D Systems). During days 1–3 of differentiation, cells were cultured with SFD medium supplemented with 10 μM Y-27632 (FUJIFILM Wako Pure Chemical), 100 ng/mL recombinant Activin A (R&D Systems) and 1% FBS. Between days 3–5 of differentiation, cells were cultured in SFD medium supplemented with 1.5 μM Dorsomorphin dihydrochloride (FUJIFILM Wako Pure Chemical) and 10 μM SB431542 (FUJIFILM Wako Pure Chemical) for 24 h, and then SFD medium supplemented with 10 μM SB431542 and 1 μM Stemolecule Wnt inhibitor IWP2 (FUJIFILM Wako Pure Chemical) for another 24 h. During days 5–12 of differentiation, cells were cultured with SFD medium supplemented with 3 μM CHIR99021 (FUJIFILM Wako Pure Chemical), 10 ng/mL human FGF10 (PeproTech, NJ, USA), 10 ng/mL human fibroblast growth factor (FGF) 10 (PeproTech), 10 ng/mL human bone morphogenetic protein (BMP), 20 ng/mL human epidermal growth factor (PeproTech), and all-trans retinoic acid (ATRA; Sigma-Aldrich). On day 12 of differentiation, cells were dissociated and embedded in the Matrigel Growth Factor Reduced Basement Membrane (Corning) to generate organoids. During days 12–20 of the differentiation, organoids were cultured in SFD medium containing 3 μM CHIR99021, 10 ng/mL human FGF10, 10 ng/mL human FGF7, 10 ng/mL human BMP4, and 50 nM ATRA. On day 20 of differentiation, organoids were recovered from the Matrigel gel, and the resulting suspension of organoids (small free-floating clumps) was seeded onto thin Matrigel-coated cell culture plates. The organoids were recovered from the Matrigel gel because the virus could not access the apical side of the epithelial cells if the organoids remained embedded in the Matrigel gel. During days 20–30 of differentiation, organoids were cultured in SFD medium containing 50 nM dexamethasone (Selleck Chemicals), 0.1 mM 8-bromo-cAMP (Tocris Bioscience), and 0.1 mM 3-isobutyl-1-methylxanthine (IBMX; FUJIFILM Wako Pure Chemical).
SARS-CoV-2 used for infectious experiments
The procedures used in this study were modified from the previous report38. The SARS-CoV-2 B.1.617.2 (Delta; GISAID accession number: EPI_ISL_9636792) or BA.1 (Omicron; GISAID accession number: EPI_ISL_9638489) variants were used isolated from the nasopharyngeal swab samples of COVID-19 patients. This study was approved by the research ethics committee of Kyoto University (approval ID: R2379-3). Viruses were replicated in TMPRSS2/Vero cells (JCRB1818, JCRB Cell Bank) and stored at −80 °C until use. TMPRSS2/Vero cells were cultured with Eagle’s Minimum Essential Media (FUJIFILM Wako Pure Chemical) supplemented with 5% FBS and P/S (Thermo Fisher Scientific). SARS-CoV-2 infection experiments were performed according to strict regulations in a biosafety level 3 facility at Kyoto University. Viral titers were measured by a median tissue culture infectious dose (TCID50) assay. TMPRSS2/Vero cells were seeded into 96-well cell culture plates (Thermo Fisher Scientific). Samples were serially diluted tenfold from 10−1 to 10−8 in cell culture medium, transferred onto the cells, and incubated at 37°C with 5% CO2 for 96 h. Cytopathic effects were evaluated under a microscope. TCID50/mL was calculated using the Reed-Muench method.
The protocol for the SARS-CoV-2 infection assay
The procedures used in this study were modified from the previous report38. The schematic protocol for the SARS-CoV-2 infection assay is shown in Fig. 6a. Briefly, hiPSC-derived lung organoids were seeded in a 96-well plate (2 × 104 cells/well). Cells were then treated with the indicated compounds, including sterilized Milli-Q water (filtered through a 0.22 µm filter) (0.5%), remdesivir (10 µM, GS-5739, MedChemExpress), tobramycin (500 µM), or sisomicin sulfate (500 µM), either alone or in combination with infection by the SARS-CoV-2 Delta or Omicron variants (MOI 0.1 each). After 6 h of incubation, the culture medium was replaced with fresh medium supplemented with Water (0.5%), remdesivir (10 μM), tobramycin (500 μM), or sisomicin (500 µM). The lung organoids were analyzed at 72 h post-infection.
Analysis cell viability in tobramycin or sisomicin-treated hiPSC-derived lung organoid by WST-8 assay
The procedures used in this study were modified from the previous report38. In Fig. 6a, cell viability in the non-infected hiPSC-derived lung organoids treated without (Mock) or with Water (0.5%), remdesivir (10 µM), tobramycin (500 µM), or sisomicin (500 µM) was determined at 72 h of incubation using a WST-8 assay with Cell Count Reagent SF (Nacalai Tesque), according to the manufacturer’s instructions.
Measuring of SARS-CoV-2 copy number grown in hiPSC-derived lung organoids
The procedures used in this study were modified from the previous report38. Cell culture supernatants from lung hiPSC-induced lung organoids in SARS-CoV-2 infection assay shown in Fig. 6a were mixed with an equal volume of 2 × RNA lysis buffer (distilled water containing 0.4 U/μL SUPERase IT™ RNase Inhibitor (Thermo Fisher Scientific), 2% Triton X-100, 50 mM KCl, 100 mM Tris–HCl [pH 7.5], and 40% glycerol) and incubated at 25°C for 10 min. The mixture was then diluted tenfold with distilled water. For the quantification of SARS-CoV-2 RNA (N-sarbeco), a One Step TB Green PrimeScript PLUS RT-PCR Kit (Perfect Real Time) (Takara Bio, Shiga, Japan) was used on a QuantStudio 1 or QuantStudio 3 real-time PCR system (Thermo Fisher Scientific). Standard curves were generated using SARS-CoV-2 RNA (105 copies/μL) purchased from Nihon Gene Research Laboratories (Miyagi, Japan). The primer sequences are following; SARS-CoV-2 RNA (N-sarbeco) Forward primer: AGC CTC TTC TCG TTC CTC ATC AC, Reverse primer: CCG CCA TTG CCA GCC ATT C.
Statistical analysis
All data are expressed as the mean ± standard error of the mean (SEM). Statistical analyses were conducted using GraphPad Prism version 10 (GraphPad Software). Differences among more than two groups were evaluated using one-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test. A p-value of less than 0.05 was considered statistically significant.
Results
Distinct glycan-binding profiles of delta and omicron S proteins
To characterize and to compare the glycan-binding properties of the SARS-CoV-2 S proteins from the Delta and Omicron variants, we analyzed their interactions with 300 glycans immobilized on a commercial glycan array. The mutation sites of the Delta and Omicron S proteins used in this study are summarized in Table S1. Different amounts of S protein (0, 1, 5, and 10 μg) were applied to the array, followed by washing and fluorescence detection. After background subtraction and normalization, the results are summarized in Table 1 and detailed in Table S2.
For the Delta variant, when 1 or 5 μg of S protein was applied, neomycin trisulfate, gentamicin sulfate, tobramycin, and sisomicin sulfate ranked highest in binding intensity (Table 1, Delta variant, columns F[1 μg–0 μg] and F[5 μg–0 μg]). When 10 μg was applied, the top 14 binding glycans included multiple aminoglycosides—sisomicin, tobramycin, kanamycin, neomycin, gentamicin, and geneticin (G418)—along with several monosaccharides and sialylated glycans (Table 1, Delta variant, column F[10 μg–0 μg]; Fig. S2).
In contrast, the Omicron variant exhibited a distinct glycan-binding pattern. The top-ranked glycan was Neu5Gc-α-2,3-Gal-β-1,3-(Fuc-α-1,4)-GlcNAc-β- [Sialyl Lewis A]-Sp, followed by two other Neu5Gc-containing glycans in ranks 3 and 4. Interestingly, Neu5Gc is absent in humans due to an irreversible mutation in the CMAH gene39, suggesting a potential evolutionary process of the Omicron variant. Neu5Ac-α-2,3-Gal-β-1,3-GlcNAc-β-Sp was commonly detected at all S protein concentrations. Among aminoglycosides, only tobramycin appeared within the top 10 binding glycans for Omicron.
These findings indicate that the glycan-binding profiles of the Delta and Omicron S proteins differ substantially (Fig. 1). Dose-dependent binding curves showed that all aminoglycosides bound the Delta S protein strongly but exhibited little or no binding to the Omicron S protein. Sisomicin displayed clear dose-dependent binding to the Delta S protein, while tobramycin bound both variants, albeit more weakly to Omicron. Interestingly, fluorescence signals for sisomicin increased proportionally with protein concentration (Fig. 1b), whereas those for tobramycin, gentamicin, and neomycin peaked at 1 μg and then declined (Fig. 1a,c,d), possibly due to steric hindrance or biotin masking at elevated protein densities.

Glycan array results. Focusing on aminoglycoside antibiotics [(a) tobramycin, (b) sisomicin sulfate, (c) gentamisin sulfate, (d) neomycin trisulfate, (e) kanamycin sulfate, and (f) geneticin disulfate salt (G418)], the change in fluorescence intensity with increasing amount of S protein of Delta and Omicron variants is shown. All values represent the average fluorescence intensity of triplet. The amount of S protein applied was 0 μg, 1 μg, 5 μg, and 10 μg, resulting in final concentrations of 0 μM, 0.17 μM, 0.83 μM, and 1.67 μM, respectively.
Surface plasmon resonance confirms binding differences between delta and omicron S proteins
To quantify binding kinetics, we examined the interactions of tobramycin and sisomicin with the Delta and Omicron S proteins by surface plasmon resonance (SPR). Sensorgrams obtained from the injection of tobramycin at various concentrations over immobilized Delta S protein (Fig. S1b) indicated rapid equilibrium binding, allowing steady-state affinity analysis. The dissociation constant (Kd) for tobramycin binding to the Delta S protein was ~ 8.3 μM (Fig. 2a), whereas the Kd values for tobramycin–Omicron, sisomicin–Delta, and sisomicin–Omicron interactions were 23 μM, 33 μM, and 63 μM, respectively (Fig. 2b–d). Thus, the Delta S protein exhibited higher affinity than the Omicron S protein, and tobramycin bound more strongly than sisomicin.

SPR Sensorgrams of tobramycin or sisomicin binding to the S protein. Top: Raw data of SPR sensorgrams. Bottom: Steady-state plot of equilibrium response versus antibiotics concentration. The solid line represents the fit derived from steady-state affinity analysis. (a) Tobramycin with the Delta variant S protein (Kd = 8.3 μM), (b) tobramycin with the Omicron variant S protein (Kd = 23 μM), (c) sisomicin with the Delta variant S protein (Kd = 33 μM), and (d) sisomicin with the Omicron variant S protein (Kd = 63 μM).
Circular dichroism analysis reveals higher binding affinity of tobramycin
To evaluate binding under solution conditions with the free form of the S protein, circular dichroism (CD) spectroscopy was performed. For the Delta S protein, titration with tobramycin produced pronounced spectral changes that saturated rapidly, suggesting a high-affinity interaction with a Kd below 1.0 × 10⁻10 M (Fig. S3 and Fig. 3a). In contrast, sisomicin induced only modest spectral changes, with a Kd of 1.3 × 10⁻⁷ M (Fig. S3 and Fig. 3c). For the Omicron S protein, tobramycin and sisomicin produced Kd values of 1.1 × 10⁻⁶ M and 4.9 × 10⁻⁶ M, respectively (Fig. S3 and Fig. 3b, d). These data indicate that both aminoglycosides bind more strongly to the Delta than to the Omicron S protein, and that tobramycin consistently exhibits higher affinity than sisomicin.

Plots of CD intensity change at 210 nm (ΔCD210) against the ligand concentration (titration curves). (a) Titration curve of the Delta S protein with tobramycin. The inset indicates the titration curve at the lower ligand concentration region. (b) Titration curve of the Omicron S protein with tobramycin. (c) Titration curve of the Delta S protein with sisomicin. (d) Titration curve of the Omicron S protein with sisomicin. In each panel, experimental data are shown as red circles, and theoretical curves are depicted as black lines. The determined Kd values are displayed in the respective panels.
Tobramycin directly binds to the receptor-binding domain of the S protein
Because ACE2 binding occurs through the receptor-binding domain (RBD) of the S protein, we investigated whether tobramycin or sisomicin interacts with the isolated RBD using NMR spectroscopy. The Wuhan RBD was used in these NMR experiments because backbone resonance assignments are available for this construct, enabling residue-level mapping of ligand-induced perturbations, and because the overall RBD fold is highly conserved among SARS-CoV-2 variants. Uniformly 15N-labeled RBD (residues 323–532, Wuhan variant) was titrated with each ligand. Sisomicin caused no detectable spectral changes even at a tenfold molar excess, indicating no interaction (Fig. S4). In contrast, tobramycin induced observable spectral changes at tenfold molar excess (Fig. 4a), indicating direct binding to the RBD. Addition of tobramycin at tenfold molar excess caused the disappearance or significant intensity reductions of 29 amide signals originating from the main-chain RBD residues (Fig. 4a). Among them, 22 signals corresponded to residues that had been previously assigned based on the RBD resonance data (covering 88% of residues; Fig. S5), allowing their location to be mapped onto the structure (Fig. 4b). The remaining 7 disappearing signals corresponded to unassigned peaks. Even at 15-fold molar excess (1.5 mM) or higher, signal intensity further decreased and ultimately many signals completely disappeared, without reaching a plateau. The residue-specific disappearance of amide signals suggests direct binding of tobramycin to the monomeric RBD. Such signal disappearance is attributable to exchange broadening caused by multiple binding states of tobramycin. Combined with the absence of detectable binding at one equivalent, the Kd was estimated to be ≥ 10⁻4 M. The affected residues were located in loop regions, including those forming the ACE2-binding interface as well as other loop regions (Fig. 4b), suggesting that multiple tobramycin molecules interact with the RBD. Seventeen out of the 22 residues are conserved among the three variants (Wuhan, Delta, and Omicron), suggesting a conserved binding mode (Fig. S6). Most of these residues were hydrophilic or glycine (16 out of 22 residues).

NMR analysis of the interaction between the RBD and tobramycin. (a) 1H-15N HSQC spectra of uniformly 15N-labeled RBD (100 μM) in the absence (black) and presence (red) of tenfold molar excess of tobramycin are overlaid. Signals that disappeared upon addition of tobramycin are indicated by boxes and labeled in blue. Asterisks indicate signals without assignments. (b) Residues with disappeared or significantly reduced signals upon addition of tenfold molar excess of tobramycin (as shown in a) were mapped on the structure of the RBD in complex with the ACE2 peptidase domain (PDB ID: 6m0j). RBD and ACE2 are shown as cartoon and surface representations, respectively. Residues not analyzed are colored black.
In silico docking predicts tobramycin binding at the inter-subunit interface of the S trimer
Molecular docking simulations were conducted using the full-length trimeric S protein (one-RBD-up model; PDB ID: 7V7O) to identify the aminoglycoside-binding site within its three-dimensional structure and to clarify the relationship between RBD binding and binding to the full-length S protein. Using AutoDock Vina34,35, both tobramycin and sisomicin were predicted to bind within the cleft between the closed B and C chains of the trimer (Fig. 5a–d), rather than at the ACE2-binding interface. This binding mode may explain the stronger affinity for the full-length trimeric S protein and lack of inhibition of viral entry.

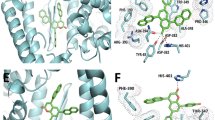
Binding sites for tobramycin and sisomicin on the S protein predicted using in silico simulation. The protein surface is displayed as a semi-transparent surface, and the ligands are shown in stick representation or as a space-filling model. (a) and (c): binding state of tobramycin, (b) and (d): binding state of sisomicin, red: chain A, blue: chain B, green: chain C, orange dashed line: RBD. Purple circles show the tobramycin and sisomicin. Interaction analysis between ligands, (e) tobramycin (orange) and (f) sisomicin (orange), and the S protein. Blue lines: hydrogen bonds, dashed lines: van der Waals interaction between hydrophobic groups, yellow lines: salt bridges.
The predicted binding energies were –7.271 kcal/mol (sisomicin) and –7.366 kcal/mol (tobramycin), indicating slightly stronger binding by tobramycin. Structural analysis using PLIP and PyMOL40,41 revealed that sisomicin forms one van der Waals interaction between hydrophobic groups, one salt bridge, and six hydrogen bonds, whereas tobramycin forms one van der Waals interaction between hydrophobic groups, one salt bridge, and ten hydrogen bonds (Fig. 5e,f). The average hydrogen bond donor–acceptor distances were 2.66 Å (sisomicin) and 2.69 Å (tobramycin), suggesting that the higher affinity of tobramycin may derive from the greater number of hydrogen bonds.
Limited antiviral activity of tobramycin or sisomicin in hiPSC-derived lung organoids
Because aminoglycosides bind to the full-length S protein and NMR data indicated binding of tobramycin to the RBD, we examined whether this interaction inhibits viral infection. Human iPSC-derived lung organoids were treated with tobramycin (500 μM) or sisomicin (500 μM), either alone or in combination with infection by the SARS-CoV-2 Delta or Omicron variants (Fig. 6a). Water (0.5%) and remdesivir (10 μM) served as negative and positive controls, respectively. At 72 h post-infection, neither tobramycin nor sisomicin affected organoid viability (Fig. 6b). When applied to the Delta variant of SARS-CoV-2, sisomicin and tobramycin reduced the average viral copy number by approximately 30% and 80%, respectively (Table S3, Fig. 6c). These drugs exhibited similar effects on the Omicron variant, reducing the average viral copy number by approximately 66% and 62%, respectively (Table S3, Fig. 6d, tobramycin: p = 0.053; sisomicin: p = 0.088 vs Water). Although tobramycin and sisomicin tended to reduce viral copy numbers (Fig. 6c, d), the statistical analysis indicated that these differences were not significant.

Analysis of the SARS-CoV-2 growth in hiPSC-derived lung organoids treated with tobramycin or sisomicin. (a) A schematic representation of the protocol for infection assay using hiPSC-derived lung organoids infected with SARS-CoV-2 B.1.617.2 (Delta) or BA.1 (Omicron) variants. (b) Cell viability in non-infected hiPSC-derived lung organoids treated without (Mock) or with Water (0.5%), remdesivir (10 µM), tobramycin (500 µM), or sisomicin (500 µM) (n = 3 each) was assessed using the WST-8 assay. Data are presented as mean ± SEM and are representative of three independent experiments. (c, d) The viral copy number in the culture supernatant of hiPSC-derived lung organoids treated with Water (0.5%), remdesivir (10 µM), tobramycin (500 µM), or sisomicin (500 µM) (n = 3 each) in combination with infection by the SARS-CoV-2 Delta (c) or Omicron (d) variants were measured by RT-qPCR. “Mock” indicates uninfected and untreated control samples. Data are presented as mean ± SEM and are representative of three independent experiments. p = 0.053, p = 0.088, and ***p < 0.001 indicates significant differences compared to the Water-treated infected group, as determined by one-way ANOVA followed by Dunnett’s post hoc test. Results were reproducible through these experiments.
Discussion
This study employed a glycan array to identify carbohydrates capable of binding to the SARS-CoV-2 S protein in an enzyme-independent manner and revealed a diverse range of glycan interactions (Table 1). Glycan arrays are widely used to profile carbohydrate–protein interactions across various viral species42. Among the identified ligands, the aminoglycosides tobramycin and sisomicin exhibited prominent binding signals and were therefore examined in detail. Tobramycin has previously been reported to inhibit HIV replication in cultured cells43. We initially hypothesized that aminoglycosides would be active against the intact viral particles and thereby inhibit viral growth. However, our findings indicate that binding to the S protein does not necessarily translate into effective viral inhibition. We therefore propose that conjugation of aminoglycosides with an additional functional domain may be required to enhance their antiviral activity, representing a potential strategy for the development of anti-SARS-CoV-2 therapeutics.
In glycan array experiments, signal intensities decrease once binding becomes saturated (Fig. 1). There are likely optimal concentrations of S protein for detecting binding to aminoglycosides, this optimal concentration appears to differ between Delta and Omicron variants, possibly due to differences in their amino acid sequences. We suggest that the optimal S protein concentrations for neomycin and tobramycin are distinct. At 10 μg, the S protein exhibited stronger binding to Neu5Ac-α-2,8-Neu5Ac-α-2,6-Gal-β-1,4-Glc-Sp than to Neomycin (Table 1), indicating that the optimal S protein concentration differs between Neomycin and sialylated glycans. In glycan arrays, the glycans are immobilized on the chip surface, whereas in SPR experiments, the S protein is immobilized. We believe that these differences in experimental configuration contribute to the discrepancies observed between the results obtained by these methods.
SPR and CD analyses demonstrated that both antibiotics bind more strongly to the Delta S protein than to the Omicron variant, with tobramycin consistently exhibiting higher affinity (Table 2, Figs. 2, 3 and 4). NMR spectroscopy confirmed direct binding of tobramycin to the RBD, and docking simulations suggested that it bridges the interface between S subunits (Fig. 5). Together, these combined results support a model in which aminoglycosides recognize interfacial or loop regions of the S protein.
The apparent discrepancy between the affinities obtained by SPR and NMR likely reflects differences in the protein constructs and interaction modes probed by each technique. SPR measurements were performed using the trimeric full-length S protein, which presents a broad range of potential binding surfaces, including inter-domain and inter-subunit regions. In contrast, NMR experiments were conducted using the isolated monomeric RBD, in which the trimeric interfaces and associated conformational dynamics are absent. The much weaker and more widely distributed perturbations observed by NMR therefore most likely represent multiple low-affinity or partially non-specific contacts rather than a single well-defined binding site. Consequently, the higher affinity observed by SPR can be interpreted as the cumulative effect of tobramycin binding to regions outside the RBD, consistent with the larger and more complex interaction surface of the trimeric S protein. In glycan array experiments, glycans are immobilized on the array surface, whereas in SPR measurements the S protein is immobilized. In contrast, in CD measurements neither the glycans nor the S protein are immobilized. These methodological differences are reflected in the experimental outcomes, as summarized in Table 2. Furthermore, NMR analysis was performed using only the RBD, allowing the identification of specific interacting residues. Although the NMR experiments were conducted using the Wuhan RBD, most of the affected residues are conserved among the Wuhan, Delta, and Omicron variants (Fig. S6), suggesting that the observed weak contacts reflect a broadly shared interaction tendency. Variant-dependent differences detected for the full-length S protein likely arise from the trimeric context and glycan shielding effects that are not captured by the isolated RBD.
Despite measurable binding, tobramycin and sisomicin showed a tendency to inhibit SARS-CoV-2 replication in hiPSC-derived lung organoids (Fig. 6, tobramycin: p = 0.053; sisomicin: p = 0.088 vs Water), and this effect did not reach statistical significance. For the Delta variant, one tobramycin data point fell significantly outside the expected range (Table S3), resulting in a lack of overall statistical significance. This discrepancy likely arises because the recombinant S proteins used in binding assays lack native glycosylation, rendering binding sites more accessible. In contrast, intact virions possess a dense glycan shield that sterically hinders aminoglycoside access to the RBD and inter-subunit clefts. Thus, glycan shielding likely limits the antiviral efficacy of these antibiotics in cellular systems.
Consistent with the binding data, docking simulations suggested that tobramycin binds within interfacial regions of the trimeric S protein that are not accessible in the monomeric RBD. Although the docking model represents a static approximation, the qualitative agreement supports the possibility of interfacial aminoglycoside binding, which warrants further validation by mutagenesis or cryo-EM studies.
The sialic acid–binding pocket has been reported to be located around residues 18–7744, which is distinct from the aminoglycoside-binding site. The Neu5Ac9NAc-binding pocket comprises three regions (residues 99–246, 200–466, and 391–983)45. Although these regions partially overlap with those identified in our study, the corresponding amino acids (Arg355, Cys391, Phe464, and Arg466) differ from those involved in aminoglycoside binding. The heme-binding pocket has been reported to be located around residues 99–22646, which is also distinct from the aminoglycoside-binding site. Similarly, the furin-binding pocket has been reported to be located around residues 682–68647, a region that does not overlap with the aminoglycoside-binding site.
In contrast, the fatty acid–binding pocket overlaps with the aminoglycoside-binding pocket (Fig. S7)48. Phe377, Arg408, and Gln409 have been identified as residues that interact with both linoleic acid and aminoglycosides in silico. In addition, Lys417 has been identified as an interacting residue for both linoleic acid49 and aminoglycosides by in silico analysis and NMR experiments. These observations suggest that the fatty acid–binding pocket and the aminoglycoside-binding pocket are closely related and functionally relevant.
In addition to tobramycin and sisomicin, the glycan array identified other aminoglycosides—neomycin, gentamicin, kanamycin, and geneticin—as potential S protein-binding compounds (Table 2). Kanamycin, in particular, exhibited strong binding at low S protein concentrations and has previously been reported to interact with the RBD50. Docking studies have also predicted that paromomycin, plazomicin, ribostamycin, and amikacin can bind to the SARS-CoV-2 RBD50,51. These were not included in our glycan array and remain to be evaluated experimentally.
Several of these aminoglycosides exhibit antiviral activity against other viruses, acting through diverse mechanisms. For example, a kanamycin derivative (1-N-eicosanoyl-3″-N-trifluoroacetyl kanamycin A) inactivates influenza virus by inhibiting uncoating52 and suppresses herpes simplex virus type 2 replication53. Neomycin inhibits HSV-1 attachment via glycoprotein C blockade54 and suppresses Megalocytivirus replication by modulating host redox state55. Geneticin inhibits bovine viral diarrhea virus56 and dengue virus57. Aminoglycosides can also target viral RNA, as demonstrated for influenza and HIV, where they inhibit RNA transcription and translation58,59. Tobramycin, neomycin, and gentamicin inhibit ribozyme self-cleavage in hepatitis delta virus60.
Collectively, these findings suggest that aminoglycosides represent a structurally versatile scaffold for antiviral drug development. Chemical modification of tobramycin or sisomicin to enhance their interaction with the S protein—potentially inducing conformational disruption—could provide a foundation for the development of novel anti-SARS-CoV-2 therapeutics.
This study has several limitations. First, our investigation primarily focused on in vitro and in silico binding assays. A comprehensive evaluation of antiviral activity, including in vivo studies, remains an important challenge for future research. Because aminoglycosides are already used clinically as antibacterial agents, their pharmacokinetics and other in vivo properties have been well documented. However, although our data demonstrate that aminoglycosides can bind to the SARS-CoV-2 S protein, their actual antiviral efficacy remains unclear. In addition, this study evaluated only the interaction between aminoglycosides and the SARS-CoV-2 S protein. Broader exploration of structure–activity relationships across a wider range of aminoglycoside derivatives will be necessary. Addressing these limitations in future studies will be essential to validate the potential use of aminoglycosides as anti–SARS-CoV-2 agents.
Data availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information files. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Holmes, E. C. et al. The origins of SARS-CoV-2: A critical review. Cell 184, 4848–4856. https://doi.org/10.1016/j.cell.2021.08.017 (2021).
Meyer, N. J., Gattinoni, L. & Calfee, C. S. Acute respiratory distress syndrome. Lancet 398, 622–637. https://doi.org/10.1016/s0140-6736(21)00439-6 (2021).
Hatakeyama, D., Masuda, T., Miki, R., Ohtsuki, S. & Kuzuhara, T. In-vitro acetylation of SARS-CoV and SARS-CoV-2 nucleocapsid proteins by human PCAF and GCN5. Biochem. Biophys. Res. Commun. 557, 273–279. https://doi.org/10.1016/j.bbrc.2021.03.173.4 (2021).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. https://doi.org/10.1038/s41586-020-2012-7 (2020).
Lu, R. et al. Genomic characterization and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 395, 565–574. https://doi.org/10.1016/s0140-6736(20)30251-8 (2020).
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454. https://doi.org/10.1038/nature02145 (2003).
Delgado, J. et al. Molecular basis for higher affinity of SARS-CoV-2 spike RBD for human ACE2 receptor. Proteins 89, 1134–1144. https://doi.org/10.1002/prot.26086 (2021).
Wang, Q. et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181, 894–904. https://doi.org/10.1016/j.cell.2020.03.045 (2020).
Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263. https://doi.org/10.1126/science.abb2507 (2020).
Acter, T. et al. Evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) as coronavirus disease 2019 (COVID-19) pandemic: A global health emergency. Sci. Total Environ. 730, 138996. https://doi.org/10.1016/j.scitotenv.2020.138996 (2020).
Kumar, A. et al. Emerging SARS-CoV-2 variants can potentially break set epidemiological barriers in COVID-19. J. Med. Virol. 94, 1300–1314. https://doi.org/10.1002/jmv.27467 (2022).
Harvey, W. T. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 19, 409–424. https://doi.org/10.1038/s41579-021-00573-0 (2021).
Chen, K. W. K. et al. SARS-CoV-2 variants: Evolution, spike protein, and vaccines. Biomed. J. 45, 573–579. https://doi.org/10.1016/j.bj.2022.04.006 (2022).
Singh, P. et al. Genomic characterization unravelling the causative role of SARS-CoV-2 Delta variant of lineage B.1.617.2 in 2nd wave of COVID-19 pandemic in Chhattisgarh, India. Microb. Pathog. 164, 105404. https://doi.org/10.1016/j.micpath.2022.105404 (2022).
Yazdanpanah, F. et al. COVID-19 Delta variation; more contagious or more pernicious?. Acta Biomed. 92, e2021454. https://doi.org/10.23750/abm.v92i6.12181 (2022).
Wang, L. & Cheng, G. Sequence analysis of the emerging SARS-CoV-2 variant Omicron in South Africa. J. Med. Virol. 94, 1728–1733. https://doi.org/10.1002/jmv.27516 (2022).
Yang, W. & Shaman, J. L. COVID-19 pandemic dynamics in South Africa and epidemiological characteristics of three variants of concern (Beta, Delta, and Omicron). Elife 11, e78933. https://doi.org/10.7554/elife.78933 (2022).
Lewnard, J. A. et al. Association of SARS-CoV-2 BA.4/BA.5 Omicron lineages with immune escape and clinical outcome. Nat. Commun. 14, 1407. https://doi.org/10.1038/s41467-023-37051-5 (2023).
Shi, H. et al. Immune escape of SARS-CoV-2 variants to therapeutic monoclonal antibodies: A system review and meta-analysis. Virol. J. 20, 266. https://doi.org/10.1186/s12985-023-01977-5 (2023).
Watanabe, Y., Allen, J. D., Wrapp, D., McLellan, J. S. & Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 369, 330–333. https://doi.org/10.1126/science.abb9983 (2020).
Xie, Y. & Butler, M. Quantitative profiling of N-glycosylation of SARS-CoV-2 spike protein variants. Glycobiology 33, 188–202. https://doi.org/10.1093/glycob/cwad007 (2023).
Campos, D., Girgis, M. & Sanda, M. Site-specific glycosylation of SARS-CoV-2: Big challenges in mass spectrometry analysis. Proteomics 22, e2100322. https://doi.org/10.1002/pmic.202100322 (2022).
Li, B. et al. Identification of potential binding sites of sialic acids on the RBD domain of SARS-CoV-2 spike protein. Front. Chem. 9, 659764. https://doi.org/10.3389/fchem.2021.659764 (2021).
Unione, L. et al. The SARS-CoV-2 spike glycoprotein directly binds exogeneous sialic acids: A NMR view. Angew. Chem. Int. Ed. Engl. 61, e202201432. https://doi.org/10.1002/anie.202201432 (2022).
Díaz-Salinas, M. A., Jain, A., Durham, N. D. & Munro, J. B. Single-molecule imaging reveals allosteric stimulation of SARS-CoV-2 spike receptor binding domain by host sialic acid. Sci. Adv. 10, eadk4920. https://doi.org/10.1126/sciadv.adk4920 (2024).
Nguyen, L. et al. Sialic acid-containing glycolipids mediate binding and viral entry of SARS-CoV-2. Nat. Chem. Biol. 18, 81–90. https://doi.org/10.1038/s41589-021-00924-1 (2022).
Kuzuhara, T., Sei, Y., Yamaguchi, K., Suganuma, M. & Fujiki, H. DNA and RNA as new binding targets of green tea catechins. J. Biol. Chem. 281, 17446–17456. https://doi.org/10.1074/jbc.m601196200 (2006).
Get a head start in SPR assay development: Application guides and handbooks. https://www.cytivalifesciences.com/en/us/solutions/protein-research/Interaction-analysis-with-Biacore-surface-plasmon-resonance-SPR/Get-started-with-surface-plasmon-resonance-SPR-interaction-analysis#Applicationguideshandbook
Bloomfield, V. A., Crothers, D. M. & Tinoco, I. Nucleic acids: structures, properties and functions (University Science Books, 2000).
Creutznacher, R. et al. NMR experiments provide insights into ligand-binding to the SARS-CoV-2 spike protein receptor-binding domain. J. Am. Chem. Soc. 144, 13060–13065. https://doi.org/10.1021/jacs.2c05603 (2022).
Schoenle, M. V. et al. NMR based SARS-CoV-2 antibody screening. J. Am. Chem. Soc. 143, 7930–7934. https://doi.org/10.1021/jacs.1c03945 (2021).
Pettersen, E. F. et al. UCSF Chimera--A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. https://doi.org/10.1002/jcc.20084 (2004).
Hanwell, M. D. et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 17. https://doi.org/10.1186/1758-2946-4-17 (2012).
Trott, O. & Olson, A. J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. https://doi.org/10.1002/jcc.21334 (2010).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61, 3891–3898. https://doi.org/10.1021/acs.jcim.1c00203 (2021).
Nakagawa, M. et al. A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci. Rep. 4, 3594. https://doi.org/10.1038/srep03594 (2014).
Hashimoto, R. et al. Human iPS cell-derived respiratory organoids as a model for respiratory syncytial virus infection. Life Sci. Alliance 8, e202402837. https://doi.org/10.26508/lsa.202402837 (2025).
Hashimoto, R. et al. Evaluation of broad anti-coronavirus activity of autophagy-related compounds using human airway organoids. Mol. Pharm. 20, 2276–2287. https://doi.org/10.1021/acs.molpharmaceut.3c00114 (2023).
Liu, Y., Li, J. & Liu, Q. Inactivation of the CMAH gene and deficiency of Neu5Gc play a role in human brain evolution. Inflamm. Regen. 45, 5. https://doi.org/10.1186/s41232-025-00368-3 (2025).
Salentin, S., Schreiber, S., Haupt, V. J., Adasme, M. F. & Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 43, W443–W447. https://doi.org/10.1093/nar/gkv315 (2015).
The PyMOL molecular graphics system, Version 3.0. Schrödinger, LLC.
Stencel-Baerenwald, J. E., Reiss, K., Reiter, D. M., Stehle, T. & Dermody, T. S. The sweet spot: Defining virus-sialic acid interactions. Nat. Rev. Microbiol. 12, 739–749. https://doi.org/10.1038/nrmicro3346 (2014).
Baker, T. J., Luedtke, N. W., Tor, Y. & Goodman, M. Synthesis and anti-HIV activity of guanidinoglycosides. J. Org. Chem. 65, 9054–9058. https://doi.org/10.1021/jo001142e (2000).
Awasthi, M. et al. The sialoside-binding pocket of SARS-CoV-2 spike glycoprotein structurally resembles MERS-CoV. Viruses 12, 909. https://doi.org/10.3390/v12090909 (2020).
Oh, L., Varki, A., Chen, X. & Wang, L. P. SARS-CoV-2 and MERS-CoV spike protein binding studies support stable mimic of bound 9-O-acetylated sialic acids. Molecules 27, 5322. https://doi.org/10.3390/molecules27165322 (2022).
Freeman, S. L. et al. Heme binding to the SARS-CoV-2 spike glycoprotein. J. Biol. Chem. 299, 105014. https://doi.org/10.1016/j.jbc.2023.105014 (2023).
Bertelli, A. et al. Role of Q675H mutation in improving SARS-CoV-2 spike interaction with the furin binding pocket. Viruses 13, 2511. https://doi.org/10.3390/v13122511 (2021).
Toelzer, C. et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 370, 725–730. https://doi.org/10.1126/science.abd3255 (2020).
Queirós-Reis, L., Mesquita, J. R., Brancale, A. & Bassetto, M. Exploring the fatty acid binding pocket in the SARS-CoV-2 spike protein: Confirmed and potential ligands. J. Chem. Inf. Model. 63, 7282–7298. https://doi.org/10.1021/acs.jcim.3c00803 (2023).
Prajapat, M. et al. Virtual screening and molecular dynamics study of approved drugs as inhibitors of spike protein S1 domain and ACE2 interaction in SARS-CoV-2. J. Mol. Graph. Model. 101, 107716. https://doi.org/10.1016/j.jmgm.2020.107716 (2020).
Tariq, A., Mateen, R. M., Afzal, M. S. & Saleem, M. Paromomycin: A potential dual targeted drug effectively inhibits both spike (S1) and main protease of COVID-19. Int. J. Infect. Dis. 98, 166–175. https://doi.org/10.1016/j.ijid.2020.06.063 (2020).
Yamana, Y. et al. Inhibition of influenza A virus replication by a kanamycin derivative. Antiviral Res. 15, 171–182. https://doi.org/10.1016/0166-3542(91)90064-x (1991).
Rahnama, S., Irani, M. A., Amininasab, M. & Ejtehadi, M. R. S494 O-glycosylation site on the SARS-CoV-2 RBD affects the virus affinity to ACE2 and its infectivity; a molecular dynamics study. Sci. Rep. 11, 15162. https://doi.org/10.1038/s41598-021-94602-w (2021).
Herold, B. C. & Spear, P. G. Neomycin inhibits glycoprotein C (gC)-dependent binding of herpes simplex virus type 1 to cells and also inhibits postbinding events in entry. Virology 203, 166–171. https://doi.org/10.1006/viro.1994.1469 (1994).
Deng, H. et al. Neomycin inhibits Megalocytivirus infection in fish by antagonizing the increase of intracellular reduced glutathione. Fish Shellfish Immunol. 127, 148–154. https://doi.org/10.1016/j.fsi.2022.06.016 (2022).
Birk, A. V., Dubovi, E. J., Zhang, X. & Szeto, H. H. Antiviral activity of geneticin against bovine viral diarrhoea virus. Antivir. Chem. Chemother. 19, 33–40. https://doi.org/10.1177/095632020801900105 (2008).
Zhang, X. G. et al. Antiviral activity of geneticin against Dengue virus. Antiviral Res. 83, 21–27. https://doi.org/10.1016/j.antiviral.2009.02.204 (2009).
Sullivan, J. M., Goodisman, J. & Dabrowiak, J. C. Absorption studies on aminoglycoside binding to the packaging region of human immunodeficiency virus type-1. Bioorg. Med. Chem. Lett. 12, 615–618. https://doi.org/10.1016/s0960-894x(01)00823-x (2002).
Kim, H. et al. Aminoglycoside antibiotics bind to the influenza A virus RNA promoter. Mol. Biosyst. 8, 2857–2859. https://doi.org/10.1039/c2mb25333j (2012).
Chia, J.-S., Wu, H.-L., Wang, H.-W., Chen, D.-S. & Chen, P.-J. Inhibition of hepatitis Delta virus genomic ribozyme self-cleavage by aminoglycosides. J. Biomed. Sci. 4, 208–216. https://doi.org/10.1007/bf02253420 (1997).
Acknowledgements
Circular dichroism (CD) measurements and analyses were conducted in collaboration with the Center for Instrumental Analysis, Faculty of Pharmaceutical Sciences, Tokushima Bunri University.
Funding
This study was supported by Tokushima Bunri University for Educational Reform and Collaborative Research (TBU2024-6-3) to D.H., Japan Agency for Medical Research and Development (AMED) (JP21gm1610005, JP23fk0108590) to K.T., and Nanken-Kyoten, Science Tokyo 2025-38 to K.T.
Author information
Authors and Affiliations
Contributions
D.H. performed the glycan array and wrote the manuscript. M.S., R.H. and K.T. performed the infectious experiment using hiPSC-derived lung organoids. Y.M., K.N. and Y.T. performed the CD titration analysis. S.U. and K.Y. performed the SPR binding analysis. M.Y and M.O. performed the NMR analysis. Y.K. and H.I. performed in silico simulation. T.K. wrote the manuscript and supervised this study.
Corresponding author
Ethics declarations
Competing interests
The authors have declared that no competing interests exist.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hatakeyama, D., Shoji, M., Miki, Y. et al. Glycan-binding properties of SARS-CoV-2 spike proteins: interactions with aminoglycoside antibiotics. Sci Rep 16, 12769 (2026). https://doi.org/10.1038/s41598-026-42404-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-42404-3
