
Abstract
Macrophage plasticity is critical for maintaining immune function and developing solid tumors; however, the macrophage polarization mechanism remains incompletely understood. Our findings reveal that Mg2+ entry through distinct plasma membrane channels is critical to macrophage plasticity. Naïve macrophages displayed a previously unidentified Mg2+ dependent current, and TRPM7-like activity, which modulates its survival. Significantly, in M1 macrophages, Mg2+ entry is facilitated by a novel Mg²-dependent current that relies on extracellular Mg2+, which was crucial for activating iNOS/NFκB pathways and cellular bioenergetics, which drives pro-inflammatory cytokines. Conversely, in M2 macrophages, Mg2+ entry occurs primarily through TRPM7 channels, pivotal for IL-4 and IL-10-mediated anti-inflammatory cytokine secretion. Notably, the Mg2+ deficient diet or addition of TRPM7 agonist Naltriben suppresses the M1 phenotype while promoting angiogenic factors and fostering tumor growth. These findings suggest that Mg2+ flux via specific channels is indispensable for macrophage polarization, with its dysregulation playing a pivotal role in tumor progression.
Similar content being viewed by others

Introduction
Modulating the immune response through changing macrophage polarization is a promising strategy to elicit beneficial outcomes such as removing pathogens and effective tissue repair1,2. The innate immune response depends on macrophage polarization that eliminate the invading pathogens directly via phagocytosis or via the production of cytokines3,4,5. Cytokines such as interferon (IFNγ), produced by lymphocytes, has been shown to transform naïve macrophages into activated macrophages (M1) that amplify the antigen-presenting capacity, complement-mediated phagocytosis, and secretes pro-inflammatory cytokines6. Release of pro-inflammatory mediators such as tumor necrosis factor (TNF)-α, IL-1, IL-6, and reactive nitrogen species from the M1 phenotype leads to a strong bactericidal and tumoricidal activity7,8,9,10.
Similarly, TH2-produced interleukin (IL)-4 could transform naïve macrophages into alternatively activated macrophages (M2). The M2 phenotype expresses molecules including Arginase1 (Arg1), which is involved in parasite infestation, tissue remodeling, and tumor progression11,12. Besides cytokines, alternative pathways have also been shown to modulate macrophage polarization; importantly, M2 polarization relies on oxidative phosphorylation and fatty acid oxidation; however, the mechanisms of how they modulate macrophage polarization are yet to be fully identified. In addition, the conventional M1/M2 classification of macrophages has been challenged13, and perhaps a quick transition between M1 and M2 phenotypes is more appropriate. Thus, understanding factors common in macrophage polarization might dictate these key events. An Mg2+-deficient diet also induced tumor growth, leading to metastatic spread14. Mg2+ plays a vital role in immune regulation and mitochondrial bioenergetics15, and low serum Mg2+ is associated with a variety of chronic diseases, such as insulin resistance and type 2 diabetes, cancer development, metabolic syndrome, hypertension, cardiovascular disease, stroke, migraine, Alzheimer’s disease, and asthma16,17. However, our understanding of its cellular regulation and clinical relevance in macrophage cells is not fully defined, as molecular components of Mg2+ homeostasis in different macrophage phenotypes remain undefined.
Magnesium (Mg2+) is the second-most abundant cation in cellular systems that plays a key role in various cellular responses. In immune cells, Mg2+ is a co-factor for immunoglobulin synthesis, C3 convertase. Similarly, Mg2+ is essential for immune cell adherence, antibody-dependent cytolysis, IgM lymphocyte binding, macrophage response to lymphokines, and T–B cell adherence18,19. Intracellular Mg2+ can also function as a second messenger in the immune activation of T and B lymphocytes16. Similarly, Mg2+ levels also regulate cytokine production, which plays a leading role in macrophage function, and loss of Mg2+ has been shown to induce autoimmunity20. Several Mg2+ permeable ion channels and transporters have been identified, and their characterization could provide new insights into the regulation of Mg2+ homeostasis and how it affects immune responses21. Importantly, deletion of Mg2+ channel TRPM7 in chicken DT40 B lymphocytes becomes growth deficient, leading to cell death within 48 hours22. TRPM7 deficiency in mice results in early embryonic lethality, and murine TRPM7−/− thymocytes experience a developmental block, suggesting a critical role of TRPM7 in cell physiology23,24.
Additionally, genetic mutations of the intracellular Mg2+ regulator, MAGT1, have been shown to elicit specific immune defects in affected individuals (XMEN—X-linked immunodeficiency), suggesting that Mg2+ regulation is essential for immune function25. Similarly, SLC41 and TMEM63 are also shown to increase Mg2+ entry, but their role in immune cells is not known26. Thus, identifying the precise function of these Mg2+ entry channels in various immune cells and establishing the mechanisms of immune regulatory networks are critical in modulating the immune response.
This study is aimed to identify and characterize the Mg2+ entry channels/transporters in various macrophage phenotypes. Our data suggests that Mg2+ homeostasis in naïve and M2 macrophages is primarily mediated through TRPM7, which is critical for tumor development. In contrast, the M1 phenotype depended on an unidentified plasma membrane Mg2+ entry channel. Mice fed a diet deficient in Mg2+ had reduced M1-type macrophages and promoted tumor growth. Finally, loss of TRPM7 specifically prevented M2 polarization without affecting the M1 phenotype, along with a decrease in critical anti-inflammatory cytokines. Moreover, activation of TRPM7 was sufficient to promote M2 polarization and promotes tumor progression. These results suggest that differential expression of Mg2+ entry channels and transporter is essential for M1/M2 function in modulating tumor development and progression.
Results
Mg2+ entry shapes cytotoxic M1 activity and inhibition of tumor development
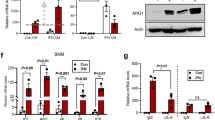
Macrophages have a dual role in tumor development, where M2 cells promote tumor progression, whereas M1 cells produce cytokines that inhibit tumor progression27. Thus, to evaluate the role of Mg2+ entry in different macrophage phenotypes and its role in solid tumor growth, we randomized nude mice in our experimentation, as they lack the T and B cells. The first cohort was given the Mg2+ supplemented diet, whereas the other cohort was put on an Mg2+ deficient diet (Fig. 1A). Interestingly, the solid tumors grew significantly more in Mg2+ deficient diet, when compared with Mg2+ supplemented diet (Fig. 1B). Increased tumor volume was observed in mice placed on a Mg2+ deficient diet (Fig. 1B, C). Using flow cytometry, we evaluated the presence of the M1 and the M2 phenotype in both Mg2+ supplemented and Mg2+ deficient diets. Interestingly, fewer M1-positive cells were observed in animals placed on the Mg2+ deficient diet when compared with Mg2+ supplemented diet (Fig. 1D, E). In contrast, the number of M2 cells was not significantly affected (Fig. 1D). Confocal microscopy further confirmed the immunohistochemical data, where an increase in M1 cells was observed in Mg2+ supplemented diet, whereas the number of M2 cells were increased in Mg2+ deficient diet (Fig. 1E). Next, we evaluated the characteristics of the Mg2+ entry channel using in vivo conditions and macrophages isolated from solid tumors showed the initiation of a Mg2+ current, which was significantly higher in macrophages isolated from Mg2+ deficient diet that shows the M2 phenotype (Fig. 1D-F). Significantly, the Mg2+ currents reversed at 0 mV, which is consistent with the previous recording observed with TRPM7 channels28,29 (Fig. 1F). Furthermore, the tumor growth factors VGEF were also decreased in Mg2+ supplemented diet (Fig. 1G), along with a decrease in pro-inflammatory cytokines, without affecting the M2-dependent cytokines (Fig. 1G). We next estimated serum Mg2+ levels, which were significantly decreased in mice subjected to Mg2+ deficient diet, even though the TRPM7 channel activity was higher (Fig. 1H). These results suggest a crucial role for Mg2+ transport in distinctly modulating the immune response.

Schematic overview of mice diet study, on day 0, mice were put on an Mg2+ deficient diet and an Mg2+ normal diet. On day 3, 3×106 Cal27 cells were injected into the mice, and tumor growth was measured every week. On day 28, mice were sacrificed for tumor collection (A). Tumor volumes were measured using a digital caliper every 7 days using a Xenograft tumor model protocol. The data shown are representative of three independent experiments with similar results. Tumor volumes are depicted in the picture. Bar graphs depict average ± SD for relative values, NS = non-significant (Student’s t-test) (B). The pictures shown below are isolated solid tumors from mice, and (C) shows microscopy analysis showing H&E staining of tumors from the Mg2+ deficient diet and Mg2+ normal diet. D Flow cytometer analysis on nude mice tumors from Mg2+ deficient diet, and Mg2+ normal diet was performed to measure the levels of surface markers to demonstrate the efficacy of cell infiltration. Data show low arginase and CD206+ M2 markers and high CD80+ and iNOS M1 markers to illustrate successful M1 infiltration on Mg2+ normal diet and low levels of both markers when mice were given Mg2+ deficient diet. Bar graphs show the percentage of M1 and M2 markers (average ± SD for relative values, ***p ≤ 0.001 (Student’s t-test)) on different diets, from three independent experiments (E). Expression analysis showing the level of expression of arginase 1 and iNOS of mice placed on Mg2+ sufficient diet (normal) or Mg2+ deficient diet. F Representative whole-cell recording showing outward currents at +100 mV from macrophages isolated from tumor cells (n = 6) in mice placed on Mg2+ sufficient diet or Mg2+ deficient diet. IV curves (acquired when currents reach peak) under various conditions are also shown. A bar graph (average ± SE) showing the quantification of the current is also shown. G VGEF in tumors from the Mg2+ deficient diet and Mg2+ normal diet. The data shown are representative of three independent experiments. ***p ≤ 0.001. Bar graphs represent quantification ( ± SD) of tumor cytokines present from the Mg2+ deficient diet and the Mg2+ normal diet. The data shown are representative of three independent experiments. ***p ≤ 0.001, NS = non-significant (Student’s t-test). H Quantification (bar graphs ± SE) of serum Mg2+ levels in Mg2+ deficient diet and Mg2+ supplemented diet. ****p ≤ 0.005 (Student’s t-test).
Identification and characterization of Mg2+ entry channels in naïve macrophages
Next, we characterized the Mg2+ entry channels in primary macrophage cells. The bone marrow of wildtype (C57) mice was used to isolate primary macrophage cells and cultured them in conditioned media. Naïve macrophages (Mφ) were analyzed by flow cytometer, and only F4/80+ and CD11b+ positive cells were used for all experiments (Supplementary Fig. 1A). To address the role of Mg2+ in the survival/proliferation of naïve Mφ, we treated these cells with low (0–0.5 mM) and high (1–20 mM) Mg2+ concentrations (Fig. 2A, B). Importantly, a decrease in the cell proliferation of naïve Mφ was observed when cells were deprived of Mg2+ (Fig. 2B; Supplementary Fig. 1B). In contrast, increasing concentrations of external Mg2+ showed higher cell proliferation/viability (Fig. 2B). Further, to establish the role of Mg2+ in Mφ function, RNA-seq analysis was performed on Mφ cultured in high and low [Mg2+], which showed a differential expression of genes under low (0.1 mM) and high (2.0 mM) Mg2+ (Fig. 2C, D; and Supplemental Table 1). Pathway analysis revealed that genes that were crucial for immune function as well as their ability to withstand stress were increased in high [Mg2+]; whereas, IL4 pathway and type 2 immune response were increased in low [Mg2+] (Fig. 2E). These results are consistent with previous findings30,31 and suggest a crucial role for Mg2+ entry in modulating Mφ proliferation/survival and assisting in their polarization.

A Flow cytometry proliferation analysis of naïve BMDM (Mφ) from WT mice (C57BL/6 J) at 24 h after treatment with 0.1 mM Mg2+ and 20 mM Mg2+. Results from immunophenotyping experiment to evaluate cell proliferation with CFDA-SE. Data are represented as histograms of AlexaFluor488 expression (mean ± SD) of different treatments from three independent experiments. B Cell viability was performed in naïve Mφ cultured with different Mg2+ concentrations by BrdU colorimetric assay. Data shown are representative of three independent experiments with similar results. C Volcano plot showing changes in gene expression in primary Mφ cultured with 0.1 mM Mg2+ and 2.0 mM Mg2+. D Heatmap showing genes that were increased in Mφ cultured at higher (2.0 mM) Mg2+ concentrations than cells growing in 0.1 mM Mg2+. E Gene ontologies (GO) and pathway analysis showing an increase in biological pathways in Mφ cultured with 2.0 mM Mg2+ and at 0.1 mM Mg2+. F Representative whole cell recording (n = 8-10) showing outward currents at +100 mV in control and 2APB treated (100 μM) Mφ cells are presented. G IV curves (acquired when currents reach peak) under various conditions (Bath application 100 μM 2APB, or 20 μM NS8593) as labeled in the figure, and quantitation of current density at +100 mV is shown in (H). I–K Bath application 5 mM Mg2+ inhibited TRPM7-like current. IV curves and quantitation of current density at +100 mV under various conditions (Bath application 5 mM Mg2+, 10 mM Mg2+, or 20 mM Mg2+) as labeled in the figure. Data shown are representative of (8-12 individual patches). L–O Increasing external Mg2+ increases Mg2+ entry, as shown in analog plots of the fluorescence ratio (340/380). Bath application of 5-20 mM Mg2+ induced an inward current (holding at −120mV). IV curves (N) and quantitation (O) of current density at -120mV under various conditions as labeled in figure. indicate significance (p < 0.05). P Immunoblotting analysis showing the level of protein expression in wildtype mice naïve BMDM in comparison with activated M1 and M2 BMDM. Data shown are representative of three independent experiments with equivalent results.
We next focused on characterizing the transporter/channels that could contribute to Mg2+ entry in naïve Mφ. Decreasing intracellular Mg2+ led to the activation of Mg2+ entry and an outward rectifying current elicited by a voltage ramp protocol ranging from −100 to +100 mV (Fig. 2F, G). The outwardly rectifying currents reversed close to 0 mV, consistent with the previous recording observed with TRPM6/7 channels28,29. To further separate if the Mg2+ currents observed are through TRPM6 or TRPM7 channels, we exploited the differential effect of 2-APB on these channels as 2-APB activates TRPM6, but inhibits TRPM7 currents16,32. Mg2+ current amplitude was substantially reduced by the addition of 2-APB (50 μM; Supplementary Fig. 1C); furthermore, a TRPM7-specific inhibitor (NS8593) also had a similar effect on the Mg2+ current amplitude (Fig. 2F–H). Furthermore, the addition of Ca2+ in the external media also showed similar current properties; however, the inward current amplitude was slightly higher in the presence of Ca2+ (Supplementary Fig. 1D, E). Pretreatment of naïve Mφ with a nonspecific Ca2+ channel blocker (SKF96365) also showed a decrease in TRPM7 currents (Supplementary Fig. 1F, G), suggesting that TRPM7 is one of the channels that initiate Mg2+ entry in naïve Mφ. As increased proliferation and survival were observed at higher external Mg2+ levels, we evaluated the TRPM7 current properties at higher Mg2+ levels. Surprisingly, increasing external Mg2+ showed a decrease in TRPM7 currents, and a dose-dependent reduction in TRPM7 currents was observed when external Mg2+ levels were gradually increased (Fig. 2I–K).
These results contrast the cell proliferation data and suggest that perhaps additional Mg2+ entry channels in naïve Mφ could modulate Mg2+ entry. Importantly, TRPM7−/− cells have been shown to have regular Mg2+ influx33, suggesting that another channel/transporter could also be critical for Mg2+ entry. Thus, we next evaluated this unknown Mg2+ entry channel. In naïve Mφ increasing external Mg2+ led to a gradual increase in intracellular Mg2+ levels (Fig. 2L; Supplementary Fig. 1H), which was not blocked by the addition of gadolinium (Gd3+), a known non-specific divalent cation inhibitor34, or the addition of either 2-APB, SKF96365, or flufenamic acid (FFA, a TRPM channel inhibitor)35 (Supplementary Fig. 1I-L). We also evaluated the current properties of this unique Mg2+ channel, which showed that a linear current is evoked by the presence of external Mg2+ and increasing external Mg2+ levels, leading to a gradual increase in Mg2+ currents (Fig. 2M–O). The IV relationship was not altered upon increasing Mg2+ concentrations, and in naïve Mφ, the currents reversed between -10 and 0 mV (Fig. 2N), which is slightly different from that observed with MagT1 or Tmem63b36. Supplementation of Ca2+ or Ba2+ did not show the activation of these currents, which is different from both MagT1 and Tmem63b currents, suggesting that it is a different Mg2+ entry channel (Supplementary Fig. 1M). To further establish the molecular identity of the channel, we silenced the known Mg2+ channels. Silencing of Slc41A/B/C or Tmem63b did not decrease Mg2+ entry, whereas, silencing of MagT1 only partially decreased Mg2+ entry in naïve Mφ (Supplementary Fig. 1N), suggesting that this Mg2+ entry channel differs from the currently known Mg2+ channels.
Lipopolysaccharide (LPS) or IFN-γ can induce M1 polarization; meanwhile, M2 polarization is achieved by the addition of IL-41,37,38,39. Treatment of naïve Mφ with LPS or IL-4 increases intracellular [Mg2+] (Supplementary Fig. 2A, B). To further evaluate this, naïve Mφ were polarized with IFN-γ (20 ng/ml) or IL-4 (20 ng/ml) for 24 h, and the expression of Mg2+ entry channels was evaluated. Immunoblot analysis of Mg2+ channels and transporters showed expression of TRPM6, TRPM7, and MagT1 was observed in naïve Mφ an increase in MagT1 expression, but not that of TRPM6/7, was observed in IFNγ-induced M1 macrophages. Similarly, a distinct elevated expression of TRPM7 was observed in IL4-induced M2 macrophages (Fig. 2P), showing an interesting distinction in the expression of individual Mg2+ entry channels between the two macrophage phenotypes.
Mg2+ influx in M1 macrophages occurs via a selective Mg2+ channel that modulates its polarization and maintains the M1 phenotype
Data presented above shows that distinct Mg2+ entry channels are present in naïve Mφ. To outline the signaling mechanism by which Mg2+ modulates the M1 effector function, we next characterized the Mg2+ entry channel critical for the M1 phenotype. Naïve macrophages were treated with IFNγ (20 ng/ml) for 24 h and M1-polarization was confirmed using flow cytometry. Increased CD80 and reduced CD206 signify M1 polarization (Fig. 3A)40,41. The analysis of intracellular Mg2+ levels showed a significant increase in Mg2+ levels in M1 cells compared with naïve Mφ (Fig. 3B, C). Evaluation of TRPM7 currents in M1 cells showed a decrease in TRPM7 channel activity compared with naïve Mφ (Fig. 3D, E). Next, an in vivo murine model of peritonitis was used, and wildtype mice were i.p. infected with Klebsiella pneumoniae (KPN) or PBS (CTRL) for 24 h before collection of peritoneal macrophages for electrophysiological analysis. Again, TRPM7 currents in KPN-induced M1 cells showed a decrease in TRPM7 activity compared with PBS-treated macrophages (Supplementary Fig. 2C–E). In contrast, increasing external Mg2+ (20 mM) showed activation of a novel channel, which was significantly higher in M1 cells as compared with naïve Mφ, without any changes to its reverse potential (Fig. 3F, G). Moreover, silencing of MagT1 only partially decreased this Mg2+-dependent channel activity (data not shown), suggesting that MagT1 is not the major Mg2+ entry channel in M1 cells.

A Flow cytometer analysis on WT mice cells was performed to measure the levels of surface markers to demonstrate the efficacy of cell polarization. Data show a high CD80+ M1 marker and low CD206+ M2 marker to illustrate successful M1 polarization when naïve M0 cells are treated with IFN-γ (20 ng/ml) for 24 h. Data shown are representative of three independent experiments with comparable results. B Mg2+ imaging was performed using Mag-Fura in M0 and M1 cells. Bath application of 20 mM Mg2+ induces Mg2+ influx, and analog plots of the fluorescence ratio (340/380 from 50-60 cells) are shown. C Quantification (mean ± SE) of intracellular Mg2+ concentration. *indicate significance (p < 0.05). IV curves of TRPM7 current in M0 and M1 cells and quantitation of current density at +100 mV (mean ± SD of 6 experiments) are shown in (D) and (E). F, G IV curves of Mg2+ entry current in M0, M1 cells and quantitation of current density at -120mV (mean ± SD of 6 experiments) are represented in this figure. H Proliferation analysis by CFDA-SE Alexa Fluor488 fluorescence on M1 activated wild type mice BMDM treated with media containing 0 mM Mg2+ and 20 mM Mg2+ versus control to show the impact of no magnesium on M1 macrophage proliferation. Data shown in histograms are representative of three independent experiments with similar results. I Flow cytometer analysis on M1 activated wild type mice BMDM treated with several Mg2+ concentrations (0 mM, 0.1 mM, 0.5 mM, 10 mM, and 20 mM) for 24 hr to show how magnesium is relevant for macrophage polarization, increasing CD80+ M1 marker expression. Data shown are representative of three independent experiments with similar results. Bar graphs depict average ± SD for relative values in (J), ***p ≤ 0.001, (Student’s t-test). K Activated BMDM M1 cells cultured in 6 wells plate (2×106 cells/well) and treated with 0 mM, 0.1 mM, 0.5 mM, 1 mM, 2 mM and 5 mM Mg2+ for 24 hr. The concentration of pro-inflammatory cytokines IL-6 released in the media was performed by ELISA and bar graphs depict average ± SD for relative values (n = 3), ***p ≤ 0.001 (Student’s t-test). L, M Activated BMDM M1 cells cultured in 6 wells plate (2×106 cells/well) and treated with 2APB (10 μM) plus 2APB + 20 mM Mg2+ and 2APB + 20 mM Ca2+; SKF (10 μM) plus SKF + 20 mM Mg2+ and SKF + 20 mM Ca2+ overnight. The concentration of pro-inflammatory cytokines (IL-6 and IL1-β) released in the media was performed by ELISA. Data shown are representative of three independent experiments with similar results. Bar graphs depict average ± SD for relative values, ***p ≤ 0.001 (Student’s t-test). N Immunoblotting analysis showing the level of expression of pNFκB, NFκB, iNOS, and β-actin in wildtype mice M1 activated BMDM with increasing Mg2+ concentrations. Data shown are representative of three independent experiments with equivalent results.
We next evaluated the function of external Mg2+, and as expected, low or removal of external Mg2+ leads to a decrease in the development/activation of the M1 phenotype. Flow cytometry assays were performed to evaluate the proliferation (with AlexaFluor488 beads) and for the development of the M1 phenotype. Notably, a significant decrease in M1 proliferation was observed in cells devoid of Mg2+; whereas a high concentration of Mg2+ induced M1 proliferation (Fig. 3H). Moreover, flow cytometry further showed that a gradual increase of external Mg2+ increases the surface markers for M1 markers (CD80), suggesting that Mg2+ is critical for developing the M1 phenotype (Fig. 3I, J). Similarly, a significant increase in the release of pro-inflammatory cytokine (IL-6) was observed in the presence of high Mg2+ concentrations (Fig. 3K). Significantly, the addition of 2APB (50 μM for 6 hr) and SKF treatment (10 μM for 6 hr) decreased the release of pro-inflammatory cytokines (IL-6 and IL-1β) (Fig. 3L, M). However, increasing external, Mg2+ concentration (20 mM) was able to overcome 2APB/SKF-mediated inhibition of the pro-inflammatory cytokines (Fig. 3L, M). Consistent with these results, increasing external Mg2+ concentration also leads to an increase in NFκB phosphorylation and iNOS expression, suggesting a positive activation by Mg2+ in regulating the M1 phenotype (Fig. 3N). Together the data presented suggest that Mg2+ influx via an unknown Mg2+-dependent channel is essential for M1 survival, as well as for the activation of downstream signaling factors that modulate cytokine releases and in the development of the activated M1 phenotype.
M2 phenotype polarization is mediated by TRPM7 activation
Activation of the M1 phenotype is critical for inhibiting infections; however, after the resolution of the infection process, these macrophages are transformed into the anti-inflammatory M2 phenotype. Thus, we next investigated if, Mg2+ would also have similar effects on the M2 phenotype. Naïve macrophages from wildtype mice were polarized by treating with IL-4 (20 ng/ml) for 24 h. Flow cytometry was utilized to confirm M2-polarization by detection of the M2 surface marker, CD206+ (Fig. 4A). IL-4 caused an increase in intracellular Mg2+ levels in M2 Mφ (Supplementary Fig. 2B), and the release of anti-inflammatory cytokines was observed in M2 cells grown in Mg2+ (1 mM) (Fig. 4B). In contrast, pretreatment with high concentrations of Mg2+ (5 mM) decreased the release of anti-inflammatory cytokine IL1RA (Fig. 4B). Moreover, 1 mM Mg2+ was sufficient to increase M2 polarization; whereas, lower (0 mM, 0.1 mM) or higher (10 mM and 20 mM) Mg2+ levels decreased the M2 phenotype (as shown by a decrease in (CD206+) expression) (Fig.4C, D). Nonspecific channel blockers such as 2APB (50 μM for 6 h) and SKF (10 μM for 6 h) showed a significantly lower release of anti-inflammatory cytokines (IL1RA and IL10) (Fig. 4E, F). Interestingly, high concentrations of Mg2+ (20 mM) failed to show any increase in the release of the same cytokines (Fig. 4E, F). These data are in contrast with the data observed in M1 cells and suggest that perhaps higher Mg2+ levels can inhibit the development of the M2 phenotype.

A Flow cytometer analysis on WT mice cells was performed to measure the levels of surface markers to demonstrate the efficacy of cell polarization. Data show a high CD206+ M2 marker and low CD80+ M1 marker to illustrate successful M2 polarization when naïve M0 cells are treated with IL-4 (20 ng/ml) for 24 h. Data shown are representative of three independent experiments with similar results. B Activated BMDM M1 cells cultured in 6 wells plate (2×106 cells/well) and treated with 1 mM and 5 mM Mg2+ and Ca2+ for 24 hr. The concentration of anti-inflammatory cytokine IL-1RA released in the media was performed by ELISA. Data shown are representative of three independent experiments with similar results. Bar graphs depict average ± SD for relative values, ***p ≤ 0.001 (Student’s t-test). C, D Flow cytometer analysis on M1 activated wild type mice BMDM treated with several Mg2+ concentrations (0 mM, 0.1 mM, 0.5 mM, 5 mM, 10 mM and 20 mM) for 24 hr to show how Mg2+ is relevant for macrophage polarization, increasing CD80 + M1 marker expression. Data shown are representative of three independent experiments with similar results. Bar graphs depict average ± SD for relative values, ***p ≤ 0.001 (Student’s t-test). E, F Activated BMDM M2 cells cultured in 6 wells plate (2×106 cells/well) and treated with 2APB (10 μM) plus 2APB + 20 mM Mg2+ and 2APB + 20 mM Ca2+; SKF (10 μM) plus SKF + 20 mM Mg2+ and SKF + 20 mM Ca2+ overnight. The concentration of anti-inflammatory cytokines (IL-10 and IL-1RA) released in the media was performed by ELISA. Data shown are representative of three independent experiments with similar results. Bar graphs depict average ± SD for relative values, NS = non-significant (Student’s t-test). G IV curves of TRPM7 current in M0, M2 cells and quantitation of current density at +100 mV (mean ± SD of 6 experiments) is shown in (H). Bath application 10 mM Mg2+, 20 mM Mg2+ induced inward current (holding at -120mV) are shown in (I). IV curves (J) and quantitation of current density at −120mV (K) in M0, M2 cells, as labeled in the figure. L Immunoblotting analysis showing the level of expression of TRPM7, pSTAT6, STAT6, and β-actin in wildtype mice M2 activated BMDM with increasing Mg2+ concentrations. Data shown are representative of three independent experiments with similar results. M Elisa quantification for VEGF in supernatants of M2 activated BMDM with 2APB treatment (10 µM overnight) and increasing Mg2+ concentrations. Data shown are representative of three independent experiments. Bar graphs depict average ±SD for relative values, ***p ≤ 0.001 (Student’s t-test). N, O Flow cytometer analysis on naïve BMDM (Mφ) with supplemented with 1 mM and 20 mM Mg2+ concentrations for 24 hr, showing the presence of M1 (CD 80) and M2 (CD 206) markers respectively. Bar graphs depict average ±SD for relative values, **p ≤ 0.005 (Student’s t-test).
To establish the identity of the Mg2+ entry channels that modulate the M2 phenotype, we again evaluated the electrophysiological properties of the channel. Similar to naïve Mφ, decreasing intracellular Mg2+ led to the development of an TRPM7-like outward rectifying current, which reversed close to 0 mV in M1 cells (Fig. 4G, H). In contrast, a significant decrease in the current properties of the novel Mg2+-dependent channel was observed (Fig. 4I–K). Naïve Mφ showed a linear Mg2+-evoked current, and increasing external Mg2+ (20 mM) led to an increase in the Mg2+ current, which was not observed in M2 cells (Fig. 4I-K). To further confirm the role of these different Mg2+ channels on M2 phenotype polarization, immunoblotting was performed on M2 cell lysates. Importantly, the expression of Arginase 1 and pSTAT6, the classical activation markers of M2 cells, was highest when cells were incubated in 1 mM external Mg2+ (Fig. 4L). Furthermore, either decreasing (0.5 mM) or increasing Mg2+ (5 mM) concentration leads to a decrease in the phosphorylation of STAT6 and the expression of Arginase1 (Fig. 4L). Consistent with these results, TRPM7 expression was also increased at 1 mM external Mg2+ but decreased drastically when increasing Mg2+ (5 mM) concentration (Fig. 4L). Importantly, a functional angiogenesis assay was also performed on the supernatants of M2 phenotype cells when treated with varying concentrations of Mg2+. Interestingly, a significant increase in the release of VEGF was directly related to Mg2+ concentrations in the range of 1-2 mM, as either decreasing or increasing Mg2+ levels further inhibited M2 function (Fig. 4M). Also, a significant increase in cell proliferation of M2 cells42,43 was observed by increasing the Mg2+ concentrations (Supplementary Fig. 2F, G). Interestingly, modulating external Mg2+ levels (independent of cytokine stimulation), lead to the polarization of naïve Mφ. Higher Mg2+ levels (20 mM), promoted the development of M1 phenotype, along with a suppression of M2 cells (Fig. 4N, O). In contrast, supplementation of 1 mM Mg2+ to naïve Mφ led to an increase in M2 markers (Fig. 4 N, O), suggesting that external Mg2+ levels are sufficient to modulate macrophage polarization.
Differential Mg2+ entry is critical for maintaining the mitochondrial bioenergetics in M1/M2 phenotypes
Metabolism has been suggested as a key regulator for immune cell (M1/M2) differentiation and its activation44,45. Importantly, both glycolysis and oxidative phosphorylation have been shown to modulate macrophage polarization, but factors that could alter oxidative phosphorylation are not known. Interestingly, Mg2+ has a stimulatory effect on several mitochondrial enzymes, thereby can significantly impact the metabolic state of immune cells. Thus, we evaluated mitochondrial Mg2+ levels in various macrophage phenotypes. Importantly, a significant difference in intracellular mitochondrial Mg2+ (mMg2+) levels was observed between naïve, M1, and M2 macrophages. Naïve macrophages from WT mice exhibited a higher level of basal mMg2+ release upon FCCP uncoupling than the M1 polarized macrophages. Conversely, the M2 macrophages exhibited a higher intrinsic mMg2+, restored to the levels of naïve macrophages, that would drive a lowering of mitochondrial metabolism and ATP synthesis (Fig. 5A, B).

A Spectrofluorimetric traces depicting permeabilized macrophages (Naïve, M1, M2) exposed to FCCP at the indicated time point to assess basal mMg2+. B Quantification of basal mMg2+ in permeabilized macrophages. C Spectrofluorimetric traces representing permeabilized macrophages (M1, M2) in the presence of 20 mM Mg2+ for 1 h exposed to FCCP at the indicated time point to assess basal mMg2+. D Quantification of basal mMg2+ in permeabilized macrophages in the presence of Mg2+. E, G, I Oxygen Consumption Rate (OCR) was analyzed in macrophages isolated from WT with and without Mg2+ for 1 hour for M0, M1, and M2 phenotypes. N = 3 performed in duplicate. F, H, J Basal, maximal respiration, and proton leak analysis of macrophages isolated from WT with and without Mg2+ for 1 hour. Mean ± SEM, n = 3, ***p < 0.001.
In contrast, higher mitochondrial Mg2+ levels were observed in naïve and M2 phenotypes, suggesting that changes in mitochondrial Mg2+ levels might be essential for macrophage polarization. Importantly, with 20 mM Mg2+ for 1 hour, we observed an inverse effect of Mg2+ retention in the mitochondria. While the M1 macrophages retained similar levels of basal mMg2+ to the absence of extracellular Mg2+, M2 macrophages exhibited a reduced level of basal Mg2+ than M1 macrophages (Fig. 5C, D).
We further evaluated the oxygen consumption rate (OCR), which is a readout for mitochondrial bioenergetics in these different conditions. Interestingly, we observed that loss of external Mg2+ exhibited lower OCR, and levels of basal, maximal, and proton leak were inhibited in all the macrophage phenotypes (Fig. 5E–J). However, increasing Mg2+ levels (20 mM) showed a significant increase in both basal and maximal respiration in M1 cells (Supplementary Fig. 2H), suggesting that high Mg2+ concentration is perhaps needed to maintain the M1 phenotype. In contrast, M2 cells failed to show any increase in the maximal respiration and the basal respiration levels at higher Mg2+ levels (20 mM) (Supplementary Fig. 2H). Together these results suggest that there is a tight regulation of Mg2+ entry in the mitochondria that is critical for regulating the metabolic programming and phenotypic plasticity in macrophage cells.
TRPM7 is sufficient for the development of the M2 phenotype and tumor growth without altering the M1 phenotype
Based on the data presented above, we assessed the physiological effect of TRPM7 on M2 polarization. Naïve Mφ (M0) or IFNγ-treated (M1) or IL-4-treated M2 cells were either transfected with non-targeting siRNA (SiC, for control) or TRPM7siRNA (siTRPM7) to evaluate its role in the development of the M2 phenotypes. Immunoblot analysis showed a decrease in TRPM7 protein in all macrophage cells (Fig. 6A). Importantly, loss of TRPM7 in M2 cells displayed low expression of the classical M2 protein marker “Arginase 1” when compared with control siRNA treated cells (Fig. 6A), Electrophysiological analysis further showed a decrease in TRPM7 currents in M2 cells that express siTRPM7, without altering the previously unidentified Mg2+-dependent channel function in M1 cells (Fig. 6B, C). Elisa assay further showed a significant decrease in the release of anti-inflammatory cytokines from M2 cells without affecting the pro-inflammatory M1 cytokines (Fig. 6D, E). Notably, silencing of TRPM7, but not MagT1, significantly decreased VGEF levels (Fig. 6F) as well as mitochondrial function (OCR) was also inhibited upon TRPM7 silencing in M2 cells (Fig. 6G, H).

A Western blots showing expression of various proteins in silenced TRPM7 in naïve M0 plus activated BMDM M1 and M2 cells cultured in 6 wells plate (2 × 106 cells/well). B, C IV curves of Mg2+ entry currents in control, siTRPM7 naïve M0 cells, and quantitation of current density at -120mV is shown as bar graph. TRPM7 current is in control, and siTRPM7 cells (C) are quantitated at current density at +100 mV. D, E The concentration of pro and anti-inflammatory cytokines (IL-1β and IL-10, respectively) released in the media. Data shown are representative of three independent experiments with similar results. Bar graphs depict average ± SD for relative values, ***p ≤ 0.001, NS = non-significant (Student’s t-test). F Quantification for VEGF in supernatants of M2-activated BMDM with siTrpm7 and siMagt1 in comparison with siControl. Data shown are representative of three independent experiments. Bar graphs depict average ±SD for relative values, NS = non-significant, ***p ≤ 0.001 (Student’s t-test). G, H Oxygen Consumption Rate (OCR) was analyzed in M2 macrophages with control siRNA or MagT1 or TRPM7 siRNA, respectively. N = 3 performed in duplicate. I IV curves of TRPM7 currents (acquired when currents reach peak) in control and 50 µM NTB groups as labeled in the figure and are shown. The quantitation of current density at -+100 mV is shown as a bar graph. *p ≤ 0.01.
We further investigated the importance of the activation TRPM7 channel in macrophage polarization by treating the cells with a known TRPM7 activator (Naltriben)46. Naïve macrophage cells treated with Naltriben (NTB) (20 μM) for 24 h showed a clear increase in TRPM7 channel activity (Fig. 6I). Flow cytometry was also performed on the naïve cells treated with different concentrations of NTB, interestingly two of the major M2 phenotype markers, EGR2 and arginase, were increased in the presence of NTB, which was similar as observed with IL-4 (Fig. 7A, B; Supplementary Fig. 2I). Similarly, the levels of M2 cytokines, IL-1Ra, and IL-10 were also increased by the treatment with NTB (Fig. 7C; Supplementary Fig. 2J). Consistent with these results, a significant increase in VEGF levels was observed in cells treated with NTB (Fig. 7D). Furthermore, increasing NTB concentration also led to a constant increase in the expression of arginase (Fig. 7E), clearly showing that the activation of the TRPM7 channel by NTB plays a vital role in M2 phenotype polarization and thus critical for immune functions. Finally, mice treated with naltriben (TRPM7 agonist), promoted tumor growth (Fig. 7F). Together the data presented here, suggest that macrophage polarization is dependent on Mg2+ entry, where high levels of Mg2+ influx through an uncharacterized high-affinity Mg2+ influx channel drive the classically activated M1 phenotype and increase pro-inflammatory cytokines. In contrast, the alternatively activated M2 phenotype requires a low threshold Mg2+ entry channel, which is mediated by TRPM7 activation that leads to anti-inflammatory phenotype and tumor induction. Furthermore, activation of TRPM7 (via Naltriben) is sufficient to induce M2 phenotype and promote macrophage plasticity and tumor progression (Fig. 7G).

A–C Flow cytometer analysis on M0 activated wild type mice BMDM treated with IL4 or Naltriben-treated cells at different concentrations (20 µM, 30 µM, 50 µM) for 24 hr to show how TRPM7 activation is relevant for macrophage polarization, increasing expression of Arginase 1 (B). Data shown are representative of three independent experiments with similar results. Bar graphs depict average ± SD for relative values, **p ≤ 0.01, (Student’s t-test). C Naïve M0 cells cultured in 6 wells plate (2×106 cells/well) and treated with IL-4 (20 ng/ml) and Naltriben treatment (20 µM, 30 µM and 50 µM) for 24 h. The concentration of anti-inflammatory cytokines (IL-10 and IL-1RA) released in the media was performed by ELISA. Data shown are representative of three independent experiments with similar results. Bar graphs depict average ± SD for relative values, ***p ≤ 0.001 (Student’s t-test). D Elisa quantification for VEGF in supernatants of naïve (M0) BMDM with and without IL-4 treatment (20 ng/ml) and Naltriben treatment (20 µM, 30 µM and 50 µM) for 24 h. Data shown are representative of three independent experiments. Bar graphs depict average ±SD for relative values, **p ≤ 0.01, ***p ≤ 0.001 (Student’s t-test). E Immunoblotting analysis showing the level of expression of arginase and β-actin of wildtype mice naïve M0 with and without IL-4 (20 ng/ml) and Naltriben treatment (20 µM, 30 µM and 50 µM) for 24 h. The data shown are representative of three independent experiments with similar results. F Tumor volumes (average ±SE) under various conditions (n = 6). G The schematic overview of macrophage polarization, where Mg2+ influx drives macrophage phenotype transformation. M1:classically activated and pro-inflammatory cytokines release (IL-6, IL1β, TNFα) is regulated by an uncharacterized Mg2+ influx channel. In contrast activation and anti-inflammatory cytokine release (IL-10, IL-1rn, and IL1RII) are controlled by TRPM7, and it can be induced by Naltriben to promote plasticity.
Discussion
In the presence of pathogens, macrophages polarize into the classical M1 phenotype, whereas the alternative M2 phenotype is essential for the resolution of inflammation and tissue repair. Similarly, macrophages can either promote or prevent tumor development. Thus, understanding factors that modulate macrophage polarization could lead to novel strategies for their reprogramming as a potential means to prevent infections, treat inflammatory diseases, and prevent cancer. In this study, we have demonstrated that Mg2+ is essential for regulating macrophage function, and sequestering external Mg2+ levels inhibited their proliferation and induced cell death. Importantly, reprogramming of naïve Mφ toward the M1 and M2 phenotype was also dependent on Mg2+ entry and inhibiting Mg2+ entry, decreased both pro- and anti-inflammatory cytokines. Moreover, we have identified distinct Mg2+ channels that increase Mg2+ entry in M1 and M2 phenotypes that are needed for maintaining their functions. Although previous studies have identified different modulators that induce transcriptional regulations along with metabolic reprogramming as the key factors that regulate macrophage polarization47, events that are upstream of mitochondrial bioenergetics/transcription regulation have not been identified.
Importantly Mg2+ entry was vital for maintaining critical macrophage functions, and naive macrophages express several of the known Mg2+ entry channels. Increasing Mg2+ levels increased cell proliferation in naïve macrophages; however, it also inhibited TRPM7 currents, suggesting that additional Mg2+ channels could be essential in modulating their function. Previous studies have identified that TRPM7 is vital for macrophage function; however, this function was linked to its ability to bring in Ca2+, which could suggest that perhaps TRPM7 modulates the basal function in naïve macrophages. Interestingly, similar to previous findings33, TRPM7 exhibited a small inward current, but higher outward currents in macrophage cells, suggesting that higher levels of Mg2+ entry could not be through TRPM7 channels. Interestingly, Mg2+-induced Mg2+ influx in thymocytes that were obtained from Trpm7−/- mice were insensitive to the deletion of TRPM7, indicating that additional Mg2+ influx channels are functional in immune cells33. To further evaluate the identity of this critical Mg2+-induced Mg2+ entry channel, we evaluated the expression and functional properties of this elusive channel. Our results show that increasing external Mg2+ leads to a dose-dependent increase in Mg2+ entry that was critical for the development of the M1 phenotype. This uncharacterized channel was not mediated via MagT1, Slc41a/b/c, or Tmem63b but had some similar current features (linear, similar reverse potential), but few distinctive channel features were also noted (selectivity towards Mg2+, not blocked by Gd3+, activated by external Mg2+). Although the molecular identity of this channel is not yet known, we did rule out that none of the recently identified Mg2+ channels are indeed the Mg2+-induced Mg2+ entry channel, and more research is needed to identify this important channel. Interestingly, an increase in plasma Mg2+ concentration was observed in leukocytes during an acute asthma attack48, which is mediated via the M1 pro-inflammatory phenotype. These results support our findings that higher external Mg2+ levels induce an Mg2+-induced Mg2+ entry channel that modulates the pro-inflammatory M1 macrophages. Interestingly silencing of MagT1 only partially decreases the Mg2+-induced Mg2+ entry channel. MagT1 is expressed both in the plasma membrane as well as in the endoplasmic reticulum, where plasma membrane MagT1 could contribute to Mg2+ entry or assist the unknown Mg2+-induced Mg2+ entry channel. Nonetheless, our data showed that the Mg2+-induced Mg2+ entry channel was critical for the development of the M1 phenotype a, which could prevent infection and cancer progression.
Classically activated M1 macrophages have long been known to be induced by IFNγ alone or in concert with microbial stimuli (e.g., LPS). Interestingly, the addition of LPS or IFNγ led to an increase in intracellular Mg2+ levels, suggesting that Mg2+ could provide the initial trigger needed for their polarization and development into the M1 phenotype. Importantly, a decrease in extracellular Mg2+ levels led to a decrease in M1 surface markers and increasing Mg2+ levels restored the M1 phenotype. Nude mice that lack T and B cells, were used for our experimentation as we used human tumor cells, which can only grow when T cells are absent. Nonetheless, nude mice on an Mg2+-deficient diet had decreased M1 cells and showed higher tumor volume, further strengthening the notion that Mg2+ levels dictate M1 polarization which is important for inhibiting tumor progression, particularly when T and B cells are absent. However, it cannot be excluded that the phenotypes observed in this study might be absent or even reversed in immunocompetent mice and more research is needed to evaluate this further. Importantly, macrophage polarization is a highly plastic and dynamic process, which could be easily modulated by Mg2+ entry. Notably, one of the important discoveries indicated here is that our results provide the spatial and temporal resolution that dictates these two opposing events. Although both M1 and M2 cells require Mg2+ entry for their function, they both have different Mg2+ thresholds, which are regulated by distinct Mg2+ entry channels. M1 cells require high Mg2+ levels, which can only be provided through the Mg2+-induced Mg2+ entry channel. Increasing extracellular Mg2+ was critical for resolving 2APB or SKF-mediated inhibition, which suggests that it can even override the Ca2+-dependent process essential for immune activation. In contrast, M2 cells had a decreased Mg2+ threshold and needed lower Mg2+ levels, which are regulated by TRPM7 channels. These results are in contrast with a recent finding which showed that higher external Mg2+ was important for bone growth but decreased M1-like phenotype36. However, these results were generated using THP1 cell lines, which differ greatly from primary macrophages. In addition, it was suggested that TRPM7 was critical for these functions, which is inhibited by higher external Mg2+ 23,49, and thereby could not contribute to these functions.
One of the fundamental questions is how low Mg2+ entry from TRPM7 modulates the M2 phenotype. Our data shows that it activates the phosphorylation of STAT6, and increasing external Mg2+ that inactivates TRPM7 channels had a negative effect on STAT6 phosphorylation. Interestingly, TRPM7 also has a kinase domain, and it is possible that it can directly phosphorylate STAT6. In addition, the cleaved TRPM7 kinase fragments have also been shown to translocate to the nucleus and phosphorylate histones that modulate gene expression50, which can also contribute to tumor development. Mg2+ entry through TRPM7 also regulated oxidative phosphorylation; therefore, an alternative explanation could be that M2 macrophages display enhanced mitochondrial oxidative phosphorylation in comparison to M1 macrophages36,51. Mg2+ levels have been shown to modulate mitochondrial function, and it is responsible for ion channels such as voltage-dependent Ca2+ channels and K+ channels19,52. Our data support that decreasing Mg2+ levels in M2 cells, but not in M1 cells, decreased oxidative phosphorylation. Importantly, a decrease in the expression of classical M2 markers and M2 cytokines was also decreased upon the downregulation of TRPM7. Interestingly, Mg2+- a deficient diet promoted solid tumor growth, which was blocked by the inhibition of TRPM7, but increased via the activation of TRPM7. These results suggest that Mg2+ entry via TRPM7 is needed for maintaining the M2 phenotype without altering the M1 phenotype, and activation of TRPM7 by Naltriben was enough to induce M2 polarization and tumor development. In conclusion, we provide evidence that distinct Mg2+ channels and transporters are present in these macrophage phenotypes, and exploiting their unique characteristics could be critical for macrophage transformation.
Methods
Mice, primary cells, reagents, silencing of Magt1 and Trpm7, and Naltriben mesylate treatment
For the primary cells, C57BL/6 mice (6 weeks old, male) were used, and bone marrow-derived macrophages (BMDMs) were isolated from the femurs of mice. Femurs were flushed out with PBS using a sterile needle, then cultivated in a DMEM medium (Life Technologies, Carlsbad, CA, USA) containing L929 conditioned media. After 5 days, fully differentiated BMDMs formed on the culture plates’ bottom. The isolated macrophage cells were then incubated at 37 °C in a DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin solution53,54. For silencing experiments, respective Magt1 siRNA (ThermoFisher, Assay ID: 183726) and Trpm7 siRNA (ThermoFisher, Assay ID:181935) were used, the transfection was performed with Lipofectamine® RNAi Reagent following the manufacturer’s protocol. Cells were seeded to be 70% confluent, and the Lipofectamine 2000 reagent was diluted in an optimal concentration (ThermoFisher, LMRNA015) 25pmol per well in a 6-well dish) in Opti-MEM medium (Gibco, Dublin, Ireland). Lipofectamine was added to the diluted siRNA in a 1:1 ratio and incubated for 5 minutes at room temperature. The complex was then layered on the cells for 5 hours, followed by adding fresh media and used for individual experiments. For the experiments utilizing differential [Mg2+] and [Ca2+], HBSS (Hank’s Balanced Salt Solution without calcium chloride, magnesium chloride, and magnesium sulfate, Gibco) was used with Ca2+ and Mg2+ added accordingly. Naltriben mesylate (Tocris, Minneapolis, MN, USA) treatment was performed on isolated naïve BMDM cells (2 × 106 cells per well) from WT mice at different concentrations (20 µM, 30 µM and 50 µM per well) in a 6 well dish for 24 h. Animal studies adhered to the international guidelines on animal welfare following the National Institute of Health Guide for the Care and Use of Laboratory Animals and complied with the ARRIVE guidelines and the 2013 AVMA euthanasia guidelines. All studies were approved by UT Health San Antonio (UT Health SA) Institutional Animal Care and Use Committee. Mg2+ and tumor studies were carried out in six- to eight-week-old female athymic nude mice given by Dr. Cara Gonzales (Department of Comprehensive Dentistry- UTHSCSA) under a laminar air-flow cabinet in pathogen-free conditions; both conditions were given under the skin of the conscious animal. Mice were kept in a 12 h light/dark schedule at controlled temperature and humidity with food and water ad libitum. Mice were acclimated for one week before the study initiation.
Magnesium deprivation diet and tumor studies
3 × 106 Cal-27 cells in saline solution were injected subcutaneously into the flank as described previously by us55,56. Animals were conscious during injection and no anesthesia was used in this section of the study. Mice were stratified into 2 experimental groups (n = 5 per group): A magnesium-deprived diet along with a respective control diet were purchased from Envigo, Madison WI, USA. Individual mice were acclimatized with respective diets for 3 days prior to the injection of the Cal27 cells, which was monitored for additional 28 days (when tumors achieved the expected size for selection). Mice were monitored daily for tumor growth using digital calipers and for cachexia. Tumor volumes were calculated using the elliptical formula: ½(length x width2). Upon conclusion of the study, mice were euthanized following the Ethics Committee guidelines using isoflurane, and tumors were fixed (on 10% neutral-buffered formalin, then transferred into 70% ethanol) and processed for histological and sequencing analysis. We ensured that the tumors were collected on week 4 to prevent any animal fatalities resulting from severe tumor growth and declining health. The Department of Histology and Immunohistochemistry Laboratory performed hematoxylin and eosin (H&E) staining and provided the unstained slides.
Cell proliferation assay
Cell viability was performed using Cell Proliferation ELISA, BrdU (colorimetric) kit from Roche (Basel, Switzerland). Cells are cultured in the presence of the respective test substances in a 96-well plate at 37 °C for 24 hours. During this labeling period, the pyrimidine analog BrdU is incorporated in place of thymidine into the DNA of proliferating cells. Subsequently, BrdU is added to the cells, and the cells are incubated again for 24 hr. After removing the culture medium, the cells are fixed, and the DNA is denatured in one step by adding FixDenat (the denaturation of the DNA is necessary to improve the accessibility of the incorporated BrdU for detection by the antibody). The anti-BrdU-POD binds to the BrdU incorporated in newly synthesized cellular DNA. The subsequent substrate reaction detects the immune complexes. The reaction product is quantified by measuring the absorbance at the respective wavelength using a scanning multiwell spectrophotometer (ELISA reader). The developed color and absorbance directly correlate to the amount of DNA synthesis. BMDM cell proliferation after treatments with Mg2+ was assessed by measuring the serial halving of cell fluorescence intensity via flow cytometry. The CFDA-SE Cell Proliferation Assays (AlexaFluor488; Bio-Rad, USA) were used according to the manufacturer’s protocol. After being placed in the appropriate conditions for cell proliferation, cells are harvested, sorted, and analyzed by LSRII (BD Pharmigen, San Jose, CA, USA).
Cell viability assay
Cell viability was measured using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) (Sigma-Aldrich, USA) according to the manufacturer’s protocol. Cell viability was measured using the MTT method. Cells were seeded in 96‐well plates at a density of 0.5×105 cells/well and grown for 24 hours. 10 μL of MTT reagent (5 mg/ml MTT in PBS) was added to each well and incubated in a CO2 incubator for 4 hours. The resulting formazan dye was extracted with 100 μL of 0.01 N HCl in isopropanol, and within an hour, the absorbance was on an ELISA plate reader with a test wavelength of 570 nm and a reference wavelength of 630 nm. Cell viability was expressed as a percentage of the control culture.
Magnesium measurement and electrophysiology
For Imaging experiments, differentiated cells that were grown on glass bottom coverslips were incubated with 2 μM Mag-Fura 2-AM (Invitrogen) for the measurement of intracellular Mg2+ or with Fura-2 (Molecular Probes for 45 min) for the measurement of intracellular Ca2+. After loading cells were washed twice with SES (Standard External Solution that includes: 10 mM HEPES, 120 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 10 mM glucose, pH 7.4) buffer. For fluorescence measurements, the fluorescence intensity of Fura-2 or Mag-FURA-loaded cells was monitored with a CCD camera-based imaging system linked with an Olympus XL70 inverted fluorescence microscope. Fluorescence traces from individual cells imaged was obtained and the data shown represent [Mg2+]i values that are average from at least 30–40 cells. Also, the data presented are representative of at least 3–4 individual experiments performed in duplicate. Mg2+ or Ca2+ concentrations in individual cells were estimated by evaluating the 340/380 ratio as described before57. The patch pipette had resistances between 3-5 m after filling with the standard intracellular solution that contained the following (mM): cesium methane sulfonate, 145; NaCl, 8; MgCl2, 10; HEPES, 10; EGTA, 10; pH 7.2 (CsOH). The maximum peak currents were calculated at a holding potential of -80 mV. The I/V curves were made using a ramp protocol where current density was evaluated at various membrane potentials and plotted. Blood was collected in heparin-containing collection tubes and separated into plasma by centrifugation at 3,000 g for 20 minutes at 4 °C. Mg2+ levels were determined with the QuantiChrom Magnesium Assay Kit (BioAssay System) by following the manufacturer’s instructions. Briefly, plasma samples and dilutions of a standard Mg2+ solution (0, 0.2, 0.4, 0.6, 0.8, 1.0, and 1.2, 1.4, 1.6 and 2 mmol/l) were mixed with detecting reagents in microplates and incubated for 2 minutes at room temperature. The optical density (OD) was read at 500 nm. Then, EDTA solution was added, and the OD was read again at 500 nm to obtain background absorbance. Mg2+ concentration was calculated as ([ODsample – ODblank]/[ODMg – ODMgblank] × 2 [mg/dL]). ODsample and ODblank were sample absorbance values before and after the addition of EDTA. ODMg and ODMgblank were OD values of the standard before and after the addition of EDTA.
Protein extractions and Western blotting
Whole-cell protein lysates were prepared in lysis buffer (10 mM Tris,140mMNaCl,1%NP-40, 0.5% SDS, and protease inhibitors, pH 8.0), protein concentrations were determined using the Bradford reagent (Bio-Rad Laboratories, USA), and 25–50 ug of protein were resolved on NuPAGE Novex 4–12% BisTris gels, transferred to PVDF membranes The membrane was blocked with skim milk in PBST solution and incubated with antibodies (Cell Signaling, MA, USA). After washing with PBST, secondary antibodies (Cell Signaling, MA, USA) were applied and detected by the Clarity Western ECL Substrate and Clarity Max Western ECL Substrate (Bio-Rad Laboratories, Hercules, CA, USA). Analysis and results were corrected for protein loading by normalization for β-actin expression. Densitometric analysis was performed using ImageJ analysis, and results were corrected for protein loading by normalization for β‐actin (Cell Signaling, 4970S) as previously described58. Blot stripping was performed whenever necessary, using Restore PLUS Western Blot Stripping Buffer (Thermo Fisher, Waltham, MA, USA). The blots were washed with PBS Tween 20 (Sigma, USA), immersed in stripping buffer, and then incubated for 5-15 min at room temperature, then the stripping buffer was removed, and the blot was washed again and blocked for another 30 minutes before incubation with new antibody. The following primary antibodies (dilution accordingly by manufacturer’s protocol) were used for western blot analysis: anti-NFκB p65 (p65) (Cell Signaling, 8242S), anti-pNFκB p65 (pp65) (Cell Signaling, 3033S), anti-STAT1 (Cell Signaling, 14994S), anti-pSTAT1 (Cell Signaling, 9167S), anti-STAT6 (Cell Signaling, 5397S), anti-p-STAT6 (Cell Signaling, 56554S), anti-iNOS (Cell Signaling, 13120S), anti-Arginase 1 (Cell Signaling, 93668S), anti-TRPM7 (Abcam, ab245408), anti-MAGT1 (Invitrogen, PAS-24978), anti-TRPM6 (Abcam, ab47017). Goat anti-rabbit IgG (Cell Signaling, 7074S), goat anti-mouse IgG (Cell Signaling, 7076S), and rabbit anti-Guinea IgG (H + L) were used as secondary antibodies. Appropriate antibody dilution was used according to the manufacturer’s protocol.
Measurement of cytokines levels
IL-6, IL-1β, IFN-γ, IL-4, and IL-10 levels in the supernatant were measured using the Enzyme-Linked ImmunoSorbent Assay (ELISA) kit (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s instructions using standard diluent buffers designed for use with mouse serum. All samples were measured on a single 96-well plate for each cytokine, the cultured medium was centrifuged at 800xg for 5 min (4 °C), and the supernatant was used as the sample. Based on that criterion, all cytokine values for the bone marrow-derived macrophage serum samples examined were above the detection limit and within the reportable range of each assay.
Macrophage polarization and flow cytometer
On day 7, the formation of mature BMDM is evaluated using flow cytometry analysis and fluorophore-conjugated antibodies. Cells were resuspended with FACS buffer (PBS supplemented with 0.2% BSA), then blocked with Anti-Mouse CD16/CD32 Fc Blocker (BD Pharmigen, San Jose, CA, USA) for 15 minutes. After blocking, cells were tested for the expression of Anti-CD11b antibody [M1/70] (PE/Cy5®) (Abcam, Burlingame, CA, USA) and F4/80 Monoclonal Antibody (BM8), Pacific Blue (Thermo Fisher, Waltham, MA, USA) for macrophage line confirmation. For M1 macrophages, cells were stained with Anti-Mouse CD80 PE (eBioscience, San Diego, CA, USA); for M2 macrophage cells were stained with Anti-Mouse CD206 APC (eBioscience, San Diego, CA, USA). Cells were incubated for 30 min at 4 °C, then quantified on a BD LSR II (BD Pharmigen, San Jose, CA, USA). For polarization stimulation, BMDM was exposed to IFN-γ (20 ng/ml, Peprotech, Rocky Hill, NJ, USA) to generate the M1 phenotype and IL4 (20 ng/ml, Peprotech, Rocky Hill, NJ, USA) to generate the M2 phenotype. Stimulated cells were removed from wells and incubated in anti-mouse BD Fc Block CD16/CD32 (BD Biosciences, San Jose, CA) for 30 minutes on ice in FACS buffer (PBS with 3% FBS), then surface stained with antibodies for surface markers CD80 (Abcam, Cambridge, UK) and CD206 (Abcam, Cambridge, UK) for 15 min at 4 °C, following manufactory’s protocol then analyzed by LSRII (BD Biosciences, San Jose, USA) and Cytek Autora (Cytek, California, USA) and FlowJo software version 9.0.
Measurement of mitochondrial oxygen consumption rate
Bone marrow-derived macrophages (BMDM) from WT mice (n = 3) were plated on 96-well Agilent Seahorse XF Cell culture microplates at a density of 4 x 105 cells per well. The macrophages were plated in their normal growth media overnight. The plated macrophage naive macrophages (M0) from WT were polarized to become pro-inflammatory macrophages (M1) and anti-inflammatory macrophages (M2) upon Interferon-γ (IFN-γ) or interleukin-4 (IL-4) stimulation for 24 hours. After polarization, the media was changed to Seahorse XF Cell Mito Stress Test Kit (Agilent) assay media supplemented with glucose, glutamine, and pyruvate concentrations equivalent to that of the growth media. Naïve Mφ, M1, and M2 macrophages were treated with no Mg2+ media and media containing 20 mM Mg2+ and with the SOCE channel inhibitor 2-ABP (10 µM) overnight, and oxygen consumption rate (OCR) was measured at 37 °C in an XF96 extracellular flux analyzer (Seahorse Bioscience, Agilent), which had been previously calibrated using Seahorse XF Calibrant solution (Seahorse Bioscience, Agilent) in a CO2-free incubator overnight. Respiratory chain inhibitors were then loaded into the XF96 flux analyzer and added sequentially to cells at indicated time points. M0, M1, and M2 macrophages received oligomycin (2 μM), FCCP (5 μM), and a mixture of antimycin A and rotenone (1 μM) sequentially59. Data were collected using Agilent Seahorse Wave 2.6.1 Desktop software and exported to GraphPad Prism version 8 for analysis.
Measurement of macrophage basal mMg2+ in the permeabilized cell system
Naive primary macrophages derived from WT mice bone marrow were polarized to mature into M1 and M2 phenotypes using IFN-γ (20 ng/mL for 24 h) and IL4 (20 ng/mL for 24 h), respectively. After 24 h of polarization, the macrophages were washed in Ca2+ and Mg2+ free DPBS, pH 7.4. An equal number of cells ( ~ 3 x 106 cells) in the presence and absence of 20 mM Mg2+ for 1 h were resuspended and permeabilized with 40 µg/mL digitonin in 1.5 mL of intracellular medium (ICM) composed of 120 mM KCl, 1 mM KH2PO4, 10 mM NaCl, 20 mM HEPES-Tris PH 7.2. The measurement of basal mMg2+ was performed by loading the permeabilized macrophages with Mag-Green (1 µM; Ex: 505 nm/Em: 535 and 595 nm)59. The spectrofluorimetric runs were performed at 37°C with constant stirring in the presence of 1.5 µM K+-ATP and 5 mM succinate. A single bolus of the mitochondrial uncoupler, FCCP (10 µM), was added at the indicated time point. Fluorescence measurements were recorded in a multi-wavelength excitation dual-wavelength emission spectrofluorometer (Delta RAM, PTI, HORIBA). m[Mg2+]out is represented as f.a.u. of Mag-Green fluorescence (ex/em 505/595)59.
Confocal microscopy
Laser confocal scanning microscopy (LASX Life Science microscope; Leica Microsystems) was used for single immunofluorescence staining to evaluate the expression of iNOS (Cell Signaling, iNOS (D6B6S) Rabbit mAb) and Arginase-1 (Cell Signaling, D4E3M, Rabbit mAb). Slides with fixed tumor cells were stained following manufacture protocol. Cells were blocked with horse serum (ThermoFisher, MA). The cells were counterstained with DAPI antifade mounting medium (2 mg/ml; ThermoFisher, MA) and observed with Laser confocal scanning microscopy. The immunofluorescence staining procedure was repeated three times.
RNA Sequencing and data analysis
Bone marrow derived macrophages (BMDM) were obtained from C57BL6/J wild-type (WT) 8-10-week-old male mice (The Jackson Laboratories, Bar Harbor, ME, USA). Naïve macrophages cells were treated with IFN-γ (20 ng/ml) and IL-4 (20 ng/ml) to activate macrophage polarization and change the naïve phenotype to M1 and M2 respectively. Cells were then treated with different concentrations of Mg2+, then snap frozen in Trizol and sent for RNA sequencing to Novogene (Sacramento, CA, USA) for RNA extraction, and library preparation. The original image data file from high-throughput sequencing platforms (such as Illumina) is transformed to sequenced reads by CASAVA base recognition (Base Calling). FastQC v0.12.1 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) tool has been used for the assessment of raw-read and then trimming of raw reads was performed using Trimmomatic version 0.36 with parameters such as “LEADING:3 TRAILING:3 HEADCROP:15 MINLEN:30”60. Cleaned and good quality reads were mapped onto mm10 reference genome using HISAT2 tool with parameters “--rna-strandness RF”61. Aligned results were further used for reading count assessment using FeatureCounts62. Finally, the edgeR tool was used for the identification of differential expressed genes using p-value cutoff 0.05 for significant genes63. WebGestalt (WEB-based Gene SeT AnaLysis Toolkit), an online server has been used for pathways as well as gene ontology analysis of differential expressed genes62.
Statistical analysis
Data analysis was performed using a one‐way ANOVA or Student t-test on GraphPad prism 8.0. Calcium measurement and electrophysiology were analyzed using Origin 9.0. Experimental values are expressed as means ± SD. * indicates significance of P < 0.05, ** of P < 0.01 and *** of P < 0.001, respectively.
Data availability
The data supporting the findings of this study have been made available within the paper and its supplementary materials.
Change history
28 February 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41698-025-00836-6
References
Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014).
Wynn, T. A., Chawla, A. & Pollard, J. W. Macrophage biology in development, homeostasis and disease. Nature 496, 445–455 (2013).
Kigerl, K. A. et al. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 29, 13435–13444 (2009).
Castro-Dopico, T. et al. GM-CSF calibrates macrophage defense and wound healing programs during intestinal infection and inflammation. Cell Rep. 32, 107857 (2020).
Lackey, D. E. & Olefsky, J. M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 12, 15–28 (2016).
Werz, O. et al. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat. Commun. 9, 59 (2018).
Kubala, M. H. et al. Plasminogen activator inhibitor-1 promotes the recruitment and polarization of macrophages in cancer. Cell Rep. 25, 2177–2191.e2177 (2018).
Van den Bossche, J., O’Neill, L. A. & Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 38, 395–406 (2017).
Tur, J., Vico, T., Lloberas, J., Zorzano, A. & Celada, A. Macrophages and mitochondria: a critical interplay between metabolism, signaling, and the functional activity. Adv. Immunol. 133, 1–36 (2017).
Weinberg, S. E., Sena, L. A. & Chandel, N. S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417 (2015).
Hamers, A. A. J. & Pillai, A. B. A sweet alternative: maintaining M2 macrophage polarization. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aav7759 (2018).
Dowling, J. K. et al. Mitochondrial arginase-2 is essential for IL-10 metabolic reprogramming of inflammatory macrophages. Nat. Commun. 12, 1460 (2021).
Mosser, D. M. & Edwards, J. P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008).
Lotscher, J. et al. Magnesium sensing via LFA-1 regulates CD8(+) T cell effector function. Cell 185, 585–602.e529 (2022).
Daw, C. C. et al. Lactate Elicits ER-Mitochondrial Mg(2+) dynamics to integrate cellular metabolism. Cell 183, 474–489.e417 (2020).
Li, F. Y. et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 475, 471–476 (2011).
Romani, A. M. Magnesium homeostasis in mammalian cells. Front Biosci. 12, 308–331 (2007).
Kanellopoulou, C. et al. Mg(2+) regulation of kinase signaling and immune function. J. Exp. Med. 216, 1828–1842 (2019).
Yamanaka, R. et al. Mitochondrial Mg(2+) homeostasis decides cellular energy metabolism and vulnerability to stress. Sci. Rep. 6, 30027 (2016).
Feske, S., Wulff, H. & Skolnik, E. Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 33, 291–353 (2015).
Feske, S., Skolnik, E. Y. & Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 12, 532–547 (2012).
Deason-Towne, F., Perraud, A. L. & Schmitz, C. The Mg2+ transporter MagT1 partially rescues cell growth and Mg2+ uptake in cells lacking the channel-kinase TRPM7. FEBS Lett. 585, 2275–2278 (2011).
Ryazanova, L. V. et al. TRPM7 is essential for Mg(2+) homeostasis in mammals. Nat. Commun. 1, 109 (2010).
Ryazanova, L. V. et al. Elucidating the role of the TRPM7 alpha-kinase: TRPM7 kinase inactivation leads to magnesium deprivation resistance phenotype in mice. Sci. Rep. 4, 7599 (2014).
Li, F. Y., Lenardo, M. J. & Chaigne-Delalande, B. Loss of MAGT1 abrogates the Mg2+ flux required for T cell signaling and leads to a novel human primary immunodeficiency. Magnes. Res. 24, S109–S114 (2011).
Mastrototaro, L., Smorodchenko, A., Aschenbach, J. R., Kolisek, M. & Sponder, G. Solute carrier 41A3 encodes for a mitochondrial Mg(2+) efflux system. Sci. Rep. 6, 27999 (2016).
Yi, L. et al. Macrophage colony-stimulating factor and its role in the tumor microenvironment: novel therapeutic avenues and mechanistic insights. Front Oncol. 14, 1358750 (2024).
Nadler, M. J. et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 411, 590–595 (2001).
Runnels, L. W., Yue, L. & Clapham, D. E. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat. Cell Biol. 4, 329–336 (2002).
Sun, Y. et al. Increase in serum Ca2+/Mg2+ ratio promotes proliferation of prostate cancer cells by activating TRPM7 channels. J. Biol. Chem. 288, 255–263 (2013).
Gotru, S. K. et al. Cutting edge: imbalanced cation homeostasis in MAGT1-deficient B cells dysregulates B cell development and signaling in mice. J. Immunol. 200, 2529–2534 (2018).
Li, M. et al. Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J. Biol. Chem. 282, 25817–25830 (2007).
Jin, J. et al. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 322, 756–760 (2008).
Zhang, Y. H. & Hancox, J. C. Gadolinium inhibits Na(+)-Ca(2+) exchanger current in guinea-pig isolated ventricular myocytes. Br. J. Pharm. 130, 485–488 (2000).
Guinamard, R., Simard, C. & Del Negro, C. Flufenamic acid as an ion channel modulator. Pharm. Ther. 138, 272–284 (2013).
Qiao, W. et al. TRPM7 kinase-mediated immunomodulation in macrophage plays a central role in magnesium ion-induced bone regeneration. Nat. Commun. 12, 2885 (2021).
Martinez, F. O., Sica, A., Mantovani, A. & Locati, M. Macrophage activation and polarization. Front Biosci. 13, 453–461 (2008).
Martinez, F. O. & Gordon, S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13 (2014).
Kimura, T. et al. Polarization of M2 macrophages requires Lamtor1 that integrates cytokine and amino-acid signals. Nat. Commun. 7, 13130 (2016).
Orecchioni, M., Ghosheh, Y., Pramod, A. B. & Ley, K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. 10, 1084 (2019).
Bosco, M. C. Macrophage polarization: Reaching across the aisle? J. Allergy Clin. Immunol. 143, 1348–1350 (2019).
Corliss, B. A., Azimi, M. S., Munson, J. M., Peirce, S. M. & Murfee, W. L. Macrophages: an inflammatory link between angiogenesis and lymphangiogenesis. Microcirculation 23, 95–121 (2016).
Diez-Tercero, L., Delgado, L. M., Bosch-Rue, E. & Perez, R. A. Evaluation of the immunomodulatory effects of cobalt, copper and magnesium ions in a pro inflammatory environment. Sci. Rep. 11, 11707 (2021).
Mills, E. L. et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470.e413 (2016).
Wang, F. et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. 28, 463–475.e464 (2018).
Starostina, I. et al. Distinct calcium regulation of TRPM7 mechanosensitive channels at plasma membrane microdomains visualized by FRET-based single cell imaging. Sci. Rep. 11, 17893 (2021).
Roy, S. et al. Redefining the transcriptional regulatory dynamics of classically and alternatively activated macrophages by deepCAGE transcriptomics. Nucleic Acids Res. 43, 6969–6982 (2015).
Mircetic, R. N., Dodig, S., Raos, M., Petres, B. & Cepelak, I. Magnesium concentration in plasma, leukocytes and urine of children with intermittent asthma. Clin. Chim. Acta 312, 197–203 (2001).
Ferioli, S. et al. TRPM6 and TRPM7 differentially contribute to the relief of heteromeric TRPM6/7 channels from inhibition by cytosolic Mg(2+) and Mg.ATP. Sci. Rep. 7, 8806 (2017).
Krapivinsky, G., Krapivinsky, L., Manasian, Y. & Clapham, D. E. The TRPM7 chanzyme is cleaved to release a chromatin-modifying kinase. Cell 157, 1061–1072 (2014).
Nadolni, W. & Zierler, S. The channel-kinase TRPM7 as novel regulator of immune system homeostasis. Cells https://doi.org/10.3390/cells7080109 (2018).
Pilchova, I., Klacanova, K., Tatarkova, Z., Kaplan, P. & Racay, P. The involvement of Mg(2+) in regulation of cellular and mitochondrial functions. Oxid. Med. Cell Longev. 2017, 6797460 (2017).
Weischenfeldt, J. & Porse, B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc 2008, pdb prot5080. https://doi.org/10.1101/pdb.prot5080 (2008).
Trouplin, V. et al. Bone marrow-derived macrophage production. J Vis Exp, e50966 (2013).
Gonzales, C. B. et al. Vanilloids induce oral cancer apoptosis independent of TRPV1. Oral. Oncol. 50, 437–447 (2014).
De La Chapa, J. J. et al. The novel capsazepine analog, CIDD-99, significantly inhibits oral squamous cell carcinoma in vivo through a TRPV1-independent induction of ER stress, mitochondrial dysfunction, and apoptosis. J. Oral. Pathol. Med. 48, 389–399 (2019).
Sun, Y., Kamat, A. & Singh, B. B. Isoproterenol-dependent activation of TRPM7 protects against neurotoxin-induced loss of neuroblastoma cells. Front Physiol. 11, 305 (2020).
Singh, B. B. et al. VAMP2-dependent exocytosis regulates plasma membrane insertion of TRPC3 channels and contributes to agonist-stimulated Ca2+ influx. Mol. Cell 15, 635–646 (2004).
Madaris, T. R. et al. Limiting Mrs2-dependent mitochondrial Mg2+ uptake induces metabolic programming in prolonged dietary stress. Cell Rep. https://doi.org/10.1016/j.celrep.2023.112155 (2023).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Acknowledgements
This work was funded by grant support from the National Institutes of Health (R01DE017102; R01DE022765) awarded to B.B.S and (R01GM109882, R01DK135179, and R01HL142673) awarded to M.M. T.R.M. is supported by the NIH (R01GM109882-S1 and T32 AG 021890).The funders had no further role in the study design, data analysis, and/or interpretation of the data. Flow Cytometry Facility is supported by UTHSCSA, NIH-NCI P30 CA054174-20, and UL1 TR001120. We also thank Dr. Cara Gonzales (Department of Comprehensive Dentistry- UTHSCSA) for providing the female athymic nude mice for the tumor model.
Author information
Authors and Affiliations
Contributions
Conceptualization, methodology, and analysis, V.C., T.R.M., B.B.M. M.M and B.B.S., Investigation, V. C., Y.S., V. D., R., J., X., C., K.R., M.V., J.D., L.C., Writing–Review and Editing, V.C., T.R.M., B.B.S. M.M.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nascimento Da Conceicao, V., Sun, Y., Venkatesan, M. et al. Naltriben promotes tumor growth by activating the TRPM7-mediated development of the anti-inflammatory M2 phenotype. npj Precis. Onc. 9, 29 (2025). https://doi.org/10.1038/s41698-025-00815-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-00815-x
