
Abstract
Some patients with polycystic ovarian syndrome (PCOS) suffered from metabolic syndrome (MetS) including dyslipidemia, hyperinsulinism, but the underlying mechanism is unclear. Although C-terminal Binding Protein 1 (CTBP1) is a transcriptional co-repressor frequently involved in hormone secretion disorders and MetS-associated diseases, the role of CTBP1 in PCOS is rarely reported. In the present study, we found that CTBP1 expression was significantly elevated in primary granulosa cells (pGCs) derived from the PCOS with MetS patients and was positively associated with serum triglyceride, but negatively correlated with serum estradiol (E2) or high-density lipoprotein. Mechanistic study suggested that CTBP1 physically bound to the promoter II of cytochrome P450 family 19 subfamily A member 1 (CYP19A1) to inhibit the aromatase gene transcription and expression, resulting in the reduced E2 synthesis. Moreover, CTBP1 interacted with the phosphorylated SREBP1a at S396 in nuclei, leading to the FBXW7-dependent protein degradation, resulting in the reduced lipid droplets formation in pGCs. Therefore, we conclude that CTBP1 in GCs dysregulates the synthesis of steroid hormones and lipids through suppression of aromatase expression and promotion of SREBP1a protein degradation in PCOS patients, which may offer some fresh insights into the potential pathological mechanism for this tough disease.
Similar content being viewed by others


Introduction
Polycystic ovary syndrome (PCOS) is clinically characterized by hyperandrogenism, polycystic ovary, long-term anovulation, and partially accompanied with dyslipidaemia, hyperinsulinism, and hormone secretion disorders1,2. PCOS also shares multiple features with metabolic syndrome (MetS) in terms of clinical symptoms such as insulin resistance, abdominal obesity, and dyslipidaemia. Patients with both PCOS and MetS may account up to 50% depending on ethnicity3. In a meta-analysis, MetS prevalence is approximately 2-fold higher in females with PCOS than in those without4, implying a reciprocal pathophysiologic relationship between PCOS and MetS5,6. Currently, the pathophysiology of PCOS still remains poorly understood, but accumulating evidence shows that PCOS is mainly resulted from the abnormalities of ovarian follicles and the dysfunctions of granulosa cells (GCs)7. The extra-ovarian factors Follicle-stimulating hormone (FSH) and luteinizing hormone (LH) stimulate the expression of the aromatase gene CYP19A1 (cytochrome P450 family 19 subfamily A member 1), which converts androgens into estrogen to generate estradiol (E2) in GCs. The pre-antral follicle is overgrown in PCOS patients due to the excessive intrinsic androgen that makes GCs hypersensitive to FSH, leading to anti-Mullerian hormone (AMH) overexpression7, whereas the premature arrest of follicle growth may be caused by the premature response of GCs (from a proportion of small antral follicles) to LH in terms of E2 synthesis8. Moreover, the intra-ovarian factor epidermal growth factor (EGF), a major member of the EGF receptor ligands, mediates the LH signaling by inducing oocyte maturation and cumulus expansion in various mammalian species including humans9. This signaling network may be disrupted in the follicles of females with PCOS10, which results in the elevated serum and follicular fluid (FF) levels of EGF in the patients11, whereas the hyperactivity of EGF may also block the stimulation of aromatase in ovarian GCs12. Thus, FSH, LH, and EGF are closely associated with PCOS.
The C terminal-binding protein (CTBP) family, consisting of two isoforms CTBP1 and CTBP2, plays a crucial role in cell differentiation, apoptosis, oncogenesis, and development13. In human cells, both isoforms share a highly conserved protein structure (78% homology) with similar functions including carbohydrate metabolism and epigenetic regulation of multiple transcription factors14. CTBP1 binds to the C-terminus of adenovirus E1A protein and acts as a transcription repressor15. Depletion of CTBP1 in the xenograft tumor with MetS derived from androgen-insensitive prostate cancer cells induces the expression of aromatase16. The elevated androgen inhibits CTBP1 expression in androgen receptor positive but not negative prostate cancer cells17. The EGF-induced micropinocytosis is associated with CTBP118. Located in the antisense region of CTBP1, lncRNA CTBP1-AS directly suppresses CTBP1 expression by recruiting the RNA-binding transcriptional repressor PSF together with histone deacetylases to abate the transcriptional repression activity of CTBP117. Because of highly expressed in the peripheral blood cells of patients with PCOS, CTBP1-AS is considered as a risk factor for the occurrence of PCOS19,20. While high expression of CTBP1 is associated with metabolic abnormalities and steroid hormone sensitivities in prostate and breast cancers21,22, PCOS is also a well-known disease highly involved in metabolic abnormalities and hormone secretion disorders. Therefore, the expression and the function of CTBP1 in GCs of PCOS patients with or without MetS may be highly worthy of investigations.
In this study, by analysis of clinical data and in vitro study, we found that the elevated CTBP1 expression in pGCs was positively associated with the occurrence of PCOS with MetS. CTBP1 not only repressed the aromatase expression and E2 synthesis, but also facilitated the ubiquitin-mediated degradation of the phosphorylated SREBP1a at S396 to inhibit the intracellular lipid accumulation and the expression of ACC1 and FASN. Thus, CTBP1 may link metabolic syndrome to PCOS to dysregulate estrogen and lipid synthesis through suppressing CYP19A1 transcription and enhancing SREBP1a degradation in cell nuclei.
Results
Decreased blastocyst formation rate in PCOS patients with MetS
Based on the analysis of the demographics, baseline profiles, and IVF-ET laboratory indicators between 47 PCOS patients and 49 eligible control subjects (Supplementary Table 1), we first found that testosterone (T), LH, LH/FSH, AMH, and MetS-associated indicators including body mass index (BMI) and low-density lipoprotein (LDL) were higher, but FSH was lower in PCOS patients than in control ones. By subdividing both PCOS patients and control subjects into four subgroups including controls without (A) or with (B) MetS and PCOS patients without (C) or with (D) MetS based on IDF criteria (Table 1), we further found that MetS-associated profiles including BMI, FBG, SBP, DBP, and TG were higher, but HDL was lower in control patients with MetS than in those without MetS, whereas the mean ages were insignificant among the four subgroups. Significant differences of BMI and TG were found among all groups, indicating that BMI and TG levels are most frequently altered variates, which may be potentially useful for targeted treatment of PCOS and prediction of pregnancy outcomes. Moreover, significant differences of T, FSH, LH/FSH, AMH, were found between A and C or A and D patients. HDL levels were significantly different between A and B, A and D, or C and D patients. However, no statistical differences of high qualified embryo rate were found between the comparable patients. A statistical difference of blastocyst formation rate was barely observed between the control patients without MetS (A) and the PCOS patients with MetS (D, p = 0.043) (Table 1), which may be attributed to the smaller sample size. These data suggest that among the MetS-associated markers, BMI, TG and HDL are most important factors contributing to PCOS, although they do not highly affect the number of mature oocytes rate, the rates of fertilization, high qualified embryo and blastocyst formation.
Association of CTBP1 with abnormal hormones and lipids
As aforementioned, CTBP1 may be associated with PCOS and MetS. To examine the role of CTBP1 in GCs, we first collected primary GCs (pGCs) from PCOS and control patients and evaluated their CTBP1expression levels. The results showed that CTBP1 mRNA was significantly higher in pGCs of PCOS patients than in those of control patients (Fig. 1A), which was supported by detection of CTBP1 expression using Western blot in 3 pGCs samples of PCOS in comparison with 3 samples of non-PCOS (Fig. 1B). Moreover, CTBP1 was also higher in pGCs of the patients with MetS than in those of patients without MetS (Fig. 1C). Among PCOS and control patients with or without MetS, the mean mRNA level of CTBP1 was the highest in PCOS patients with MetS but was the lowest in control patients without MetS (Fig. 1D). Next, we analyzed the correlation between CTBP1 expression and hormones, IVF-ET laboratory indicators, or metabolic factors, and found that CTBP1 was negatively associated with LH and E2 only in PCOS patients (Fig. 1E, F), but positively or negatively associated with TG or HDL in both control and PCOS patients (Fig. 1G, H). No statistical significance was found between CTBP1 and BMI, T, AMH, TC, FSH, LDL, high qualified embryo rate, or blastocyst formation rate in both control and PCOS patients (Supplementary Fig. 1). These data suggest that high expression of CTBP1 is considerably associated with hormonal and lipid abnormalities in granulosa cells of PCOS patients, and may therefore indicate an important role of CTBP1 in PCOS development.

A Relative levels of CTBP1 mRNA detected by real-time PCR in primary granulosa cells (pGCs) from controls (N = 49) and PCOS (N = 47) patients. B CTBP1 proteins detected by western blot in pGCs from control (N1, N2, N3) and PCOS patients (P1, P2, P3). C, D CTBP1 mRNA levels detected by real-time PCR in pGCs from females without MetS (N = 63) and with MetS (N = 33) (C), or from control patients with or without MetS (N = 49) and PCOS patients with or without MetS (N = 47) (D). GAPDH is used as an internal control. E Correlation between CTBP1 and serum basal LH in all control and PCOS patients. F Correlation between CTBP1 and serum basal E2 in control and PCOS patients. G. Correlation between CTBP1 and TG in control and PCOS patients. H Correlation between CTBP1 and HDL in control and PCOS patients. Quantification data are presented as mean ± SD and analyzed using two-tailed t-test (A, C, D). Spearman correlation is used for continuously variable data (E–H). **p < 0.05, **p < 0.01, ***p < 0.001 are considered significant. NS not significant. MetS metabolic syndrome, PCOS polycystic ovary syndrome.
High CTBP1 expression is an independent risk factor for PCOS with MetS
Because CTBP1 was highly expressed in pGCs of PCOS patients with MetS, we investigated whether the high expression of CTBP1 is correlated with PCOS and MetS occurrence. By using multivariable logistic regression, we first analyzed the data of PCOS patients to seek independent predictors for PCOS (Supplementary Table 2). In Model 1 (M1), we found that high expression of CTBP1 was statistically correlated with PCOS incidents (p = 0.010) under the adjustments of age and BMI. In Models 2 (M2) and 3 (M3), high expression of CTBP1 is still an independent risk factor for PCOS occurrence under further adjustments of hormonal disturbance and diagnosed MetS (p = 0.001). Moreover, the correlations were also found between PCOS incidents and high levels of T (p = 0.005, p = 0.01) and AMH (p = 0.002, p = 0.002) in M2 and M3. The sensitivity, specificity, AUC and p values of above models were enumerated in Supplementary Table 3. We then used the same statistical method to seek independent predictors for MetS from both PCOS and control patients, and found that high levels of TG, FBG, and BMI, but low level of HDL were significantly associated with MetS incidents as indicated in Supplementary Tables 4, 5.
Finally, by multinomial logistic regression, we analyzed the independent predictors from above factors for PCOS incidents with or without MetS (Table 2). The results showed that CTBP1 (p = 0.002) was positively correlated with the occurrence of PCOS without MetS compared with the control ones without MetS. Levels of CTBP1 (p = 0.02), T (p = 0.005), and TG (p = 0.006) were also confidently associated with the incidence of PCOS with MetS, but HDL (p = 0.023) was negatively correlated with the incidence of PCOS with MetS compared with controls without MetS. Furthermore, the association between CTBP1 and PCOS with MetS (p = 0.02) was less significant than that between CTBP1 and PCOS without MetS (p = 0.002). These data reveal that patients diagnosed with MetS are statistically associated with PCOS incidence, and high expression of CTBP1 is closely correlated with PCOS patients with or without MetS.
Inverse regulation of CTBP1 and aromatase expression by FSH/LH and EGF
The signal pathways involved with FSH, LH and EGF are closely associated with follicular differentiation and selection as well as oocyte meiosis and maturation23,24,25. Any abnormalities such as FSH deficiency, hypersecretion of LH, and hyperandrogenemia may interrupt the crosstalk between granulosa cells and oocytes, which reduces the oocyte maturation and embryonic development and subsequently leads to unsuccessful outcomes of PCOS patients undergoing assisted reproduction26. To test whether CTBP1 expression is regulated by FSH, LH, and EGF, we directly treated primary GCs from PCOS with MetS with these factors in different concentrations. The results showed that treatment of pGCs with FSH and LH for 24 h highly downregulated the mRNA and protein levels of CTBP1 in a dose dependent manner (Fig. 2A, B), whereas treatment of the cells with EGF steadily increased CTBP1 expression, compared with those treated with diluent (0 ng/ml) (Fig. 2C). These results demonstrate that CTBP1 is conversely regulated by FSH/LH and EGF, respectively in pGCs, giving us some hints to target CTBP1 in treatment of PCOS patients with MetS. Because the conversion of androgen into estrogen in GCs is catalyzed by aromatase encoded by CYP19A1, we first silenced the expression of CTBP1 in pGCs by specific siRNAs and found that the expression of aromatase was markedly upregulated compared within cells expressing control siRNA (siNC) (Fig. 2D, E), which resulted in an increased E2 secretion tested in the conditioned medium of cells (Fig. 2F), whereas the expression of aromatase and E2 was downregulated following the transient transfection of CTBP1 cDNA in the pGCs transfected with CTBP1 siRNAs (Fig. 2G–I). The similar data were obtained in CTBP1-silencing/overexpressing KGN cells (Fig. 2J–O). To ensure the physiological response induced by FSH, LH, EGF and CTBP1 silencing/overexpression, the relative levels of GAPDH mRNA corresponding to Fig. 2A–D, G, J, M were shown in Supplementary Fig. 2, indicating that GAPDH is a proper internal control for our study. These data demonstrate that FSH/LH and EGF conversely control the expression of CTBP1 and aromatase/E2, and that CTBP1 may negatively regulate the expression of aromatase/E2, which may need further investigations.

A–C CTBP1 expression detected by real-time PCR (upper panel) and WB (lower panel) in pGCs treated with FSH (A), LH (B), or EGF (C) at different concentrations for 24 h. D–F Silencing of CTBP1 enhances aromatase (D, E) and E2 (F) expression in pGCs. G-I. Re-introduction of CTBP1 cDNA inhibits aromatase (G, H) and E2 (I) expression in pGCs. J–O Silencing of CTBP1 (J–L) or reintroduction of CTBP1 increases or inhibits (M–O) aromatase and E2 expression in KGN cells. Quantification data are presented as mean ± SD and analyzed using two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 are considered significant.
CTBP1 directly inhibits aromatase transcription and expression
To investigate whether CTBP1 directly inhibits the expression of aromatase at the transcriptional level, we constructed a luciferase reporter plasmid containing the CYP19A1 promoter II sequence for specific aromatase expression in ovarian tissue and performed luciferase assays in KGN cells according to the reference27. The results showed that CTBP1 markedly reduced the transcription level of luciferase (Fig. 3A). To further confirm this result, we used Chromatin Immunoprecipitation (ChIP) assay with either an antibody to CTBP1 or IgG, followed by qPCR examination of ChIP products using the specifically designed two primer pairs spanning the whole promoter II sequence (381 bp) of CYP19A1. The results demonstrated that the specific qPCR product was only conceived by primer pairs 1 covering +109 \(\sim\) + 304 bp but not by primer pairs 2 covering +306 ~ +460 bp (starting from the first nucleotide of exon 1) (Fig. 3B, C), which was further identified by sequencing of the ChIP-PCR products after purified in agarose gel and ligated into a T-vector (Fig. 3D). These results suggest that CTBP1 binds to the core sequence between +108 and +304 nt of CYP19A1 promoter II as a repressor to inhibit the transcription of CYP19A1, leading to the reduced expression of aromatase and E2.

A Firefly/Renilla luciferase activity ratios tested by co-transfection of vector or CTBP1 cDNA along with pGL3 basic vector and pGL3-CYPT19A1-II plasmid in HEK293T cells. B CYP19A-II promoter fragments detected by ChIP-qPCR using CTBP1 antibody or isotypical IgG, and the primer pairs 1-2. C, D PCR products detected by agarose gel electrophoresis (C) and sequencing (D). E–J CTBP1, aromatase, and E2 expression detected by qRT-PCR (E, H), WB (F, I) or ELISA (G, J) in KGN cells stably expressing either vector or CTBP1 cDNA after treatment with FSH (E–G) or LH (H–J) at 400 ng/ml. Quantification data are presented as mean ± SD and analyzed using two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 are considered significant. pGCs Primary granulosa cell, WB Western blot.
To explore whether gonadotropin regulates aromatase expression through CTBP1, we stably transfected CTBP1 cDNA in KGN cells, followed by FSH and LH treatment at 400 ng/ml. The data showed that FSH upregulated the expression of aromatase, but downregulated CTBP1 expression in vector expressing cells (Fig. 3E, F). Introduction of CTBP1 cDNA partially blocked the FSH-induced aromatase expression (Fig. 3F), and the levels of E2 in culture medium was upregulated by FSH treatment although inhibited by CTBP1 overexpression (Fig. 3G). Similar data were obtained by LH treatment of cells with or without CTBP1 overexpression (Fig. 3H–J). These data indicate that excessive FSH and LH inhibit the expression of CTBP1, and the absence of CTBP1 triggers CYP19A1 transcription, which in turn promotes estrogen synthesis.
CTBP1 represses lipid synthesis
As we know, lipid metabolism has been extensively reported to be indispensable for regulating the function of GCs and maintaining follicular development28,29. On the one hand, mammalian ovarian GCs can synthesize fatty acids from lipogenesis to accumulate TG essential for oocyte and ovarian development30. On the other hand, ovarian GCs can also synthesize steroid hormones from cholesterol31,32. Therefore, the intracellular TG and cholesteryl ester existing in lipid droplets (LDs) are imperative for hormone synthesis and cellular function of GCs. Thus, we investigated whether CTBP1 regulates lipid droplet formation in GCs. By oil-red O staining, we found that CTBP1 knockdown promoted the LDs formation in pGCs and KGN cells, and whereas reintroduction of CTBP1 cDNA in these cells expressing si-CTBP1-2 markedly inhibited the formation of intracellular LDs compared with in cells expressing vector (Fig. 4A, B), which were further confirmed by Nile red and BODIPY staining in KGN cells expressing si-CTBP1-2 followed by introduction of vector or CTBP1 cDNA (Fig. 4C, D;Supplementary Fig. 3A, C). Given that the main component of LDs is TG, we tested and found that the intracellular TG level was distinctively promoted or repressed by CTBP1 siRNAs or CTBP1 cDNA in HEK293T, pGCs, and KGN cells (Fig. 4E). We then used InBio-Discover™ kit (https://inbio-discover.com/) to evaluate the interconnected signaling networks between different proteins to gain some valuable biological functions. The results showed that CTBP1 might mainly control the fatty acid metabolism (GO: 0009892, GO: 0009893, GO: 0080090) through interaction with SREBF1 (Sterol Regulatory Element Binding Transcription Factor 1), ACC1 (ACACA, Acetyl-CoA Carboxylase Alpha), FASN (Fatty Acid Synthase), CPT1A (Carnitine Palmitoyltransferase 1A), etc. (Fig. 4F). By western blot, we found that silencing of CTBP1 in KGN cells stimulated the expression of ACC1, FASN, and SREBP1 (protein of SREBF1), but downregulated the expression of CPT1A, whereas reintroduction of CTBP1 cDNA in above cells reversed the expression of ACC1, FASN, SREBP1, and CPT1A (Fig. 4G, H). These data reveal that CTBP1 may dysregulate the lipid metabolism through above molecules to contribute to PCOS with MetS. However, the detailed mechanism may need to be further investigated.

A Detection of lipid droplets by oil red O staining in pGCs and KGN cells treated with or without CTBP1-siRNA (control siRNA as si-NC) (upper panel), and in cells further transfected with CTBP1 cDNA or vector. Scale bars, 20 µm. B Quantification of lipid droplets in cells from A. C. Detection of lipid droplets by Nile red staining in cells pretreated with CTBP1 siRNA and further transfected with CTBP1 cDNA. Scale bars, 20 µm. D Quantification of Nile red/cell from (C). E Relative TG levels detected in of 293T cells, pGCs and KGN cells expressing control (si-NC)/CTBP1 siRNA (upper panel) and vector/CTBP1 cDNA (lower panel). F Signal network analyzed by the inbio-discover protocol from https://inbio-discover.com/#search to predict the interconnecting signal networks between proteins. G, H Expression of ACC1, FASN, CPT1A, and SREBP1 detected by WB in KGN cells overexpressing control or CTBP1 siRNA (G), and in cells expressing vector or CTBP1 cDNA following control/CTBP1 siRNA treatment (H). Quantification results are presented as mean ± SD and analyzed by using two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 are considered statistically significant. LD Lipid droplet, pGCs Primary granulosa cell, TG Triglyceride, WB Western blot.
CTBP1 mediates the EGF-induced inhibition of lipid droplets and lipid synthesis through downregulation of SREBP1
Increased EGF in the follicular fluid of PCOS patients inhibits estrogen synthesis in GCs, blocks antral follicle growth, and results in follicular arrest, indicating that EGF is involved in the maintenance of PCOS33. Whether an elevated level of EGF in follicular fluid controls GCs function, oocyte quality and embryonic developmental competence through regulation of the lipid droplet formation is still unclear. Thus, we treated GCs with EGF and stained the cells with oil red O to detect the intracellular lipid droplet formation. The results showed that LDs per cell were significantly decreased after EGF treatment in both pGCs and KGN cells (Fig. 5A, B). The significant inhibition of intracellular LDs formation in KGN cells treated with EGF was also confirmed by Nile red and BODIPY staining (Fig. 5C, D; Supplementary Fig. 3B, D). Consistently, EGF also repressed intracellular TG levels (in both KGN and pGCs cells) (Fig. 5E) and the expression of lipid metabolism-related proteins ACC1, FASN and SREBP1 in KGN cells (Fig. 5F). To test whether the above data were associated with CTBP1 expression, we used siRNAs to silence the CTBP1 expression and found that silencing of CTBP1 rescued the reduced LDs (Fig. 5G, H), intracellular TG level (in both KGN and pGCs cells) (Fig. 5I), and the expression of ACC1, FASN and SREBP-1 caused by EGF inhibition (Fig. 5L), which was also confirmed by Nile red staining (Fig. 5J, K) and BODIPY staining (Supplementary Fig. 3E, G) in KGN cells, showing that EGF treatment reduced LD formation, and silencing of CTBP1 enhanced LD formation. Thus, CTBP1 may mediate the EGF-induced inhibition of intracellular accumulation of lipid droplets and lipid synthesis through downregulation of SREBP1. This brought us to explore how SREBP1 is inhibited by CTBP1.

A Lipid droplet detection by oil red O staining after EGF treatment for 24 h. Scale bars, 20 µm. B Quantification analysis of lipid droplets from A. C Lipid droplet formation determined by Nile red staining after EGF treatment for 24 h. Scale bars, 50 µm. D Quantification analysis of Nile red/cell from (C). E Relative TG levels detected in KGN and pGCs treated with EGF for 24 h. F Detection of FASN, ACC1 and SREBF1 by WB in KGN cells after EGF treatment. G, H Lipid droplets detected by oil red O staining (G) and quantification (H) in KGN cells expressing control/CTBP1 siRNA after treatment with 100 ng/mL EGF for 24 h. Scale bars, 20 µm. I. Detection of TG in pGCs and KGN cells expressing CTBP1 siRNA and treated with EGF. J–K Detection of lipid droplets by Nile red staining (L) and quantification (K) in KGN cells expressing control/CTBP1 siRNA after treatment with 100 ng/mL EGF for 24 h. L Detection of ACC1, FASN and SREBP1 in KGN cells from (G). The quantification results are presented as mean ± SD and analyzed using two-tailed t-test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 are considered significant. LD, lipid droplet; pGCs, primary granulosa cell; WB, western blot.
CTBP1 facilitates SREBP1 ubiquitination and protein degradation
To investigate how SREBP1 is suppressed by CTBP1, we first tested and found that the mRNA levels of SREBP1 were not significantly altered between cells with and without CTBP1 overexpression compared with those of CTBP1 (Fig. 6A). As SREBP1 includes two isoforms a and c generated from a single gene through the alternative transcription start sites, a semi-quantitative RT-PCR assay was performed to identify the major isoform in pGCs and KGN cells by using two 5’ primers specifically recognizing SREBP1a and SREBP1c respectively, and one 3’ primer recognizing both isoforms. The size of PCR products showed that the major isoform in pGCs and KGN cells is SREBP1a (263 bp) rather than SREBP1c (191 bp) (Fig. 6B, C), which was further confirmed by sequencing.

A mRNA levels of CTBP1 and SREBP1 mRNA detected by real time qPCR in KGN cells stably expressing vector or exogenous CTBP cDNA. B, C Semi-quantitative analysis of SREBP1a and SREBP1c showing that both pGCs and KGN cells express isoform SREBP1a other than SREBP1c. D Detection of pre-SREBP1, mature SREBP1, phosphorylated SREBP1, CTBP1 and FBXW7 in CTBP1 overexpressing KGN cells with or without MG-132 treatment, showing that SREBP1, phosphorylated SREBP1 at S396, and FBXW7 are increased following MG-132 treatment. E, F Co-IP and WB assays showing that overexpression of CTBP1 promotes SREBP1 high ubiquitination and lower binding to CTBP1. G–J Co-localization and fluorescent intensity of CTBP1 with SREBP1 (G, H) (Scale bars: 15 um) or with pS396-SREBP1 (I–J) (Scale bars: 10 um) in cells overexpressing CTBP1 with or without MG132 treatment.
Although the precursor of SREBP1a includes 1147aa, the mature protein containing only 490aa (around 50 kDa) may translocate into cell nuclei to activate the transcription of target genes including those for cholesterol and fatty acid synthesis34. However, the phosphorylation of SREBP1a at T426/S430 facilitates the degradation of nuclear SREBP1a through interacting with the ubiquitin ligase FBXW735, whereas the phosphorylation of SRBEP1a at S439 by CDK1 mediates the PLK-induced further phosphorylation of SREBP1a at T424, S467 and S486 during cell mitosis, which prevents the protein from the FBXW7-mediated proteolytic degradation36. The other study demonstrates that SREBP-1c may be phosphorylated at S372 by AMPK to suppress the cleavage and nuclear translocation of mature protein, resulting in reduced lipogenesis37, however whether SREBP1a is also phosphorylated at S396 is unreported. Thus, we first treated cells with MG132 and found both precursor and mature proteins (mSREBP1) of SREBP1a were markedly increased even in cells expressing exogenous CTBP1 compared in cells without MG132 treatment (Fig. 6D), suggesting that the expression level of SREBP1a may be controlled at the ubiquitin-mediated protein degradation. Based on commercially available antibodies, we found that S396-SREBP1a was remarkably reduced in cells expressing exogenous CTBP1, but was largely detected in cells treated with MG132, whereas the change of S439-SREBP1a was not conceived between cells with and without CTBP1 overexpression, or between parallel cells with and without MG132 treatment (Fig. 6D), indicating that the degradation of SREBP1a may be mainly associated with the phosphorylation site S396 other than S439 of SREBP1a. Treatment of cells with MG-132 also increased the expression of FBXW7 (Fig. 6D). By co-IP assay, we found that the ubiquitination of SREBP1 was increased in cells treated with MG-132, and that binding of CTBP1 to SREBP1 was highly reduced in cells with CTBP1 overexpression without regard to MG-132 treatment (Fig. 6E), whereas the mature/phosphorylated SREBP1 (S396) mainly bound to CTBP1 (Fig. 6F). To further confirm these findings, we examined the expression patterns of CTBP1 and SREBP1 by immunofluorescence and found that the nuclear expression of SREBP1 was reduced with CTBP1 overexpression, while treatment of cells with MG-132 mainly increased the cytoplasmic expression of SREBP1, and co-localization of CTBP1 and SREBP1a was clearly found in nuclei of the cells treated with MG-132 (Fig. 6G, H). The phosphorylated SREBP1a at S396 was located in cell nuclei either with or without MG-132 treatment, overexpression of CTBP1 reduced the nuclear expression of S396-SREBP1a in cells treated without MG-132 but not in cells treated with MG-132. The expression levels of both CTBP1 and S396-SREBP1a were consistent with the results detected by western blot, and the co-localization of CTBP1 and S396-SREBP1a was mostly detected in CTBP1 overexpression cells treated with MG-132 (Fig. 6I, J). Therefore, we conclude that CTBP1 induces the protein degradation of nuclear S396-SREBP1a through ubiquitination, which may negatively control the synthesis and uptake of cholesterol and TG.
Discussion
Granulosa cells are involved in the steroid synthesis and metabolism during the development of female gamete in the ovary38, while abnormal functions may alter GCs normal ‘dialogue’ with oocytes in early growing follicles with PCOS39. In this study, by analysis of the clinically recruited 49 PCOS patients and 47 control subjects during pre-IVF examination, we found that some of hormones and lipids were highly dysregulated in PCOS patients with MetS, and were significantly associated with the high expression of CTBP1 in primary GCs. High expression of CTBP1 was an independent risk factor for the patients with both PCOS and MetS. On the contrary, some studies reported that lncRNA CTBP1-AS, an antagonist of CTBP1, is increased in PCOS, but those studies were performed only by quantitative realtime PCR using total RNAs isolated from the peripheral venous blood samples of PCOS patients19,40, which does not reflect any changes of CTBP1-AS or CTBP1 in PCOS granulosa cells. Based on our data, we conclude that CTBP1 overexpression in GCs may contribute to human PCOS with or without MetS.
GCs cultured from the follicles of an ovulatory female with PCOS are highly responsive to FSH and prematurely responsive to LH in terms of E2 synthesis8,41,42. High reactivity to FSH may result from the overexpression and high activity of FSHR in GCs from patients with PCOS43. Massillo et al. reported in 2019 that CYP19A1 gene expression is inhibited by CTBP1 in prostate cancer cells, but the aromatase isoform in prostate cancer cells is different from that in ovarian granulose cells because the promoters binding to CTBP1 are quite different between prostate cancer cells and ovarian granulose cells21,27. By in vitro experiments, we next revealed that gonadotropins FSH and LH reduced the expression of CTBP1 but induced the expression of aromatase, which in turn promoted oestrogen synthesis because CTBP1 repressed the CYP19A1 transcription through binding to the CYP19A1 promoter II, which is different from in prostate cancer cells21. This finding may shed a light on potentially utilizing CTBP1 as a therapeutic target to treat PCOS.
SREBP1a/c can directly activate the expression of multiple genes involved in the synthesis and uptake of cholesterol, fatty acids and TG as a master transcription factor44. The phosphorylated SREBP1a/c may translocate into the nucleus to promote the transcription of downstream target genes including those encoding the key enzymes ACLY, ACC1, FASN, and SCD1 in the adipogenesis pathway45,46. Ferrarese et al. reported that CTBP1/2 may activate the transcription of SREBP genes47, which is opposite to the finding that SREBP2 is transcriptionally repressed by CTBP2 alone with ZEB148. In our study, we revealed that CTBP1 does not regulate SREBP1 expression at transcription level, but stimulates the ubiquitination-associated protein degradation of SREBP1a through the enhanced phosphorylation of SREBP1 at S396 and the FBXW7-mediated pathway, resulting in a sustained inhibition of lipid synthesis shown as the reduced number of lipid droplets. However, the detailed mechanism how CTBP1 interacts with the phosphorylated SREBP1 and other molecules such as FBXW7 needs further investigations.
Based on Table 1, LH level is not significantly changed between different patient groups, whereas FSH levels are significantly altered between non-MetS control (A) and non-MetS PCOS patients (C) (p = 0.013), or between non-MetS control (A) and MetS PCOS patients (D) (p = 0.009). Although the levels of LH are increased around 2 mIU/mL in PCOS (8 mIU/mL) compared with in control patients (6 mIU/mL), the units of serum LH and FSH are mIU/mL, but the concentration used to induce a significant alteration of CTBP1 in both pGCs and KGN cells (Fig. 2A, B, Fig. 3F, I) is much higher (100–400 ng/ml), which is consistent with other studies performed in granulosa cells49,50. Nevertheless, the increase of LH in PCOS compared with in control patients may indicate a native feedback from PCOS patients. On the other hand, numerous studies have shown that the alteration of hormones including FSH and LH is highly associated with PCOS51,52, and that EGF is positively associated with CTBP118, although no clearly associated studies between CTBP1 and FSH or LH are reported at present. Therefore, in order to explore the function of CTBP1 in human ovarian granulosa cells in terms of hormonal changes, we designed the in vitro experiments shown in Fig. 2 and the resulting data were basically consistent with what we found from patients (Tables 1, 2, and Supplementary Tables 1–5).
Previous studies have shown a relationship between EGF and lipid metabolism53,54. EGF can significantly attenuate chronic lipid droplets accumulation induced by ethanol and decrease the TG level in mice liver55. In patients with PCOS, the EGF levels in serum and FF are significantly elevated56, thereby considered as a factor causing the accumulation of multiple small antral follicles57 and an independent risk factor inhibiting pregnancy outcomes among patients11. Our data suggested that EGF positively regulated CTBP1 expression in GCs, whereas knockdown of CTBP1 rescued the descending of LD accumulation and intracellular TG levels inhibited by EGF treatment through activating the function of nuclear phosphorylated SREBP1.
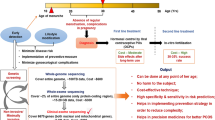
Many studies are focused on the effects of PCOS on woman’s health in terms of reproduction, and the patients diagnosed with PCOS is usually associated with clustered metabolic features including insulin resistance, obesity, hyperandrogenism etc. 13,14, whereas other studies indicate these characteristics unnecessarily connected with PCOS [15]. Thus, the metabolic disorders may be differentially presented in different PCOS patients, indicating the differences between PCOS and Metabolic Syndrome, while the common mechanisms between PCOS and Metabolic Syndrome may be truly connected with obesity as indicated by BMI and TG in our study. Specifically, BMI and TG are significantly associated with MetS and/or PCOS (Table 1, whereas CTBP1 is associated with PCOS with or without MetS (Table 2). By in vitro experiments we proved a mechanism that CTBP1 promotes the degradation of the phosphorylated SREBP1 to inhibit lipid metabolism and suppresses E2 secretion through blocking aromatase transcription in human granulosa cells. Based on our clinical data analyses and in vitro experimental data, we conclude that CTBP1 links metabolic syndrome to PCOS through inhibiting CYP19A1 transcription and facilitating SREBP1 degradation in nucleus. (Fig. 7), which might provide a new perspective to further understand the pathogenesis of PCOS.

CTBP1 expression can be suppressed by FSH/LH, but induced by EGF through binding to their receptors. CTBP1 not only blocks the transcription of CYP19A1 to suppress aromatase expression and E2 synthesis, but also promotes the ubiquitination-mediated protein degradation of the nuclear pS396-SREBP1 to induce metabolic syndrome including abnormal lipid droplet formation, triglyceride and lipid synthesis.
Materials and Methods
Participants
The study protocols were approved by the ethics committees of the First Affiliated Hospital of Zhengzhou University and Fudan University Shanghai Cancer Centre. All patients were provided with an informed written consent before participation. 96 females including 47 patients with PCOS and 49 individuals without PCOS (control) were recruited and further classified as Mets and non-MetS subgroups, among which the number of MetS subgroups was 18 and 15 in PCOS patients and controls, respectively.
The inclusive criteria for PCOS patients were with the indication of in vitro fertilization and fulfilled the diagnostic criteria of PCOS based on the revised 2003 Rotterdam consensus58, including oligo ovulation or anovulation combined with either hyperandrogenism or polycystic ovaries and exclusion of other aetiologies (e.g., androgen-secreting tumours, congenital adrenal hyperplasia, Cushing’s syndrome). Control patients were infertility caused by fallopian tube factors or male factors. In additional, the exclusion criteria were either subjects with ovarian dysfunction, unilateral oophorectomy history, endometriosis, or other medical conditions that contraindicate the assisted reproductive technology.
Patients were diagnosed as MetS with three or more of the following criteria: (1) waist circumference ≥ 80 cm, (2) high-density lipoprotein-cholesterol (HDL-C) ≤ 1.29 mmol/L, (3) triglyceride (TG) ≥ 1.7 mmol/L, (4) systolic/diastolic blood pressure (SBP/DBP) ≥ 130/85 mmHg or specific medication treatment for hypertension, (5) fasting blood-glucose (FBG) ≥ 5.6 mmol/L (100 mg/ dL) or specific medication treatment for diabetes59,60,61,62. As the previously described, the ovarian stimulation regimen, oocyte retrieval, and fertilization were performed according to the standardized protocol recommended by the American Society of Reproductive Medicine and Chinese guidelines63.
Statistics and reproducibility
Data were analyzed using IBM SPSS Statistics version 22. Continuous variables expressed as mean ± SD were examined by independent sample t-test for normally distributed data and Mann-Whitney U test for non-normally distributed data. For categorical variables, the differences of frequency (percentage) or the number of cases between groups were investigated by Chi-squared test. Comparisons among three groups were performed by ANOVA analysis. A baseline univariable comparison among different groups was conducted in order to select a group of potential predictors for PCOS and MetS in these women, and these variables were included for the logistic regression analyses. The predicted probability of logistic regression models was used to construct a receiver operating characteristic (ROC) curve. In vitro data from cell experiments were analyzed using student t-test. All statistical significance was defined as a P value < 0.05. The experiments were repeated three times, and representative data were shown.
Ovarian stimulation and human ovarian GCs collection
The participants were treated with a GnRH agonist on the 2nd or 3rd day of menstruation. After 30–42 days, ovarian stimulation was performed using gonadotropin. When one dominant follicle diameter ≥ 20 mm, three follicles diameter ≥ 17 mm or two-thirds follicles diameter ≥ 16 mm, 250 μg of Azer (Meker, Italy) and 2000 IU of human chorionic gonadotropin (Livzon, China) were administered. After 36-37 hours of human chorionic gonadotropin administration, transvaginal ultrasound-guided needle puncture was performed to retrieve oocytes. The oocyte maturation rate was calculated as a percentage of the number of MII stage oocytes in the total number of oocytes. Oocytes were fertilized by either IVF or ICSI. Fertilization was confirmed on day 1 by the presence of two pronuclei (18–19 h after insemination or ICSI). Fertilization rate was calculated through this formula: normal fertilization rate in IVF = (2PN/the total number of oocytes) ×100%; normal fertilization rate in ICSI = (2PN/the number of MII oocytes) ×100%. The quality of the cleavage-stage embryo was assessed on day 2 and/or on day 3 considering the number of blastomeres, the degree of fragmentation and the uniformity of blastomeres (according to Veeck’s criteria). Cleavage rate was calculated through this formula: Cleavage rate = (the number of cleaved embryos / 2PN) × 100%. Grade 1(blastomeres of equal size and no fragmentation) and grade 2(blastomeres of equal size with minor fragmentation) embryos were defined as high-quality embryos, as reported previously64,65. Blastocyst formation was assessed on day 5 and 6 according to the expansion of the blastocele cavity and the number and integrity of both the inner cell mass and trophectoderm cells, as described previously66. High qualified embryo rate = (number of high-quality embryos/ 2PN) × 100%. Blastocyst formation rate= (number of formed blastocysts / the number of cultured blastocysts) × 100%.
Human ovarian primary granulosa cells (pGCs) were collected from follicular fluid on the day of oocytes retrieval. The aspirated follicular fluid was collected in sterile tubes, and centrifuged at 400 × g for 10 minutes to pellet follicular cells. Next, the pGCs were purified by density gradient centrifugation with a Percoll gradient as previously described67.The GC pellets used to detect the CTBP1 expression were then stored in liquid nitrogen within a single tube per participant, and RNA extraction and quantitative real-time PCR were conducted in the same batche after all samples were collected.
Cell cultures and reagents
KGN, a human granulosa-like tumor cell line, was generously gifted by Henan Key Laboratory of Reproduction and Genetics in the First Affiliated Hospital of Zhengzhou University. KGN and primary GCs (pGCs) were cultured in DMEM/F12 (Sigma) supplemented with 10% fetal bovine serum (Biological Industries, ISR) and 1% penicillin–streptomycin (Invitrogen) and then placed in the humidified atmosphere containing 5% CO2 at 37 °C. Recombinant human LH was purchased from Solarbio in Beijing, China, and recombinant human FSH and EGF were purchased from MCE in Shanghai, China.
RNA isolation and quantitative reverse transcription polymerase chain reaction (qRT-PCR)
To extract the total RNAs of cells including KGN and primary GCs, 1 ml TRNzol reagent (DP424, TIANGEN, Beijing, China) was added per 10 cm2 area/5 × 106–1 × 107 cells, and 0.2 ml chloroform was added for every 1 ml TRNzol reagent. The mixture was shakeed vigorously for 15 s, and placed at room temperature at least 3 minutes, and then total RNA was extracted according to the manufacturer’s protocol. NanoDrop (Thermo Fisher Scientific, USA) was used to determine the RNA concentration, and the purified RNA was then reversely transcribed using the PrimeScript RT Master Mix (RR036A, Takara, Japan) to create a cDNA library. Control of genomic contamination was measured for each RNA and cDNA sample. Real-time PCR was performed on a 7500 Real-Time PCR System (Bio-Rad Laboratories) by using a SYBR Premix Ex TaqTM kit (RR420A, Takara, Japan). The relative quantitative RNA was normalized to GAPDH with the ΔΔCt method. All primers used in this study were listed in Supplementary Table 6.
Semi-quantitative RT-PCR
RNA isolation and reverse transcription were performed as described above. To identify the isoforms a and c of SREBP1 expressed in pGCs and KGN cells, specific primers designed to amplify the 5´ cDNAs of SREBP1a and SREBP1c were performed with the following conditions 98 °C for 5 min; 98°C 10 s, 58 °C 5 s, 72 °C 2 min for 25 cycles; 72 °C10 min. Internal control primers were designed to amplify GAPDH. The PCR products were run in 1.5% agarose gel and quantified using the Image J software, a statistical significance was calculated from the results of 3 independently repeated assays. Specific primers are listed in Supplementary Table 6.
Plasmid constructions
CTBP1 cDNA was amplified with primers (Supplementary Table 6) by PCR with the following conditions 94 oC 2 min, 94 °C 30 sec/54 °C 30 sec/72 °C 2 min for 35 cycles, and 72oC for 10 min, from a cDNA library prepared by reverse transcription of total RNA isolated from KGN cells. The purified cDNA fragments were digested with Xba I and EcoR I and inserted into either pcDNA 3.1- for transient transfection or pCDH-CMV-MCS-EF1-Puro vector for stable transfection mediated by lentivirus delivery system as described before68. The cloned cDNA was used for this study after sequencing identification. The ovarian tissue specific promoter II (381nt) of CYP19A1 was amplified by PCR according to the reference69, from genomic DNA of KGN cells and cloned into pGL3 basic vector by Kpn I and Xho I digestion designed in primers sequences (Supplementary table 6).
Transient and stable transfection of siRNAs and/or cDNA
CTBP1 siRNAs including SASI_Hs01_00243819 (designed as si-CTBP1-1) and SASI_Hs01_00243820 (designed as siCTBP1-2) targeting 738-756nt and 949-967nt of CTBP coding region (NM_001012614) were purchased from Merck-Sigma Aldrich Company. Control siRNA-A (sc-37007) was from Santa Cruz Biotechnology, Inc. To transiently silence CTBP1 expression, human pGCs and KGN cells were seeded into six-well plates. When cell density reached 80% confluence, CTBP1 siRNAs or control siRNA were transfected into cells by using Lipofectamine 3000 reagent according to the manufacturer’s instruction. Forty-eight hours later, the cells were treated with different drugs including FSH, LH, EGF in different concentrations (0, 10, 100, or 200 ng/ml) at the indicated times, and the supernatant and/or cells were collected after additional 24 h. To transiently overexpress CTBP1, pcDNA-CTBP1 or empty vector was similarly transfected into pGCs or KGN cells expressing CTBP siRNAs or control siRNA. To stably overexpress CTBP1 protein, CTBP1 cDNA in pCDH-CMV-MCS-EF1-Puro or empty vector was first co-transfected with packing (pSPAX2) and envelope (pMD2.G) plasmids into 293 T cells by using Lipofectamine 3000 reagent. The virus-containing medium was harvested after 48–72 h post transfection and used to infect KGN cells after filtered with 0.22 μm MCE sterile filters (Millipore products GSWP04700). The infected cells were then selected in the medium containing 2 μg/ml puromycin for 2–3 days according to our previously published method68.
Immunoprecipitation (IP) and western blot (WB)
IP assay was performed using the methods described previously68. Protein extracts were prepared by a kit from Beyotime Biotechnology (Shanghai, China) and quantified by BCA method. A total of 500 mg proteins was used to incubate with 10 μg of antibodies to CTBP1 (ab129181, Abcam or sc-398945, anti-mouse, Santa Cruz), SREBP1 (ab28481, anti-rabbit, Abcam / sc-365513, anti-mouse, Santa Cruz / SREBP1 (PA1-337, anti-rabbit, ThermoFisher), Phospho-SREBP1 (Ser439/415) (PA5-99371, anti-rabbit, ThermoFisher), Phospho-SREBP1 (Ser396/372) (PA5-105169, anti-rabbit, ThermoFisher), Phospho-SREBP1 (Ser338/314) (PA5-105567, anti-rabbit, ThermoFisher) or normal rabbit/mouse IgG (negative control) at 4 °C for 14–16 h. Thereafter, the samples were incubated with Protein A/G PLUS-Agarose beads (Santa Cruz, sc-2003) at 4 °C for 5–6 h. The conjugated agarose beads containing antibody-antigen mixture were then washed with IP buffer at 4 °C for five times. The immunoprecipitated products were then treated with 2 × SDS loading buffer at 95 °C for 5 minutes. Protein samples were analyzed by western blot (WB) after centrifugation at the speed of 10,000/min for 2 min.
WB was performed according to the previously described method68. Cells were lysed with RIPA buffer, and the protein concentrations were normalized with a BCA assay kit (Beyotime Biotechnology, China). Antibodies to GAPDH (ab9482, Abcam, 1:5000), CTBP1 (ab129181, anti-rabbit, Abcam, 1:1000/sc-398945, Santa Cruz, anti-mouse, 1:500), aromatase (ab18995, Abcam, 1:1000), SREBP1 (ab28481, anti-rabbit, Abcam, 1:1000/ sc-365513, anti-mouse, Santa Cruz, 1:500), Phospho-SREBP1 (Ser439/415) (PA5-99371, anti-rabbit, ThermoFisher, 1:2000), Phospho-SREBP1 (Ser396/372) (PA5-105169, anti-rabbit, ThermoFisher, 1:2000), ACC1 (4190S, CST, 1:2000), FASN (3180S, CST, 1:1000), and CPT1A (97361S, CST, 1:2000) were purchased from the different companies and used at the indicated dilutions. Antibody to ubiquitin was from Abcam company (ab134953, 1:2000 dilution), and the secondary antibodies were from Cell Signaling Technology, Inc.: HRP-linked anti-mouse IgG (#7076, 1:4000) and HRP-linked anti-rabbit IgG (#7074, 1:4000).
E2 and TG measurement
For E2 production assessment, the culture medium of cells from different treatment groups was collected and divided into three tubes. Of note, KGN requires the addition of testosterone as the substrate for 24 h before culture medium collection. E2 concentration was measured using Electrochemiluminescence Immunoassay Kit (Roche Diagnostics, Rotkreuz, Switzerland) by Roche Diagnostics Cobas 6000 (Roche Diagnostics).
Quantitative analysis of triglyceride content in KGN and pGCs was conducted using an adipogenesis assay kit (Abcam ab102513) according to the manufacturer’s instructions. Cells were re-suspended and homogenized in 1 ml of 5% NP-40 solution, then slowly heated in a water bath to 80–100 °C for 2–5 min, or until the NP-40 solution becomes cloudy, and cooled to room temperature. Samples were centrifuged at maximum speed for 2 minutes with micro-centrifugation to remove any insoluble material. The supernatant was transferred to the new tube, and diluted with ddH2O to 4-fold. The 50 µL samples were added with 2 µL Cholesterol Lipase and incubated at room temperature for 20 min to convert triglyceride to glycerol and fatty acid. Each sample was added with 50 µL of Reaction Mix (including background controls), and mixed and incubated at room temperature for 60 min protected from light before measuring the absorbance at 570 nm using the ELISA reader. Each sample were triply tested.
Oil red O staining, Nile red staining, and BODIPY staining
The lipid droplets in pGCs and KGN cells were measured by oil red O staining, Nile red staining, and BODIPY 493/503 staining. Oil red O staining was performed as previously described in ref. 70. Briefly, cell slides were fixed with 4% paraformaldehyde for 15 min and washed twice with PBS. The working solution for oil red O was prepared by dilute 6 mL of 0.5% oil red O stock solution in 4 mL of PBS and followed by filtration with a 0.45 um filter. Next, the cells were stained for 30 min at room temperature, and then washed with PBS thrice, and counterstained with 1:10 dilution of hematoxylin. Images were acquired by optical microscope. Quantitative analysis of LD/cell was performed from 20 cells per view, total 3 views were accounted.
Nile red staining was performed as previously described in ref. 71. In brief, cell seeded on slides were fixed with 4% paraformaldehyde for 15 min and then washed twice with PBS. Thereafter, the cells were stained with 2 μg/mL Nile red for 30 minutes. After washing with PBS at least thrice, cell nuclei were stained with DAPI for 15 minutes at room temperature. Images were acquired by fluorescence confocal microscopy (Nikon N1CCD). Quantitative analysis of Nile red/cell was performed from 20 cells per view, total 3 views were accounted.
For BODIPY 493/503 staining, living cells pretreated with or without EGF treatment / CTBP1 siRNA transfection were seeded on cover slides or 60 mm dishes overnight and washed with PBS and incubated with BODIPY 493/503 staining dye (5 μM, HY-D1614, MedChemExpress) for 15 min at 37 °C in dark. Then, cells were washed with culture medium and observed by fluorescence microscopy (Axio Imager M2, Carl Zeiss) after addition of DAPI (Sigma-Aldrich. St. Louis, MO, USA). Besides, 3 × 104 cells were collected, washed with PBS and incubated with BODIPY 493/503 staining dye (5 μM, HY-D1614, MedChemExpress) and detected by flow cytometry (CytoFLEX LX, Beckman Coulter) to analyze the ratio of LDs. The data of flow cytometry were analyzed with FlowJo software. The gating strategy was shown in Supplementary Fig. 5.
All above individual images (n = 3 for each sample) were analyzed using ImageJ software evaluating Red or Green staining intensity72,73.
Chromatin immunoprecipitation (ChIP) and luciferase reporter assay
The ChIP assay was performed using Pierce Agarose CHIP Kit (Thermo scientific) in accordance with the manufacturer’s instructions, and chromatin was precipitated with anti-CTBP1 (ab129181, Abcam) antibody in KGN cells. The specific binding sequences were identified by qPCR. DNA fragments of CYP19A1-II promoter immunoprecipitated with CTBP1 antibody were detected by PCR and purified by agarose gel electrophoresis and subsequently ligated to a T vector for sequencing. The primers are listed in Supplementary Table 6.
Luciferase reporter assay was performed using Dual-Luciferase Reporter Assay system (Promega, WI, USA). CTBP1 cDNA in pcDNA3.1- and empty vector, pGL3 plasmid containing CYP19A1 PII promoter and control vector were differentially co-transfected into 293 T cells by using Lipofectamine 3000 reagent. 48 hours post transfection, cells were collected to detect Firefly luciferase activities and normalized with Renilla activities.
Immunofluorescence
Cells cultured overnight (around 60% confluence) on cover slides were used for IF staining using our published methods68. Primary antibodies were used to detect CTBP1 (sc-398945, Santa Cruz, anti-mouse, 1:50), SREBP1a (ab28481, anti-rabbit, Abcam, 1:100) and S396-SREBP1a (PA5-105169, anti-rabbit, ThermoFisher, 1:200). The secondary antibodies were from Jackson ImmunoResearch (West Grove, PA, USA): Goat anti-rabbit IgG (H + L) Alexa Fluor® 594 (111-585-003, red, 1:2000), goat anti-mouse Alexa Fluor® 488 (115-545-003, green, 1:2000). Nuclei were stained with DAPI (Sigma-Aldrich. St. Louis, MO, USA). The images were photographed with OLYMPUS FV3000 confocal fluorescence microscope (OLYMPUS, Japan), and processed with FV31S-SW software. Quantification of SREBP1 and CTBP1 was performed by evaluating Red or Green staining intensity of individual images (n = 3 for each sample) using ImageJ software as above.
Cell treatment with MG-132
Because a pilot study showed that the treatment of KGN cells with MG-132 at 20 μM (Sigma Aldrich) for 8 and 12 h apparently blocked the ubiquitinated protein degradation (Supplementary Fig. 4), thus KGN cells expressing vector or CTBP1 were treated with diluent or MG-132 at 20 μM for 8 h. The treated cells were collected for western blot analysis or seeded for immunofluorescent assay described above.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Numerical source data for all graphs in the manuscript can be found in supplementary data 1 file. All other data are available from the corresponding author on reasonable request.
Abbreviations
- AMH:
-
anti-Mullerian hormone
- CHIP:
-
chromatin immunoprecipitation
- CI:
-
confidence interval
- CTBP1:
-
C-terminal Binding Protein 1
- DBP:
-
Diastolic blood pressure
- E2:
-
estradiol
- EGF:
-
Epidermal growth factor
- FSH:
-
follicle-stimulating hormone
- FT3:
-
Free Triiodothyronine
- FT4:
-
free thyroxine
- FBG:
-
fasting blood-glucose
- Gn:
-
gonadotropin
- HDL:
-
high-density lipoprotein
- LDL:
-
low-density lipoprotein
- LDs:
-
lipid droplets
- LH:
-
luteinizing hormone
- MetS:
-
metabolic syndrome
- OR:
-
odds ratio
- P:
-
progesterone
- PCOS:
-
polycystic ovary syndrome
- pGCs:
-
primary granulosa cells
- PRL:
-
prolactin
- RT-qPCR:
-
reverse transcription quantitative real-time PCR
- ROC:
-
receiver operating characteristic
- SBP:
-
Systolic blood pressure
- T:
-
testosterone
- TC:
-
serum total cholesterol
- TG:
-
triglycerides
- TSH:
-
Thyroid-stimulating hormone
- IF:
-
immunofluorescence.
References
Macut, D., Bjekić-Macut, J., Rahelić, D. & Doknić, M. Insulin and the polycystic ovary syndrome. Diabetes Res. Clin. Pract. 130, 163–170 (2017).
Zhang, R. et al. Oxidative stress status in Chinese women with different clinical phenotypes of polycystic ovary syndrome. Clin. Endocrinol. 86, 88–96 (2017).
Naghshband, Z., Kumar, L., Mandappa, S., Niranjana Murthy, A. S. & Malini, S. S. Visceral Adiposity Index and Lipid Accumulation Product as diagnostic markers of Metabolic Syndrome in South Indians with Polycystic Ovary Syndrome. J. Hum. Reprod. Sci. 14, 234–243 (2021).
Moran, L. J., Misso, M. L., Wild, R. A. & Norman, R. J. Impaired glucose tolerance, type 2 diabetes and metabolic syndrome in polycystic ovary syndrome: a systematic review and meta-analysis. Hum. Reprod. update 16, 347–363 (2010).
Kandaraki, E., Christakou, C. & Diamanti-Kandarakis, E. Metabolic syndrome and polycystic ovary syndrome and vice versa. Arquivos brasileiros de. endocrinologia e metabologia 53, 227–237 (2009).
Teede, H., Deeks, A. & Moran, L. Polycystic ovary syndrome: a complex condition with psychological, reproductive and metabolic manifestations that impacts on health across the lifespan. BMC Med. 8, 41 (2010).
Dewailly, D. et al. Interactions between androgens, FSH, anti-Müllerian hormone and estradiol during folliculogenesis in the human normal and polycystic ovary. Hum. Reprod. update 22, 709–724 (2016).
Willis, D. S. et al. Premature response to luteinizing hormone of granulosa cells from anovulatory women with polycystic ovary syndrome: relevance to mechanism of anovulation. J. Clin. Endocrinol. Metab. 83, 3984–3991 (1998).
Richani, D. & Gilchrist, R. B. The epidermal growth factor network: role in oocyte growth, maturation and developmental competence. Hum. Reprod. update 24, 1–14 (2018).
Ambekar, A. S. et al. Proteomics of follicular fluid from women with polycystic ovary syndrome suggests molecular defects in follicular development. J. Clin. Endocrinol. Metab. 100, 744–753 (2015).
Gao, J., Song, Y., Huang, X., Wang, D. & Wang, H. The expression of platelet-derived growth factor, epidermal growth factor, and insulin-like growth factor-II in patients with polycystic ovary syndrome and its correlation with pregnancy outcomes. Ann. Palliat. Med. 10, 5671–5678 (2021).
Homburg, R. Involvement of growth factors in the pathophysiology of polycystic ovary syndrome. Gynecol. Endocrinol. : Off. J. Int. Soc. Gynecol. Endocrinol. 12, 391–397 (1998).
Corda, D., Colanzi, A. & Luini, A. The multiple activities of CtBP/BARS proteins: the Golgi view. Trends Cell Biol. 16, 167–173 (2006).
He, Y. et al. CtBP1/2 differentially regulate genomic stability and DNA repair pathway in high-grade serous ovarian cancer cell. Oncogenesis 10, 49 (2021).
Chinnadurai, G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. cell 9, 213–224 (2002).
Moiola, C. P. et al. Prostate tumor growth is impaired by CtBP1 depletion in high-fat diet-fed mice. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 20, 4086–4095 (2014).
Takayama, K. et al. Androgen-responsive long noncoding RNA CTBP1-AS promotes prostate cancer. EMBO J. 32, 1665–1680 (2013).
Haga, Y., Miwa, N., Jahangeer, S., Okada, T. & Nakamura, S. CtBP1/BARS is an activator of phospholipase D1 necessary for agonist-induced macropinocytosis. EMBO J. 28, 1197–1207 (2009).
Nabi, M. et al. Androgen receptor coregulator long noncoding RNA CTBP1-AS is associated with polycystic ovary syndrome in Kashmiri women. Endocrine 75, 614–622 (2022).
Liu, Z. et al. Androgen Receptor Coregulator CTBP1-AS Is Associated With Polycystic Ovary Syndrome in Chinese Women: A Preliminary Study. Reprod. Sci. (Thousand Oaks, Calif.) 22, 829–837 (2015).
Massillo, C. et al. CTBP1/CYP19A1/estradiol axis together with adipose tissue impacts over prostate cancer growth associated to metabolic syndrome. Int. J. cancer 144, 1115–1127 (2019).
De Luca, P. et al. CtBP1 associates metabolic syndrome and breast carcinogenesis targeting multiple miRNAs. Oncotarget 7, 18798–18811 (2016).
Kawamura, K. et al. Pre-ovulatory LH/hCG surge decreases C-type natriuretic peptide secretion by ovarian granulosa cells to promote meiotic resumption of pre-ovulatory oocytes. Hum. Reprod. 26, 3094–3101 (2011).
Rossi, G., Macchiarelli, G., Palmerini, M. G., Canipari, R. & Cecconi, S. Meiotic spindle configuration is differentially influenced by FSH and epidermal growth factor during in vitro maturation of mouse oocytes. Hum. Reprod. 21, 1765–1770 (2006).
Coticchio, G. et al. Mouse oocyte meiotic resumption and polar body extrusion in vitro are differentially influenced by FSH, epidermal growth factor and meiosis-activating sterol. Hum. Reprod. 19, 2913–2918 (2004).
Qiao, J. & Feng, H. L. Extra- and intra-ovarian factors in polycystic ovary syndrome: impact on oocyte maturation and embryo developmental competence. Hum. Reprod. update 17, 17–33 (2011).
Hashemain, Z. et al. CYP19A1 Promoters Activity in Human Granulosa Cells: A Comparison between PCOS and Normal Subjects. Cell J. 24, 170–175 (2022).
Chen, X. et al. FASN-Mediated Lipid Metabolism Regulates Goose Granulosa Cells Apoptosis and Steroidogenesis. Front. Physiol. 11, 600 (2020).
Elis, S. et al. Cell proliferation and progesterone synthesis depend on lipid metabolism in bovine granulosa cells. Theriogenology 83, 840–853 (2015).
Hu, S. et al. Differential actions of diacylglycerol acyltransferase (DGAT) 1 and 2 in regulating lipid metabolism and progesterone secretion of goose granulosa cells. J. steroid Biochem. Mol. Biol. 202, 105721 (2020).
Shen, W. J., Azhar, S. & Kraemer, F. B. Lipid droplets and steroidogenic cells. Exp. cell Res. 340, 209–214 (2016).
Khor, V. K. et al. The proteome of cholesteryl-ester-enriched versus triacylglycerol-enriched lipid droplets. PLoS One 9, e105047 (2014).
Giovanni Artini, P. et al. Growth factors and folliculogenesis in polycystic ovary patients. Expert Rev. Endocrinol. Metab. 2, 215–223 (2007).
Shimano, H. et al. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J. Clin. Investig. 98, 1575–1584 (1996).
Sundqvist, A. et al. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 1, 379–391 (2005).
Bengoechea-Alonso, M. T. & Ericsson, J. The phosphorylation-dependent regulation of nuclear SREBP1 during mitosis links lipid metabolism and cell growth. Cell cycle 15, 2753–2765 (2016).
Li, Y. et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-induced Insulin Resistant Mice. Cell Metab. 13, 376–388 (2011).
Pan, J. X. et al. Aberrant expression and DNA methylation of lipid metabolism genes in PCOS: a new insight into its pathogenesis. Clin. Epigenetics 10, 6 (2018).
Franks, S., Stark, J. & Hardy, K. Follicle dynamics and anovulation in polycystic ovary syndrome. Hum. Reprod. update 14, 367–378 (2008).
Liu, Z. et al. Androgen Receptor Coregulator CTBP1-AS Is Associated With Polycystic Ovary Syndrome in Chinese Women: A Preliminary Study. Reprod. Sci. 22, 829–837 (2015).
Mason, H. D. et al. Estradiol production by granulosa cells of normal and polycystic ovaries: relationship to menstrual cycle history and concentrations of gonadotropins and sex steroids in follicular fluid. J. Clin. Endocrinol. Metab. 79, 1355–1360 (1994).
Erickson, G. F., Magoffin, D. A., Garzo, V. G., Cheung, A. P. & Chang, R. J. Granulosa cells of polycystic ovaries: are they normal or abnormal? Hum. Reprod. 7, 293–299 (1992).
Catteau-Jonard, S. et al. Anti-Mullerian hormone, its receptor, FSH receptor, and androgen receptor genes are overexpressed by granulosa cells from stimulated follicles in women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 93, 4456–4461 (2008).
Shimano, H. & Sato, R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol. 13, 710–730 (2017).
Horton, J. D., Goldstein, J. L. & Brown, M. S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 109, 1125–1131 (2002).
Yi, J., Zhu, J., Wu, J., Thompson, C. B. & Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl Acad. Sci. USA 117, 31189–31197 (2020).
Ferrarese, R. et al. ZBTB18 inhibits SREBP-dependent lipid synthesis by halting CTBPs and LSD1 activity in glioblastoma. Life Sci. Alliance 6, https://doi.org/10.26508/lsa.202201400 (2023).
Zhao, Z. et al. CtBP promotes metastasis of breast cancer through repressing cholesterol and activating TGF-beta signaling. Oncogene 38, 2076–2091 (2019).
Fujinaga, H., Yamoto, M., Shikone, T. & Nakano, R. FSH and LH up-regulate epidermal growth factor receptors in rat granulosa cells. J. Endocrinol. 140, 171–177 (1994).
Murphy, B. D. & Dobias, M. Homologous and heterologous ligands downregulate follicle-stimulating hormone receptor mRNA in porcine granulosa cells. Mol. Reprod. Dev. 53, 198–207 (1999).
Yang, J. & Chen, C. Hormonal changes in PCOS. J. Endocrinol. 261, https://doi.org/10.1530/JOE-23-0342 (2024).
Pratama, G. et al. Mechanism of elevated LH/FSH ratio in lean PCOS revisited: a path analysis. Sci. Rep. 14, 8229 (2024).
Berrahmoune, H. et al. Association between EGF and lipid concentrations: a benefit role in the atherosclerotic process? Clin. Chim. acta; Int. J. Clin. Chem. 402, 196–198 (2009).
Huang, J. et al. Epidermal Growth Factor Stimulates Fatty Acid Synthesis Mainly via PLC-γ1/Akt Signaling Pathway in Dairy Goat Mammary Epithelial Cells. Animals (Basel) 10, https://doi.org/10.3390/ani10060930 (2020).
Guo, F. F. et al. Downregulation of mitogen-activated protein kinases (MAPKs) in chronic ethanol-induced fatty liver. Toxicol. Mech. Methods 30, 407–416 (2020).
Huang, J. C. et al. HB-EGF induces mitochondrial dysfunction via estrogen hypersecretion in granulosa cells dependent on cAMP-PKA-JNK/ERK-Ca(2+)-FOXO1 pathway. Int. J. Biol. Sci. 18, 2047–2059 (2022).
Homburg, R. & Amsterdam, A. Polysystic ovary syndrome-loss of the apoptotic mechanism in the ovarian follicles? J. Endocrinol. Investig. 21, 552–557 (1998).
Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertility Sterility 81, 19–25 (2004).
Eckel, R. H., Grundy, S. M. & Zimmet, P. Z. The metabolic syndrome. Lancet 365, 1415–1428 (2005).
Alberti, K. G. et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 120, 1640–1645 (2009).
Zhou, B. F. Predictive values of body mass index and waist circumference for risk factors of certain related diseases in Chinese adults-study on optimal cut-off points of body mass index and waist circumference in Chinese adults. Biomed. Environ. Sci.: BES 15, 83–96 (2002).
He, Y. et al. Influence of metabolic syndrome on female fertility and in vitro fertilization outcomes in PCOS women. Am. J. Obstet. Gynecol. 221, 138.e131–138.e112 (2019).
Shi, Y. et al. Live birth after fresh embryo transfer vs elective embryo cryopreservation/frozen embryo transfer in women with polycystic ovary syndrome undergoing IVF (FreFro-PCOS): study protocol for a multicenter, prospective, randomized controlled clinical trial. Trials 15, 154 (2014).
Tamura, I. et al. Effects of Melatonin on the Transcriptome of Human Granulosa Cells, Fertilization and Blastocyst Formation. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms23126731 (2022).
Nomura, M. et al. Preferable correlation to blastocyst development and pregnancy rates with a new embryo grading system specific for day 3 embryos. J. Assist. Reprod. Genet. 24, 23–28 (2007).
Gardner, D. K., Lane, M., Stevens, J., Schlenker, T. & Schoolcraft, W. B. Blastocyst score affects implantation and pregnancy outcome: towards a single blastocyst transfer. Fertil. Steril. 73, 1155–1158 (2000).
Xu, X. et al. Impaired telomere length and telomerase activity in peripheral blood leukocytes and granulosa cells in patients with biochemical primary ovarian insufficiency. Hum. Reprod. 32, 201–207 (2017).
Yin, H. et al. Extracellular matrix protein-1 secretory isoform promotes ovarian cancer through increasing alternative mRNA splicing and stemness. Nat. Commun. 12, 4230 (2021).
To, S. Q., Knower, K. C., Cheung, V., Simpson, E. R. & Clyne, C. D. Transcriptional control of local estrogen formation by aromatase in the breast. J. Steroid Biochem. Mol. Biol. 145, 179–186 (2015).
Zhu, Z., Guo, L., Nurzat, Y., Xu, H. & Zhang, Y. CCL2-Enhanced Adipogenesis and Angiogenesis of hADSC and HUVEC co-culture system in adipose tissue engineering. J. Tissue Eng. Regen. Med. https://doi.org/10.1002/term.3264 (2021).
Hsu, J. Y., Lin, H. H., Chyau, C. C., Wang, Z. H. & Chen, J. H. Aqueous Extract of Pepino Leaves Ameliorates Palmitic Acid-Induced Hepatocellular Lipotoxicity via Inhibition of Endoplasmic Reticulum Stress and Apoptosis. Antioxidants (Basel) 10, https://doi.org/10.3390/antiox10060903 (2021).
Creff, A. et al. Evidence that endosperm turgor pressure both promotes and restricts seed growth and size. Nat. Commun. 14, 67 (2023).
Tsai, K. et al. Differential expression of mTOR related molecules in the placenta from gestational diabetes mellitus (GDM), intrauterine growth restriction (IUGR) and preeclampsia patients. Reprod. Biol. 21, 100503 (2021).
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (81402153for Chunxia Ren, and 81772789, 82473079 for Gong Yang).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Yong Wang, Haiyang Wang and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Jun Wei Pek and Joao Valente.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kong, Y., Yang, G., Feng, X. et al. CTBP1 links metabolic syndrome to polycystic ovary syndrome through interruption of aromatase and SREBP1. Commun Biol 7, 1174 (2024). https://doi.org/10.1038/s42003-024-06857-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06857-4
