
Abstract
Gastric cancer (GC), of which cases with peritoneal metastasis are particularly challenging, retains its position of being highly complex and remarkably resistant to therapy. Understanding the spatial heterogeneity and leveraging recent technologies such as machine learning to uncover explanatory patterns remains critical to truly understanding this disease. Here, we conducted spatial transcriptomics analysis to identify distinct niches within GC tissues. Among these, a niche enriched with fibroblasts and macrophages exhibited a striking spatial co-infiltration pattern with tumor cells dominant niches. Further validation by multiplex immunofluorescence highlighted the coordinated cellular interactions that characterize the TME. Through integration of sc-RNA with bulk RNA sequencing, we identified DAB2⁺ TAMs and ACTA2⁺ myCAFs as the main contributors to this co-infiltration pattern. NicheNet analysis further revealed that the PLAU-PLAUR signaling axis holds a central regulatory role in the communication between macrophages, fibroblasts and tumor cells. Given the prognostic value of this spatial pattern, we additionally applied transfer learning based on an ImageNet pre-trained ResNet-50 model to develop a machine learning framework that can accurately recognize the macrophage-fibroblast-malignant cell co-infiltration pattern, called Gastric-Discovery. Potentially, Gastric-Discovery could be a tool for precise patient stratification and provides novel insights into the dynamic architecture of the TME.
Similar content being viewed by others


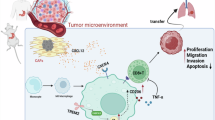
Introduction
Gastric cancer (GC) ranks among the main cancer-related causes of mortality globally and demonstrates a marked elevated incidence in China and other East Asian countries, posing a considerable burden on regional healthcare systems. A range of underlying factors have been identified as contributing to this complicated situation, among them the striking tendency of metastatic potential, with peritoneal metastasis being both the most prevalent and the most challenging form of the disease to manage1. Peritoneal metastasis uniquely resists systemic chemotherapy due to the drug-blocking peritoneal barrier1,2. Commonly, GCPM exhibit extensive proliferation of fibrous connective tissue and an unusually rich matrix composition. Not only does this increase the aggressiveness of the lesions, but it also promotes a highly immunosuppressive TME. Consequently, traditional treatment approaches that focus on the direct destruction of tumor cells have emerged as inadequate in this context3,4. Faced with this therapeutic impasse, clinical researchers persistently sought out novel approaches. Several treatment strategies have been proposed and refined through continuous practice. For example, S-1-based chemotherapies gained widespread acceptance in recent years due to their relative ability to penetrate the peritoneum. Targeted therapies such as apatinib likewise demonstrated a certain degree of efficacy in specific cases of GCPM. However, due to the peritoneal barrier and the unique fibrotic microenvironment characteristic of “hard cancers”, drugs oftentimes struggle to penetrate the dense fibrous matrix to reach tumor cells, resulting in overall suboptimal efficacy5. Notably, a previous study led by our research group, the DRAGON-01 trial, proposed the intraperitoneal normal-temperature combination systemic therapy (NIPS) regimen as a strategy to overcome these barriers, which significantly prolonged patient survival and improved one- and two-year survival rates6. Nevertheless, although the NIPS regimen boosts intraperitoneal drug concentrations via optimized drug delivery routes, there still call for further breakthroughs in overcoming the high-intensity therapeutic resistance barrier erected by stromal sclerosis in PM2,3,6.
Furthermore, marked spatial heterogeneity is evident between primary GC and GCPM. For certain therapeutic targets, like FGFR2b, CLDN18.2, pronounced expression discrepancies exist between primary tumors and peritoneal metastases, rendering targeted therapeutic regimens developed exclusively based on primary tumor clinically ineffective7,8. Additionally, fibroblasts, identified as the principal driver of stromal sclerosis in GC management, constitute the dominant stromal fraction within the TME4. It has been established that these cells facilitate a nurturing environment conducive to tumor proliferation and therapeutic resistance through multifarious mechanisms9. Of particular note is the observation that an intimate intercellular crosstalk network exists among CAFs, TAMs and tumor cells, where these populations reinforce one another via elaborate signaling crosstalk. Macrophages have been evidenced to activate fibroblasts through the secretion of factors like IL-6, thereby endowing the latter with an enhanced pro-tumorigenic potential. Then, activation of tumor-promoting fibroblasts could further influence the polarization and function of tumor-supporting macrophages, creating an ongoing positive feedback loop10. Understanding how these cells collaborate to each other and create a tumor-supportive niche is crucial for developing creative therapies. In order to confront the aforementioned challenges of heterogeneity and TME regulatory mechanisms, multi-omics has established itself as a cornerstone tool in recent GC TME research, leveraging its defining strength of preserving spatial positional information while decoding transcriptional profiles at a single-cell level11,12. For instance, a research team from Singapore utilized a combination of scRNA-seq and DSP analysis, complemented by in vitro and in vivo functional validation assays. Their integrated approach led to the prominent finding of a substantial increase in plasma cell abundance in patients with diffuse-type GC. Further mechanistic investigations identified that the underlying regulatory mechanism of this phenomenon is orchestrated by the transcription factor KLF2, which is specifically derived from epithelial cells within the GC microenvironment13.
Despite the fact that scRNA-seq technology revolutionized the deciphering of the complexity and heterogeneity of cellular composition in the TME of GC and highlighted the decisive role of a multitude of stromal cells, including macrophages and fibroblasts, current technical limitations do not yet allow for the preservation of information about the spatial location of cells in the original tissue. These limitations compromise our in-depth understanding of intercellular interactions, particularly those between macrophages, fibroblasts, and tumor cells, a potently functional co-infiltrating unit. The intercellular interactions, in particular the proximity and contact of these cells in physically shared space, are essential for their biological effects8. Spatial transcriptomics techniques developed in recent years attempt to fill this gap. In GC research, spatial transcriptomics technology was initially used to visualize the structure of tissue regions within tumors, such as tumor cores and invasion fronts, and to validate certain known cellular communication events. However, there is still a lack of powerful and general computational biology frameworks to systematically mine the unidentified but biologically significant cellular co-localization patterns or co-infiltration patterns from massive and high-dimensional spatial molecular data14. Against this backdrop, deep learning strategies offer a promising path forward. Here, we introduce an integrative deep learning framework leveraging multi-dimensional evidence15,16. Core to the framework is the integration of scRNA-seq and spatial transcriptomics techniques to yield finer estimates of cell type assembly proportions for each spatial location or region in spatial transcriptomics data. Then, we construct the entire tissue section or region into a complex spatial cellular graph, on which a deep learning model is trained with the goal of unsupervised or semi-supervised learning of feature representations that characterize this spatial organization pattern. With this approach, the identification of co-infiltration patterns defined and functionalized with single-cell precision is of great clinical value for determining the prognosis of patients with GC. In summary, our work identifies a prognostically relevant MFM co-infiltration signature in GC, characterizes its constituent cellular states and communication networks, based on these evidences, we also developed a deep learning working framework. These efforts may not only complement existing prognostic prediction tools, but may also point the way to the development of precise combination therapies targeting specific malignant microenvironmental ecological niches.
Results
Comparison of stromal characteristics and classification of spatial niches in GC patients
GC remains a highly challenging clinical problem due to its unique biological characteristics and therapeutic resistance. To address this, our study developed a deep learning-based biomarker detection workflow that leverages multi-omics data to deeply analyze the spatial heterogeneity features of primary and metastatic lesions in GC. (Fig. 1). To more intuitively assess stromal activation characteristics in GC samples and peritoneal metastasis samples, we applied the ESTIMATE algorithm to normal and tumor samples in the TCGA-STAD cohort, as well as to tumor and peritoneal metastasis samples in the ACRG cohort. We evaluated the tumor microenvironment from four perspectives: Stromal Score, Immune Score, ESTIMATE Score, and Tumor Purity. We found that the Stromal Score was higher in the tumor group than in the normal group, and even higher in peritoneal metastasis samples compared to primary tumor samples. Moreover, patients with high Stromal Scores had a poorer prognosis (Fig. 2A–C). These findings preliminarily suggest that enhanced stromal activity is a factor promoting tumor malignancy and may also facilitate peritoneal metastasis in GC. To further investigate this, we collected GC specimens and performed Masson staining to define tumor stromal sclerosis characteristics (Fig. 2D, E). We observed that GC patients with high stromal scores tended to have a worse prognosis (Fig. 2F). We also evaluated the relationship between TNM staging and this score. Although the results did not show a significant p-value, a notable correlation was observed (Fig. 2G). The gene expression-based Stromal Score is a significant prognostic indicator. However, the underlying spatial tissue basis for this poor prognosis remains unclear. Moreover, the Stromal Score reliably reflects the relative abundance of fibroblasts and indicates their mediated stromal remodeling and sclerosis activity. Therefore, we next aimed to investigate, at spatial resolution, the cellular composition of high stromal score regions and the spatial interaction patterns formed between fibroblasts and other core cell types. Subsequently, this study utilized spatial transcriptomic data to more intuitively and accurately support this conclusion. Referring to single-cell transcriptomic data from the same batch of studies, we performed deconvolution annotation using cell2location, initially identifying common microenvironment cell types in spatial slices, such as epithelial cells, fibroblasts, macrophages, and immune cells (Supplementary Fig. S1A, B). Next, we employed CCA to efficiently and robustly integrate eight spatial transcriptomic datasets. Hierarchical clustering was performed on the cell abundance of each region and its adjacent areas in the GC spatial transcriptomic slices. At a resolution of 0.2, eight distinct niches were identified and labeled with different numerical identifiers. After calculating the normalized distribution of cell deconvolution matrix abundances, the average abundance of each cell type was computed by niche grouping, and a composition ratio matrix of cell types within each niche was obtained. Following malignant cell scoring via infercnv, the following niche cellular compositions were specifically determined: Niche_01 (malignant epithelial cells, mal_Epi), Niche_02 (normal epithelial cells, Epi_normal), Niche_03 (T/NK and B cells), Niche_04 (macrophages and fibroblasts, Mph and Fib), Niche_05 (mast cells), Niche_06 (T cells and endothelial cells, T and EC), Niche_07 (Pericyte) and Niche_08 (endothelial cells, EC) (Fig. 2H and I, Supplementary Fig. S1C, D, G). The distribution of niches across different spatial samples was relatively uniform yet distinct. Niches 4 and Niches 1 accounted for a relatively large proportion in each slice (Supplementary Fig. S1E, F). To quantitatively assess the spatial organizational relationships between different niches, we conducted systematic neighborhood enrichment analysis. The results revealed a high-intensity neighborhood localization signal between Niches 4 (co-existing Mph and Fib) and Niches 1 (dominated by malignant cells). This localization signal is highly unique, and we preliminarily speculate that it may be the reason for the high stromal characteristics (Fig. 2J–L). Specifically, at the 2-ring neighborhood scale (Fig. 2J), we observed a strong spatial co-occurrence trend between Niches 1 and Niches 4, with a neighborhood enrichment score as high as 0.3. This pattern remained consistent across different spatial scales, demonstrating the robustness of their spatial interaction (Fig. 2K, L).

First, through spatial transcriptomics analysis, this study identified niches enriched in fibroblasts and macrophages in GC tissues; these niches exhibited a significant spatial neighborhood co-infiltration pattern with tumor cell-dominant niches. This pattern was further validated by multiplex immunofluorescence experiments and referred to as the “MFM” (macrophage-fibroblast-tumor cell) co-infiltration pattern. Subsequent Celltrek spatial localization analysis confirmed that DAB2⁺ tumor-associated macrophages (DAB2⁺TAMs) and ACTA2⁺ myofibroblasts (ACTA2⁺myCAFs)—the core participants in this co-infiltration pattern—exhibited a significant spatial co-localization relationship. Notably, patients with this co-infiltration pattern had significantly poorer prognosis. Given the important prognostic value of this co-infiltration pattern, this study conducted transfer learning based on the ImageNet-pre-trained ResNet-50 model. Using pathological sections from the TCGA cohort for training, a deep learning framework capable of accurately identifying the “macrophage-fibroblast-tumor cell” co-infiltration pattern was constructed. At the mechanistic level, the study further revealed that the PLAU-uPAR signaling axis plays a key regulatory role in the tripartite crosstalk among macrophages, fibroblasts, and tumor cells.

A Bar charts showing ESTIMATE algorithm-derived scores based on transcriptomic data of GC patients from the TCGA-STAD cohort, including four metrics: Tumor Purity, Stromal Score, Immune Score, and ESTIMATE Score. B Bar charts showing ESTIMATE algorithm-derived scores based on transcriptomic data of GC patients from the ACRG cohort, including four metrics: Tumor Purity, Stromal Score, Immune Score, and ESTIMATE Score. C Kaplan–Meier (KM) curves showing that GC patients with high Stromal Scores had significantly worse clinical prognostic outcomes than those with low Stromal Scores. D, E Representative Masson-stained images of GC pathological sections from our center. Top: 10× magnification; Bottom: 40× magnification. Scale bars: 200 μm (10×) and 50 μm (40×), respectively. F KM survival curves showing that patients with high Stromal Scores (quantified from Masson staining) had poorer prognosis. G Correlation scatter plot showing the correlation between CVF% (Stromal Component Proportion in Visual Field, i.e., the proportion of stromal components in the visual field) and TNM stage of GC patients. H Niche classification of spatial transcriptomic data from 8 GC cases; a total of 8 niches were identified, with each color representing a distinct niche. I InferCNV scoring of spatial transcriptomic data from 8 GC cases to delineate tumor regions. J–L Heatmaps revealing the neighborhood localization relationships among the 8 niches, with neighborhood localization signals observed at distances ranging from 55–55×2 μm (left, ring = 2) to 55–55 × 3 μm (middle, ring = 3) and further to 55–55 × 5 μm (right, ring = 5).
Regional features of the macrophage-fibroblast-malignant cell colocalized region
Building upon the previously described stable spatial architecture between cellular niches delineated through neighborhood analysis, this study further investigated the spatially proximate localization signals of Niches 4 and Niches 1 observed across each section and the interactions between their principal cellular components and the adjacent TME. We analyzed spatial transcriptomics data from eight GC sections included in this study. Using MISTYR to compute co-localization patterns of major TME cells from the cell2location deconvolution results, including intra-spot co-localization and neighboring-spot co-localization, we aimed to elucidate functional interactions significantly contributing to gene expression. The results demonstrated varying degrees of intra-spot and inter-neighborhood spot colocalization of macrophages and fibroblasts across all eight analyzed ST sections (Fig. 3A). We further delineated ROIs in areas with high infiltration of these cells. Under high resolution, examination of different ST sections revealed that macrophages and fibroblasts envelop tumor cells, forming an armor-like structure that impedes the infiltration of T/NK cells. For descriptive convenience, we termed this spatial organization the MFM localization pattern (Macrophage-Fibroblast-Malignant cell) (Fig. 3B–D). To validate this characteristic structure, we performed multiplex immunofluorescence staining on independent pathological paraffin-embedded sections. The results confirmed that CD68-labeled macrophages and α-SMA-labeled fibroblasts consistently aggregate around tumor cells, forming a barrier-like structure that partially excludes CD8 + T cells, thereby substantiating our hypothesis (Fig. 3E, F).

A Heatmaps showing the colocalization of cells from cell2location-derived deconvolution results (calculated via the Mistyr algorithm) across 8 GC spatial transcriptomic sections. The upper panel represents intra-spot colocalization, and the lower panel represents inter-neighborhood spot colocalization. B–D Visualization of regions of interest (ROI) for the spatial localization of macrophages, fibroblasts, and tumor cells. Distinct colors correspond to different cell types, and the intensity of each color reflects the relative abundance of the respective cell type. E, F Multiplex immunofluorescence images depicting the spatial localization of macrophages, fibroblasts, and tumor cells.
myCAF as a key component of the MFM region and its contribution to GC progression
To analyze the aforementioned cell distribution pattern in GC at the single-cell level, we integrated our in-house data, including gene expression profiles from 19 primary GC samples, 10 normal gastric tissue samples, and 12 GCPM samples. After batch effect removal using the “harmony” package and quality control filtering, high-quality cells from these 41 samples were initially classified into 9 major cell types (Fig. 4A, B). These include B cells (marked by MS4A1, CD79A, etc.), plasma cells (marked by IGKC, IGHG1, etc.), T/NK cells (marked by CD3D, etc.), epithelial cells (marked by KRT8, EPCAM, etc.), endothelial cells (marked by VWF, etc.), fibroblasts (marked by COL1A1, etc.), myeloid cells (marked by CSF1R, CD68, etc.), and mast cells (marked by TPSAB1, etc.) (Supplementary Fig. S2A, B, D). The heterogeneity of the TME is the fundamental cause of various therapeutic challenges in tumors. Although the above major cell types exist in both tumor tissues and adjacent normal tissues, their proportions vary significantly across different tissue types. As mentioned earlier, spatial transcriptomics indicated that tumor cells, macrophages, and fibroblasts are highly enriched in tumor tissues and peritoneal metastases, a distribution pattern we also observed in single-cell datasets. Specifically, the numbers of fibroblasts and myeloid cells (with macrophages accounting for the majority) gradually increased in tumor tissues and peritoneal metastases compared to normal samples (Fig. 4C, Supplementary Fig. S2C). This quantitative distribution pattern may reflect the “scirrhous characteristics” of GC, prompting us to further explore such cellular heterogeneity and its underlying mechanisms. Fibroblasts are one of the main components contributing to stromal reinforcement in the TME, and studies have confirmed their role in promoting tumorigenesis and metastasis. However, the high consistency of CAFs and their distribution patterns with surrounding cells across different tissues, as mentioned in this study, remain poorly understood. In this research, fibroblasts were re-clustered into 6 subpopulations, named c1-CFD-Fib, c2-ACTA2-Fib, c3-CCL5-Fib, c4-CCL11-Fib, c5-ACTG1-Fib, and c6-CD37-Fib (Fig. 4D), with characteristic markers for each subpopulation shown in Fig. 4G. Furthermore, we used the Velocity algorithm to infer the developmental trajectory of these fibroblast subpopulations. The results showed that c1-CFD fibroblasts have a greater potential to differentiate into c2-ACTA2 fibroblasts, while c3-CCL5 and c4-CCL11 fibroblasts are more likely to differentiate into c4-CCL11 fibroblasts (Fig. 4E, F). Functional enrichment analysis of each fibroblast subpopulation revealed distinct functions, reflecting the high heterogeneity of the tumor microenvironment (Supplementary Fig. S3A). We observed that fibroblasts are dominant in PM samples; specifically, c2-ACTA2-Fib and c1-CFD-Fib are the most abundant in PM samples (Fig. 4H–K). Previous studies have classified fibroblasts based on their functions, including lipid process-related CAFs (lpCAFs), inflammatory CAFs (iCAFs), myofibroblast-like CAFs (myCAFs), vascular CAFs (vCAFs), and proliferative CAFs (pCAFs). The types of fibroblasts vary across different tumors or even different samples of the same tumor. We scored the functional tendencies of each fibroblast subpopulation based on the expression of key molecules. The results showed that c2-ACTA2-Fib exhibits a strong correlation with myCAFs, while c1-CFD-Fib is strongly associated with iCAFs (Fig. 4M). Here, we introduced the Scissor algorithm to investigate the relationship between cell subpopulations and clinical characteristics. We successfully identified Scissor+ cells (most relevant to distant metastasis) among the fibroblast subpopulations. Based on cell proportion inference, ACTA2⁺ myCAFs accounted for the largest proportion of Scissor+ cells, suggesting they may be the core component of “MFM pattern” in GCPM and the TME (Fig. 4L). We further evaluated the correlation between ACTA2⁺ myCAF infiltration and clinical indicators using bulk RNA-seq data. High infiltration of ACTA2⁺ myCAFs was more common in tumor patients and correlated with poorer OS and advanced staging in GC patients (Fig. 4N). Validation in other GC transcriptome cohorts confirmed that high infiltration of ACTA2⁺ myCAFs is a risk factor for GC patients (Fig. 4O, P; Supplementary Fig. S3B). Functional enrichment analysis of populations with high vs. low ACTA2⁺ myCAF infiltration indicated that the infiltration level of ACTA2⁺ myCAFs is significantly associated with multiple features of the HALLMARK pathways (Supplementary Fig. S3C).

A, B UMAP plots showing the distribution of 41 single-cell samples (involved in spatial transcriptomic annotation) across different groups, and 12 major cell clusters after clustering. AD: normal samples; PT: primary tumor samples; PM: peritoneal metastasis samples. C Bar charts showing the proportions of myeloid cells and fibroblasts in different groups. The y-axis represents cell abundance. D t-SNE plot showing further sub-clustering of fibroblasts into 6 fibroblast subpopulations. E, F RNA Velocity analysis indicating the differentiation direction of different cancer-associated fibroblasts (CAFs); the start of the arrow represents the subpopulation with low differentiation potential, and the end of the arrow represents the subpopulation with high differentiation potential. F Kaplan–Meier (KM) survival curve showing that patients with high Stromal Scores (quantified from Masson staining) had poorer prognosis. G Bubble plot showing the typical markers of different fibroblast (Fib) subpopulations; the size and color of the bubbles represent the significance of the markers (bubble size indicates gene expression proportion, color intensity indicates differential significance). H, I Circular pie charts showing the proportion of each fibroblast subpopulation in different sample groups. J, K Pie chart and heatmap showing the abundance of each fibroblast subpopulation in samples of different groups, calculated using the R/oe method. L Identification of fibroblast subpopulations most associated with peritoneal metastasis using the Scissor algorithm; the proportion plot below shows the percentage of cells with Scissor+ characteristics. M Radar chart showing CAF functional scoring of different fibroblast subpopulations to identify the properties of inflammatory CAFs (iCAFs), myo-fibroblastic CAFs (myCAFs), and angiogenic CAFs (apCAFs). N Box plot showing the differences in CAF scores between groups with different clinical characteristics. O, P KM survival curves showing the survival prognosis of patients with high ACTA2⁺CAF expression in different GC transcriptomic cohorts.
The role of DAB2⁺ TAMs in GC progression
Furthermore, we investigated another component of the ecological niche of MFM: macrophages. To identify the primary macrophage subpopulations most closely associated with ACTA2⁺ myCAFs, we identified macrophages from myeloid cells and conducted subpopulation annotation. Six cell subpopulations were identified, including monocytes, which were designated c1-AIF1-Mph, c2-APOE-Mph, c3-SPP1-Mph, c4-DAB2-Mph, c5-FN1-Mph, and c6-FCN1-Mph, each expressing its specific marker (Fig. 5A, B). We then analyzed the expression levels, enrichment preferences and infiltration proportions of the macrophage subpopulations in different samples. The c4-DAB2-Mph subpopulation attracted particular attention as it displayed progressively increased expression in both PT and PM derived samples. Moreover, it showed dominant infiltration in both PT and PM samples, suggesting its potential as a key subpopulation in GCPM. The c3-SPP1-Mph subpopulation also exhibited dominant expression, although it predominantly infiltrated PT samples and showed reduced expression during PM progression (Fig. 5C, D). Furthermore, these macrophage subpopulations displayed different preferences across sample types, potentially reflecting the inherent complexity of macrophage differentiation. The complexity of macrophages lies in their ability to undergo M1/M2 switching in response to different TME. As a result, we then compared the M1/M2 propensity of the above macrophage subpopulations. Due to the diversity of macrophages, we observed that subpopulations such as c1-AIF1-Mph and c2-APOE-Mph exhibited ambiguous differentiation characteristics, consistent with the complex functional nature of macrophages. Conversely, cells within the c4-DAB2-Mph subpopulation exhibited prototypical M2 characteristics (Fig. 5E, F). We endeavored to elucidate the evolutionary dynamics within these macrophage subpopulations via RNA velocity analysis. The c4-DAB2-Mph subpopulation occupies an evolutionary endpoint and most likely differs from the c5-FN1-Mph, c2-APOE-Mph, and c6-FCN1-Mph subpopulations. Indicating that intermediate macrophage states have the potential to differentiate into M2-type macrophages (Fig. 5G). We then performed functional comparisons between the immune, metabolic, signaling, and proliferation pathways of these different macrophage subpopulations. The results show that DAB2⁺ TAMs exert a more significant influence on immune functions, while other subpopulations predominantly converge on signaling pathways (Fig. 5H). The aforementioned findings center on DAB2⁺ TAM, leading us to hypothesize that it may constitute a core component of the MFM niche. Subsequently, we scored key markers of DAB2⁺ TAM to assess their association with immune checkpoint-related indicators. Results demonstrated that the TAM score derived from DAB2⁺ TAM markers exhibited positive correlations with PDCD1, CD274, LAG3, and IDO1, aligning with the established understanding that M2 macrophages promote tumor progression by inducing immune suppression (Fig. 5I).

A UMAP plot showing the further subdivision of monocyte/macrophage populations, with a total of 6 macrophage subpopulations identified. B Bubble plot showing the expression levels of typical markers in different macrophage subpopulations. The size of the bubble represents the proportion of cells expressing the marker in the corresponding subpopulation, and the color intensity indicates the significance of the marker’s differential expression. C Stacked bar chart showing the proportion of each macrophage subpopulation across different sample groups (normal/primary tumor/peritoneal metastasis). D Heatmap showing the abundance of each macrophage subpopulation in samples of different groups, calculated using the R/OE method. Box plot (E) and heatmap (F) showing the M1/M2 polarization scores of different macrophage subpopulations. G RNA Velocity-based analysis indicating the differentiation direction of different macrophage subpopulations: the start of the arrow represents the subpopulation with low differentiation potential, and the end of the arrow represents the subpopulation with high differentiation potential. H Bubble plot showing the functional enrichment results of different macrophage subpopulations. The color intensity and size of the bubbles represent the activation level and enrichment significance of the functional pathways, respectively. I Correlation scatter plot showing the correlation between the DAB2⁺TAM signature score (TAM score) and the expression levels of common immune checkpoint molecules.
Multi-omics analysis of correlative features underlying ACTA2⁺ myCAF and DAB2⁺ TAM colocalization
Based on the above analytical results, we hypothesized that an interactive communication network forms between ACTA2⁺ myCAFs and DAB2⁺ TAMs. This network largely also acts upon tumor cells, potentially playing a key role in stromal remodeling during the malignant progression of GC and promoting the formation of an immunosuppressive microenvironment in GC. To gather more evidence supporting this hypothesis, we first performed a correlation analysis between the infiltration level of ACTA2⁺ myCAFs and macrophage infiltration levels based on transcriptomic data from different GC cohorts. The results indicated that in the vast majority of GC transcriptomic cohorts, the expression of ACTA2⁺ myCAFs correlated with macrophages (especially M2 macrophages). A typical example is the cohort from Pasini et al., where this correlation was particularly significant (R = 0.55) (Fig. 6A, B). Next, we co-embedded ST and scRNA-seq data using the CellTrek algorithm, and subsequently mapped ACTA2⁺myCAFs and different types of macrophages onto spatial tissue images via a random forest model prediction. Stable spatial co-localization of ACTA2⁺ myCAFs and DAB2⁺ TAMs was observed in both GC PT and PM samples (Fig. 6C), suggesting to some extent that these two cell types are important components constituting the MFM armor-like TME structure. Subsequently, to better interpret this phenomenon, we collected samples, hoping to provide crucial evidence through multiplex immunofluorescence demonstrating co-localization (Fig. 6D–G). Our fluorescence evidence revealed varying degrees of infiltration of DAB2⁺ TAMs and ACTA2⁺ myCAFs in primary GC lesions and peritoneal metastases. In peritoneal metastases, the spatial distance between DAB2⁺ TAMs and ACTA2⁺ myCAFs was closer, and the degree of interaction was stronger (Fig. 6D–G).

A Heatmap showing the association between CAF score and macrophage infiltration assessed by different immune infiltration algorithms across various GC cohorts. B Scatter plot demonstrating the correlation between CAF score and macrophage infiltration. C CellTrek-based deconvolution analysis reveals the distribution of DAB2⁺ TAMs and ACTA2⁺ CAFs. The heatmap displays the degree of co-localization between different CAF subtypes and macrophage subtypes in various GC spatial transcriptomics (ST) sections. D–G Multiplex immunofluorescence images showing the co-localization features of DAB2⁺ TAMs and ACTA2⁺ CAFs. Scale bars represent 200 μm and 100 μm, respectively.
PLAU signaling tends to be a critical driver in the formation of the MFM niche
In our previous work, we sought to identify evidence of fibroblast-macrophage-tumor cell tripartite co-localization in GC. Through mIF and multi-omics evidence, we confirmed the co-localization phenomenon between ACTA2⁺ myCAFs and DAB2⁺ TAMs within these structures. We then attempted to explore the potential mechanisms underlying this phenomenon via functional enrichment analysis. Performing GO functional enrichment on the previously mentioned different niches, we found that Niches 1 was primarily enriched in functions such as “Mitotic Sister Chromatid Segregation,” while Niches 4 was mainly enriched in the “Antigen Receptor-Mediated Signaling Pathway” and “B Cell Receptor Signaling Pathway,” among others (Supplementary Fig. S4B). Hallmark pathway scoring further underscored pronounced stromal activation within both niche1 and niche4. Specifically, niche1 exhibited engagement of established oncogenic pathways, including EMT, while niche4 displayed activation of myogenesis-related programs, consistent with the prevalence of myCAFs in MFM niches (Supplementary Fig S4C). Unbiased clustering identified three transcriptional programs in GC, with PM samples predominantly clustering within an EMT-enriched module (Fig. 7A; Supplementary Fig S4A). These findings imply a pivotal role for EMT within tri-cellular ecological niches. As EMT is largely executed by tumor cells, we employed the NicheNet algorithm to identify fibroblast- and macrophage-derived ligands that most strongly modulate EMT-related gene expression in tumor cells. Fibroblast-secreted ligands, including CCL2, CTGF, PLAU, and DKK1, were predicted to upregulate EMT markers such as VCAN, VIM, and CD44. Similarly, macrophage-derived ligands, notably PLAU, NODAL, INHBA, and WNT5A, were associated with upregulation of VIM and OCD1 in tumor cells (Fig. 7B, C). We also evaluated the contribution of partial EMT (p-EMT), a metastable intermediate state augmenting canonical EMT, within these niches. Cross-compartment cytokine analysis identified several ligands, including PLAU, PTHLH, CTGF, THBS1, and TFF1, within the tri-cellular communicative network (Supplementary Fig S4D, E). Taking the intersection of the aforementioned ligands, PLAU emerged prominently, leading us to identify it as a key cytokine in this multicellular ecosystem. The PLAU-PLAUR axis may hold potential therapeutic value. Subsequently, we used the COMMOT algorithm to assess signaling tendencies within the ST sections. The results suggested spatial flux of EMT signals across different locations in the ST sections, indicating the presence of EMT signal fluctuations within the GC microenvironment in a spatial context (Fig. 7D, Supplementary Fig. S4F). Furthermore, based on this, we further investigated the spatial distribution dynamics of ligands and receptors in the PLAU signaling pathway. The results showed significant spatial co-localization of PLAU-PLAUR in both PT and PM tissues. Referencing ST spatial annotations, this phenomenon was particularly evident within the MFM ecological regions (Fig. 7E; Supplementary Fig. S4G) and coincided with areas dense in EMT signals. In contrast, ligand-receptor interactions were significantly weaker in the surrounding regions (Fig. 7E, Supplementary Fig. S4G). These findings suggest potential for targeting this pathway for GC treatment. Consequently, we proceeded with laboratory experiments, treating GC cells with rh-PLAU. We observed that supplementation with exogenous rh-PLAU enhanced the proliferation, colony formation, and scratch wound healing capacity of GC cells, with effects strengthening as the dose increased (Fig. 7F–I).

A Unbiased clustering revealed 3 distinct programs in GC. The heatmap depicts pairwise correlations of these programs across different spatial transcriptomic datasets. B Heatmap showing the expression of genes associated with tumor cell EMT pathway activation induced by ligands secreted from CAFs, as analyzed by NicheNet. C Heatmap showing the expression of genes associated with tumor cell EMT pathway activation induced by ligands secreted from TAMs, as analyzed by NicheNet. D Interactions inducing EMT-related pathway alterations in spatial transcriptomic sections. Red arrow size indicates signal strength, while arrow direction reflects the trajectory of signal evolution. E Expression and colocalization of the PLAU-uPAR ligand-receptor axis across different GC ST sections. “Exp” denotes the relative expression levels of the ligand (PLAU) and receptor (uPAR); the red gradient represents the colocalization level, which indicates the interaction strength between the ligand and receptor. CCK-8 proliferation curves showing the proliferative capacity of MKN45 cells (F) and HGC27 cells (G) after treatment with exogenous recombinant human PLAU (rh-PLAU). H Colony formation assay showing the clonogenic capacity of MKN45 cells and HGC27 cells after treatment with exogenous rh-PLAU. I Wound healing assay showing the migratory (wound closure) capacity of MKN45 cells and HGC27 cells after treatment with exogenous rh-PLAU.
The incorporation of the deep learning model to highlight the predictive value of the MFM pattern
As mentioned above, there exists a multicellular ecosystem in the GC, where fibroblasts and macrophages gather around tumor cells. This structure effectively prevents the infiltration of T cells, confers a protective niche to tumor cells, and leads to the progression of these cells. Here, we evaluated the impact of different infiltration levels of these three types of cells on the prognosis of GC patients. We used ConsensusClusterPlus to identify the expression signature modules in the transcriptomic data of TCGA-STAD patients. When k = 3, GC patients was clustered into three subtypes: C1, C2, and C3 (Fig. 8A). We used a deconvolution algorithm to map the expression data of ACTA2⁺ myCAFs, DAB2⁺ TAMs, and Mal-epi to bulk RNA-seq data. The results showed that patients in the C3 subgroup had high ACTA2⁺ myCAF infiltration, high DAB2⁺ TAM infiltration, and high tumor cell infiltration, exhibiting typical MFM pattern (Fig. 8A). Subsequently, we performed KM survival analysis on these three groups of patients. The results showed that patients in the C3 subgroup had the worst prognosis (Fig. 8B), which indicates that the MFM pattern may be a potential prognostic biomarker. Next, we evaluated the immune microenvironment characteristics of these three subgroups of patients, retaining only significant results. The findings revealed that the three subgroups classified by the MFM infiltration pattern exhibited distinct TIME characteristics. Specifically, the TIME of patients in the C3 subgroup (which we focused on) tended to be stroma-enriched and immune-excluded. For example, the infiltration of CAFs showed high levels across all algorithms. Meanwhile, this subgroup was accompanied by the enrichment of macrophages, while the infiltration of lymphocytes, including various T cell subsets and NK cells were the lowest. This is consistent with our overall conclusion and suggests that one reason for the poor prognosis of patients with a high MFM infiltration pattern may be immune exhaustion (Supplementary Fig. S5A). Subsequently, pathway enrichment analysis was conducted among patients of high MFM infiltration. Principal observations revealed that individuals with a high MFM infiltration pattern frequently exhibited more distinctive characteristics of EMT, stromal generation, tumor metastasis, and hypoxia (Supplementary Fig. S5B–D). Considering the economic and time costs required for IHC staining, as well as the fact that the quantification of staining intensity varies among individuals, we trained and tested a deep learning model framework to predict the expression of the MFM pattern in H&E-stained images, naming it “Gastric-discovery” (Fig. 8C). The specific experimental workflow is as follows: We selected WSIs from TCGA-STAD patients. For the accurate identification of pathological regions corresponding to the MFM pattern, we invited senior pathologists to perform ROIs annotation on all sections. At least 5 ROI regions were outlined for each section to ensure annotation reliability. Next, we used the FindAllMarkers function in the Seurat package to screen out the highly variable gene set related to the multicellular niche. After inputting this gene set into the bulk RNA-seq data of TCGA-STAD, we calculated the enrichment score for each sample using the ssGSEA algorithm. We defined samples with the top 25% of enrichment scores as the positive group and those with the bottom 25% as the negative group. From these samples, 400 positive image patches and 400 negative image patches were randomly selected to form the training set, and the remaining samples served as the independent validation set. During the model training phase, we conducted transfer learning based on the ResNet-50 model. The model was trained for 100 epochs using the AdamW optimizer with a batch size of 32. An early stopping strategy (patience = 10) was introduced during training: training was stopped when the performance of the validation set did not improve for 10 consecutive epochs, and the model weights with the highest validation accuracy were finally retained. After training, the model showed good overall performance in fitting the training data and generalizing to new data. However, the results of the validation set fluctuated, and there was a certain gap between the performance of the validation set and the training set, indicating that there is still room for optimizing the model’s generalization ability (Fig. 8D, E). The test results showed that the optimal model achieved significant discriminative performance on the validation set (precision = 0.961, specificity = 0.9494, recall = 0.864, F1-score = 0.9099) (Fig. 8F, G). The model exhibited excellent classification performance in distinguishing between high and low MFM patterns and could reliably differentiate between different risk groups (Fig. 8H, I). To further test the accuracy of our Gastric-discovery workflow, we imported pathological sections with ST data into the system. The results showed that the MFM pattern in the ST sections was highly consistent with the MFM pattern identified by our system (Fig. 8J, K).

(A) ConsensusClusterPlus was used to identify expression modules within TCGA-STAD. Matrix clustering was performed with k = 3 (top). Ridgeline plots depict the infiltration levels of ACTA2⁺ myCAFs, DAB2⁺ TAMs, and malignant epithelial cells (Mal-epi) across patient subgroups. B Kaplan–Meier survival curves illustrating prognostic outcomes among different patient subgroups. C Architectural details of the Gastric-Discovery workflow. D, E Trends in loss and accuracy across training epochs for both the training and validation sets. F, G Velocity analysis inferring differentiation trajectories of cancer-associated fibroblast (CAF) subpopulations. Arrows indicate directionality from low-differentiation (start) to high-differentiation (end) states. F Confusion matrix evaluating the binary classification performance (“High Risk” vs. “Low Risk”) of the model. G Heatmap comparing key classification metrics (Accuracy, Precision, Recall, Specificity) during the identification process. H, I Receiver operating characteristic (ROC) curves for the multi-class task, assessing model performance in classifying high and low MFM patterns. J, K Comparison between MFM patterns identified by spatial transcriptomics and those predicted by the Gastric-Discovery deep learning framework.
Discussion
Patients suffering from GC and concomitant peritoneal metastasis harbor an extremely poor overall prognosis. Once peritoneal metastasis occurs, the median overall survival time is typically between 7 and 15 months, with a five-year survival rate of only 2%17. Conventional treatment strategies present only limited efficacy in those patients, primarily as a result of the long-standing neglect of the extensive stromal fibrosis characteristic of peritoneal metastases18. In GCPM, hyper-deposition of the ECM contributes to the formation of a dense physical barrier. At the same time, activated CAFs participate in the formation of a protective shell around the tumor cells through immunosuppression and dysregulated signaling pathways. Consequently, this structural arrangement renders therapies that exclusively target cancer cells largely ineffective and represents a significant biological factor contributing to the poor prognosis of patients with peritoneal metastasis19. To overcome this bottleneck, a paradigm shift in therapeutic strategy is needed, moving away from attacking tumor cells directly to reshaping the TME, with a focused effort on addressing the characteristic problem of matrix fibrosis in peritoneal metastases, this conceptual change completely new directions for the treatment of GCPM5. The present investigation was conducted in alignment with this line of inquiry. In light of the findings, it can be concluded that the fibroblast density, measured in accordance with the stromal scoring device, functions as an independent prognostic factor. Of particular significance is the observation that these fibroblasts tend to congregate with macrophages and aggressive tumor cells, forming a defined spatial structural community which has come to be termed the MFM pattern. It is hypothesized that such a stable three-partite structure might form a functional niche, thereby facilitating tumor invasion and immune evasion20.
In the context of breast cancer, FAP⁺ CAFs fulfill a regulatory function by means of the secretion of FN1, thereby activating the FAK-AKT-STAT3 pathway in macrophages via the Integrin α5β1 axis. This process leads to the induction of M2-like polarization and the reinforcement of an immunosuppressive environment21. In colorectal cancer, a defining feature is the close spatial co-localization of SPP1⁺ TAMs and FAP⁺ CAFs. Chemokines, like TGF-β and IL-1 signaling, drive the cooperative generation of immune-excluding fibrotic stroma by these cells, which physically limits T-cell infiltration and a low response rate to immunotherapy22. In HCC, a predominant focus on CD36⁺ CAFs has emerged, whose secretion of MIF promotes expansion of MDSCs, thereby suppressing T-cell activity. For instance, the targeting of CD36 with ASOs has been demonstrated to enhance the efficacy of immunotherapeutic interventions23. Beyond cytokine signaling and pathway crosstalk, TAMs and CAFs have been observed to attract each other through direct cellular reprogramming or metabolic interplay. Regardless of the route, the coexistence of these cells typically results in the formation of stromal structures that resemble barriers around tumors that not only protect malignant cells from immune attack but also amplify oncogenic signaling through cellular feedback24,25. The present study lends support to these concepts in relation to GC. Stromal bands composed of macrophages and fibroblasts were identified in both primary and peritoneal metastases, when lie on the microscopic level, the focus was on myofibroblasts and DAB2-rich TAMs as dominant cellular players. Contrary to the findings of previous studies, which examined these populations in isolation, emphasis is placed on the integrated ecosystem formed by macrophages, fibroblasts and tumor cells. This structure is not merely a spatial landscape, but rather a cooperative cellular alliance built from specific spatial relationships. The formation of such units appears to be crucial for shaping the pre- and post-metastatic microenvironment and promoting resistance to conventional therapies20.
Changes in intracellular signaling within the TME are likely to be the primary drivers of this stability phenomenon, whereby signaling molecules representing a primary underlying mechanism constitute the most common form of communication between cells26. Our goal is to identify signaling molecules that are secreted jointly by CAFs and TAMs and selectively target tumor cells in order to optimize translational therapeutic value. Concentrating on p-EMT and EMT, we have further clarified our investigation on the PLAU and previous extensive research have supported that the PLAU-PLAUR pathway fulfills a crucial role in tumor invasion and metastatic spread27. The PLAU-PLAUR interplay has been observed to demonstrate high avidity and specificity, resulting in the effective activation of the plasminogen system and the subsequent initiation of multiple intracellular signaling responses28. Currently, in our research, this axis not only functions as a central signaling pathway mediating tumor invasion and ECM remodeling in GCPM but is also intimately integrated with the aforementioned MFM pattern crosstalk network. Its translational potential offers a means to overcome the limitations of current therapeutic strategies29. For angiogenesis, GCPM neovascularization relies on VEGF and other factors secreted by CAFs and TAMs, with amplification via this axis. This explains the limited efficacy of current anti-angiogenic agents in subsets of patients, due to these agents target only the factors, not the PLAU-PLAUR axis and its mediated ECM degradation cascade, thus failing to fully abrogate core drivers of neovascularization30,31.
The fundamental strength of NIPS therapy lies in its remarkable capacity to augment the concentration of drug in the peritoneal space. This enhanced effect is attributable to the synergistic interaction between intraperitoneal administration of paclitaxel and oral S-1. However, the dense ECM structure of peritoneal metastases affects drug penetration, thereby reducing the efficacy of the drug on deep tissue tumor targets. In the future, the combination of a PLAU-PLAUR axis inhibitor with NIPS therapy is expected to enhance the effectiveness of the latter in the treatment of peritoneal metastases. Furthermore, the continuous advancement of multi-omics technologies is resulting in PLAUR acquiring novel biological significance in this context. For instance, in the context of HCC, PLAUR⁺ neutrophils have been observed to be particularly abundant among patients who demonstrate non-responsiveness to anti-PD-1 therapy. Such patients exhibit a suppression of CD8⁺ T-cell function through the secretion of immunosuppressive factors, including ARG1 and IL-10, thereby facilitating immunotherapy resistance32. Conversely, PLAUR monoclonal antibodies have been shown to competitively impede uPA from binding to uPAR, thereby inducing ADCC/CDC effects that culminate in the destruction of tumor cells. Consequently, direct intervention in this axis may be highly relevant for the treatment of GCPM33.
This structural protective biologic impact of the microenvironment translates into prognostic challenges in GC patients. Based on data from TCGA and several other cohorts of GC patients, we have found that patients with both ACTA2⁺ myCAF and DAB2⁺ TAM-heavily infiltrated solid tumors carry the worst prognosis, possibly due to the highly aggressive and immunosuppressive effects of these cells in the armor-like niche. Moreover, preliminary studies in our laboratory have demonstrated that intervention in the PLAU axis effectively inhibits the viability of GC cells. In view of the cumbersome nature of current clinical-pathological work and the fact that IHC staining is highly subjective, we thus developed the Gastric-discovery system, which employs the colocalized surrounding areas of these three cell types in combination with deep learning technology as a novel biomarker. The system enables accurate prediction of the prognosis of GC patients by quantifying the characteristics of the distribution of the three cells in routinely HE-stained sections It is hoped that this approach could bridge the gap between research into the mechanisms of the tumor microenvironment and clinical prognosis assessment.
In developing the Gastric-discovery system, we considered the balance between image recognition accuracy and computational efficiency, particularly the problem of gradient vanishing in deep neural networks, and we chose the classic ResNet-50 model as the core basis of the system. Compared to other models such as VGG-16, ResNet-50 enables a significant reduction in the number of parameters through residual connections in a deeper network structure. Although there have been many advances in emerging imaging analysis models, such as EfficientNet, which achieves a better balance between performance and efficiency by scaling composite models, and ViT, which introduces the Transformer architecture to the field of image processing, the mature ecosystem of ResNet-50 strengthened our confidence in choosing it as the underlying framework for the Gastric-discovery system. Overall, the present study reveals the spatial localization characteristics and cellular interaction information between macrophages, fibroblasts and tumor cells. Drawing conclusions from these findings, the study establishes promising clinical prognostic indicators. These endeavors have the potential to offer novel avenues for the enhancement of patient prognoses.
Methods
Data retrieval and acquisition
The Cancer Genome Atlas-Stomach Adenocarcinoma (TCGA-STAD) sequencing data utilized in this study were retrieved from the public domain via the TCGA database (https://portal.gdc.cancer.gov/). Concurrently, whole-slide images (WSIs) were acquired from the same repository for deep learning model training. For supplementary analyses, gene expression data corresponding to datasets GSE84426, GSE62254, GSE57303, GSE15459, GSE14208, GSE29272, GSE34942, GSE84437, GSE84433, GSE26899, GSE26901, GSE38749, GSE28541, and GSE26253 were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). Public scRNA-seq datasets included in this study were curated from multiple repositories to construct the final research cohort. Specifically, this cohort comprised different scRNA-seq samples: HRA004702, GSE183904, GSE163558, and GSE308231. Spatial transcriptomics data used herein were obtained from publicly accessible datasets within GSE251950.
Single-cell data processing
The R package Seurat (v4.4.0) was employed for downstream processing and analysis of scRNA-seq data. First, to mitigate batch effects and confounding influences of individual patient-specific variation, the Harmony algorithm was applied to integrate the scRNA-seq datasets. For QC, cells were filtered out by default if they met any of the following criteria: <5000 unique molecular identifiers (UMIs), >15% mitochondrial gene content, or <250 expressed genes. Only genes detected in at least 3 cells were retained for subsequent analyses. Additionally, the QC-passed data were evaluated using the DoubletFinder (v2.0.3) algorithm to eliminate doublets. Finally, uniform manifold approximation and projection (UMAP) was applied for dimensionality reduction and visualization of cell clusters. The annotation and identification of cell types in this study were based on known classical markers, and detailed information about these gene markers is described below. The FindAllMarkers function in the Seurat package was used for differential expression analysis between cell clusters to identify specific marker genes for each cell cluster. Genes with adj. p < 0.05 were considered statistically significant differentially expressed genes. Genes with an avg_log2FC > 0.25 were regarded as having sufficient expression differences. In addition, each gene should be expressed in at least 10% of the cells in the cluster to be identified (min.pct = 0.1), and its expression proportion in the control cluster cells should be lower than 10% or its expression level should be lower. The identification of malignant cells was performed using the inferCNV algorithm to infer copy number variation (CNV) profiles and identify tumor-specific chromosomal alterations. Velocyto was used to infer cell differentiation of different cell clusters, and to deduce developmental trajectories and differentiation states.
Functional enrichment analysis and immune infiltration analysis
DEGs that were upregulated in each cell subset were identified using the ‘FindAllMarkers‘ function in Seurat, and these DEGs were subjected to functional enrichment analysis. The R package clusterProfiler was employed for this enrichment analysis, with pathways exhibiting a false discovery rate (FDR) < 0.05 deemed significantly enriched. Fifty key cancer hallmark gene sets utilized in the enrichment analysis were obtained from the MSigDB. The R package irGSEA was utilized to compute UCell scores for samples analyzed via scRNA-seq and ST-RNA. For TCGA expression profile data, the R package ESTIMATE was used to estimate the abundance of tumor stromal and immune cells, along with tumor purity.
Cell type distribution bias across samples
The R package STARTRAC was employed to compute the Ro/e index (ratio of observed to expected cell numbers), which was used to assess the relationship between cell types and tissues, alongside cell type distribution biases. Ro/e > 1 indicates that the cell subset exhibits a higher distribution frequency in a specific tissue than would be expected by random chance, reflecting enrichment of the subset in that tissue. Conversely, Ro/e < 1 indicates that the subset is depleted in the target tissue.
Spatial transcriptomics data processing and Niche partitioning
Initially, the R package stCancer was used for quality control and normalization of the downloaded spatial transcriptomics data. A standardization and integration process based on SCTransform was adopted: each sample was first independently standardized by SCTransform34, and then 2000 highly variable genes were used as anchors to integrate multiple samples through the CCA-based FindIntegrationAnchors and IntegrateData functions to eliminate batch effects, resulting in an integrated Seurat object. PCA analysis was performed on the integrated data, and graph clustering was conducted based on the PCA results. To determine the optimal clustering resolution, the Clustree tool was used to visualize the clustering stability at different resolutions (0.2 to 1.2). To identify the cell types of spatial transcriptomics spots, cell2location was used to train cell type feature signatures from paired single-cell data, which were then applied to spatial data for deconvolution to obtain the absolute abundance of each cell type35. The integrated ST samples were then subjected to hierarchical clustering, further generating 8 categories, which we labeled as “niches”. Squidpy was used to construct a spatial adjacency network, and the sq.gr.nhood_enrichment function was used to calculate the enrichment scores of each cell type at the 1-ring, 2-ring, and 3-ring neighborhood scales. This score evaluates whether the degree of spatial proximity between two cell types is higher than randomly expected. Using the MistyR package, we established a multi-view spatial model. Specifically, the cell abundance matrix was input into the MistyR framework to build an intracellular view and an intercellular view, so as to quantify the contribution of cell intrinsic composition and local microenvironment interactions to specific molecular phenotypes.
CellTrek co-localization analysis
Single-cell-level spatial co-localization was conducted utilizing the CelltrekR package36. First, according to the single-cell file with completed subpopulation annotation, the traint function was used to integrate single-cell RNA data and spatial transcriptomics data, and the celltrek function was used for co-embedding analysis. Then, the scoloc method was used to calculate the spatial co-localization map between cell types, and finally a minimum spanning tree (MST) was generated to reveal the spatial relationship between cell populations.
Cell-Cell communication analysis
NicheNet was used to analyze cell-cell communication in the study. Specifically, precurated ligand-receptor pairing information, signaling pathway data, and gene regulatory network data were integrated to construct ligand-signaling networks and gene regulatory networks. The Personalized PageRank algorithm was employed to compute the influence coefficients between ligands (derived from macrophages, tumor cells, and fibroblasts) and downstream signaling molecules, as well as target genes. COMMOT was used here as a method to infer ligand-receptor cell communication in spatial transcriptomics37, Specifically, the data were first preprocessed, with the expression matrix normalized and transformed, followed by further filtering of ligands and receptors from the CellChatDB ligand-receptor database. Spatial communication inference was then performed on the filtered ligand-receptor pairs with statistical significance, with the spatial distance limit set to 500μm.
Consensus non-negative matrix factorization (cNMF) analysis
Consensus non-negative matrix factorization (cNMF) was used to identify potential expression programs in each set of 10 tumor samples38. By systematically testing k values from 3 to 12, and based on the stability of the consensus matrix and the inflection point of reconstruction error, k = 6 was finally determined as the optimal number of clusters. After calculating the Pearson correlation coefficients of these programs, they were classified into corresponding meta-programs. To conduct functional annotation of the expression programs identified by cNMF, we screened the genes that contributed the most to each program. Specifically, we sorted the genes according to the gene weights generated by cNMF, and selected the top 30 genes with the highest weights in each program as the signature genes of that program, which were used for subsequent gene set enrichment analysis.
Data preparation and preprocessing for deep learning framework
A total of 376 hematoxylin-eosin (H&E)-stained whole-slide images (WSIs) from TCGA-STAD patients were included in this study, and all images were obtained from the TCGA platform. The scanning magnification of WSIs was extracted and verified using Python’s openslide package. The annotation work of the mentioned WSIs was completed under the guidance of at least two experienced pathologists. They made judgments based on the World Health Organization classification criteria and cell morphological and tissue structural characteristics. First, typical tumor regions and normal epithelial regions were identified under a low-power microscope. Subsequently, the cellular and tissue structural characteristics of the above regions were confirmed under a high-power microscope, and digital pathology software was used to accurately outline these regions of interest (ROIs). For controversial regions, the two pathologists discussed together until a consensus was reached; if still undetermined, the region was excluded from the study and not used for subsequent image cutting and model training. Through the above process, we outlined ROIs containing the morphological structure of “MFM” infiltration pattern. Then, within these ROI regions, we cut and uniformly cropped the images into 224 × 224-pixel image patches, with at least 5 image patches generated from each WSI. To obtain the category labels of the image patches, we performed feature analysis and label assignment at the gene level. With the help of the FindAllMarkers function in the Seurat package, we identified differentially expressed genes related to the “MFM” infiltration pattern. This gene set was input into the RNA-seq expression matrix of the TCGA-STAD cohort, and the enrichment score of each sample was calculated using the ssGSEA algorithm. Each patient sample was divided into different MFM infiltration pattern categories according to the median of this score or other thresholds. Finally, all image patches extracted from each patient’s WSI inherited the category label of their corresponding sample, which were used for subsequent model training.
Training and deployment of deep learning framework
We input the preprocessed and annotated image patches into the ResNet-50 model for image classification training. Torchvision was used to build a data loading pipeline, which included image preprocessing steps such as normalization and data augmentation steps. We conducted 100 training epochs with a batch size set to 32. In each epoch, the AdamW optimizer was used for parameter updates. After each training epoch, we evaluated the model performance on the reserved validation set, calculated and output the training loss, validation loss, training accuracy, and validation accuracy. We adopted an early stopping strategy: if the validation accuracy did not improve for 10 consecutive epochs, the training was terminated, and the model weights with the best performance on the validation set were saved. Based on the obtained optimal ResNet-50 model weights, we developed a clinically practical UI using the Streamlit framework. The entire training process was completed on an NVIDIA GeForce RTX 4090 GPU.
Cell proliferation, colony formation, and scratch assays
The MKN-45 and HGC-27 GC cell lines were obtained from the Cell Bank of the Chinese Academy of Sciences. The cells were maintained under standard culture conditions. Cells in the logarithmic growth phase were seeded into 96-, 24-, and 12-well plates. After 24 h, recombinant human PLAU (rh-PLAU) was administered at concentrations of 2 ng/mL and 10 ng/mL. Subsequently, functional assays, including CCK-8 spectrophotometry, colony formation, and scratch wound healing, were performed. The absorbance at 450 nm was measured daily for a period of six days. For the colony formation assay, cells were fixed with paraformaldehyde and stained with Giemsa after 14 days. Scratch closure was monitored at two time points: 0 h and 24 h. All experiments were conducted in biological triplicate.
Multiplex immunofluorescence
Immunofluorescence experiments were performed on samples of GC primary tumors and peritoneal metastatic lesions. Following deparaffinization, tissue sections underwent antigen retrieval through heat-mediated epitope unmasking and subsequent blocking procedures. Permeabilization was accomplished using TBST, whereupon sections were incubated overnight at 4 °C with a panel of primary antibodies specific to Pan-CK, CD68, CD8, DAB2, FAP, and α-SMA. Species-appropriate secondary antibodies conjugated with distinct fluorophores were then applied in sequential order. Finally, nuclei were counterstained with DAPI prior to cover-slipping. This study was approved by the Ethics Committee of Ruijin Hospital, Shanghai Jiao Tong University School of Medicine (2021-336) and conducted in strict compliance with the ethical principles of the World Medical Association’s Declaration of Helsinki.
Statistical analysis
Current research was carried by R software (version 4.2.1) for all statistical analyses. For data that followed a normal distribution, we used the Student’s t-test to compare two groups. For data that did not follow a normal distribution, we used the Wilcoxon rank-sum test. When comparing multiple groups, one-way ANOVA was used for normally distributed data and the Kruskal-Wallis test for non-normally distributed data.
The relationship between continuous variables was assessed using Spearman’s rank correlation. Kaplan–Meier analysis was applied to show survival outcomes and the log-rank test to compare them. A p-value less than 0.05 was considered statistically significant.
Data availability
All datasets involved in this study have been made publicly available. The TCGA-STAD sequencing data used in this article can be accessed openly via the TCGA database. Datasets with accession numbers GSE84426, GSE62254, GSE57303, GSE15459, GSE14208, GSE29272, GSE34942, GSE84437, GSE84433, GSE26899, GSE26901, GSE38749, GSE28541, and GSE26253 are openly available through the GEO database. The single-cell RNA sequencing (scRNA-seq) data were obtained from the GEO database (accession numbers: GSE183904, GSE163558, GSE308231) and the GSA database (accession number: HRA004702). GSE308231 secure token: ohcvkuuglrctvip. The spatial transcriptomics data involved in this study were derived from the publicly available spatial transcriptomics information in GEO accession GSE251950. All data used in the study are fully accessible within this article.
Code availability
The analytical codes involved in this study can be obtained by contacting the corresponding author upon reasonable request.
References
Foster, J. M., Zhang, C., Rehman, S., Sharma, P. & Alexander, H. R. The contemporary management of peritoneal metastasis: a journey from the cold past of treatment futility to a warm present and a bright future. CA Cancer J. Clin. 73, 49–71 https://doi.org/10.3322/caac.21749 (2023).
Guchelaar, N. A. D. et al. Intraperitoneal chemotherapy for unresectable peritoneal surface malignancies. Drugs 83, 159–180 https://doi.org/10.1007/s40265-022-01828-7 (2023).
Adil, M. et al. Extensive intraoperative peritoneal lavage with surgery, compared to surgery alone, for patients diagnosed with gastric cancer: a systematic review and meta-analysis of randomized controlled trials. Int J. Surg. 110, 8073–8082 https://doi.org/10.1097/js9.0000000000002090 (2024).
Yasuda, T. & Wang, Y. A. Gastric cancer immunosuppressive microenvironment heterogeneity: implications for therapy development. Trends Cancer 10, 627–642 https://doi.org/10.1016/j.trecan.2024.03.008 (2024).
Rietveld, P. C. S. et al. A clinical pharmacological perspective on intraperitoneal chemotherapy. Drugs 85, 931–943 https://doi.org/10.1007/s40265-025-02195-9 (2025).
Zhu, Z. et al. Asian consensus on normothermic intraperitoneal and systemic treatment for gastric cancer with peritoneal metastasis. Gastric Cancer 28, 731–748 https://doi.org/10.1007/s10120-025-01631-9 (2025).
Gwee, Y. X. et al. Integration of genomic biology into therapeutic strategies of gastric cancer peritoneal metastasis. J. Clin. Oncol. 40, 2830 https://doi.org/10.1200/jco.21.02745 (2022).
Smyth, E. C. et al. FGFR2b protein overexpression: an emerging biomarker in gastric and gastroesophageal junction adenocarcinoma. Cancer Treat. Rev. 139, 102971 https://doi.org/10.1016/j.ctrv.2025.102971 (2025).
Ramos, C., Gerakopoulos, V. & Oehler, R. Metastasis-associated fibroblasts in peritoneal surface malignancies. Br. J. Cancer 131, 407–419 https://doi.org/10.1038/s41416-024-02717-4 (2024).
Dadgar, N. et al. Targeting interleukin-6 as a treatment approach for peritoneal carcinomatosis. J. Transl. Med. 22, 402 https://doi.org/10.1186/s12967-024-05205-8 (2024).
Huang, Y., Zhang, M. & Gao, Q. Mapping the tumor immune landscape: single-cell RNA sequencing in cancer immunotherapy. Cancer Lett. 633, 218012 https://doi.org/10.1016/j.canlet.2025.218012 (2025).
Zhu, L. & Wang, J. Quantifying landscape and flux from single-cell omics: unraveling the physical mechanisms of cell function. JACS Au 5, 3738–3757 https://doi.org/10.1021/jacsau.5c00620 (2025).
Kumar, V. et al. Single-cell Atlas of lineage states, tumor microenvironment, and subtype-specific expression programs in gastric cancer. Cancer Discov. 12, 670–691 https://doi.org/10.1158/2159-8290.Cd-21-0683 (2022).
Zhang, Y. & Liu, Z. Insights into the mechanisms of immune-checkpoint inhibitors gained from spatiotemporal dynamics of the tumor microenvironment. Adv. Sci. e08692. https://doi.org/10.1002/advs.202508692 (2025).
Li, Z., Wang, J., Wang, Z. & Xu, Y. Towards an optimal model for gastric cancer peritoneal metastasis: current challenges and future directions. EBioMedicine 92, 104601 https://doi.org/10.1016/j.ebiom.2023.104601 (2023).
Zhang, J., Che, Y., Liu, R., Wang, Z. & Liu, W. Deep learning-driven multi-omics analysis: enhancing cancer diagnostics and therapeutics. Brief Bioinform. 26, https://doi.org/10.1093/bib/bbaf440 (2025).
Kim, H. I. & Badgwell, B. D. Peritoneal oligometastasis in gastric cancer: diagnostic strategies, patient selection, and emerging therapeutic approaches. J. Gastric Cancer 25, 409–423 https://doi.org/10.5230/jgc.2025.25.e36 (2025).
Li, G. Z., Doherty, G. M. & Wang, J. Surgical management of gastric cancer: a review. JAMA Surg. 157, 446–454 https://doi.org/10.1001/jamasurg.2022.0182 (2022).
Cortés-Guiral, D. et al. Primary and metastatic peritoneal surface malignancies. Nat. Rev. Dis. Prim. 7, 91 https://doi.org/10.1038/s41572-021-00326-6 (2021).
Sun, H., Wang, X., Wang, X., Xu, M. & Sheng, W. The role of cancer-associated fibroblasts in tumorigenesis of gastric cancer. Cell Death Dis. 13, 874 https://doi.org/10.1038/s41419-022-05320-8 (2022).
Chen, W. et al. Fibroblast Activation Protein (FAP)(+) cancer-associated fibroblasts induce macrophage M2-like polarization via the Fibronectin 1-Integrin α5β1 axis in breast cancer. Oncogene 44, 2396–2412 https://doi.org/10.1038/s41388-025-03359-3 (2025).
Qi, J. et al. Single-cell and spatial analysis reveal interaction of FAP(+) fibroblasts and SPP1(+) macrophages in colorectal cancer. Nat. Commun. 13, 1742 https://doi.org/10.1038/s41467-022-29366-6 (2022).
Zhu, G. Q. et al. CD36(+) cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 9, 25 https://doi.org/10.1038/s41421-023-00529-z (2023).
Long, F. et al. DAB2 (+) macrophages support FAP (+) fibroblasts in shaping tumor barrier and inducing poor clinical outcomes in liver cancer. Theranostics 14, 4822–4843 https://doi.org/10.7150/thno.99046 (2024).
Wang, H. et al. POSTN(+) cancer-associated fibroblasts determine the efficacy of immunotherapy in hepatocellular carcinoma. J. Immunother. Cancer 12, https://doi.org/10.1136/jitc-2023-008721(2024).
Zavros, Y. & Merchant, J. L. The immune microenvironment in gastric adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 19, 451–467 https://doi.org/10.1038/s41575-022-00591-0 (2022).
Kisling, S. G. et al. Implications of prognosis-associated genes in pancreatic tumor metastasis: lessons from global studies in bioinformatics. Cancer Metastasis Rev. 40, 721–738 https://doi.org/10.1007/s10555-021-09991-1 (2021).
Wan, L. et al. Hypoxia-induced tumor cell-intrinsic PLAU activation drives immunotherapy resistance in collagenic lung adenocarcinoma. Int Immunopharmacol. 162, 115161 https://doi.org/10.1016/j.intimp.2025.115161 (2025).
Ferreira, G. M. et al. N-Glycosylation as a key requirement for the positive interaction of integrin and uPAR in glioblastoma. Int. J. Mol. Sci. 26. https://doi.org/10.3390/ijms26115310 (2025).
Liu, L. et al. Revealing the role of cancer-associated fibroblast senescence in prognosis and immune landscape in pancreatic cancer. iScience 28, 111612 https://doi.org/10.1016/j.isci.2024.111612 (2025).
Sun, M. et al. Cuproptosis-related lncRNA JPX regulates malignant cell behavior and epithelial-immune interaction in head and neck squamous cell carcinoma via miR-193b-3p/PLAU axis. Int. J. Oral. Sci. 16, 63 https://doi.org/10.1038/s41368-024-00314-y (2024).
Liu, S. et al. PLAUR(+) Neutrophils Drive Anti-PD-1 therapy resistance in patients with hepatocellular carcinoma by shaping an immunosuppressive microenvironment. Adv. Sci. e07167. https://doi.org/10.1002/advs.202507167 (2025).
Qin, L. et al. Therapeutic strategies targeting uPAR potentiate anti-PD-1 efficacy in diffuse-type gastric cancer. Sci. Adv. 8, eabn3774 https://doi.org/10.1126/sciadv.abn3774 (2022).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, e1821 https://doi.org/10.1016/j.cell.2019.05.031 (2019).
Kleshchevnikov, V. et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol. 40, 661–671 https://doi.org/10.1038/s41587-021-01139-4 (2022).
Wei, R. et al. Spatial charting of single-cell transcriptomes in tissues. Nat. Biotechnol. 40, 1190–1199 https://doi.org/10.1038/s41587-022-01233-1 (2022).
Cang, Z. et al. Screening cell-cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218–228 https://doi.org/10.1038/s41592-022-01728-4 (2023).
Kotliar, D. et al. Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. Elife 8, https://doi.org/10.7554/eLife.43803 (2019).
Acknowledgements
This work was supported by the National Natural Sciences Foundation of China (82273126). We express our gratitude to all those who provided consultation for this study.
Author information
Authors and Affiliations
Contributions
Q.W., Y.N., and S.L. contributed equally to this work. Q.W. performed data analysis and figure preparation. Q.W., Y.N., S.L., and B.Z. contributed to data collection and figure preparation. Y.Z., J.J., C.Y., and F.Q. conceptualized the study and revised the manuscript. Q.C. and J.J. edited the manuscript and revised the manuscript. M.S. and J.Z. supervised the project. All authors contributed to the article and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Q., Ni, Y., Lu, S. et al. Multi-dimensional omics integrated machine learning framework identifies macrophage-fibroblast-tumor co-infiltration patterns to predict prognosis in gastric cancer. npj Digit. Med. 9, 10 (2026). https://doi.org/10.1038/s41746-025-02179-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41746-025-02179-9
