
Abstract
Primary Immune Thrombocytopenia (ITP) is an autoimmune disease characterized by thrombocytopenia and bleeding tendency. Exosomes mediate abnormal crosstalk between immune cells and megakaryocytes in ITP, suggesting that exosome-related genes may serve as potential candidates for understanding disease pathogenesis. ITP transcriptome data and exosome-related genes (ERGs) were retrieved from public databases. Potential candidate genes were preliminarily identified by intersecting ITP’s differentially expressed genes (DEGs) with exosome-related key module genes, followed by exploratory screening via machine learning and the construction of a preliminary predictive model. Multi-dimensional analyses (enrichment, immune infiltration [subsequently removed due to methodological concerns], drug prediction) and RT-qPCR validation were performed. Four candidate genes (GABARAPL1, SLC39A14, HIBADH, GSR) were identified through bioinformatic analysis, involved in spliceosome and other pathways (P < 0.05, |NES| > 1). GABARAPL1, SLC39A14, and HIBADH showed exploratory correlations with specific functional T-cell subsets (|cor| > 0.3, P < 0.05). Molecular docking simulations suggested potential binding feasibility between SLC39A14/nortriptyline and GSR/oxiglutatione (binding free energy < -5 kcal/mol). RT-qPCR confirmed the significant downregulation of GABARAPL1, SLC39A14, and GSR in ITP patients (P < 0.05), while HIBADH did not show statistically significant changes (P > 0.05). In this exploratory study, GABARAPL1, SLC39A14, and GSR were identified as potential candidate biomarkers with experimental support from clinical samples. HIBADH, while predicted by bioinformatic analysis, requires further investigation to determine its clinical relevance. These findings provide exploratory insights and a preliminary basis for future hypothesis-driven research on the role of exosome-related genes in ITP.
Similar content being viewed by others

Introduction
Primary immune thrombocytopenia (ITP), an acquired autoimmune hematologic condition, is defined by two key features: a significant decline in platelet levels and an increased propensity for bleeding12. Epidemiological studies report an annual incidence rate of 2–10 cases per 100,000 population3. ITP can occur at any age, with pediatric cases typically presenting as acute forms often secondary to infections, while adult cases predominantly manifest as chronic forms, showing a slightly higher incidence in females than males45. The pathogenesis of ITP involves complex mechanisms, with three primary contributing factors: autoantibody production against platelets, impaired megakaryocyte maturation, and T-lymphocyte dysfunction67. Current therapeutic strategies primarily employ corticosteroids and intravenous immunoglobulin to rapidly elevate platelet counts, though these interventions demonstrate limited long-term efficacy89. The diagnosis of ITP remains exclusion-based due to the absence of specific biomarkers, potentially leading to misdiagnosis in some cases and consequent exacerbation of bleeding risks. Therefore, the identification of novel biomarkers holds significant value for improving ITP diagnosis and treatment.
Exosomes are nanosized extracellular vesicles containing nucleic acids, proteins, and lipid components that mediate intercellular communication and participate in diverse biological processes10,11,12. Emerging evidence demonstrates their pivotal role in autoimmune diseases including multiple sclerosis and systemic lupus erythematosus, where their expression levels serve as valuable biomarkers for disease progression while also exhibiting potential as drug delivery vehicles1314. A seminal study revealed that exosomes derived from ITP patients carry miR-363-3p, which disrupts immune homeostasis by significantly impairing the immunosuppressive function of regulatory T cells through modulation of the TBX21/ARID3A/SPI1 signaling axis15. Furthermore, specific proteins and miRNAs within exosomes have been implicated in ITP pathogenesis16. These molecular components are important drivers of disease progression, yet their regulatory potential pathways are not fully understood and need more research.
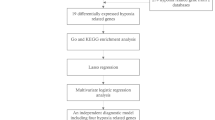
We utilized a multi-omics framework that combined transcriptome profiling and bioinformatics to explore ITP-related exosomal genetic signatures. Machine learning was subsequently employed to preliminarily screen potential candidate genes. The comprehensive analytical framework encompasses exploratory clinical prediction modeling through nomogram construction, functional enrichment analysis of biological pathways, molecular network mapping to elucidate regulatory potential pathways, and computational drug prediction with molecular docking simulations for hypothesis generation. Experimental validation was subsequently performed using reverse transcription quantitative polymerase chain reaction (RT-qPCR) on peripheral blood samples from ITP patients. This exploratory study aims to provide potential candidate genes and generate testable hypotheses regarding exosome-related mechanisms in ITP, offering a preliminary basis for future investigations into diagnostic or therapeutic strategies.
Materials and methods
Data source
In this study, the primary ITP-related dataset GSE43179 (sequencing type: microarray) was obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). This dataset contains molecular expression data based on two different sequencing platforms. Among them, mRNA expression data were derived from the GPL570 platform, including 9 peripheral blood T-cell samples from ITP patients (ITP group) and 10 peripheral blood T-cell samples from normal controls (control group). miRNA expression data were obtained from the GPL14613 platform, comprising 9 peripheral blood T-cell samples from ITP patients (ITP group) and 9 peripheral blood T-cell samples from normal controls (control group). In addition, a total of 121 exosome-related genes (ERGs) were retrieved from the ExoBCD (https://exobcd.liumwei.org/) and used for subsequent analyses (Supplementary Table S1)17. To further explore the reproducibility of the identified candidate genes, two additional independent transcriptomic datasets, GSE46922 and GSE205495, were retrieved from the GEO database as external validation cohorts. GSE46922 includes ITP patients and healthy controls; GSE205495 contains transcriptomic profiles of CD4 + T cells from 4 ITP patients and 4 healthy controls. These external cohorts were utilized to assess the expression consistency of the final candidate genes.
Differential expression analysis
For identifying ITP-related differentially expressed genes (DEGs), differential expression analysis was initially performed on samples of the ITP and control groups in dataset GSE43179 (Sequencing platform: GPL570) via the R package “limma” (v 3.54.0)18, identifying DEGs (P < 0.05, |log ~ 2 ~ fold change (FC)| > 0.25). Visualization of the differential analysis was generated using “ggplot2” (v 3.4.1)19. Visualization of the differential analysis included a heatmap and a volcano plot. The heatmap, created with “pheatmap” (v 1.0.12)20, depicted DEG expression between ITP and control groups, with the top 10 upregulated and downregulated genes (by |log2FC|) highlighted. The volcano plot was generated using “ggplot2” (v 3.4.1)19. Genes were sorted identically by |log2FC| (descending), and the top 10 DEGs mentioned above were labeled in the plot.
WGCNA
Subsequently, weighted gene co-expression network analysis (WGCNA) was employed to explore co-expression modules associated with exosome-related genes. Although the discovery cohort size (n= 19) is modest—a limitation discussed further below—WGCNA was applied as an exploratory screening tool following established guidelines for initial network construction in hypothesis-generating studies [Cite: Langfelder & Horvath21,. To further explore co-expression modules highly associated with exosomes, WGCNA was performed based on DE-ERG enrichment scores. The “hclust” function was employed for hierarchical clustering of all samples in dataset GSE43179 (sequencing platform: GPL570), aiming to identify and exclude outlier samples and guarantee data quality for subsequent co-expression network building. A sample clustering tree was also generated to visualize inter-sample relationships. Next, the optimal soft threshold (power value) was selected within the 1–20 parameter range. The best soft threshold that satisfied the scale-free network fitting index R² > 0.85 was selected. A co-expression network was built based on the optimal soft threshold, and the dynamic tree cutting algorithm in the R package “WGCNA” (v 1.73)21 was employed with the following parameter settings: minimum number of module genes set to 200, method = “tree”, and cutHeight = 0.99, to divide genes into modules labeled with different colors. To identify modules highly correlated with exosome traits, the Spearman correlation coefficient between module eigengenes and DE-ERG scores was calculated (|correlation coefficient (cor)| > 0.3 and P < 0.05). Meanwhile, a module-trait correlation heatmap was plotted using the labeledHeatmap function in the R package “WGCNA” (v 1.73)21 for visualization. Finally, we selected the modules most strongly correlated with DE-ERG scores. Using the “WGCNA” R package (v 1.73)21, Module Membership (MM) and Gene Significance (GS) were calculated for these module genes. Genes meeting the thresholds of |MM| > 0.6 and |GS| > 0.4 were retained. The GS-MM correlation was visualized in scatter plots via “ggplot2” (v 3.4.1)21, and genes fulfilling these criteria were defined as exosome-related key module genes. WGCNA utilized 121 ERGs as ‘seeds’ to identify potential genes synergistic with exosomal processes, suggesting candidate regulatory partners within the ITP exosomal signaling axis. The thresholds of |MM| > 0.6 and |GS| > 0.4 were employed to balance sensitivity and specificity in this exploratory context.
Acquisition of candidate genes and functional enrichment analysis
We next sought to identify genes linked to both ITP and exosomes. Using the R package “ggvenn” (v 1.7.1), we found the intersection of DEGs and exosome-related key module genes, thereby obtaining candidate genes for follow-up exploratory analysis. The R package “clusterProfiler” (v 4.2.2)22 was employed to perform KEGG and GO enrichment analyses on the candidate genes (P < 0.05), aiming to explore their potential biological functions and underlying pathways23,24,25. GO consisted of three parts, namely biological process (BP), cellular component (CC) and molecular function (MF). Additionally, STRING (https://string-db.org/) was utilized to examine interactions of proteins encoded by candidate genes (confidence score > 0.15). Finally, the results were visualized by constructing a PPI network diagram using Cytoscape software (v 3.8.2)26.
Acquisition of candidate genes
For the exploratory screening of potential ITP-related genes, a two-step algorithm approach was utilized: Boruta followed by LASSO regression. Feature selection via the “Boruta” package (v 8.0.0)27 on the GSE43179 dataset (GPL570) identified genes with importance scores exceeding all shadow feature benchmarks (shadowMin, shadowMean, shadowMax), yielding Feature Gene Set 1. Subsequent analysis involved applying LASSO regression to the same dataset with the “glmnet” package (v 1.1.10)28. Via 5-fold cross-validation, the optimal regularization parameter (lambda) (minimum mean squared error) was determined. Genes with non-zero regression coefficients were screened to generate Feature Gene Set 2, and regression coefficient plots plus cross-validation error curves were generated for visualization. Finally, the two feature gene sets’ intersection was calculated using “ggvenn” (v 1.7.1) to obtain the final candidate genes.
Construction and evaluation of nomogram
To explore the potential of the candidate genes in distinguishing ITP from controls, we established an exploratory nomogram prediction model based on these genes using the R package “rms” (v 6.8.1)29 and all samples from GSE43179 (Sequencing platform: GPL570). The scale range of the line corresponding to each gene indicates its predicted score interval (Points). The total score (Total Points) results from summing the scores of each gene, and a higher value corresponds to a higher probability of ITP. Preliminary predictive performance of the model was assessed through two methods: calibration curves plotted using the “rms” R package (v 6.8.1)29 and the Hosmer-Lemeshow test for assessing prediction-actuality consistency. In these curves, prediction accuracy was considered higher when the plot more closely followed the reference line (slope = 1). The nomogram showed acceptable calibration in this exploratory context, as evidenced by an HL test P value > 0.05. The exploratory predictive ability was further assessed by constructing a receiver operating characteristic (ROC) curve with the “pROC” R package (v 1.18.5)30. The performance of the identified candidate genes was evaluated using the area under the ROC curve (AUC). It should be noted that due to the limited sample size, these results are considered exploratory and hypothesis-generating, providing preliminary evidence rather than a definitive assessment of diagnostic utility.
GSEA of candidate genes
To investigate the regulatory pathways and biological functions linked to the candidate genes, GSEA was performed for each gene on ITP vs. control samples from dataset GSE43179 (Sequencing platform: GPL570). Employing the R package “psych” (v 2.1.6)31, we computed and sorted Spearman correlation coefficients between the candidate genes and other genes. GSEA was subsequently performed with the R package “clusterProfiler” (v 4.2.2)32 using the MsigDB gene set “c2.cp.kegg_legacy.v2024.1.Hs.symbols.gmt” as reference, under the criteria |NES| > 1 and adj.P < 0.05. Visualization of the top 5 significant pathways was achieved with the R package “enrichplot” (v 1.18.3)33.
GeneMANIA analysis and of candidate genes
Subsequently, related genes interacting with the candidate genes and their involved functions were explored via the GeneMANIA database (http://genemania.org/), with “Homo sapiens” set as the analysis species. From the outcomes, the candidate genes and the top 20 related genes were selected as key display nodes, and the top 5 most significant enriched pathways (P < 0.05) and functions (FDR < 0.05) were presented. A gene interaction network diagram was constructed to visualize results, showing potential correlations between the candidate genes and their functionally similar genes.
Molecular regulatory network analysis of candidate genes
Transcription factors (TFs) are key protein molecules that can specifically recognize and bind to specific DNA sequences in regulatory regions such as gene promoters or enhancers, thereby regulating the transcriptional activity of genes. First, the TRRUST database was used to explore TFs that may interact with the candidate genes. Next, based on the GSE43179 dataset (sequencing platform: GPL14613), the R package “limma” (v 3.54.0)18 was used to identify differentially expressed microRNAs (DE-miRNAs) between the ITP group and the control group. MiRNAs in the heatmap were sorted by |log2FC| values in descending order, showing the top 10 upregulated and 10 downregulated miRNAs with the most significant expression differences. A miRNA volcano plot was also created using the R package “ggplot2” (v 3.4.1)20. Subsequently, miRNet was used to predict miRNAs potentially targeting the candidate genes. The final integrative visualization of the exploratory regulatory network, built from the screened TFs, key miRNAs, and candidate genes, was achieved using Cytoscape software (v 3.8.2)26. These analyses are intended to generate hypotheses about possible regulatory interactions and should be interpreted with caution.
Drug prediction analysis of candidate genes and molecular docking
To explore potential compounds that might interact with the candidate genes, drug prediction for each gene was performed via DGIdb (https://dgidb.org/). Construction and visualization of the drug-mRNA interaction network were performed with Cytoscape software (v 3.8.2)26. To computationally assess potential drug-gene binding, we selected drugs per gene (highest interaction scores, PubChem-retrievable 2D structures) for molecular docking simulations. Drug 2D structures (SDF) were downloaded from PubChem, and protein 3D structures (PDB) encoded by the candidate genes were obtained from RCSB PDB. These were uploaded to CB-Dock for docking, with binding affinity evaluated by Vina score (binding free energy). Generally, Vina score < −5 kcal/mol suggests potential binding feasibility in silico. It is important to note that these computational simulations indicate only theoretical binding feasibility; the actual biological activity, safety, and therapeutic efficacy of these candidates remain to be determined through experimental and clinical validation.
Expression level validation of candidate genes
Differences in expression of the candidate genes within the GSE43179 dataset (GPL570) were assessed using the Wilcoxon rank-sum test (P < 0.05) and subsequently visualized in box plots constructed with the R package “ggplot2” (v 3.4.1)20.
RT-qPCR
Experimental validation of candidate gene expression in clinical samples was performed by RT-qPCR. An independent clinical cohort consisting of 20 ITP patients and 20 healthy controls was enrolled from Fengdu County Hospital of Traditional Chinese Medicine. Total RNA was extracted using TRIzol reagent (Ambion, USA) per the manufacturer’s protocol, and its concentration was determined with a NanoPhotometer N50. cDNA was synthesized by reverse transcription employing the SureScript First-Strand cDNA Synthesis Kit on a Bio-Rad S1000™ Thermal Cycler (USA). Using primers listed in Supplementary Table S2, qPCR was run on a Bio-Rad CFX Connect Real-Time PCR Instrument (USA) with the cycling profile: 95 °C for 1 min; 40 cycles of 95 °C for 20 s, 55 °C for 20 s, and 72 °C for 30s. The 2-ΔΔCT method was applied for relative quantification, and data were analyzed and visualized in GraphPad Prism 5. These clinical samples were collected de novo and are entirely separate from the samples used in the discovery dataset (GSE43179), ensuring the independence of the experimental validation.
Statistical analysis
Bioinformatics analyses utilized R software (v 4.2.2) for statistical processing. Statistical significance was set at P < 0.05. The Wilcoxon rank-sum test was used for between-group comparisons, while the t-test was employed for between-group analyses of RT-qPCR results. Correlation analyses were performed using the Spearman rank correlation method (via the ‘psych’ R package). The correlation coefficient (r) ranges from − 1 to 1. A positive r value indicates a positive linear relationship, while a negative r value indicates an inverse relationship. We defined the strength of correlation as follows: |r| > 0.3 as a potential meaningful correlation, and |r| > 0.5 as a potential strong correlation, with a P-value < 0.05 considered statistically significant. Given the exploratory nature of this study, all correlation findings should be interpreted as hypothesis-generating.
Ethics approval and consent to participate
The human subjects research component of this study was reviewed and approved by the Ethics Committee of Fengdu County Hospital of Traditional Chinese Medicine, Chongqing, China (Approval No: FDYY−2024−0729), with documented informed consent obtained from all participants. All procedures were conducted in strict compliance with the ethical principles set forth in the Declaration of Helsinki (1964) and its subsequent amendments.
Results
Acquisition and functional enrichment analysis of 23 candidate genes
A total of 105 DEGs were identified from the GSE43179 dataset (GPL570) through a comparative analysis between ITP and control groups, using thresholds of P < 0.05 and |log2FC| > 0.25. The ITP-associated DEGs comprised 48 upregulated and 57 downregulated genes (Figs. 1. A-B, Supplementary Table S3). Expression levels of 121 ERGs were then compared between ITP and control groups in the GSE43179 dataset (GPL570) via Wilcoxon rank-sum test. This screening identified seven DE-ERGs: POLR2K, IGF1R, NANOG, RAB13, ALDH9A1, HDC, and TLR5 (P < 0.05) (Figs. 1.C). Further, DE-ERG scores were calculated based on all samples from the dataset GSE43179 (Sequencing platform: GPL570). The Wilcoxon rank-sum test showed a significant difference in DE-ERG scores between the ITP group and the control group (P = 0.011) (Figs. 1.D). No obvious outlier samples were identified by hierarchical clustering analysis (Figs. 1E). A co-expression network was constructed by WGCNA with a soft-thresholding power of 19, resulting in a topological fit (R² = 0.85) (Figs. 1.F-G). After constructing the weighted co-expression network, the dynamic tree cutting method was used to identify gene modules. The minimum number of genes in each module was set to 200, and after merging highly similar modules (cutHeight = 0.99), a total of 6 co-expression modules were identified (excluding the grey module) (Fig. 1.H). Next, the correlations between each module and DE-ERG scores were analyzed, and 4 modules were found to have a potential correlation with DE-ERG scores (|cor| > 0.3, P < 0.05). Among the modules, the brown module showed the strongest positive correlation with DE-ERG scores (cor = 0.67, P < 0.05), whereas the red and blue modules were most negatively correlated (cor = −0.64, P < 0.05) (Fig. 1I). In addition, correlations between GS and MM were observed in the brown module (cor = 0.581, P < 0.05), red module (cor = 0.784, P < 0.05), and blue module (cor = −0.723, P < 0.05) (Fig. 1J). Finally, genes were screened according to the criteria of |MM| > 0.6 and |GS| > 0.4. A total of 3788, 767, and 159 genes were obtained from the blue, brown, and red modules respectively, resulting in a total of 4714 exosome-related key module genes (Fig. 1 K, Supplementary Table S4). The intersection of DEGs and exosome-related key module genes yielded 23 candidate genes (Fig. 1K, Supplementary Table S5). To investigate their potential functions, these candidate genes were subjected to GO and KEGG enrichment analyses, which identified 212 GO terms (P < 0.05), including 151 BP terms such as response to starvation and transcription by RNA polymerase III (Fig. 1L, Supplementary Table S6); 11 CC terms such as transferase complex and RNA polymerase II holoenzyme (Fig. 1L, Supplementary Table S7); and 50 MF terms such as NADP binding and Tat protein binding (Fig. 1L, Supplementary Table S8). KEGG enrichment analysis revealed that the candidate genes were enriched in 7 signaling pathways (P < 0.05), including FoxO signaling pathway, mTOR signaling pathway, and autophagy (Fig. 1M, Supplementary Table S9). Subsequently, the interaction relationships between proteins encoded by the candidate genes were explored via the STRING database with a confidence score > 0.15. From the analysis, a network containing 18 core protein nodes was identified. Among these nodes, connections were observed between SIMCI and POLR2K, TCF20, as well as EIF4ENIF1, suggesting potential functional interactions (Fig. 1N). These exploratory findings provide candidate genes and enriched pathways for future investigations into the mechanisms of ITP.



(A)Volcano plot of differentially expressed genes; (B)Heatmap displaying the expression levels of the top 10 up- and down-regulated genes, with higher expression levels shown in red and lower levels in blue; (C)Box plot of seven significantly differentially expressed ERGs; (D)Distribution of ssGSEA scores for gene sets with differential directionality in disease samples versus healthy controls in the training set; (E)Cluster analysis dendrogram of samples from the training set GSE43179; (F)Screening of the soft-thresholding power for the WGCNA co-expression network (threshold β value set to 19). (G)Screening of the soft-thresholding power for the WGCNA co-expression network (threshold β value set to 19);(H–J)Module assignment of the gene co-expression network, distinguished by different colors, identifying a total of six co-expression modules; (K)Venn diagram showing the intersection of differentially expressed genes (DEGs) from the transcriptome and ERG module genes, resulting in 23 differentially expressed ERGs; (L)GO and KEGG enrichment analysis of the 23 candidate genes23. (M)GO and KEGG enrichment analysis of the 23 candidate genes2425; (N)Protein–protein interaction (PPI) network.
Identification of 4 candidate genes and construction of nomogram
To further explore potential candidate genes from the 23 candidate genes, two machine learning algorithms—Boruta analysis and LASSO regression analysis—were applied for exploratory screening. Based on all samples from the dataset GSE43179 (Sequencing platform: GPL570), first, Feature Gene Set 1—comprising 5 genes (GABARAPL1, SLC39A14, HIBADH, FUT11, and GSR)---was obtained via Boruta analysis (Fig. 2A). Next, for the LASSO regression analysis, the optimal regularization parameter was determined through 5-fold cross-validation. When lambda.min = 0.175, the model achieved the minimum Mean Squared Error (MSE); subsequently, feature gene set 2—consisting of 5 genes (GABARAPL1, SLC39A14, HIBADH, IGF1R, and GSR)---was obtained (Figs. 2B-C). By taking the intersection of these two feature gene sets, 4 final candidate genes were identified for subsequent analysis, namely GABARAPL1, SLC39A14, HIBADH, and GSR (Fig. 2D). Subsequently, an exploratory nomogram model was constructed based on these 4 genes. For example, when the total points reached 229, the predicted probability of ITP was 89% within this cohort (Fig. 2E). The model’s calibration was assessed using the Hosmer-Lemeshow test (P = 0.168) (Fig. 2F). The model demonstrated an AUC of 0.878 in this exploratory analysis, providing a preliminary indication of the genes’ ability to differentiate ITP from controls within this specific cohort (Fig. 2G). Given the limited sample size, these findings should be considered exploratory and hypothesis-generating, serving as an initial indication of discriminatory capacity rather than a definitive diagnostic model. The candidate genes identified in this study provide a preliminary basis for future investigations into their potential role in ITP.


(A) The Boruta algorithm was employed to perform feature selection on the 23 differentially expressed ERGs, ultimately identifying 5 feature genes (highlighted in green). (B–C) Screening of the 23 differentially expressed ERGs by LASSO regression selected 5 feature genes. (D)The intersection of two machine learning algorithms revealed 4 shared genes (Venn diagram). (E–G)The resulting nomogram was validated with calibration curves and ROC analysis, indicating its diagnostic efficacy. The discriminatory power of the model was assessed by the AUC, where numerical values closer to 1 signify superior model accuracy and reliability.
GSEA and GeneMANIA analysis of candidate genes
GSEA of the candidate genes was performed to explore relevant signaling pathways and biological processes underlying ITP (|NES| > 1, adj.p < 0.05). GABARAPL1 was enriched in 30 pathways, including spliceosome and neuroactive ligand receptor interaction (Fig. 3 A, Supplementary Table S10); SLC39A14 was enriched in 29 pathways, including ubiquitin-mediated proteolysis and neuroactive ligand receptor interaction (Fig. 3B, Supplementary Table S11); HIBADH was enriched in 35 pathways, including spliceosome and neuroactive ligand receptor interaction (Fig. 3 C, Supplementary Table S12); GSR was enriched in 26 pathways, including ribosome and spliceosome (Fig. 3D, Supplementary Table S13). Subsequently, genes interacting with the four candidate genes and the biological processes they may participate in were explored using the GeneMANIA database. The results showed that the top 20 genes interacting with the four candidate genes included HIBCH, GSTO2, ATG4D, ALDH6A1, PGD, ATG7, GLRX, ATP6V1C1, ATG4A, G6PD, DLD, FOLR1, TXN, SLC4A4, HSD17B10, GLYR1, STBD1, AIFM1, RETREG3, and SLC25A20. Among these, HIBADH, together with HIBCH, ALDH6A1, and HSD17B10, was suggested to be involved in branched-chain amino acid metabolic process and catabolic process. GSR, along with TXN and GLRX, was suggested to be involved in oxidoreductase activity and disulfide oxidoreductase activity. The associations between the four candidate genes and their interacting genes were mainly based on physical interactions, suggesting potential functions in protein complex assembly or direct binding. Taken together, GSEA and GeneMANIA analyses suggest that the four candidate genes may be involved in ITP-related pathways, potentially through their influence on biological processes such as spliceosome function, amino acid metabolism, redox equilibrium, and the ubiquitin-proteasome system. These exploratory findings provide hypotheses for subsequent research to investigate the molecular mechanisms of these genes in ITP.

Molecular regulatory network analysis of candidate genes
Potential regulatory pathways of the candidate genes were further explored by molecular regulatory network analysis. Upstream TFs that may regulate the candidate genes were first predicted, and the results showed that GABARAPL1, GSR, HIBADH, and SLC39A14 were predicted to be regulated by 20, 47, 19, and 35 TFs, respectively (Fig. 4 A). This finding suggests that these candidate genes may be subject to complex regulatory networks at the transcriptional level. To further explore post-transcriptional regulatory pathways, differentially expressed miRNAs (DE-miRNAs) between the ITP group and the control group were analyzed based on the GSE43179 dataset (sequencing platform: GPL14613). A total of 45 DE-miRNAs were identified, among which 35 miRNAs were downregulated and 10 miRNAs were upregulated in the ITP group, with the screening criteria of |log2FC| > 0.25 and P < 0.05 (Figs. 4B-C, Supplementary Table S14). Meanwhile, screening of the miRNet database yielded 490 miRNAs predicted to target the candidate genes (Figs. 4D, Supplementary Table S15). The integrative bioinformatic analysis suggested a potential interaction between miR-484 and SLC39A14. Predictive screening across multiple databases identified SLC39A14 as a potential target of miR-484, with conserved binding sites in the 3’ untranslated region (3’UTR). In the GSE43179 dataset, the expression levels of miR-484 and SLC39A14 exhibited an inverse correlation, where upregulation of miR-484 was accompanied by downregulation of its potential target mRNA. This inverse relationship provides an exploratory hypothesis that the reduced expression of SLC39A14 observed in ITP may be associated with elevated miR-484 levels. These results offer preliminary insights and generate hypotheses for future research into the molecular regulatory networks (including miRNA and TF levels) of GABARAPL1, GSR, HIBADH, and SLC39A14 in ITP.

Regulatory landscape of identified candidate genes involving upstream transcription factors and microRNAs in ITP. (A)TF–mRNA regulatory network illustrating predicted transcription factors (red nodes) interacting with target candidate genes (blue nodes) based on the TRRUST database. (B)Volcano plot of differentially expressed miRNAs (DE-miRNAs) between the ITP group (n = 9) and control group (n = 9) using the “limma” package. (C)Heatmap displaying the expression profiles of the top 10 upregulated and top 10 downregulated DE-miRNAs. (D)miRNA–mRNA regulatory network depicting the targeting relationships between differential miRNAs and candidate genes predicted via miRNet. Statistical significance for differential analysis was defined as |log2FC| > 0.25 and P < 0.05.
Drug prediction analysis of candidate genes and molecular docking
To explore potential compounds that might interact with the candidate genes, drug prediction analysis identified 22 and 1 candidate compounds for GSR and SLC39A14, respectively, from the DGIdb database (Fig. 5 A). Subsequent molecular docking simulations suggested potential binding feasibility between the candidate genes and their respective compounds: SLC39A14 with nortriptyline showed a predicted binding free energy of −7.9 kcal/mol (Figs. 5B-C; Table 1), while GSR with oxiglutatione showed a predicted binding free energy of −17.1 kcal/mol (Fig. 5D; Table 1), both below the − 5 kcal/mol threshold often used to indicate potential binding in silico. It is essential to clarify that these computational results indicate only theoretical binding feasibility; the actual biological activity, safety, and therapeutic efficacy of these candidates remain to be determined through experimental and clinical validation. These findings provide a computational rationale for future pharmacological studies. The drug prediction analysis was conducted by screening all 23 candidate genes against the DGIdb database. To focus on the most promising candidates for future research, selection criteria included target-specific interactions and a predicted binding free energy lower than − 5.0 kcal/mol in molecular docking simulations. The predicted binding affinities observed in these models suggest that these compounds are promising candidates for further in vitro and in vivo studies to evaluate their actual modulatory effects on ITP-related pathways.

Molecular docking models and drug-target interaction networks. (A)Integrative network of potential therapeutic drugs and their corresponding candidate genes targets. (B-D)Detailed 3D molecular docking visualisations showing the binding poses of nortriptyline with SLC39A14 and oxiglutathione with GSR. Predicted hydrogen bonds are indicated by dashed lines, with specific amino acid residues and bond lengths labeled. The binding free energy was calculated using Vina score, with values below − 5.0 kcal/mol indicating high binding affinity.
Expression level validation of candidate genes and RT-qPCR
Analysis of the GSE43179 dataset (Sequencing platform: GPL570) revealed that all four candidate genes were significantly downregulated in ITP samples (P < 0.05) (Fig. 6 A). Experimental validation in clinical samples by RT-qPCR confirmed the significant downregulation of GABARAPL1, SLC39A14, and GSR in ITP patients (P < 0.05) (Figs. 6B-D). However, the expression levels of HIBADH showed no statistically significant difference between ITP patients and healthy controls (P > 0.05).
(Fig. 6E), suggesting that, despite its identification in the initial screening, it may be a less robust marker in this clinical context. These results provide experimental support for the association of GABARAPL1, SLC39A14, and GSR with ITP, indicating that their decreased expression may play a role in disease pathogenesis, while HIBADH remains a bioinformatic candidate requiring further investigation.

Validation of candidate genes expression levels in the discovery dataset and an independent clinical cohort. (A) Expression profiles of SLC39A14, HIBADH, GABARAPL1, and GSR in the discovery dataset GSE43179, consisting of 9 ITP patients and 10 healthy controls. (B–E) Relative mRNA expression levels of GABARAPL1, SLC39A14, GSR, and HIBADH in a de-novo clinical validation cohort (n = 20 ITP patients and n = 20 healthy controls), respectively. For the clinical cohort, relative quantification was performed using the 2 − ΔΔCt method, with GAPDH serving as the internal reference. Data are presented as mean ± SD. Statistical differences in the discovery dataset were assessed using the Wilcoxon rank-sum test, while an unpaired Student’s t-test was employed for inter-group comparisons in the clinical cohort. Statistical significance is indicated as follows: *P < 0.05, **P < 0.01, ns: not significant.
External validation of candidate genes in independent datasets
To further explore the reproducibility of the identified candidate genes, two independent ITP-related transcriptomic datasets (GSE205495 and GSE46922) were retrieved from the GEO database. Analysis within these external cohorts indicated that the expression trends of GABARAPL1, SLC39A14, and GSR remained consistent with the discovery dataset (P < 0.05). For HIBADH, significant differential expression was observed in GSE46922 (P < 0.05) but not consistently across all datasets, further supporting the need for cautious interpretation of its role. This cross-dataset consistency provides additional support for the reproducibility of the findings for GABARAPL1, SLC39A14, and GSR, helping to mitigate concerns regarding the potential instability of results derived from a small discovery sample size. These results offer cross-platform evidence for the selected candidate genes, with GABARAPL1, SLC39A14, and GSR showing the most consistent expression patterns across datasets.
Discussion
ITP is an autoimmune disorder characterized by both humoral and cellular immune-mediated platelet destruction coupled with impaired platelet production34. Emerging evidence indicates that exosomes may play a role in ITP pathogenesis through immunomodulation and intercellular communication35. Nevertheless, the precise etiological pathways underlying ITP remain incompletely understood, and specific diagnostic biomarkers are still lacking. In this exploratory study, we employed bioinformatics approaches to investigate potential exosome-associated candidate genes and explore their functional pathways in ITP. Experimental validation in peripheral blood samples from ITP patients confirmed dysregulated expression patterns of three core genes - GABARAPL1, SLC39A14, and GSR - suggesting their potential involvement in ITP pathogenesis. These findings indicate that GABARAPL1, SLC39A14, and GSR may be considered as potential candidate biomarkers for ITP. In contrast, the inconsistent validation of HIBADH in clinical samples (P > 0.05) suggests that its role may be more complex. Several factors could contribute to this discrepancy, including the high background expression from abundant cell populations masking subtle T-cell-specific signals, heightened sensitivity of this metabolic enzyme to transient physiological states, dietary factors, or pharmacological interventions, as well as disparities in clinical subtypes, disease stages, and treatment histories between discovery and validation cohorts.
GABARAPL1 serves as a regulatory protein in autophagy processes, playing a role in maintaining intracellular homeostasis and cellular survival36. Research by Lei Li et al.37 has suggested that GABARAPL1 may modulate platelet production through its regulation of autophagy levels in megakaryocytes. Ahmad Reza Panahi Meymandi et al.38 further proposed that GABARAPL1 dysfunction may promote the survival and proliferation of autoreactive lymphocytes, potentially disrupting immune tolerance to platelet antigens. The autophagy process exhibits crosstalk with exosome biogenesis, which could potentially influence autoimmune responses against platelets39. Notably, studies have reported significantly reduced GABARAPL1 expression in ITP patients40, suggesting its possible contribution to disease progression through multiple interconnected pathways, including impairment of ubiquitin phosphorylation, dysregulation of PPAR signaling pathways, compromised mitophagy, and suppression of ferroptosis. Our current findings are consistent with these observations, showing markedly lower GABARAPL1 expression in ITP patients compared to healthy controls. These findings suggest that GABARAPL1 may influence ITP pathogenesis through its potential effects on megakaryocyte function and immune cell homeostasis.
SLC39A14 functions as a zinc transporter that plays a role in maintaining immune homeostasis41. Zinc deficiency or metabolic dysregulation can promote inflammatory responses and may contribute to autoimmune pathogenesis. Alterations in SLC39A14 function or expression could disrupt zinc homeostasis, thereby potentially participating in the immune dysregulation observed in ITP42. Zhizhao Deng et al.43 reported a possible association between exosomes and SLC39A14, showing that bone marrow mesenchymal stem cell-derived exosomes deliver miR-16-5p to hepatocytes, leading to post-transcriptional suppression of SLC39A14 expression. Exosomes released from cells with low SLC39A14 expression may modify intracellular zinc levels and could exacerbate inflammatory responses44. Activated immune cells might secrete exosomes carrying specific miRNAs or autoantigens, which upon uptake by other immune cells, could hypothetically influence ITP progression45. In our current study, we observed significantly reduced SLC39A14 expression in ITP patients compared to healthy controls. These findings suggest that SLC39A14 may contribute to ITP pathogenesis through its potential involvement in zinc metabolism dysregulation.
HIBADH serves as an enzyme in valine catabolism, catalyzing the oxidation of 3-hydroxyisobutyrate to methylmalonate semialdehyde, thereby playing a role in energy metabolism and amino acid degradation46,47,48,49. In our study, although HIBADH was identified as a candidate gene through bioinformatic screening, its differential expression was not consistently validated in peripheral blood samples from ITP patients by RT-qPCR (P > 0.05). This discrepancy highlights the importance of experimental validation for bioinformatic predictions. Several factors may explain this inconsistency, including sample heterogeneity, technical variations between microarray and RT-qPCR platforms, or the possibility that HIBADH transcript levels in purified T-cells are influenced by transient physiological states. Therefore, unlike GABARAPL1, SLC39A14, and GSR which gained experimental support, HIBADH should be regarded as a purely bioinformatic candidate whose role in ITP, if any, remains to be clarified in future studies with larger sample sizes.
GSR serves as an enzyme in the cellular antioxidant defense system, playing a role in maintaining redox homeostasis50. Patients with ITP exhibit a state of oxidative stress. Yanxia Zhan et al.51 reported that elevated reactive oxygen species (ROS) levels can promote inflammatory cytokine release through immune cell activation while simultaneously causing direct damage to both platelets and megakaryocytes. Complementary research by Yuquan Xie et al.52 suggested that excessive ROS may directly attack platelet membrane phospholipids, thereby triggering platelet apoptosis. Further investigations have shown that exosomes secreted by immune cells under oxidative stress conditions may carry increased loads of inflammatory non-coding RNAs53. When internalized by megakaryocytes, these exosomes could amplify oxidative stress and inflammatory signaling, potentially exacerbating autoimmune responses. Our current study identified significantly reduced GSR expression in ITP patients compared to healthy controls. These findings suggest that GSR may contribute to ITP pathogenesis through its potential roles in antioxidant defense and possible involvement in exosome-mediated intercellular communication pathways.
The spliceosome, a macromolecular complex composed of small nuclear ribonucleoproteins and auxiliary proteins, plays a role in mRNA processing, and its dysfunction has been implicated in various hematological disorders54. Emerging evidence suggests that aberrant spliceosome regulation may lead to abnormal alternative splicing of platelet production-related genes in megakaryocytes55. Furthermore, spliceosome components might contribute to autoimmune responses through dysregulated activation of T and B lymphocytes, potentially resulting in immune-mediated platelet destruction56. Andrea Pellagatti et al.57 reported that exosomes can transport spliceosome-associated non-coding RNAs, thereby potentially modulating gene expression in recipient cells through intercellular communication. Our GSEA indicated that GABARAPL1, SLC39A14, HIBADH, and GSR are enriched in the spliceosome signaling pathway. These findings raise the possibility that exosomes derived from aberrant immune cells may transmit splicing-related signals, potentially influencing megakaryocyte differentiation and platelet maturation. Notably, these four candidate genes also showed enrichment in the neuroactive ligand-receptor interaction pathway and ubiquitin-mediated proteolysis signaling pathway, suggesting their possible involvement in multiple regulatory pathways in ITP pathogenesis.
miR-484 has been studied in various cancers and metabolic disorders, where it has been suggested to play a regulatory role in apoptosis and mitochondrial function signaling pathways58. Existing research has indicated that miR-484 may modulate apoptotic processes in immune cells59, raising the hypothesis that its dysregulated expression might contribute to T-cell homeostasis imbalance in ITP. Another study60 suggested that miR-484 may interfere with megakaryocyte differentiation and maturation by modulating the Wnt/MAPK signaling pathway and downregulating β-catenin expression. Through potential targeting of SLC39A14—a zinc transporter—miR-484 could hypothetically exacerbate cellular zinc deficiency in the hematopoietic microenvironment. Zinc ions are essential for both T-cell receptor signaling and terminal maturation of megakaryocytes; consequently, the miR-484/SLC39A14 axis may exert dual effects. Our bioinformatic analyses suggest that miR-484 may target and potentially regulate SLC39A14 expression. This potential regulatory interaction could hypothetically contribute to impaired T-cell function and disrupted megakaryocyte development, ultimately possibly influencing platelet homeostasis in ITP. However, these findings are based on computational predictions and require experimental validation.
Our study conducted exploratory drug prediction analyses for the four candidate genes, indicating that SLC39A14 and GSR may have predicted interactions with candidate compounds. The downregulation of SLC39A14 may disrupt cellular zinc homeostasis, subsequently potentially inducing T-cell dysfunction - a core pathological mechanism in ITP. Beyond its conventional role in monoamine neurotransmitter modulation, nortriptyline has been reported to exhibit immunomodulatory properties61. Emerging evidence suggests that nortriptyline might interact with SLC39A14 to regulate zinc ion flux in either megakaryocytes or T-cells, thereby hypothetically improving the immune microenvironment or promoting megakaryocyte maturation62. This provides a computational rationale for exploring nortriptyline as a candidate for drug repurposing, although its actual immunomodulatory efficacy in ITP requires rigorous experimental testing. Our study further identified significant downregulation of GSR in ITP patients. GSR deficiency leads to glutathione (GSH) depletion, rendering platelets vulnerable to ROS-mediated damage, which triggers premature apoptosis and may exacerbate platelet destruction. As the direct substrate of GSR, oxidized glutathione (GSSG) showed predicted binding affinity to the enzyme in silico. This suggests that therapeutic interventions enhancing GSR activity could hypothetically scavenge excessive ROS in ITP patients, thereby potentially preserving platelet membrane integrity, though this remains to be tested experimentally.
In this exploratory study, we investigated exosome-related genes in ITP using a combination of transcriptomic analysis and preliminary experimental validation. Our findings identified GABARAPL1, SLC39A14, and GSR as potential candidate genes with experimental support from clinical samples, as their downregulation was confirmed by RT-qPCR in an independent cohort. HIBADH, while identified through bioinformatic screening, did not show statistically significant differential expression in clinical validation (P > 0.05) and should therefore be regarded as a purely bioinformatic candidate requiring further investigation. Computational drug predictions suggested potential binding feasibility between SLC39A14/nortriptyline and GSR/oxiglutatione, providing a theoretical basis for future pharmacological studies, though these findings require experimental confirmation.
Several important limitations should be considered. The modest sample size of the discovery cohort (n = 19) represents a major constraint, and the findings should be interpreted as hypothesis-generating rather than definitive. The lack of validation for HIBADH underscores the necessity of experimental confirmation for all bioinformatic predictions. Additionally, the computational drug predictions indicate only theoretical binding feasibility and do not demonstrate biological activity or therapeutic efficacy.
In summary, this study provides preliminary evidence for the involvement of GABARAPL1, SLC39A14, and GSR in ITP and offers candidate genes and testable hypotheses for future research. Further investigations with larger, multi-center cohorts and functional studies are warranted to validate these findings and explore their clinical relevance.
Data availability
The dataset (GSE43179) supporting the conclusions of this article is available in the [GEO] repository, [https://www.ncbi.nlm.nih.gov/geo/]. The ERGs data are available in the [ExoBCD] repository, [https://exobcd.liumwei.org/].Source of the validation set: GEO database (https://www.ncbi.nlm.nih.gov/geo), with the accession number GSE205495.
References
Schramm, T. et al. Fibrinolysis is impaired in patients with primary immune thrombocytopenia. J. Thromb. Haemost. 22 (11), 3209–3220 (2024).
Cao, J. et al. MST4 kinase regulates immune thrombocytopenia by phosphorylating STAT1-mediated M1 polarization of macrophages. Cell. Mol. Immunol. 20 (12), 1413–1427 (2023).
Schifferli, A. et al. Adolescents and young adults with newly diagnosed primary immune thrombocytopenia. Haematologica 108 (10), 2783–2793 (2023). Published 2023 Oct 1.
Author, R. M. et al. Erratum to: 36th International Symposium on Intensive Care and Emergency Medicine: Brussels, Belgium. 15–18 March 2016. Crit Care. ;20:347. Published 2016 Oct 24. (2016).
Hu, Y. et al. Efficacy and safety of sovleplenib (HMPL–523) in adult patients with chronic primary immune thrombocytopenia in China (ESLIM–01): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Haematol. 11 (8), e567–e579 (2024).
Zhuang, X. et al. Decreased cyclooxygenase–2 associated with impaired megakaryopoiesis and thrombopoiesis in primary immune thrombocytopenia. J. Transl Med. 21 (1), 540 (2023). Published 2023 Aug 12.
Huang, Y. et al. Plasma Exosomes Derived from Patients with Primary Immune Thrombocytopenia Attenuate TBX21 + Regulatory T Cell-Mediated Immune Suppression via MiR–363–3p. Inflamm. Published online March 4, (2025).
Larue, M. et al. Efficacy and safety of dapsone in adult primary immune thrombocytopenia. Blood Adv. 9 (8), 1976–1983 (2025).
Yan, S. et al. A phase 2 trial of orelabrutinib showing promising efficacy and safety in patients with persistent or chronic primary immune thrombocytopenia. Am. J. Hematol. 99 (7), 1392–1395 (2024).
Goodman, S. R. The exosome collection. Cell. Mol. Biol. Lett. 30 (1), 33 (2025). Published 2025 Mar 24.
Arya, S. B., Collie, S. P. & Parent, C. A. The ins-and-outs of exosome biogenesis, secretion, and internalization. Trends Cell. Biol. 34 (2), 90–108 (2024).
Isaac, R., Reis, F. C. G., Ying, W. & Olefsky, J. M. Exosomes as mediators of intercellular crosstalk in metabolism. Cell. Metab. 33 (9), 1744–1762 (2021).
Fang, Y. et al. Exosomes as biomarkers and therapeutic delivery for autoimmune diseases: Opportunities and challenges. Autoimmun. Rev. 22 (3), 103260 (2023).
Shen, Z. et al. Effects of Mesenchymal Stem Cell-Derived Exosomes on Autoimmune Diseases. Front. Immunol. 12, 749192 (2021). Published 2021 Sep 27.
Huang, Y. et al. Plasma Exosomes Derived from Patients with Primary Immune Thrombocytopenia Attenuate TBX21 + Regulatory T Cell-Mediated Immune Suppression via MiR–363–3p. Inflamm. Published online March 4, (2025).
Sun, Y. et al. Proteomic analysis and microRNA expression profiling of plasma-derived exosomes in primary immune thrombocytopenia. Br. J. Haematol. 194 (6), 1045–1052 (2021).
Wang, J., Qing, M., Gui, J., Zhong, P. & Hua, H. Identification of exosome-related genes associated with prognosis and immune infiltration features in pancreatic cancer. Discov Oncol. 16 (1), 192 (2025). Published 2025 Feb 17.
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43 (7), e47 (2015).
Shi, Y. et al. Crosstalk of ferroptosis regulators and tumor immunity in pancreatic adenocarcinoma: novel perspective to mRNA vaccines and personalized immunotherapy. Apoptosis 28 (9–10), 1423–1435 (2023).
Gustavsson, E. K., Zhang, D., Reynolds, R. H., Garcia-Ruiz, S. & Ryten, M. ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics 38 (15), 3844–3846 (2022).
Horvath, S. & Dong, J. Geometric interpretation of gene coexpression network analysis. PLoS Comput. Biol. 4 (8), e1000117 (2008). Published 2008 Aug 15.
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (Camb). 2 (3), 100141 (2021). Published 2021 Jul 1.
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–D677 (2025).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28 (11), 1947–1951 (2019).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L. & Ideker, T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27 (3), 431–432 (2011).
Mahmoudian, M., Venäläinen, M. S., Klén, R. & Elo, L. L. Stable Iterative Variable Selection Bioinf. ;37(24):4810–4817. (2021).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 33 (1), 1–22 (2010).
Li, M. et al. Recognition of refractory Mycoplasma pneumoniae pneumonia among Myocoplasma pneumoniae pneumonia in hospitalized children: development and validation of a predictive nomogram model. BMC Pulm Med. 23 (1), 383 (2023). Published 2023 Oct 10.
Robin, X. et al. pROC: an open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011). Published 2011 Mar 17.
Nazari-Ghadikolaei, A., Fikse, F., Gelinder Viklund, Å. & Eriksson, S. Factor analysis of evaluated and linearly scored traits in Swedish Warmblood horses. J. Anim. Breed. Genet. 140 (4), 366–375 (2023).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (Camb). 2 (3), 100141 (2021). Published 2021 Jul 1.
Qi, W. Y. et al. Immune cells in metabolic associated fatty liver disease: Global trends and hotspots (2004–2024). World J. Hepatol. 17 (3), 103327 (2025).
González-López, T. J. & Provan, D. The new era of primary immune thrombocytopenia management in adults: A narrative review of current and emerging treatments. Blood Rev. 73, 101300 (2025).
Li, G. et al. The therapeutic potential of exosomes in immunotherapy. Front. Immunol. 15, 1424081 (2024). Published 2024 Jul 8.
Guo, H. et al. Dysfunction of astrocytic glycophagy exacerbates reperfusion injury in ischemic stroke. Redox Biol. 74, 103234 (2024).
Li, L. et al. Beclin 1 of megakaryocytic lineage cells is locally dispensable for platelet hemostasis but functions distally in bone homeostasis. Bone Res. 13 (1), 32 (2025). Published 2025 Mar 3.
Panahi Meymandi, A. R. et al. Crosstalk between autophagy and metabolic regulation of (CAR) T cells: therapeutic implications. Front. Immunol. 14, 1212695 (2023). Published 2023 Aug 22.
Yan, J. et al. Platelet Pharmacytes for the Hierarchical Amplification of Antitumor Immunity in Response to Self-Generated Immune Signals. Adv. Mater. 34 (23), e2109517 (2022).
Mo, J. et al. Comprehensive analysis and prediction model of mitophagy and ferroptosis in primary immune thrombocytopenia. Br. J. Haematol. 204 (6), 2429–2441 (2024).
Yang, Y. et al. SOX4-ZIP14-zinc metabolism mediates oncogenesis and suppresses T cell immunity in nasopharyngeal carcinoma. Cell. Rep. Med. 6 (9), 102300 (2025).
Shao, Y. et al. SLC39A10 is a key zinc transporter in T cells and its loss mitigates autoimmune disease. Sci. China Life Sci. 68 (7), 1855–1870 (2025).
Deng, Z. et al. Mesenchymal Stem Cells Prevent SLC39A14-Dependent Hepatocyte Ferroptosis through Exosomal miR–16–5p in Liver Graft. Adv. Sci. (Weinh). 12 (6), e2411380 (2025).
Sauer, A. K. et al. Zinc is a key regulator of gastrointestinal development, microbiota composition and inflammation with relevance for autism spectrum disorders. Cell. Mol. Life Sci. 79 (1), 46 (2021). Published 2021 Dec 22.
Ma, L. et al. lncRNA, miRNA, and mRNA of plasma and tumor-derived exosomes of cardiac myxoma-related ischaemic stroke. Sci Data. ;12(1):91. Published 2025 Jan 16. (2025).
Tukacs, V. et al. Chronic Cerebral Hypoperfusion-Induced Disturbed Proteostasis of Mitochondria and MAM Is Reflected in the CSF of Rats by Proteomic Analysis. Mol. Neurobiol. 60 (6), 3158–3174 (2023).
Chen, W. et al. Mitochondrial enzyme HIBADH protects against calcium oxalate nephrolithiasis by modulating oxidative stress and apoptosis. Arch. Biochem. Biophys. 770, 110452 (2025).
Bishop, C. A. et al. Detrimental effects of branched-chain amino acids in glucose tolerance can be attributed to valine induced glucotoxicity in skeletal muscle. Nutr. Diabetes. 12 (1), 20 (2022). Published 2022 Apr 13.
Huang, Y. et al. Plasma Exosomes Derived from Patients with Primary Immune Thrombocytopenia Attenuate TBX21 + Regulatory T Cell-Mediated Immune Suppression via MiR–363–3p. Inflamm. Published online March 4, (2025).
Zhang, R. et al. Pleiotropic effects of a mitochondrion-targeted glutathione reductase inhibitor on restraining tumor cells. Eur. J. Med. Chem. 248, 115069 (2023).
Zhan, Y. et al. Impaired mitochondria of Tregs decreases OXPHOS-derived ATP in primary immune thrombocytopenia with positive plasma pathogens detected by metagenomic sequencing. Exp. Hematol. Oncol. 11 (1), 48 (2022). Published 2022 Sep 1.
Xie, Y. et al. PM2.5 promotes platelet activation and thrombosis via ROS/MAPKs pathway-mediated mitochondrial dysfunction. Environ. Res. 283, 122116 (2025).
Wang, C., Xu, M., Fan, Q., Li, C. & Zhou, X. Therapeutic potential of exosome-based personalized delivery platform in chronic inflammatory diseases. Asian J. Pharm. Sci. 18 (1), 100772 (2023).
Wang, J. et al. Second-generation antipsychotics induce cardiotoxicity by disrupting spliceosome signaling: Implications from proteomic and transcriptomic analyses. Pharmacol. Res. 170, 105714 (2021).
Sun, T. et al. Proteomics landscape and machine learning prediction of long-term response to splenectomy in primary immune thrombocytopenia. Br. J. Haematol. 204 (6), 2418–2428 (2024).
Karginov, T. A., Ménoret, A. & Vella, A. T. Optimal CD8 + T cell effector function requires costimulation-induced RNA-binding proteins that reprogram the transcript isoform landscape. Nat. Commun. 13 (1), 3540 (2022). Published 2022 Jun 20.
Pellagatti, A. et al. Impact of spliceosome mutations on RNA splicing in myelodysplasia: dysregulated genes/pathways and clinical associations. Blood 132 (12), 1225–1240 (2018).
Zhao, Z. et al. Targeted delivery of exosomal miR–484 reprograms tumor vasculature for chemotherapy sensitization. Cancer Lett. 530, 45–58 (2022).
Li, H. et al. Mir–484 contributes to diminished ovarian reserve by regulating granulosa cell function via YAP1-mediated mitochondrial function and apoptosis. Int. J. Biol. Sci. 18 (3), 1008–1021 (2022). Published 2022 Jan 1.
Lin, T. et al. Identification of exosomal miR–484 role in reprogramming mitochondrial metabolism in pancreatic cancer through Wnt/MAPK axis control. Pharmacol. Res. 197, 106980 (2023).
Guo, Y. et al. Rbms1 promotes pulmonary fibrosis by stabilising Sumo2 mRNA to facilitate Smad4-SUMOylation and fibroblast activation. Eur. Respir J. 66 (3), 2401667 (2025). Published 2025 Sep 25.
De Luca, B. et al. Efficacy and tolerability of antidepressants in individuals suffering from physical conditions and depressive disorders: network meta-analysis. Br. J. Psychiatry. 227 (2), 553–566 (2025).
Acknowledgements
The authors gratefully acknowledge the technical support and facilities provided by the Department of Hematology at the First Affiliated Hospital of Chongqing Medical University and Fengdu County Traditional Chinese Medicine Hospital.
Funding
This study was supported by grants from the Chongqing Medical Scientific Research Project (Grant No. 2025MSXM065) and the Chongqing Natural Science Foundation (Grant No. CSTB2025NSCQ-GPX1218).
Author information
Authors and Affiliations
Contributions
F.F.L.completed the bioinformatics analysis component of this study, drafted the initial manuscript, and participated in subsequent revisions. Z.Y.C.and Z.H.Y.systematically organized the research data and refined all figures and tables. J.Y.S.and J.P.collected clinical specimens and performed analytical processing of the research outco mes. P.H.and Z.S.Y.were responsible for the conceptual framework and methodological rigor of this study, additionally conducting critical reviews of the revised manuscript for academic coherence.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lou, F., Chen, Z., Yuan, Z. et al. Exploring exosome-related genes as candidate biomarkers in primary immune thrombocytopenia through transcriptomics and preliminary experimental validation. Sci Rep 16, 14322 (2026). https://doi.org/10.1038/s41598-026-43618-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-43618-1
