
Abstract
Using a large real-world database with matched genomic and transcriptomic data, we characterized clinical and molecular differences between patients with early-onset esophagogastric cancer (EOEGC; <50 years), intermediate-onset esophagogastric cancer (IOEGC; 50-65 years), and average-onset esophagogastric cancer (AOEGC; >65 years). We analyzed clinicopathologic, whole transcriptome, and DNA-sequencing data from 5175 patient samples (EOEGC, n = 530; IOEGC, n = 1744; AOEGC, n = 2901) from the Caris Life Sciences database. Immune deconvolution was performed with quanTIseq and pathway enrichment with Gene Set Enrichment Analysis (GSEA). Real-world overall survival was estimated from insurance claims data. Prevalence of EOEGC was higher in patients who were Black, Asian, Hispanic/Latino, and female. Patients with EOEGC had higher proportion of CDH1 mutations; FGFR2, CCNE1, MYC copy number alterations; and ARHGAP26 fusions. Patients with EOEGC had decreased prevalence of immune-oncology markers of microsatellite instability-high, tumor mutation burden-high, and PD-L1 positivity. Immune microenvironment analysis identified significant enrichment of M2 macrophages and decreased M1 macrophages in patients with EOEGC. GSEA identified enrichment of epithelial mesenchymal transition and coagulation pathways in patients with EOEGC. This large real-world characterization of age-stratified esophagogastric cancer found that EOEGC was associated with significant racial, ethnic, and gender differences, and notable molecular differences that may have prognostic and therapeutic implications.
Similar content being viewed by others

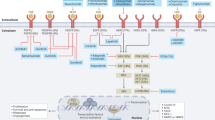

Introduction
Esophagogastric cancer (EGC) is a highly prevalent cancer globally with over 1.5 million new diagnoses each year1,2. The average age of onset for EGC in the United States is 68 years of age. While there has been a decline in EGC rates overall, the incidence of early-onset esophagogastric cancer (EOEGC), defined as age of initial diagnosis less than 50 years old, has strikingly increased over 30% in recent decades3,4,5,6,7,8. Additional studies are needed to understand the reasons for this trend in early-onset disease and to better characterize EOEGC as a disease entity from a molecular perspective.
Known clinical risk factors for EGC include smoking, alcohol consumption, chronic gastroesophageal reflux, Helicobacter pylori infection, and obesity9,10. The increased incidence of EOEGC may be related to corresponding rising rates of obesity, gastroesophageal reflux disease, and certain environment exposures in the younger population5,11,12. Studies to date analyzing germline testing in EOEGC cohorts have identified pathogenic variants in only approximately 25% of patients13,14. A recent large single-center study found that compared to patients with EGC diagnosed at age ≥ 50 years old, tumors from patients with EOEGC were more likely to have the genomically stable molecular phenotype, have diffuse-type histology, and unlikely to be microsatellite-instability-high (MSI-H)15. Prior studies examining the molecular alterations in EOEGC have been limited to single center or small multi-institutional cohort analyses. The purpose of this study is to compare the distinct molecular and immune microenvironment characteristics of EOEGC and later onset esophagogastric cancer in a large real-world dataset.
Results
Patient clinicopathologic characteristics
The study population was composed of 5175 EGC samples with annotated molecular data. 530 (10.2%) patients with EOEGC had a median age of 43 (range 14–49) [Table 1]. In terms of cancer subtype of esophagogastric cancer, patients with EOEGC were characterized by higher proportion of gastric adenocarcinoma compared to patients with IOEGC and AOEGC (53.4% vs 34.8% vs 33.6% respectively; p < 0.05) [Table 1]. In terms of tumor specimen site, patients with EOEGC had a higher portion of metastatic site compared to patients with IOEGC or AOEGC (39.4% vs 34.5% vs 24.3% respectively; p < 0.05) [Table 1].
The Laurén subtype was limited as most samples were classified as mixed/unclear, but there was a higher proportion of diffuse subtype in patients with EOEGC compared to patients with IOEGC and AOEGC (12.8% vs 6.1% vs 5.3% respectively; p < 0.05). While the self-reported history of smoking was limited, in the available data there was no significant difference between patients with EOEGC, IOEGC, and AOEGC (94.6% vs 94.4% vs 95.5% respectively; p = 0.757) [Table 1].
Patients with EOEGC had a higher proportion of female gender compared to patients with IOEGC and AOEGC (34.0% vs 25.1% vs 26.3% respectively; p < 0.05) [Table 1]. There was a significantly higher proportion of Black race in patients with EOEGC compared to patients with IOEGC and AOEGC (19.2% vs 16.6% vs 11.3% respectively; p < 0.05) [Table 1]. Furthermore, there was an increased proportion of Asian race in patients with EOEGC compared to patients with IOEGC and AOEGC (8.1% vs 6.6% vs 5.2% respectively; p < 0.05) [Table 1]. Additionally, patients with EOEGC had a higher proportion of Hispanic/Latino ethnicity compared to patients with IOEGC and AOEGC (27.5% vs 17.7% vs 12.1% respectively; p < 0.05) [Table 1]. Overall, these data highlight significant disparities related to race, ethnicity, and gender in EOEGC.
Comparative molecular landscape of EOEGC, IOEGC, and AOEGC
To determine if EOEGC is associated with distinctive molecular features in a real-world dataset, we analyzed clinically relevant pathogenic/likely pathogenic mutations, copy number alterations, and fusions across the three age cohorts. TP53 mutations were the most prevalent somatic alterations in patients with EOEGC, IOEGC, and AOEGC. Fewer patients with EOEGC had TP53 mutations compared to patients with IOEGC (65.9% vs 74.5%; p < 0.001, q = 0.006) and AOEGC (65.9% vs 74.4%; p < 0.001, q = 0.004) [Fig. 1A, Supplementary Table 1]. CDH1 mutations were more frequent in patients with EOEGC compared to patients with IOEGC (18.9% vs 7.2%; p < 0.001, q < 0.001) and AOEGC (18.9% vs 6.1%; p < 0.001, q < 0.001). The most common copy number alterations in patients with EOEGC compared to AOEGC were in FGFR2 (3.5% vs 1.5%; p = 0.002, q = 0.07), CCNE1 (6.8% vs 3.8%; p = 0.002, q = 0.06), and MYC (6.1% vs 3.9%, p = 0.02, q = 0.33) [Fig. 1B, Supplementary Table 2]. ARHGAP26 fusions were more prevalent in patients with EOEGC compared to patients with IOEGC (5.7% vs 3.7%; p = 0.04, q = 0.53) and AOEGC (5.7% vs 1.3%; p < 0.001, q < 0.001) [Fig. 1C, Supplementary Table 3]. The ARHGAP26 fusion components are in Supplementary Table 4. There were no major differences between cohorts in HER2 positivity by RNA, IHC, copy number alteration, and chromogenic in situ hybridization, except for increased HER2 positivity by CISH in patients with IOEGC compared to patients with EOEGC [Supplementary Fig. 1, Supplementary Table 5].

A Bar graph of 21 most common mutational alterations. B Bar graph of 11 most common copy number alterations. C Bar graph of 3 most common fusions. *p < 0.05; **p < 0.01; ***p < 0.001 when compared to EOEGC by chi-square or Fisher’s exact tests.
Patients with EOEGC had decreased positivity of several immune-oncology markers that predict response to immune checkpoint inhibition. Patients with EOEGC had decreased rates of TMB-high compared to patients with IOEGC (3.8% vs 7.8%; p = 0.002, q = 0.05) and AOEGC (3.8% vs 11.5%; p < 0.001, q < 0.001) [Fig. 2A, Supplementary Table 6]. Patients with EOEGC also had decreased prevalence of dMMR/MSI-H compared to patients with IOEGC (1.3% vs 3.3%; p = 0.02, q = 0.28) and AOEGC (1.3% vs 7.1%; p < 0.001, q < 0.001) [Fig. 2B, Supplementary Table 6]. Although patients with EOEGC had no difference in PD-L1 positivity compared to patients with IOEGC (68.1% vs 70.5%; p = 0.32, q = 1.00), they demonstrated decreased PD-L1 positivity compared to patients with AOEGC (68.1% vs 73.2%; p = 0.02, q = 0.34) [Fig. 2C, Supplementary Table 6]. Taken together, these data highlight several notable differences in somatic mutations, copy number alterations, fusions, and common immune-oncology markers in patients with EOEGC.

A Bar graph for TMB-High status (≥10 mutations/MB). B Bar graph for dMMR/MSI-H status. C Bar graph for PD-L1 positive status as determined by immunohistochemistry from the 22c3 assay. *p < 0.05; **p < 0.01; *** p < 0.001 when compared to EOEGC by chi-square or Fisher’s exact tests.
Characterization of the immune tumor microenvironment and immune checkpoint gene expression
Given the decreased positivity of immune-oncology biomarkers in patients with EOEGC, we performed further analyses to quantify immune cell infiltration and immune gene expression.
There was enrichment of B-cells (p < 0.001, q < 0.001), M2 macrophages (p < 0.001, q < 0.001), natural killer cells (p < 0.001, q < 0.001), dendritic cells (p = 0.001, q = 0.004), monocytes (p = 0.0004, q = 0.002), and CD4 + T cells (p = 0.01, q = 0.03) in tumors from patients with EOEGC compared to patients with AOEGC [Fig. 3A, B, Supplementary Table 7]. In contrast, there was decreased relative enrichment of M1 macrophages (Fold change 0.93; p = 0.002, q = 0.006) in tumors from patients with EOEGC compared to patients with AOEGC. A similar trend of immune cell infiltrates was observed when comparing between tumors from patients with EOEGC and IOEGC.

A Computationally inferred intratumoral immune population. The heatmap indicates fold change of IOEGC relative to EOEGC in median immune fraction according to quanTIseq. B The heatmap indicates fold change of AOEGC relative to EOEGC in median immune fraction according to quanTIseq. Tumor microenvironment cell fractions were analyzed among cohorts using nonparametric Kruskal-Wallis testing. The Benjamini-Hochberg method was utilized to adjust p-values for multiple comparisons (*q < 0.05; **q < 0.01; ***q < 0.001). C For cell types with median values of “0”(Ex: monocytes and CD4+ T cells), the percentage of tumors with nonzero immune infiltrates were compared. D Fold change gene expression levels in transcripts per million (TPM) of immune checkpoint genes IOEGC relative to EOEGC. E Fold change gene expression levels in TPM of immune checkpoint genes AOEGC relative to EOEGC. Mann-Whitney U test used to determine statistically significant differences in immune gene expression. The Benjamini-Hochberg method was utilized to adjust p-values for multiple comparisons (*q < 0.05; **q < 0.01; ***q < 0.001).
For immune checkpoint gene expression analysis, there was increased HAVCR2 expression in tumors of patients with EOEGC compared to patients with IOEGC (Fold change 1.03; p = 0.02, q = 0.07) and AOEGC (Fold change 1.03; p = 0.02, q = 0.07) [Fig. 3D, E, Supplementary Table 8]. Conversely, there was decreased CD274 and FOXP3 expression in tumors of patients with EOEGC compared to patients with IOEGC and AOEGC. There were no significant differences in other key immune checkpoint genes such as LAG3, CTLA4, and IDO1.
Differentially regulated signaling pathways in tumor transcriptomes in EOEGC, IOEGC, and AOEGC
To complement the mutational profiling, immuno-oncology markers, and immune microenvironment analyses in this study, we performed gene set enrichment analysis to identify potential underlying pathways in patients with EOEGC. A positive normalized enrichment score (NES) was indicative of enhanced pathway signalling in the given GSEA gene set.
Patients with EOEGC had significant enrichment of epithelial-to-mesenchymal transition (EMT) genes compared to patients with IOEGC (NES = 1.83, false discovery rate (FDR) = 0.00) and AOEGC (NES = 1.82, FDR = 0.00) [Fig. 4, Supplementary Tables 9, 10]. There was also significant enrichment of coagulation genes in patients with EOEGC compared to patients with IOEGC (NES = 1.60, FDR = 0.02) and AOEGC (NES = 1.64, FDR = 0.04). Conversely, there was significant overrepresentation of the genes related to E2F targets, G2M checkpoint pathway, MYC targets, and mTORC1 signaling targets in both patients with IOEGC and AOEGC compared to patients with EOEGC [Fig. 4, Supplementary Table 10].

A GSEA differences in pathways based on normalized enrichment scores (NES) in EOEGC versus IOEGC. B GSEA differences in pathways based on NES in EOEGC versus AOEGC. Positive NES would imply higher values in EOEGC. All pathways with false discovery rate (FDR) <0.25.
Additionally, the MAPK pathway activity score was decreased in patients with EOEGC compared to patients with IOEGC (MPAS score 0.04; p = 0.04, q = 0.06) and AOEGC (MPAS score 0.18; p = 0.001, q = 0.004) [Supplementary Fig. 2, Supplementary Table 11].
Clinical outcomes of patients with EOEGC, IOEGC, and AOEGC
Real world overall survival (rwOS) was calculated with insurance claims-related follow-up. We observed significantly reduced rwOS after chemotherapy in patients with EOEGC compared to patients with IOEGC and AOEGC (Median rwOS 17.6 months vs 20.1 months vs 20.4 months respectively; p < 0.001) [Fig. 5A]. When further stratified by CDH1 mutation status, patients with EOEGC who were CDH1-mutated had the worst survival after chemotherapy of all subgroups, including patients with EOEGC who were CDH1-wildtype, patients with AOEGC whose tumors were CDH1-mutated, and patients with AOEGC with CDH1-wildtype tumors (Median rwOS 15.1 months versus 18.7 months versus 20.4 months versus 20.6 months respectively; p < 0.001) [Fig. 5B]. For tumors that were HER2-positive, there was no significant difference in rwOS for patients with EOEGC treated with trastuzumab compared to patients with IOEGC or AOEGC [Supplementary Fig. 3C].

A Kaplan-Meier curves of rwOS of patients with EOEGC, IOEGC, AOEGC after treatment with chemotherapy (Defined as Carboplatin, Cisplatin, Docetaxel, Fluorouracil, Oxaliplatin, Paclitaxel, FOLFIRI, FOLFOX, FOLFIRINOX). B rwOS stratified by CDH1-mutation status after treatment with chemotherapy. C rwOS after treatment with immune checkpoint inhibitor with pembrolizumab or nivolumab. Log-rank test was performed with significance determined as p < 0.05.
Although biomarkers associated with clinical response to immune checkpoint inhibition, such as TMB-High and dMMR/MSI-H status, were less prevalent in patients with EOEGC, there was no significant difference in rwOS after immune checkpoint inhibitor treatment with pembrolizumab or nivolumab in patients with EOEGC compared to patients with IOEGC and AOEGC (Median rwOS 11.4 months vs 12.8 months vs 12.0 months respectively; p = 0.255) [Fig. 5C]. When stratified by dMMR/MSI-H status, there remained no difference in rwOS in patients after immune checkpoint inhibitor treatment in patients with EOEGC compared to patients with IOEGC and AOEGC (Median rwOS not reached vs 30.9 months vs 21.2 months respectively; p = 0.116) [Supplementary Fig. 3A]. Moreover, when stratified by TMB High status, there remained no difference in rwOS in patients after immune checkpoint inhibitor treatment in patients with EOEGC compared to patients with IOEGC and AOEGC (Median rwOS 18.7 months vs 19.8 months vs 16.9 months respectively; p = 0.541) [Supplementary Fig. 3B].
Discussion
To our knowledge, this study is the largest real-world investigation of 5175 patients with esophagogastric cancer and associated comprehensive analyses of molecular and immunologic features. We identified several differences in demographics, molecular characteristics, and immunologic features between EOEGC and older-onset esophagogastric cancer.
The largest single institution study at Memorial Sloan Kettering Cancer Center analyzing clinical and molecular features of EOEGC (n = 219) provided useful insight into several differing molecular and clinical characteristics15. Our data is consistent with previously observed findings including increased incidence in female patients, increased CDH1 and CCNE1 alterations, and decreased prevalence of MSI-H. Our study expands upon existing knowledge by leveraging a large and diverse real-world cohort with highly annotated molecular and transcriptomic data.
Prior studies characterizing early versus average onset cancers utilize age of diagnosis at 50 as a discriminatory cutoff, which may be limiting as the average age of onset for EGC is approximately 68 years old7,15. Our study provides additional granularity by stratifying patients with EGC who are were diagnosed between age ≥50 and <65 (IOEGC) and diagnosed at age ≥ 65 years (AOEGC). Our data highlight racial, ethnic, and gender disparities in EOEGC with higher incidence in Black, Asian/Pacific Islander, Hispanic/Latino, and Female populations. We postulate that these disparities may be potentially related to rising incidence of diffuse-type gastric cancer in these populations16,17,18,19,20,21.
Key genomic alterations that were enriched in patients with EOEGC included CDH1, FGFR2, CCNE1, MYC, and ARHGAP26 fusions. CDH1 alterations are commonly found in diffuse-type gastric cancer and germline mutations are strongly associated with hereditary diffuse gastric cancer syndrome22,23. FGFR2 alterations are relatively uncommon in gastric cancer, but have important clinical implications given the promising efficacy of FGFR2-targeting therapeutics such as bemarituzumab24,25. CCNE1 amplifications have been linked to worse prognosis after treatment with trastuzumab26,27. There are ongoing clinical trials utilizing Wee1 kinase inhibitor adavosertib and protein kinase membrane associated tyrosine/threonine 1 (PKMYT1) inhibitor lunresertib to target tumors with CCNE1 amplifications28,29. MYC has traditionally been thought to be undruggable, but there are several ongoing preclinical and clinical studies directly and indirectly targeting MYC30. ARHGAP26 fusions are structural rearrangements that often involve Claudin 18 (CLDN18) and are also enriched in diffuse-type gastric cancer31,32. Taken together, there is significant enrichment of molecular alterations associated with diffuse-type gastric cancer and also alterations that may be targetable with emerging treatments being tested in ongoing clinical trials.
In terms of immune-oncology markers, patients with EOEGC had decreased prevalence of dMMR/MSI-H, TMB-High, and PD-L1 positivitity, which are each biomarkers associated with response to immune checkpoint inhibition. To provide further insight into the immune microenvironment, we analyzed immune cell infiltrate and observed increased B-cells, M2 macrophages, natural killer cells, dendritic cells, monocytes, and CD4 + T cells in tumors from patients with EOEGC. There was also decreased M1 macrophages in tumors from patients with EOEGC. Broadly speaking, M1 macrophages are considered pro-inflammatory and M2 macrophages are considered anti-inflammatory33. Further analyses in gastric cancer have identified increased M1 macrophages associated with improved prognosis and increased M2 macrophages associated with worse prognosis, diffuse-type histology, and more advanced stage34. Additionally in esophagogastric cancer, a lower M1/M2 ratio has been associated with poorer prognosis35,36. There have been preclinical efforts to target tumor associated macrophages and induce polarization from M2 macrophages to M1 macrophages37. Pexidartinib, a colony-stimulating factor 1 receptor (CSF-1R) inhibitor, has shown synergy with PD-1 inhibition to induce macrophage repolarization towards M1 macrophages in a preclinical model of esophageal adenocarcinoma38,39. Another target includes Dickkopf-related protein (DKK1) for which elevated expression is associated with worsened survival in gastric cancer, increased M2 macrophages, and an immunosuppressive microenvironment40. DKN-01, a humanized IgG4 monoclonal antibody against circulating DKK1, is able to repolarize macrophage subsets towards M1 macrophages and showed promising clinical results in a phase II trial in combination with tislelizumab (anti-PD-1) and chemotherapy in the first line setting for advanced HER2-negative gastric/gastroesophageal adenocarcinoma41. Overall, these data represent a potentially unfavorable tumor immune microenvironment with relative decreased M1 macrophages and increased M2 macrophages in patients with EOEGC and a potential therapeutic opportunity.
GSEA identified genes related to EMT and coagulation to be significantly enriched in patients with EOEGC compared to both IOEGC and AOEGC. EMT is enriched in diffuse-type and plays an important role in its pathogenesis42,43,44,45. A study analyzing high expression of coagulation genes from the TCGA in gastric cancer identified an association with increased tumoral angiogenesis, increased EMT gene expression, low tumor mutation burden, and worse patient prognosis46. These differences in GSEA genes may be potentially related to enrichment of diffuse-type gastric cancer in the early onset group.
Our survival data identified decreased rwOS in patients with EOEGC after chemotherapy compared to patients with IOEGC and AOEGC. This worsened survival was most pronounced in patients with EOEGC with CDH1-mutated tumors. This decreased survival may be related to chemotherapy resistance associated to enrichment of EMT, which is frequently seen in patients with CDH1 mutations47. While there was decreased prevalence of key biomarkers related to response to immune checkpoint inhibition such as dMMR/MSI-H and PD-L1 positivity in patients with EOEGC, there was no corresponding decreased rwOS in patients with EOEGC who receive immune checkpoint inhibitors. We hypothesize that patients who received immune checkpoint inhibitors were likely prescribed treatment based on biomarkers, exhibiting tumor characteristics such as dMMR/MSI-H or PD-L1 positivity, and that these biomarkers may have contributed to a favorable response to treatment across cohorts. Nonetheless, since there was decreased prevalence of biomarkers related to ICI response in patients with EOEGC, there were less patients eligible to receive beneficial treatment with ICI.
There are several limitations of our study. The Caris CODEai database lacks clinical annotation of certain clinicopathologic variables such as such as performance status, method of tissue acquisition, and stage of diagnosis and had limited information on the Laurén histological subtype. There was very limited Claudin 18.2 immunohistochemistry testing in this study cohort (<2 samples per age cohort), but this assay has been recently incorporated into standardized profiling for EGC, which will enable assessment in the future. Patients with advanced disease are more likely to undergo molecular testing and may not be representative of the entire population of EGC. Bulk tumor sequencing limits the ability to fully characterize cell-specific molecular features and future studies may utilize single-cell RNA sequencing, spatial transcriptomics, or quantitative immunofluorescence to provide further insight into tumor microenvironment specific interactions and features.
In summary, EOEGC is characterized by racial, ethnic, and gender disparities, increased CDH1 mutations, decreased prevalence of markers of response to ICI, and enrichment of pathways related to EMT and coagulation. Our data appear largely concordant with other published reports studying EOEGC and adds important insight into immune cell infiltrate and pathway alterations. We hypothesize that the rise of EOEGC may be related to the ongoing rise in diffuse-type gastric cancer given similar trends in mutations, molecular biomarkers, and pathway enrichment. There remains an ongoing need to further elucidate the impact of environment factors on increased EOEGC incidence. Overall these unique differential characteristics present therapeutic opportunities, but also demonstrate the limitations of currently approved therapies in patients with EOEGC.
Methods
Patient samples
5175 EGC samples were identified in the Caris Life Sciences database (Phoenix, AZ) with matched DNA sequencing, whole-transcriptome sequencing (WTS), and immunohistochemistry (IHC) data. We defined EGC esophageal squamous cell carcinoma, esophageal adenocarcinoma, gastric adenocarcinoma, and other/mixed histologies occurring in the esophagus, esophagogastric junction, or stomach.
We defined EOEGC as younger than 50 years old at diagnosis (n = 530). While prior studies have defined AOEGC as age ≥ 50 years old, we utilized two age cohorts given the average age of diagnosis of EGC is approximately 68 years old7,8. We defined intermediate-onset EGC (IOEGC) as age ≥50 and <65 at diagnosis (n = 1744) and AOEGC as age ≥ 65 years at diagnosis (n = 2901). Histology and Laurén subtype were included when available. We classified site of tumor specimen as primary or metastatic. Metastatic tumor sites included liver, peritoneum/retroperitoneum, lymph node, or other (sites not classified as primary or one of the specified metastatic sites). Smoking history and self-reported race and ethnicity were included when provided.
Next Generation Sequencing (NGS)
NGS was performed on genomic DNA isolated from formalin fixed paraffin-embedded (FFPE) tumor samples using the NextSeq or NovaSeq 6000 platforms (Illumina, Inc., San Diego, CA) as previously described48. For NextSeq-sequenced tumors, a custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). For NovaSeq-sequenced tumors, a hybrid pull-down panel of baits designed to enrich for >700 clinically relevant genes at high coverage and read depth was used, along with a separate panel to enrich for an additional >20,000 genes at lower depth. Genetic variants were detected with >99% confidence and were categorized by board-certified molecular geneticists. Only mutations previously classified as “pathogenic” and “presumed pathogenic” were included in the mutation frequency analysis.
Immunohistochemistry (IHC)
IHC was performed using automated staining techniques on slides obtained from FFPE tumor blocks as previously described (Benchmark XT; Ventana; and AutostainerLink 48; Dako, Carpinteria, CA)49. PD-L1 (22c3) and HER2 (4B5) was stained and scored according to previous criteria50. Staining was scored for intensity (0: negative; 1: weak; 2: moderate; 3: strong) and staining percentage (0–100%). Chromogenic in situ hybridization (CISH) was used for HER2/neu amplification (INFORM HER2 Dual ISH DNA Probe Cocktail)50. A HER2-CEP17 ratio of ≥2 was defined as positive for HER2 amplification.
Tumor Mutational Burden (TMB)
TMB was measured (592 genes and 1.4 megabases [MB] sequenced per tumor) as previously described by counting all non-synonymous missense mutations found per tumor that had not been previously described as germline alterations according to dbSNP and 1KG databases51. TMB-High was defined as ≥10 mutations/MB per the KEYNOTE-158 trial52.
Mismatch repair deficiency and microsatellite instability
A combination of multiple test platforms was used to determine the mismatch repair deficiency (dMMR)/MSI-H status of the tumors, including fragment analysis (FA, Promega), IHC [MLH1, M1 antibody; MSH2, G2191129 antibody; MSH6, 44 antibody; and PMS2, EPR3947 antibody (Ventana Medical Systems, Inc.)], and NGS. The three platforms generated highly concordant results as reported previously53. In the rare cases of discordant results, dMMR/MSI-H status was determined in the order of IHC, FA, and NGS.
Whole Transcriptomic Sequencing (WTS)
Tumor-specific mRNA was isolated with a Qiagen RNeasy FFPE Kit (Germantown, MD) from manually microdissected areas of FFPE sections with a tumor content of at least 10%. Quality and quantity were determined with the Agilent TapeStation (Santa Clara, CA). Library preparation and whole-transcriptome sequencing were performed on the NovaSeq 6500 platform (Illumina, Inc, San Diego, CA) to an average of 60 M reads as previously described48. Gene fusions were detected using the ArcherDX fusion assay (ArcherDX, Boulder, CO) and Illumina MiSeq platform (Illumina MiSeq, San Diego, CA) as previously described54. Gene set enrichment analysis (GSEA) was performed on WTS data55. MAPK pathway activation score was calculated using a 10-gene set (SPRY2, SPRY4, ETV4, ETV5, DUSP4, DUSP6, CCND1, EPHA2, and EPHA4)56. Immune checkpoint gene expression was analyzed for CD274, FOXP3, HAVCR2, LAG3, PDCD1, PDCD1LG2, CTLA4, and IDO1. Immune cell infiltrate fractions were calculated from transcriptomic data using quanTIseq57.
Clinical outcomes
Real-world overall survival (rwOS) data was obtained from insurance claims data and calculated from the date of tissue collection or treatment start as a surrogate for diagnosis until date of last contact. Patients without contact/claims data for a period of at least 100 days were presumed deceased. Conversely, patients with a documented clinical activity within 100 days prior to the latest data update were censored in the analysis. Kaplan-Meier estimates were calculated for EOEGC, IOEGC, and AOEGC cohorts within the broader Caris CODEai clinicogenomic database. Hazard ratios (HR) were determined by Cox Proportional Hazards model and rwOS differences were assessed with the log-rank test, with significance defined as p < 0.05.
Statistical analysis
Statistical significance was determined using chi-square, Fisher exact and Mann-Whitney U test as appropriate. The Benjamini-Hochberg method was utilized to adjust p-values for multiple comparisons (q < 0.05). Tumor immune microenvironment cell fractions were analyzed among cohorts using nonparametric Kruskal-Wallis testing. RwOS utilized the log-rank test with significance defined as p < 0.05.
Compliance statement
This study was conducted in accordance with guidelines of the Declaration of Helsinki, Belmont report, and U.S. Common rule. Per 45 CFR 46.101(b), this study was performed utilizing retrospective, deidentified clinical data. Therefore, this study is considered Institutional Review Board exempt and informed consent was not required.
Data availability
The data presented in this study are not publicly available due to data size and patient privacy but are available on reasonable request from the corresponding author. The NGS raw data are owned by Caris Life Sciences and cannot be publicly shared because of the data usage agreement signed by Dr. Ryan H. Moy. Qualified researchers can apply for access to these data by contacting Joanne Xiu (jxiu@carisls.com) and signing a data usage agreement.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Ferlay, J. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J. Cancer 144, 1941–1953 (2019).
Islami, F., DeSantis, C. E. & Jemal, A. Incidence Trends of Esophageal and Gastric Cancer Subtypes by Race, Ethnicity, and Age in the United States, 1997-2014. Clin. Gastroenterol. Hepatol. 17, 429–439 (2019).
Bergquist, J. R. et al. Early-onset gastric cancer is a distinct disease with worrisome trends and oncogenic features. Surgery 166, 547–555 (2019).
Codipilly, D. C. et al. Epidemiology and Outcomes of Young-Onset Esophageal Adenocarcinoma: An Analysis from a Population-Based Database. Cancer Epidemiol. Biomark. Prev. 30, 142–149 (2021).
Sung, H., Siegel, R. L., Rosenberg, P. S. & Jemal, A. Emerging cancer trends among young adults in the USA: analysis of a population-based cancer registry. Lancet Public Health 4, e137–e147 (2019).
SEER Cancer Stat Facts: Esophageal Cancer, https://seer.cancer.gov/statfacts/html/esoph.html (2024).
SEER Cancer Stat Facts: Stomach Cancer, https://seer.cancer.gov/statfacts/html/stomach.html (2024).
Domper Arnal, M. J., Ferrandez Arenas, A. & Lanas Arbeloa, A. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J. Gastroenterol. 21, 7933–7943 (2015).
Karimi, P., Islami, F., Anandasabapathy, S., Freedman, N. D. & Kamangar, F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol. Biomark. Prev. 23, 700–713 (2014).
Murphy, C. C., Yang, Y. C., Shaheen, N. J., Hofstetter, W. L. & Sandler, R. S. An age-period-cohort analysis of obesity and incident esophageal adenocarcinoma among white males. Dis. Esophagus 30, 1–8 (2017).
Li, J. Gastric Cancer in Young Adults: A Different Clinical Entity from Carcinogenesis to Prognosis. Gastroenterol. Res Pr. 2020, 9512707 (2020).
Ku, G. Y. et al. Prevalence of Germline Alterations on Targeted Tumor-Normal Sequencing of Esophagogastric Cancer. JAMA Netw. Open 4, e2114753 (2021).
Pocurull, A. et al. Clinical, Molecular and Genetic Characteristics of Early Onset Gastric Cancer: Analysis of a Large Multicenter Study. Cancers (Basel) 13, https://doi.org/10.3390/cancers13133132 (2021).
Lumish, M. A. et al. Clinical and Molecular Characteristics of Early-Onset versus Average-Onset Esophagogastric Cancer. J. Natl. Cancer Inst. https://doi.org/10.1093/jnci/djad186 (2023).
Chen, Y. C. et al. Clinicopathological Variation of Lauren Classification in Gastric Cancer. Pathol. Oncol. Res 22, 197–202 (2016).
Setia, N. et al. Morphologic and molecular analysis of early-onset gastric cancer. Cancer 127, 103–114 (2021).
Omofuma, O. O., Cook, M. B., Abnet, C. C. & Camargo, M. C. Race and Ethnicity, Stage-Specific Mortality, and Cancer Treatment in Esophageal and Gastric Cancers: Surveillance, Epidemiology, and End Results (2000-2018). Gastroenterology 164, 473–475 e474 (2023).
Tavakkoli, A. et al. Ethnic Disparities in Early-Onset Gastric Cancer: A Population-Based Study in Texas and California. Cancer Epidemiol. Biomark. Prev. 31, 1710–1719 (2022).
Jin, H., Pinheiro, P. S., Callahan, K. E. & Altekruse, S. F. Examining the gastric cancer survival gap between Asians and whites in the United States. Gastric Cancer 20, 573–582 (2017).
Wu, L. W. et al. Diffuse Gastric Cancer: A Comprehensive Review of Molecular Features and Emerging Therapeutics. Target Oncol. https://doi.org/10.1007/s11523-024-01097-2 (2024).
Hansford, S. et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 1, 23–32 (2015).
Becker, K. F. et al. E-cadherin gene mutations provide clues to diffuse type gastric carcinomas. Cancer Res 54, 3845–3852 (1994).
Klempner, S. J. et al. FGFR2-Altered Gastroesophageal Adenocarcinomas Are an Uncommon Clinicopathologic Entity with a Distinct Genomic Landscape. Oncologist 24, 1462–1468 (2019).
Wainberg, Z. A. et al. Bemarituzumab in patients with FGFR2b-selected gastric or gastro-oesophageal junction adenocarcinoma (FIGHT): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 23, 1430–1440 (2022).
Kim, J. et al. Preexisting oncogenic events impact trastuzumab sensitivity in ERBB2-amplified gastroesophageal adenocarcinoma. J. Clin. Invest 124, 5145–5158 (2014).
Rustgi, N. et al. Molecular Landscape and Clinical Implication of CCNE1-amplified Esophagogastric Cancer. Cancer Res Commun. 4, 1399–1409 (2024).
Fu, S. et al. Multicenter Phase II Trial of the WEE1 Inhibitor Adavosertib in Refractory Solid Tumors Harboring CCNE1 Amplification. J. Clin. Oncol. 41, 1725–1734 (2023).
Wainberg, Z. A. et al. 504MO Phase I study of the PKMYT1 inhibitor lunresertib (lunre) in combination with FOLFIRI in advanced gastrointestinal (GI) cancers (MINOTAUR study). Ann. Oncol. 35, S201 (2024).
Llombart, V. & Mansour, M. R. Therapeutic targeting of “undruggable” MYC. EBioMedicine 75, 103756 (2022).
Cancer Genome Atlas Research, N Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209 (2014).
Shu, Y. et al. Prognostic significance of frequent CLDN18-ARHGAP26/6 fusion in gastric signet-ring cell cancer. Nat. Commun. 9, 2447 (2018).
Sica, A. et al. Macrophage polarization in tumour progression. Semin Cancer Biol. 18, 349–355 (2008).
Yin, S. et al. The Prognostic and Clinicopathological Significance of Tumor-Associated Macrophages in Patients with Gastric Cancer: A Meta-Analysis. PLoS One 12, e0170042 (2017).
Pantano, F. et al. The role of macrophages polarization in predicting prognosis of radically resected gastric cancer patients. J. Cell Mol. Med 17, 1415–1421 (2013).
Zhang, J. et al. Tumor associated macrophages in esophageal squamous carcinoma: Promising therapeutic implications. Biomed. Pharmacother. 167, 115610 (2023).
Wang, S. et al. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. NPJ Precis Oncol. 8, 31 (2024).
Wesolowski, R. et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Ther. Adv. Med. Oncol. 11, 1758835919854238, https://doi.org/10.1177/1758835919854238 (2019).
Omstead, A. N. et al. CSF-1R inhibitor, pexidartinib, sensitizes esophageal adenocarcinoma to PD-1 immune checkpoint blockade in a rat model. Carcinogenesis 43, 842–850 (2022).
Shi, T. et al. DKK1 Promotes Tumor Immune Evasion and Impedes Anti-PD-1 Treatment by Inducing Immunosuppressive Macrophages in Gastric Cancer. Cancer Immunol. Res 10, 1506–1524 (2022).
Klempner, S. J. DKN-01 in Combination With Tislelizumab and Chemotherapy as First-Line Therapy in Advanced Gastric or Gastroesophageal Junction Adenocarcinoma: DisTinGuish. J. Clin. Oncol. 43, 339–349 (2025).
Huang, L., Wu, R. L. & Xu, A. M. Epithelial-mesenchymal transition in gastric cancer. Am. J. Transl. Res 7, 2141–2158 (2015).
Oh, S. C. et al. Clinical and genomic landscape of gastric cancer with a mesenchymal phenotype. Nat. Commun. 9, 1777 (2018).
Lei, Z. et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology 145, 554–565 (2013).
Monster, J. L., Kemp, L. J. S., Gloerich, M. & van der Post, R. S. Diffuse gastric cancer: Emerging mechanisms of tumor initiation and progression. Biochim Biophys. Acta Rev. Cancer 1877, 188719 (2022).
Oshi, M. et al. Higher intra-tumoral expression of pro-coagulation genes is a predictor of angiogenesis, epithelial mesenchymal transition and worse patient survival in gastric cancer. Am. J. Cancer Res 12, 4001–4014 (2022).
Pan, G., Liu, Y., Shang, L., Zhou, F. & Yang, S. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun. (Lond.) 41, 199–217 (2021).
In, G. K. et al. Multi-omic profiling reveals discrepant immunogenic properties and a unique tumor microenvironment among melanoma brain metastases. NPJ Precis Oncol. 7, 120 (2023).
Tokunaga, R. et al. Molecular Profiling of Appendiceal Adenocarcinoma and Comparison with Right-sided and Left-sided Colorectal Cancer. Clin. Cancer Res 25, 3096–3103 (2019).
Salem, M. E. et al. Comparative Molecular Analyses of Esophageal Squamous Cell Carcinoma, Esophageal Adenocarcinoma, and Gastric Adenocarcinoma. Oncologist 23, 1319–1327 (2018).
Wang, J. et al. Large-scale analysis of KMT2 mutations defines a distinctive molecular subset with treatment implication in gastric cancer. Oncogene 40, 4894–4905 (2021).
Marabelle, A. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365 (2020).
Vanderwalde, A., Spetzler, D., Xiao, N., Gatalica, Z. & Marshall, J. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med 7, 746–756 (2018).
Ferguson, S. D. et al. Targetable Gene Fusions Associate With the IDH Wild-Type Astrocytic Lineage in Adult Gliomas. J. Neuropathol. Exp. Neurol. 77, 437–442 (2018).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Wagle, M. C. et al. A transcriptional MAPK Pathway Activity Score (MPAS) is a clinically relevant biomarker in multiple cancer types. NPJ Precis Oncol. 2, 7 (2018).
Finotello, F. et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med 11, 34 (2019).
Acknowledgements
L.W.W. was supported by the NIH/NCI Molecular Oncology Training Program (5T32CA203703-09). R.H.M. is supported by grants from the NIH (K08CA263304), Gastric Cancer Foundation, the 2023 AACR-Debbie’s Dream Foundation Innovation and Discovery Grant (Grant Number 23-80-41-MOY), and the US Department of Defense (HT9425-24-1-0419). This publication was supported by the NIH/NCI Cancer Center Support Grant P30CA013696.
Author information
Authors and Affiliations
Contributions
R.H.M was responsible for conceptualization, investigation, and supervision. L.W.W., S.K.D., R.H.M. were responsible for the development of methodology. L.W.W. and R.H.M. were responsible for writing – original draft/review/revising. L.W.W., S.K.D., S.W., J.X., V.K.L., E.L., S.G., R.T.S., R.H.M. contributed to data acquisition. L.W.W., S.K.D., S.W., J.X., S.J., J.P., V.K.L., E.L., S.G., R.T.S., R.H.M. contributed to data analysis and interpretation. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
L.W.W., S.J., J.P., and S.G. declare no competing interests. S.K.D., S.W., and J.X. are employees of Caris Life Sciences. V.K.L. has served in a consultant/advisory role for Pfizer, Genentech/Roche, Iovance Biotherapeutics, Anheart Therapeutics, Takeda, Seattle Genetics, Bristol Myers Squibb, AstraZeneca and Guardant Health, and has received research funding from GlaxoSmithKline, Bristol Myers Squibb, AstraZeneca, Merck and Seattle Genetics. R.T.S. has served in a consult/advisory role for AstraZeneca, Boehringer Ingelheim Pharma, Clovis, Genentech, Incyte, Merck, QED Therapeutics, Servier, Taiho, Zymeworks Biopharm, Syros, Astellas, Natera, Hoopika Pharma, Abbvie, Duo Oncology, and has received research funding from Bayer, Bristol Myers Squibb, Exelixis Pharmaceuticals, IMV Inc, LOXO, Novocure, NUCANA, Pieris, Rafael Pharmaceuticals, Seagen. R.H.M. has served in a consult/advisory role for Puretech Health, IDEAYA Biosciences, Nimbus Therapeutics, and Amgen; and has received research funding from Nimbus Therapeutics and Repare Therapeutics.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, L.W., Deshmukh, S.K., Wu, S. et al. Multi-omic characterization of early-onset esophagogastric cancer. npj Precis. Onc. 9, 241 (2025). https://doi.org/10.1038/s41698-025-01030-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01030-4
