
Abstract
Relieving renal fibrosis are promising therapeutic strategies for chronic kidney disease (CKD). Here we showed that decreased homeobox A2 (HOXA2) expression with excessive ER stress activation were observed in the renal tissues of UUO mice established on male C57BL/6 mice and TGF-β1-induced human proximal tubular epithelial cells (HK-2 cells). After transfected HOXA2 overexpression plasmids into HK-2 cells and administered adeno-associated virus (AAV) containing HOXA2 to UUO mice, the expression of ER stress markers such as ATF6, GRP78 and CHOP decreased, renal dysfunction and fibrosis were significantly attenuated. Mechanistically, HOXA2 binds to the SIRT1 promoter, enhancing SIRT1 transcription and deacetylase activity, which led to ATF6 deacetylation and downregulation. The protective effect of HOXA2 was confirmed against the ER stress agonist thapsigargin. Moreover, DNMT1-mediated promoter methylation was identified as a potential mechanism for HOXA2 suppression in fibrosis. Targeting HOXA2 maybe an innovative therapeutic strategy for fibrosis treatment in CKD.

Similar content being viewed by others


Introduction
The incidence of chronic kidney disease (CKD), which is a serious threat to public health, is increasing because of the increasing prevalence of hypertension, diabetes and obesity, as well as the surge of population ageing1,2. Patients with CKD have a high risk of acute kidney injury (AKI), and the repeated development of AKI also promotes the progression of CKD (AKI-to-CKD transition), contributing to irreversible tubulointerstitial injury and fibrosis and eventually leading to end-stage renal failure3. The contribution of CKD to global morbidity and mortality is increasing rapidly4, highlighting the urgency of finding new targets and treatments.
Renal interstitial fibrosis (RIF), a hallmark of different types of progressive CKD, is characterized by the deposition of excessive amounts of extracellular matrix (ECM) components (such as fibronectin and collagen), the accumulation of fibroblasts and the loss of renal tubular cells5. In the progression of RIF and CKD, epithelial‒mesenchymal transition (EMT) is a vital biological process and is characterized by renal tubular epithelial cells (TECs) undergoing the transformation of the mesenchymal phenotype and acquiring myofibroblast function6. Studies have shown that the transforming growth factor-beta (TGF-β1)/Smad and Wnt/β-catenin pathways are the major factors involved in triggering EMT and leading to RIF7,8. However, there is currently no effective treatment for RIF.
Notably, studies have confirmed the close relationship between RIF and endoplasmic reticulum (ER) stress9,10. In response to environmental stimuli such as reactive oxygen species (ROS), ER stress is a cytoprotective mechanism that involves the accumulation of misfolded proteins in the ER; ER stress triggers the unfolded protein response (UPR) to relieve protein misfolding stress in the ER through upregulation of the ER chaperone glucose-related protein 78 (GRP78) via the activation of 3 major sensors: PKR-like ER kinase (PERK), inositol requiring enzyme-1 (IRE1) and activating transcription factor 6 (ATF6)11. When ER stress induces the UPR, ATF6 is activated and translocated into the nucleus to transcriptionally regulate the transcription of ER proteins, including GRP78 and C/EBP homologous protein (CHOP)12,13. However, studies have suggested that chronic or prolonged ER stress leads to apoptosis, inflammation, EMT, and renal fibrosis10,14. For example, excessive ER stress stimulates apoptosis via the ATF6-CHOP pathway in human renal TECs under ischaemia‒reperfusion injury-induced AKI conditions13. The inhibition of ATF6 results in the suppression of ER stress and EMT in high-glucose-treated HK2 cells15. Importantly, ER stress causes various problems in the kidneys during the development of AKI and CKD; in particular, it contributes to renal fibrosis through multiple signalling pathways, such as EMT, TGF-β1 and oxidative stress, in CKD, and ERS inhibition can alleviate the progression of renal fibrosis16,17,18,19. On the basis of the close relationship between AKI and CKD, focusing on ERS inhibition may be a promising therapeutic strategy against renal fibrosis in patients with CKD.
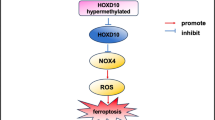
Homeobox A2 (HOXA2), a member of the extremely conserved HOX gene family, plays an important role in coding transcription factors with well-documented functions in embryonic development and organogenesis20,21, and HOXA2 participates in the regulation of multiple cell behaviours, such as proliferation, invasion, metastasis and EMT22,23,24. An increasing number of studies have recently revealed that the HOX gene family is involved in fibrosis of multiple organs, including the heart, lung, liver and kidney25,26,27,28. Our latest study revealed that HOXD10 is dedicated to relieving renal fibrosis through ferroptosis induced by NOX429. Other studies have reported that the loss of HOXA5 via abnormal DNA methylation contributes to kidney fibrosis through the JAG1/NOTCH signalling pathway30, indicating the potential role of the HOX gene family in renal fibrosis. Interestingly, the severe chronic liver fibrosis induced by hepatitis B is closely associated with the hypermethylation of HOXA231. HOXA2 appears to be a key regulator of fibrosis, yet to our knowledge, the precise role of HOXA2 in renal fibrosis in CKD remains unexplored.
Sirtuins are a family of nicotinamide adenine dinucleotide (NAD+)-dependent protein deacetylases that participate in multiple biological processes by deacetylating various types of proteins, including histones and nonhistone proteins, and seven different sirtuins, named SIRT1-7, have been identified32. Sirtuin 1 (SIRT1), which is the most widely known sirtuin in the kidney, plays a key protective role against the progression of renal interstitial fibrosis through controlling many processes, such as oxidative stress, ageing, lipid metabolism, EMT, and inflammation32. SIRT1 has been confirmed to alleviate ER stress through the deacetylation of key ER stress molecules, such as GRP78, PERK, IRE1, and ATF633. Studies have reported that SIRT1 upregulation can inhibit ER stress-mediated renal fibrosis in TGF-β1-induced human proximal tubular cells (HK-2 cells) and kidneys of mice with unilateral ureteral obstruction (UUO)34,35,36. We predicted the presence of HOXA2 binding sites within the promoter region of SIRT1. Surprisingly, nothing is known about the mechanistic link between HOXA2 and SIRT1. It is therefore interesting to explore whether HOXA2 could regulate ER stress-mediated renal fibrosis via SIRT1 in CKD.
In this study, we explored the hypothesis that HOXA2 acts on the SIRT1 promoter to promote its transcriptional ability, hence exerting a protective effect on renal fibrosis induced by ER stress in both UUO-induced animal models and TGF-β1-treated HK-2 cell models, and attempted to elucidate the underlying molecular mechanism. Furthermore, we examined the relationship between the expression level of HOXA2 and hypermethylation. This study provides initial evidence suggesting that HOXA2 may be a potential target for the diagnosis and treatment of renal fibrosis in patients with CKD.
Results
HOXA2 expression was low in renal biopsies from CKD patients with renal fibrosis
HOXA2 expression was measured in renal tissues collected from CKD patients and normal control renal tissues from nephrectomies performed on tumour patients. First, we performed H&E staining, and the results revealed renal pathological alterations, such as interstitial oedema, tubular dilatation, epithelial desquamation and loss of brush border, with markedly increased tubular injury scores in CKD patients (Fig. 1A). Prosirius red and Masson’s trichrome staining revealed a significant increase in collagen deposition, which indicated renal fibrosis in CKD patients (Fig. 1B, C). Next, IHC staining was performed and revealed that HOXA2 expression was low in renal tissues from patients with CKD and that compared with the glomeruli, HOXA2 was distributed mainly in the tubulointerstitial area (Fig. 1D). We further utilized dual immunofluorescence using an antibody against the proximal tubule marker Lotus tetragonolobus lectin (LTL) to bind to HOXA2 in kidney tissues. The IF results revealed that HOXA2 was expressed in proximal tubule areas in renal tissues from normal controls and CKD patients (Supplementary Fig. S2). These results indicate that HOXA2 may be related to CKD renal fibrosis.

A H&E staining of renal tissues from normal controls and CKD patients and relative tubular injury score. B Picro-sirius red stainings of renal tissues from normal controls and CKD patients and semiquantitative analysis of average optical density of fibrotic area. C Masson’s trichrome staining of renal tissues from normal controls and CKD patients and semiquantitative analysis of average optical density of fibrotic area. D IHC staining of HOXA2 in renal tissues from normal controls and CKD patients and average optical density of HOXA2. The scale bar represents 50 µm. Magnification is 400×. Normal control, normal kidney biopsies (n = 10). CKD, kidney biopsies from chronic kidney disease patients (n = 10). **P < 0.01.
TGF-β1 induces decreased HOXA2 expression by upregulating promoter hypermethylation in HK-2 cells
To further explore the role of HOXA2 in CKD renal fibrosis, we used TGF-β1 to stimulate HK-2 cells to establish an in vitro renal fibrosis model as described by others and our group previously29,37. The results of the qRT‒PCR and Western blot analyses revealed that both the mRNA and protein expression of HOXA2 decreased significantly in the TGF-β1-stimulated HK-2 cells and that the expression of HOXA2 did not differ between the control group and the vehicle group (Fig. 2A, B). The IF results also revealed that HOXA2 was expressed in both the cytoplasm and nucleus of HK-2 cells (Supplementary Fig. S3). BSP analysis revealed notable hypermethylation of the HOXA2 promoter in the TGF-β1-treated HK-2 cells compared with the vehicle-treated cells (Fig. 2C). DNA methyltransferase 1 (DNMT1) is crucial for maintaining the DNA methylation pattern, and studies have reported that DNMT1 can bind to the HOXA2 promoter and regulate DNA methylation levels in dental follicle stem cells38. To verify whether HOXA2 was regulated by DNA methylation in TGF-β1-stimulated HK-2 cells, the activity of DNMT1 was tested, and the results revealed that DNMT activity was upregulated significantly after TGF-β1 treatment and was effectively inhibited by the DNA methylation transferase inhibitor drug 5-Aza in HK-2 cells (Fig. 2D). The ChIP results further confirmed that DNMT1 was enriched in the HOXA2 promoter region and that TGF-β1 enhanced DNMT1 enrichment in HK-2 cells (Fig. 2E). To further verify the role of DNMT1 in HOXA2 promoter methylation, DNMT1 siRNA was used to transfect HK-2 cells. The results of the qRT‒PCR analysis revealed that the mRNA expression of DNMT1 significantly increased after treatment with TGF-β1 and markedly decreased after treatment with DNMT1 siRNA (Fig. 2F). In TGF-β1-stimulated HK-2 cells, Western blot analysis also revealed that the decreased protein expression of HOXA2 increased significantly after DNMT1 siRNA transfection (Fig. 2G). Moreover, 5-Aza also increased the mRNA and protein expression of HOXA2 in a dose-dependent manner (Fig. 2H, I). These results suggest that TGF-β1 promotes HOXA2 promoter hypermethylation, followed by the inhibition of HOXA2 expression in HK-2 cells.

A qRT-PCR shows that the mRNAs of HOXA2 are downregulated in TGF-β1-induced HK-2 cells. B Western blot analysis shows that the proteins of HOXA2 are downregulated in TGF-β1-induced HK-2 cells. C BSP results of the HOXA2 promoter methylation in HK-2 cells treated with or without TGF-β1. D DNMT1 activity detection. E The enrichment of DNMT1 on the HOXA2 promoter region in HK-2 cells treated with or without TGF-β1 by ChIP assays. F The mRNA expression levels of DNMT1 were assessed using qRT-PCR after forty-eight hours treatment with DNMT1 siRNA in TGF-β1-induced HK-2 cells. G The protein expression levels of HOXA2 were assessed using Western blot analysis after forty-eight hours treatment with DNMT1 siRNA in TGF-β1-induced HK-2 cells. H qRT-PCR shows that the HOXA2 mRNA is recovered after treatment with demethylation drug 5-Aza for 48 h in TGF-β1-induced HK-2 cells. I Western blot analysis shows that the HOXA2 protein is recovered after treatment with demethylation drug 5-Aza for 48 h in TGF-β1-induced HK-2 cells. In all panels, the data are representative of three independent experiments (n = 3). Data are presented as the mean ± SD. **P < 0.01, NS not significant.
HOXA2 directly promotes SIRT1 transcriptional activity
With the UCSC Genome Browser and JASPAR tools, we predicted 2 HOXA2 binding sites in the SIRT1 promoter, indicating that HOXA2 may target SIRT1 (Fig. 3A). To determine the role of HOXA2 in SIRT1 expression, a HOXA2 overexpression plasmid was constructed and used to transfect HK-2 cells. The results of the qRT‒PCR and Western blot analyses revealed that both the mRNA and protein expression of HOXA2 and SIRT1 significantly decreased after TGF-β1 treatment but markedly increased after treatment with the HOXA2 overexpression plasmid (Fig. 3B–D). On the basis of the predicted binding sites, we performed a luciferase reporter assay to confirm the correlation between HOXA2 and SIRT1. After treatment with the HOXA2 overexpression plasmid, the activity of the SIRT1 promoter increased in the WT group, and mutations at site 1, but not site 2, did not significantly affect promoter activity, indicating that HOXA2 targeted the SIRT1 promoter at site 1 in HK-2 cells stimulated with or without TGF-β1 (Fig. 3E). Mechanistically, we established HOXA2-FLAG-tagged fusion expression vectors to detect the binding of exogenous HOXA2 to the SIRT1 promoter by ChIP‒qPCR. The IF results revealed that the vectors were expressed successfully in HK-2 cells (Supplementary Fig. S4). The ChIP results suggested that HOXA2 bound to the promoter of SIRT1 (Fig. 3F). These data reveal that HOXA2 binds directly to the SIRT1 promoter to transcriptionally activate SIRT1 gene expression in HK-2 cells.

A JASPAR tool predicted HOXA2 binding sites on the promoter of SIRT1. B The mRNA expression levels of HOXA2 were assessed using qRT-PCR after forty-eight hours treatment with HOXA2 overexpression plasmids in TGF-β1-induced HK-2 cells. C The mRNA expression levels of SIRT1 were assessed using qRT-PCR after forty-eight hours treatment with HOXA2 overexpression plasmids in TGF-β1-induced HK-2 cells. D The proteins expression levels of HOXA2 and SIRT1 were assessed using western blot analysis after forty-eight hours treatment with HOXA2 overexpression plasmids in TGF-β1-induced HK-2 cells. E Luciferase reporter analysis elucidated the influence of HOXA2 overexpression on the luciferase activity of the SIRT1 promoter reporter in HK-2 cells treated with or without TGF-β1. F ChIP analysis of HOXA2-FLAG tagged fusion vector levels at the SIRT1 promoter in HK-2 cells. ChIP results were analyzed via qRT–PCR using SIRT1 promoter-specifc primers and are expressed as the percentage of the input. In all panels, the data are representative of three independent experiments (n = 3). Data are presented as the mean ± SD. **P < 0.01, NS not significant.
HOXA2 inhibits oxidative stress, ECM accumulation and EMT in TGF-β1-induced HK-2 cells by suppressing ER stress
To investigate the role of HOXA2 in ER stress, ECM accumulation and EMT, we measured the expression of ER stress marker proteins, such as CHOP and GRP78; ECM components, such as fibronectin (FN) and collagen I (COL I); and the EMT markers α-SMA and E-cadherin. Western blot analysis revealed that the expression of CHOP, GRP78, FN, COL I and α-SMA significantly increased and that the expression of E-cadherin markedly decreased in TGF-β1-treated HK-2 cells, whereas the expression of these proteins was partly restored by HOXA2 overexpression (Fig. 4A). A CCK-8 assay demonstrated that HOXA2 overexpression suppressed the increase in HK-2 cell proliferation induced by TGF-β1 (Fig. 4B). Moreover, the TEM results revealed that the mitochondrial and ER morphological structures were obviously abnormal in the TGF-β1 group, as indicated by a notable decrease in ER integrity and an increase in the number of fragmented mitochondria. However, HOXA2 overexpression improved ER and mitochondrial damage in TGF-β1-induced HK-2 cells (Fig. 4C). Additionally, H2-DCFDA staining revealed that the TGF-β1-induced overproduction of ROS was significantly inhibited by HOXA2 overexpression in HK-2 cells (Fig. 4D). We further applied an agonist of ER stress, thapsigargin, to establish an ER stress model39 in HK-2 cells to explore the relationship between HOXA2 and ER stress and fibrosis. Western blot analysis revealed that the expression of ER stress markers, including CHOP and GRP78; the expression of the ECM components FN and COL I; and the expression of the EMT marker α-SMA and E-cadherin (another EMT marker) was markedly decreased in TGF-β1-treated HK-2 cells, whereas the expression of these proteins was partly restored by HOXA2 overexpression (Fig. 4E). These findings suggest that HOXA2 may inhibit ECM accumulation and EMT by suppressing ER stress in HK-2 cells.

A The proteins expression levels of CHOP, GRP78, E-cadherin, α-SMA, FN and COL I were assessed using Western blot analysis after forty-eight hours treatment with HOXA2 overexpression plasmids in TGF-β1-induced HK-2 cells. B Cell proliferation was detected with a CCK-8 assay after transfection with the HOXA2 overexpression plasmidsat 0, 24, 48 and 72 h. C Representative images of HOXA2 overexpression improved ER and mitochondrial injury in TGF-β1-induced HK-2 cells were observed by TEM (×20.0k). Bar = 500 nm. D H2-DCFDA staining showed that HOXA2 overexpression plasmids alleviated TGF-β1-induced ROS generation in HK-2 cells (×200). Bar = 100 μm. E The proteins expression levels of CHOP, GRP78, E-cadherin, α-SMA, FN and COL I were assessed using Western blot analysis after forty-eight hours treatment with HOXA2 overexpression plasmids in thapsigargin-induced HK-2 cells. In all panels, the data are representative of three independent experiments (n = 3). Data are presented as the mean ± SD. **P < 0.01, NS not significant.
HOXA2 promotes SIRT1-induced deacetylation of ATF6, followed by the suppression of ATF6 expression and activation
SIRT1, a histone deacetylase, has been confirmed to attenuate ER stress via the ATF6 pathway in diabetic cardiomyopathy40. An ER stress sensor, ATF6 is cleaved from its full-length (~90 kDa) form to its active cleaved form (~50 kDa) in a thapsigargin-induced ER stress environment41. To explore the mechanism through which HOXA2 regulates ER stress, we first detected ATF6 expression by Western blot analysis, and the results revealed that HOXA2 overexpression inhibited the TGF-β1-induced increase in total ATF6 protein levels and the subsequent nuclear translocation and activation of ATF6 (Fig. 5A). A thapsigargin-induced ER stress HK-2 cell model was used to further verify the relationship between HOXA2 and ER stress. Western blot analysis revealed that HOXA2 overexpression inhibited the thapsigargin-induced increase in total ATF6 protein levels and the subsequent nuclear translocation and activation of ATF6 (Fig. 5B). These results suggest that HOXA2 may inhibit ATF6 expression and activation during ER stress.

A The proteins expression levels of total ATF6, nuclear ATF6, and cytoplasmic ATF6 were assessed using Western blot analysis after forty-eight hours treatment with HOXA2 overexpression plasmids in TGF-β1-induced HK-2 cells. B The proteins expression levels of total ATF6, nuclear ATF6, and cytoplasmic ATF6 were assessed using Western blot analysis after forty-eight hours treatment with HOXA2 overexpression plasmids in thapsigargin-induced HK-2 cells. C Co-IP assay confimed an interaction between SIRT1 and active form ATF6 in HK-2 cells nucleus. D The mRNA expression levels of SIRT1 were assessed using qRT-PCR after forty-eight hours treatment with SIRT1 siRNA in control HK-2 cells. E The protein expression levels of SIRT1 were assessed using Western blot analysis after forty-eight hours treatment with SIRT1 siRNA in control HK-2 cells. F SIRT1 deacetylase activity in each HK-2 cells group. G Acetylation of active form ATF6 in nucleus of each HK-2 cells group was determined by IP with an anti-ATF6 antibody, followed by the immunoblotting analysis with an acetylated-lysine and ATF6 antibody. In all panels, the data are representative of three independent experiments (n = 3). Data are presented as the mean ± SD. **P < 0.01, NS not significant.
Next, we explored the possible mechanism underlying the interaction between HOXA2 and ATF6. We could not predict possible HOXA2 binding sites in the ATF6 promoter with the UCSC Genome Browser and JASPAR tool. To more accurately explore the relationship between HOXA2 and ATF6, we used HEK293 cells for subsequent detection. Western blot analysis revealed that the expression of HOXA2 in HEK293 cells was negligible (Supplementary Fig. S5A). One study reported that STAT6 could bind to the ATF6 promoter and positively induce ATF6 mRNA transcription42. Thus, a STAT6 overexpression plasmid was used as a positive control. The results of the qRT‒PCR analysis revealed that the mRNA expression of STAT6 significantly increased after transfection with the STAT6 overexpression plasmid (Supplementary Fig. S5B). A luciferase reporter assay was performed to confirm the correlation between HOXA2 and ATF6 expression, and the results revealed that compared with that in the empty vector group, the activity of the ATF6 promoter increased in the STAT6 overexpression plasmid group, whereas no significant difference in promoter activity was detected in the HOXA2 overexpression plasmid group, indicating that HOXA2 could not bind directly to the ATF6 promoter (Supplementary Fig. S5C). These results suggest that HOXA2 may indirectly affect the activation of ATF6.
Studies have also shown that SIRT1 can control the deacetylation of ATF6, which is an important posttranslational mechanism responsible for regulating the activity and stability of the protein of ATF6 in HepG2 cells43. Co-IP revealed an interaction between SIRT1 and the active form of ATF6 in the HK-2 cell nucleus (Fig. 5C). Next, we established a SIRT1 siRNA to determine the relationship between SIRT1 and ATF6. The results of the qRT‒PCR and Western blot analyses revealed that both the mRNA and protein expression of SIRT1 significantly decreased after SIRT1 siRNA treatment in HK-2 cells (Fig. 5D, E). We found that SIRT1 deacetylase activity was significantly decreased in TGF-β1-induced HK-2 cells but markedly increased after transfection with the HOXA2 overexpression plasmid. After cotransfection with both SIRT1 siRNA and the HOXA2 overexpression plasmid, SIRT1 deacetylase activity decreased significantly compared with that after treatment with the HOXA2 overexpression plasmid alone (Fig. 5F). We measured the acetylation levels of the active form of ATF6 by IP and subsequent Western blotting with an anti-acetyl-lysine antibody in the HK-2 cell nucleus and found that the acetylation levels of the active form of ATF6 significantly increased after TGF-β1 treatment and markedly decreased after transfection with the HOXA2 overexpression plasmid. However, a rescue experiment revealed that after cells were cotransfected with SIRT1 siRNA and the HOXA2 overexpression plasmid, the degree of ATF6 acetylation increased significantly compared with that after cells were transfected with the HOXA2 overexpression plasmid alone. The acetylation levels of ATF6 in each group were consistent with the trend of ATF6 activation and opposite to the trend of SIRT1 activity in all groups of HK-2 cells (Fig. 5G). Thus, these findings imply that HOXA2 indirectly inhibits the expression and activation of ATF6 by promoting SIRT1-induced deacetylation of ATF6.
HOXA2 overexpression relieves kidney injury and collagen accumulation in UUO mice
Next, we verified the function of HOXA2 in mice. UUO was performed to establish a renal fibrosis model as we reported previously29. In 8-week-old mice, AAV vectors encoding HOXA2 were injected via the tail vein to induce the overexpression of HOXA2, followed by UUO surgery at 10 weeks of age, and all blood and kidney samples were collected at 12 weeks of age (Fig. 6A). Fluorescence microscopy revealed that AAV-mediated GFP expression was successful in the tubulointerstitium (Supplementary Fig. S6). HOXA2 overexpression significantly reversed the deterioration of mouse kidney function, as indicated by the marked inhibition of the increased serum creatinine and BUN levels in the UUO group after AAV-HOXA2 (+) treatment (Fig. 6B, C). H&E staining revealed that AAV-HOXA2(+) mice exhibited markedly improved UUO-induced tubular atrophy and dilation. Masson’s trichrome and picrosirius red staining indicated that the UUO-induced notable accumulation of collagen deposition and fibrosis was significantly inhibited after AAV-HOXA2 (+) treatment (Fig. 6D). These data suggest that HOXA2 overexpression may alleviate the deterioration of kidney function and reduce kidney collagen deposition caused by UUO in mice.

A Schematic of the mouse experimental protocol including AAV-HOXA2 (+) treatment and UUO model establishment. B Serum creatinine and (C) Serum BUN in each group at 12 weeks of age. D Representative images of H&E, Masson’s trichrome and picro-sirius red stainings and relative tubular injury score and relative collagen deposition area of each group (×200). Bar = 100 μm. Data are presented as the mean ± SD of each mouse (n = 6 mice in each group). **P < 0.01, NS not significant.
HOXA2 overexpression transcriptionally promotes renal SIRT1 expression in UUO mice
To clarify the role of HOXA2 in SIRT1 activity, we detected SIRT1 expression. The mRNA and protein levels of HOXA2 and SIRT1 were markedly decreased and were notably increased, respectively, in renal tissues from UUO mice compared with those from sham mice after AAV-HOXA2 (+) treatment (Fig. 7A, B). IHC staining revealed decreased expression levels of HOXA2 and SIRT1 and increased expression levels in renal tissues from UUO mice after AAV-HOXA2 (+) treatment. Moreover, we observed that HOXA2 was expressed in both the tubulointerstitial and glomerulus areas by IHC, but more HOXA2 was distributed in the tubulointerstitial area (Fig. 7C). We further utilized dual immunofluorescence using antibodies against segment-specific tubular cell markers, which are markers for collecting duct epithelium Dolichos biflorus agglutinin (DBA), a marker for proximal tubule Lotus tetragonolobus lectin (LTL) and a marker for distal convoluted tubule-peanut agglutinin (PNA), to costain with HOXA2 in kidney tissues from sham mice. The IF results revealed that HOXA2 was expressed in all of the above areas (Fig. 7D). These results indicate that HOXA2 may be enriched in the renal tubulointerstitial area and that HOXA2 overexpression may transcriptionally promote SIRT1 expression in the kidneys of mice.

A qRT-PCR analysis detected the mRNA expressions of HOXA2 and SIRT1 in the mice renal tissues of each group. B Western blot analysis detected the protein expression of HOXA2 and SIRT1 in the mice renal tissues of each group. C IHC detection of HOXA2 and SIRT1 in the mice renal tissues of each group and average optical density of each index (×200). Bar = 100 μm. D Dual immunofluorescent staining of HOXA2 and segment-specific tubular markers in the mice renal tissues from sham group. LTL, lotus tetragonolobus lectin; PNA, peanut agglutinin; DBA, dolichos biflorus agglutinin. Arrows indicate positive tubules with colocalization of HOXA2 and specific tubular markers (×200). Bar = 100 μm. Data are presented as the mean ± SD of each mouse (n = 6 mice in each group). **P < 0.01, NS not significant.
HOXA2 overexpression attenuated renal ER stress and oxidative stress in UUO mice
Studies have shown that chronic ER stress promotes a secondary increase in reactive oxygen species (ROS) production, leading to cell death44. We measured ER stress and oxidative stress conditions in mice and found that the protein expression of the ER stress-related markers ATF6, GRP78 and CHOP significantly increased in the renal tissues of UUO mice but partly decreased after AAV-HOXA2 (+) treatment (Fig. 8A). Consistent with the protein expression changes, the IHC results also revealed that the high expression of ATF6, GRP78 and CHOP in the renal tissues of UUO mice decreased after AAV-HOXA2 (+) treatment (Fig. 8B). DHE staining revealed increased ROS generation, which was attenuated by AAV-HOXA2 (+) treatment in the UUO group (Fig. 8C). Both the GSH/GSSG ratio and the SOD level were significantly lower in the UUO group than in the sham group and were markedly increased after the administration of AAV-HOXA2 (+) (Fig. 8D, E). In contrast, MDA levels were markedly elevated in the UUO group, and AAV-HOXA2 (+) treatment successfully blocked this increase (Fig. 8F). These results suggest that HOXA2 overexpression can reduce ER stress and oxidative stress in the kidneys of UUO mice.

A Western blot analysis detected the protein expressions of ATF6, GRP78 and CHOP in the mice renal tissues of each group. B IHC detection in the mice renal tissues of each group and average optical density of each index. ATF6, GRP78 and CHOP (×200, Bar = 100 μm). C DHE staining showed that AAV-HOXA2 (+) treatment alleviates ROS generation in UUO mice kidneys (×400). Bar = 50 μm. D IGSH/GSSG ratio, (E) SOD and (F) MDA levels in the mice renal tissues of each group. Data are presented as the mean ± SD of each mouse (n = 6 mice in each group). **P < 0.01, NS not significant.
HOXA2 overexpression attenuated renal EMT and fibrosis in UUO mice
Finally, we detected the expression of EMT and fibrosis markers to investigate the antifibrotic function of HOXA2 in UUO mice. In the renal tissues of UUO mice, the protein expression of E-cadherin decreased and was notably elevated by AAV-HOXA2 (+) treatment; moreover, the protein expression of α-SMA, COL I and FN were all high and were markedly decreased by AAV-HOXA2 (+) treatment (Fig. 9A). Consistent with the Western blot results, the IHC results also revealed that the high expression of α-SMA, COL I and FN in the renal tissues of UUO mice decreased after AAV-HOXA2 (+) treatment, whereas the low expression of E-cadherin in the renal tissues of UUO mice increased after AAV-HOXA2 (+) treatment (Fig. 9B). These findings suggest that UUO-induced renal EMT and fibrosis progression is prevented by HOXA2 overexpression.

A Western blot analysis detected the protein expressions of E-cadherin, α-SMA, COL I and FN in the mice renal tissues of each group. B IHC detection of E-cadherin, α-SMA, COL I and FN in the mice renal tissues of each group and average optical density of each index (×200). Bar = 100 μm. Data are presented as the mean ± SD of each mouse (n = 6 mice in each group). **P < 0.01, NS not significant.
Discussion
CKD is the main cause of end-stage renal failure, and renal fibrosis is a common pathological condition in CKD patients; however, to date, there is no effective treatment available for renal fibrosis. Accumulating studies have shown that ER stress plays a vital role in the occurrence and development of renal fibrosis in CKD44,45. The inhibition of ER stress can alleviate renal fibrosis; thus, new avenues of research and therapeutic advances in renal fibrosis management have been identified16. However, the underlying mechanism of ER stress in renal fibrosis remains largely unknown. In the current study, we provide the first evidence that HOXA2 plays a protective role against renal fibrosis caused by excessive ER stress in UUO model mice and TGF-β1-induced HK-2 cells.
ER stress is often accompanied by the overproduction of ROS, causing cell death and renal fibrosis46,47. In this study, we established UUO mice, which exhibited severe tubulointerstitial injury (such as brush border loss, interstitial oedema, renal tubular dilation and tubular epithelial cell necrosis/loss), as reported by others and our group19,29. Activation of renal ER stress and oxidative stress, shown as the upregulation of ATF6, CHOP, GRP78 and ROS production, were accompanied by EMT and fibrosis, as indicated by the upregulation of COL I, FN, and α-SMA expression and the downregulation of E-cadherin expression in the kidneys of UUO mice. Similar changes in the expression of the above markers of ER stress, oxidative stress, EMT and ECM accumulation were also observed in HK-2 cells stimulated with TGF-β1, accompanied by ER and mitochondrial damage, as shown by TEM. HOXA2 overexpression inhibited the activation of ER stress and alleviated EMT and fibrosis in the kidneys of UUO mice and HK-2 cells treated with TGF-β1. This effect was accompanied by a significant upregulation of SIRT1 expression. The results of luciferase reporter assays and ChIP assays suggested that HOXA2 binds to the promoter of SIRT1 and positively regulates its expression. SIRT1, which is a histone deacetylase (HDAC), is abundantly expressed in renal tubules, participates in the regulation of kidney physiology and protects against renal fibrosis, oxidative damage, inflammation and apoptosis through the deacetylation of numerous transcription factors48,49,50. Studies have shown that SIRT1 can control the stability of the protein and activity of ATF6 through regulation of the deacetylation of ATF6 and subsequent inactivation of ATF6 in nonalcoholic fatty liver disease43. TGF-β1 can increase the total protein level of ATF6, promote the nuclear translocation of ATF6 in HK-2 cells, and subsequently increase the expression of the ER stress-responsive genes GRP78 and CHOP51. In the present study, we found that HOXA2 restored reduced SIRT1 activity in TGF-β1-treated HK-2 cells. Co-IP results also revealed that SIRT1 interacted with the active form of ATF6 and promoted its deacetylation, resulting in the downregulation of ATF6 expression in TGF-β1-treated HK-2 cells. Moreover, our results revealed that the TGF-β1-induced upregulation of total ATF6 protein levels and the subsequent nuclear translocation and activation of ATF6, as well as the increased expression of CHOP and GRP78, were inhibited by HOXA2 overexpression plasmids. We speculated that ATF6 may be activated into the nucleus (cleaved form) under ER stress and subsequently promote the transcription of ATF6, further leading to an increase in the expression of the full-length form of ATF6. However, our results also revealed that HOXA2 could not directly regulate ATF6 expression or activation but indirectly inhibited the expression and activation of ATF6 by promoting SIRT1-induced deacetylation of ATF6. These findings further indicated that ER stress may be involved in the mechanism through which the HOXA2/SIRT1 axis exerts an antifibrotic effect in CKD.
Members of the HOX gene family are homologous domain transcription factors that play key roles in animal embryo shape and organogenesis control52. HOX genes are divided into four genomic clusters, namely, HOX A, B, C, and D. The HOXA and HOXD families are critical for the development of renal fibrosis28,29,30. HOXA2 belongs to the HOXA cluster, whose members play crucial roles in cell proliferation, embryonic development, and cancer53. Recent studies have revealed that HOXA2 is a tumour suppressor in breast cancer that may be epigenetically regulated by DNA methylation and lead to low expression54. The hypermethylation of HOXA2 has been confirmed to be related to liver fibrosis31; however, the relationship between HOXA2 methylation and CKD-related fibrosis remains unknown. In this study, we first found notable hypermethylation of the HOXA2 promoter in an in vitro model of CKD renal fibrosis via BSP analysis. DNMT1 mediates gene suppression by increasing promoter methylation55, and we discovered that DNMT1 siRNA could increase the protein expression of HOXA2 in TGF-β1-induced HK-2 cells. In terms of the underlying mechanism, ChIP assays confirmed that DNMT1 is enriched in the HOXA2 promoter region and that TGF-β1 enhances DNMT1 enrichment in HK-2 cells. Moreover, 5-Aza inhibited DNMT activity and increased the mRNA and protein expression of HOXA2 in TGF-β1-induced HK-2 cells. These data indicate that HOXA2 is regulated by methylation during the progression of fibrosis in CKD.
In this study, we first demonstrated that the HOXA2 promoter is hypermethylated and that its expression is low in the context of CKD-related fibrosis. HOXA2 positively regulates SIRT1 expression transcriptionally and thus promotes the deacetylation and inactivation of ATF6, ultimately alleviating tubular injury and tubulointerstitial fibrosis by inhibiting ER stress (see the graphical abstract). Although we provide the first evidence that HOXA2 plays a vital role in renal fibrosis, this work has several limitations. For example, there are three major ER stress sensors, namely, PERK, IRE1 and ATF6, and we only detected ATF6 in this study. Further work needs to be performed to determine whether the other two sensors are related to the role of HOXA2 in ER stress. Moreover, to explore the function of HOXA2 in CKD, other types of cell injury, such as podocyte injury and mesangial cell proliferation, need to be investigated. Additionally, we found only the antifibrotic effect of HOXA2 in animal and cell models; the specific treatment effects should be further fully explored, and the related mechanism also needs to be verified in human subjects through clinical trials. In the future, other new methods, including sequencing techniques, such as transcriptomics and metabolomics, should be used to explore in depth the mechanisms of HOXA2. These methods may provide a direction for the clinical application of HOXA2.
Conclusion
Our research provided strong evidence to support the conclusion that HOXA2 expression was inhibited by aberrant hypermethylation, leading to decreased SIRT1 levels, which could increase ATF6 acetylation and activation as well as subsequent ER stress, resulting in renal damage in CKD-related fibrosis. When HOXA2 is overexpressed, SIRT1 transcription is increased, which in turn promotes the deacetylation and inactivation of ATF6, eventually alleviating renal fibrosis by inhibiting ER stress in CKD. These findings provide a mechanistic link between HOXA2 and ER stress in CKD and suggest that HOXA2 may function as a therapeutic target in CKD-related fibrosis.
Methods
Recruitment of clinical renal biopsy samples
All procedures were approved by the medical ethics committee of the Fourth Affiliated Hospital of China Medical University (number: EC-2024-KS-182) and followed the tenets of the Declaration of Helsinki of 1975, as revised in 1983. Residual portions of diagnostic renal biopsies were from 10 CKD patients, and normal kidney tissues were from 10 tumour patients who underwent nephrectomies. All ethical regulations relevant to human research participants were followed. The related biochemical data were retrieved from the hospital management system. All subjects were informed of the use of their renal biopsy samples, and written informed consent was obtained. In accordance with international guidelines, CKD is defined as a glomerular filtration rate (GFR) of less than 60 ml/min per 1.73 m2, a marker of kidney damage, or both for a duration of at least 3 months. Clinical subjects were recruited at The Fourth Hospital of China Medical University from August 2023 to July 2024. For CKD patients, the average eGFR was 14.9 ± 10.2 ml/min/1.73 m2, while the average serum creatinine concentration was 521.3 ± 120.1 µmol/L.
Animals
We acquired C57BL/6 mice (males, n = 24) aged 7 weeks from the Model Animal Research Center of Nanjing University (Nanjing, China). Adeno-associated virus (AAV) was used to overexpress HOXA2 genes in the mice during the 8th week. AAV2/9-HOXA2 (pAAV-ITR-CAG-Hoxa2 (mouse)-IRES-EGFP-WPRE-Sv40 polyA-ITR) and normal control AAV2/9-Vector (pAAV-ITR-CAG-MCS-IRES-EGFP-WPRE-Sv40 polyA-ITR) were from purchased from HanBio Technology (Shanghai, China). Recombinant AAV vectors or vehicle (PBS) were injected via the tail vein at the 8th week. The volume of the AAV recombinant vectors that was used for the mice experiments was 200 μL [1011 genome copies (GCs)/animal]. We used qRT–PCR and Western blotting to measure the transfection efficiency and fluorescence microscopy to investigate the green fluorescent protein (GFP) expression of AAV-Vector and AAV-HOXA2(+) in mice renal tubular interstitial area.
RIF models were established by UUO in the mice during the 10th week56. Mice were given general anesthesia intraperitoneally with pentobarbital sodium (30 mg/kg) and the left ureter was exposed through the left incision, and two points were ligated with 4-0 silk sutures and cut between the 2 ligation points. The mice in the sham group underwent surgery involving kidney exposure without ligation. We randomly divided the mice into four groups as follows: (1) the sham group (n = 6); (2) the UUO group (n = 6); (3) the UUO + AAV-Vector group (n = 6); and (4) the UUO + AAV-HOXA2 group (n = 6). The mice were raised at the Laboratory Animal Centre of China Medical University under a 50% ± 20% humidity environment, which included a 12 h:12 h light/dark cycle at 22 ± 2.0 °C, and we provided food and water ad libitum to the mice. All mice were euthanized during the 12th week by intraperitoneal injection of pentobarbital sodium (50 mg/kg)57, and we collected blood and renal samples for biochemical and pathological analyses. All procedures were performed according to the approved protocol of the Institutional Animal Care and Use Committee (IACUC) of China Medical University and approved by the Ethics Committee of China Medical University (number: KT20240170). We have complied with all relevant ethical regulations for animal use.
Histological examination
Haematoxylin and eosin (H&E), picrosirius red and Masson’s trichrome staining were used to stain sections (4 μm thick) cut from the paraffin-embedded renal tissues of mice and humans. Each staining was performed according to the instructions of the relevant kit, and each slide was observed under a Leica microscope as described in our previous study29. We randomly selected sections of at least 6 fields of each sample for the quantification of each positive signal for histopathological changes and fibrosis, which is indicated by the red or blue area based on the amount of collagen by Image-Pro Plus 6.0 software (Version X; Media Cybernetics, Silver Springs, MD, USA). The tubular injury scoring and interstitial collagen deposition scoring were performed as previously described in refs. 58,59.
Biochemistry detection
Serum creatinine and blood urea nitrogen (BUN) levels were measured using kits (Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer’s instructions.
Immunohistochemical (IHC) staining
IHC staining of sections from mouse and human kidney tissues was performed as described in previous studies by our group29,60. For IHC analysis, the sections (4 μm) cut from paraffin-embedded renal tissues of mice and humans were deparaffinized with xylene and gradient ethanol. Antigen retrieval was performed in 10 mM citric acid buffer in a microwave oven for 10 min. Internal peroxides were inactivated using 3% hydrogen peroxide for 30 min. Next, 10% goat serum was used to block these sections and then the sections were incubated overnight at 4 °C with primary antibodies against HOXA2, SIRT1, ATF6, GRP78, CHOP, FN, COL I, E-cadherin and α-SMA. Next, the sections were incubated with secondary antibodies for 2 h. DAB was added for visualization. After restaining the nuclei, we sealed the sections with neutral gum and obtained the images under light microscopy. Positive signals were quantified using Image-Pro Plus 6.0 software (Version X; Media Cybernetics, Silver Springs, MD, USA). The dilution ratios of the antibodies are provided in the Supplementary Table S1.
Immunofluorescence (IF) staining
IF staining of sections from mouse and human kidney tissues was performed as previously reported by our group29,60. For IF analysis, the sections (4 μm) cut from paraffin-embedded renal tissues of mice and humans were deparaffinized, rehydrated and then heated in sodium citrate buffer (0.01 mol/L, pH 6.0). After treatment with 3% H2O2 for 20 min and permeabilized with 0.2% Triton-X-100 for 10 min, the sections were blocked with 3% bovine serum albumin (BSA) and subsequently incubated with primary antibodies overnight at 4 °C. Next, the slides were incubated with fluorescently labeled secondary antibodies at room temperature for 1 h, and the nuclei were stained with Hoechst. We used a fluorescence microscope to capture images and analyzed images by the LAS AF Lite (Leica, Wetzlar, Germany). For HK-2 cells, cells were fixed with 4% paraformaldehyde, followed by blocking with goat serum and incubated with primary antibody overnight at 4 °C. After washing with PBS, fluorescent-labeled secondary antibody was added. Hoechst was used to stain the nuclei. A widefield fluorescence microscope was used to capture images, and analyzed images by the LAS AF Lite (Leica, Wetzlar, Germany) and ThermoFIsher EVOS M7000 Imaging System (Massachusetts, USA). The dilution ratios of the antibodies are provided in the Supplementary Table S2.
Measurement of ROS generation
For HK-2 cells, we used H2-DCFDA dye to detect ROS production. Briefly, cells were stained with 10 µM H2-DCFDA (Thermo Fisher, D399, Waltham, MA, USA) dissolved in PBS at room temperature for 30 min. In mouse renal tissue sections, dihydroethidium (DHE) staining was used to measure ROS levels as described in our group’s previous study29. After the sections were deparaffinized and rehydrated, DHE (10 µM; Beyotime Biotechnology, S0063; Shanghai, China) was added to the sections, which were incubated at room temperature for 30 min. The nuclei were stained with Hoechst dye, and fluorescence microscopy was used to assess the sections.
Measurement of the renal glutathione (GSH)/oxidized glutathione (GSSG) ratio and malondialdehyde (MDA) and superoxide dismutase (SOD) levels
GSH and GSSG levels were measured by kits purchased from Beyotime (S0053, Shanghai, China), and MDA (thiobarbituric acid method) and SOD (hydroxylamine method) levels were measured by kits purchased from Jiancheng Bioengineering Institute (A003-1 and A001-1, Nanjing, China) in fresh mouse renal tissues according to the manufacturer’s instructions, as we described previously29. A microplate reader (BioTek, Vermont, USA) was used to measure the absorbance.
Cell culture
HK-2 cells (American Type Culture Collection (ATCC), Manassas, VA, USA) were cultured in DMEM/F12 (1:1; Grand Island, NY, USA) supplemented with 10% foetal bovine serum (Grand Island, NY, USA), 100 U/mL penicillin (Grand Island, NY, USA) and 100 µg/mL streptomycin (Grand Island, NY, USA) at 37 °C with 5% CO2. HEK293 cells (American Type Culture Collection (ATCC), Manassas, VA, USA) were cultured in DMEM (Grand Island, NY, USA) supplemented with 10% foetal bovine serum (Grand Island, NY, USA) and 1% penicillin/streptomycin at 37 °C in 5% CO2.
HK-2 cells were divided into three groups: (a) the basal medium group (Control group); (b) the 4 mM HCl-containing 0.1% BSA group (Vehicle group); and (c) the TGF-β1 group (10 ng/mL; 7754-BH, R&D Systems, Minneapolis, MN, USA). Stock: 10 µg/mL in 4 mM HCl containing 0.1% BSA (1:1000 dilution of stock). TGF-β1 was reconstituted according to previous methods61. The cells from each group were cultured with vehicle or TGF-β1 for 48 h.
Plasmids, the demethylation drug 5-aza-2′-deoxycytidine (5-Aza), and thapsigargin (an ER stress agonist)
HOXA2 and STAT6 overexpression plasmids and empty plasmid vectors were synthesized by HanBio Technology (Shanghai, China). HK-2 cells were transfected with the HOXA2 overexpression plasmid or empty vector in the TGF-β1 group and named the TGF-β1 + HOXA2 (+) or TGF-β1+Vector group. HEK293 cells were transfected with a STAT6 overexpression plasmid or empty plasmid vector and named the STAT6 (+) or Vector group, respectively. HK-2 cells were transfected with SIRT1 siRNA or control siRNA (synthesized by HanBio Technology, Shanghai, China) and named the siSIRT1 or siNC group, respectively. The sequences of the siRNAs targeting SIRT1 are provided in the supplementary Table S3. HK-2 cells were transfected with DNMT1 siRNA (sc-35204; Santa Cruz Biotechnology, USA) or control siRNA (sc-37007; Santa Cruz Biotechnology, USA) in the TGF-β1 group and named the TGF-β1+siDNMT1 or TGF-β1+siNC group, respectively. All the plasmids were transfected at a dose of 2000 ng using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA), and all the siRNAs were transfected at a final concentration of 50 nmol/L with jetPRIME® according to the manufacturer’s instructions. For demethylation treatment, HK-2 cells were divided into the 5-Aza group (Sigma‒Aldrich, St. Louis, MO, USA), and 5-Aza was dissolved in DMSO at concentrations of 1, 2, or 5 µM for 48 h. HK-2 cells were exposed to the ER stress agonist thapsigargin (HY-13433; MedChemExpress, New Jersey, USA) dissolved in DMSO at a concentration of 0.2 μM at 37 °C at 2 h prior to HOXA2 overexpression plasmid treatment to induce ER stress to explore the regulatory mechanism of HOXA2. The DMSO group was named the Vehicle group. The concentrations of these drugs were chosen according to previous studies29,39.
Chromatin immunoprecipitation (ChIP)
The SIRT1 promoter sequence was obtained from the UCSC Genome Browser (https://genome.ucsc.edu/index.html), and the potential binding relationship of HOXA2 to the SIRT1 promoter was predicted by the JASPAR tool. We purchased HOXA2-flag-tag fusion expression vectors from Genechem (Shanghai, China) to detect the binding of exogenous HOXA2 to the SIRT1 promoter via ChIP‒qPCR. ChIP assays were carried out using a SimpleChIP Plus Enzymatic Chromatin IP Kit (9003; Cell Signaling). In brief, after formaldehyde was used in Flag-tagged HOXA2-overexpressing HK-2 cells to cross-link the protein to the DNA, glycine was added to stop the reaction. Cells were suspended in ChIP lysis buffer, and the cell lysate was sonicated to generate DNA fragments and then immunoprecipitated with ChIP-grade anti-Flag monoclonal antibody (F1804; Sigma) or normal mouse IgG (12–371; Sigma). Subsequently, PCR was performed using specific primers to observe the binding of HOXA2 to the SIRT1 promoter in precipitated DNA (forward: 5ʹ-GTTCCAGGATACAGGTGCAGA-3ʹ; reverse: 3ʹ-GAGCGAAACTCCGTCTCAAA-5ʹ). For the ChIP assays to detect the binding of endogenous DNMT1 to endogenous HOXA2, normal rabbit IgG (#2729; Cell Signaling Technology, USA) and anti-DNMT1 monoclonal antibody (ab320817; Abcam, USA) were used. PCR was performed with primers against the HOXA2 promoter region to observe the binding of DNMT1 to the HOXA2 promoter (forward: 5ʹ-CAGAGCGACAAGGACAAGAA-3ʹ; reverse: 3ʹ-GAAGTTACCAGCAGGGAAAGA-5ʹ), as reported previously38.
Dual-luciferase reporter assay
The process was described in our group’s previous report29,60. After predicting the potential HOXA2 binding sites in the SIRT1 promoter region using the JASPAR tool, HK-2 cells were cultured in 24-well plates. SIRT1 wild-type (WT)/mutant (Mut) luciferase plasmids were constructed by inserting SIRT1 promoter fragments containing WT or Mut HOXA2 binding sites into the pGL3.0 reporter vector (Genechem, Shanghai, China). After 48 hours of cotransfection with HOXA2 overexpression plasmids or empty vectors, a reporter gene assay was performed using a Dual Luciferase Assay System (Promega, Madison, WI, USA).
The dual luciferase reporter assay was used to explore the direct binding between STAT6, HOXA2 and ATF6. The promoter region of ATF6 were cloned into the pGL3.0 plasmid (Genechem, Shanghai, China). Subsequently, HEK293 cells were co-transfected with ATF6 promoter with STAT6 or HOXA2 overexpression plasmid or empty vectors. After 48 h, luciferase activities were tested with the Dual Luciferase Reporter Assay System (Promega, Madison, WI, USA). Renilla luciferase activity was normalized to firefly luciferase activity.
Co-immunoprecipitation (Co-IP)
Co-IP experiments were performed as we described previously62. The primary antibodies used were as follows: anti-SIRT1 (sc-135792; Santa Cruz Biotechnology, USA), anti-ATF6 (24169-1-AP; Proteintech Co., Ltd., USA) and normal rabbit IgG control (#2729; Cell Signaling Technology, USA). The acetylation of ATF6 in each HK-2 cell group was determined using an anti-ATF6 antibody (24169-1-AP; Proteintech Co., Ltd., USA) by IP, followed by immunoblotting analysis with an acetylated-lysine antibody (#9441; Cell Signaling Technology, USA). For the IP experiments, we used TrueBlot secondary antibodies (Rockland, USA) to exclude antibody interference and used chemiluminescence for band detection.
Bisulfite sequencing PCR (BSP) assay
The process was described in our group’s previous report29. Briefly, a DNeasy Blood & Tissue Kit (QIAGEN, Duesseldorf, Germany) was used to isolate DNA from HK-2 cells that were treated with or without TGF-β1. From NCBI, we downloaded sequences that included 2,000 bp upstream of the transcription initiation site to downstream of the transcription initiation site in the promoter regions. We employed the EMBOSS tool for predicting promoter-related CpG islands. The genomic DNA was exposed to sodium bisulfite, and specific primers were used for PCR (HOXA2-Forward 5’-TGGTAATTTTTAATAGATTTTTTGG-3’, HOXA2-Reverse 5’-CTCCCAATATCCAACCCAA-3’). We purified and recovered the amplified bisulfate PCR products with a SanPre Column PCR Purification Kit (Sangon, Shanghai, China) and then subcloned them and inserted them into the pUC18-T vector system. Ten individual clones were used for DNA sequencing. We provided the predicted CpG islands and the results of agarose gel electrophoresis to confirm the PCR products in Supplementary Fig. S1.
qRT‒PCR
qRT‒PCR was performed as we described previously29. Total RNA was extracted from mice kidneys and HK-2 cells by a TRIzol Reagent Kit (Invitrogen, Carlsbad, CA, USA). RNA was reverse transcribed into cDNA using a GoScriptTM Reverse Transcription System (Promega, Madison, WI, USA) according to the manufacturer’s protocol. We used GoTaq® qPCR Master Mix (Promega, Madison, WI, USA) to detect the mRNA expression levels on the CFX96 PCR System (Bio-Rad, Hercules, CA, USA). Relative mRNA expression was normalized to that of β-actin via the 2-ΔΔCt method. The sequences of the primers are provided in the Supplementary Table S4.
Western blotting analysis
Western blotting analysis was performed as we described previously29. Proteins were extracted from mice kidneys and HK-2 cells by RIPA buffer containing a protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). The BCA kit was used to detect the protein contents (P0011, Beyotime Biotechnology, Shanghai, China). SDS‒PAGE was employed to separate the proteins and then the proteins were transferred to PVDF membranes (Millipore, Boston, USA). 5% skim milk powder-TBST was used to block the membranes for 1.5 h at room temperature, followed by incubation with primary antibodies overnight at 4 °C. The next day, the membranes were incubated with HRP-conjugated secondary antibodies at room temperature for 1 h. ECL (Millipore, Boston, USA) was used to visualize the levels of the proteins on the membranes. ImageJ software was used to semiquantify protein expression, and β-actin was used as the loading control. The antibody dilution ratios are provided in the Supplementary Table S5.
Cell Counting Kit-8 (CCK-8) analysis
CCK-8 analysis was performed as we described previously29. After transfecting cells with either empty plasmids or HOXA2 overexpression plasmids, cells were seeded in 96-well plate and were treated with TGF-β1 for 24, 48, or 72 h. Subsequently, we added CCK-8 reagent (CK04-13; Dojindo,Kumamoto, Japan) to the medium, and incubated the cells for 2 h at 37 °C. A microplate reader (BioTek, Vermont, USA) was employed to detected the absorbance.
SIRT1 deacetylase activity
The process was performed as reported previously63. In brief, we used a fluorometric SIRT1 assay kit (ab156065; Abcam, USA) to quantify SIRT1 deacetylase activity in HK-2 cells from each group. We measured the fluorescence intensity at an excitation wavelength of 350 nm and an emission wavelength of 460 nm every 2 min. Next, the data were normalized to the amount of protein to evaluate the overall SIRT1 protein deacetylase activity. The overall SIRT1 deacetylase activity of each group was compared with that of the control group.
Transmission electron microscopy (TEM)
TEM was used to investigate mitochondrial damage and ER changes in HK-2 cells. In brief, we used 3% glutaraldehyde to prefix the samples and then employed 1% osmium tetroxide to fix the samples. After gradual dehydration with acetone, the samples were embedded in Epon 812 (GP2001, Servicebio Technology, Wuhan, China) overnight at room temperature. Ultrathin sections (60 nm) were prepared and stained with uranyl acetate/lead citrate. Finally, a Hitachi H7700 electron microscope was used to observe the sections and for image acquisition.
DNMT activity assay
In accordance with the manufacturer’s instructions, we extracted nuclear proteins with an Active Motif Nuclear Extraction Kit (Active Motif, Carlsbad, USA) and evaluated DNMT activity with an EpiQui DNA Methyltransferase Activity/Inhibition Assay Kit (Epigentek, Farmingdale, USA) as reported previously64.
Cytoplasmic and nuclear protein extraction
In accordance with previously reported methods65, we used an NE-PER nuclear and cytoplasmic isolation kit (78833; Invitrogen, USA) to extract cytoplasmic and nuclear proteins from HK-2 cells, and the purity of nuclear proteins was assessed using Histone H3 as a marker protein.
Statistics and Reproducibility
Statistical analysis was performed with SPSS 26.0 software (IBM, Armonk, New York, USA) and GraphPad Prism 9.0 software (San Diego, CA, USA). All the data are presented as the means ± standard deviations of at least three independent experiments. Student’s t-test and one-way ANOVA analysis of variance with post hoc tests were used to compare groups. A P-value < 0.05 indicated a statistically significant difference.
Data availability
The source data and the statistical data can be found in Supplementary Data 1 and Supplementary Materials and DNA-sequencing results can be found in NCBI database (SRA: PRJNA1377853). The data that support the findings of this study are available on request from the corresponding author.
References
Fountoulakis, N., Miyamoto, Y., Pavkov, M. E., Karalliedde, J. & Maltese, G. Pathophysiology of vascular ageing and the effect of novel cardio-renal protective medications in preventing progression of chronic kidney disease in people living with diabetes. Diabet. Med 42, e15464 (2025).
Bhayana, S., Schytz, P. A., Bisgaard Olesen, E. T., Soh, K. & Das, V. Single-Cell Advances in Investigating and Understanding Chronic Kidney Disease and Diabetic Kidney Disease. Am. J. Pathol. 195, 55–68 (2025).
Maeda, S. et al. MondoA and AKI and AKI-to-CKD Transition. J. Am. Soc. Nephrol. 35, 1164–1182 (2024).
Huang, R., Fu, P. & Ma, L. Kidney fibrosis: from mechanisms to therapeutic medicines. Signal Transduct. Target Ther. 8, 129 (2023).
Wang, Y. et al. Irisin ameliorates UUO-induced renal interstitial fibrosis through TGF-β1/periostin/MMP-2 signaling pathway. PLoS One 19, e0299389 (2024).
Hadpech, S. & Thongboonkerd, V. Epithelial-mesenchymal plasticity in kidney fibrosis. Genesis 62, e23529 (2024).
Jiang, W. et al. Eucommiae cortex extract alleviates renal fibrosis in CKD mice induced by adenine through the TGF-β1/Smad signaling pathway. J. Nat. Med 79, 170–179 (2025).
Cohen, C. et al. WNT-dependent interaction between inflammatory fibroblasts and FOLR2+ macrophages promotes fibrosis in chronic kidney disease. Nat. Commun. 15, 743 (2024).
Wu, Y. et al. Renalase Prevents Renal Fibrosis by Inhibiting Endoplasmic Reticulum Stress and Down-Regulating GSK-3β/Snail Signaling. Int J. Med Sci. 20, 669–681 (2023).
Andrade-Silva, M. et al. The critical role of endoplasmic reticulum stress and the stimulator of interferon genes (STING) pathway in kidney fibrosis. Kidney Int 107, 302–316 (2025).
Chen, X., Shi, C., He, M., Xiong, S. & Xia, X. Endoplasmic reticulum stress: molecular mechanism and therapeutic targets. Signal Transduct. Target Ther. 8, 352 (2023).
Chen, L. W. et al. The Epstein-Barr Virus Lytic Protein BMLF1 Induces Upregulation of GRP78 Expression through ATF6 Activation. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22084024 (2021).
Yue, L. L. & Du, X. Thrombospondin 1 Promotes Endoplasmic Reticulum Stress and Apoptosis in HK-2 Cells by Upregulating ATF6-CHOP. Curr. Med Sci. 42, 341–347 (2022).
Sankrityayan, H., Kale, A., Shelke, V. & Gaikwad, A. B. Cyproheptadine, a SET7/9 inhibitor, reduces hyperglycaemia-induced ER stress alleviating inflammation and fibrosis in renal tubular epithelial cells. Arch. Physiol. Biochem 130, 411–419 (2024).
Chen, Y. Y. et al. Protein arginine methyltranferase-1 induces ER stress and epithelial-mesenchymal transition in renal tubular epithelial cells and contributes to diabetic nephropathy. Biochim Biophys. Acta Mol. Basis Dis. 1865, 2563–2575 (2019).
Zhu, L. R., Cui, W. & Liu, H. P. Molecular mechanisms of endoplasmic reticulum stress-mediated acute kidney injury in juvenile rats and the protective role of mesencephalic astrocyte-derived neurotrophic factor. J. Pharm. Pharm. 77, 609–620 (2025).
Guo, S. et al. Endoplasmic Reticulum Stress-Mediated Cell Death in Renal Fibrosis. Biomolecules 14, https://doi.org/10.3390/biom14080919 (2024).
Qian, X. et al. SMN haploinsufficiency promotes ischemia/ reperfusion-induced AKI-to-CKD transition by endoplasmic reticulum stress activation. Faseb J. 37, e23276 (2023).
Li, L. et al. Tauroursodeoxycholic acid inhibits TGF-β1-induced renal fibrosis markers in cultured renal mesangial cells by regulating endoplasmic reticulum stress. Exp. Ther. Med. 23, 432 (2022).
Zhao, H. et al. Poricoic acid A attenuates renal fibrosis by inhibiting endoplasmic reticulum stress-mediated apoptosis. Braz. J. Med Biol. Res. 57, e14249 (2024).
Smith, T. M., Wang, X., Zhang, W., Kulyk, W. & Nazarali, A. J. Hoxa2 plays a direct role in murine palate development. Dev. Dyn. 238, 2364–2373 (2009).
Bridoux, L., Deneyer, N., Bergiers, I. & Rezsohazy, R. Molecular Analysis of the HOXA2-Dependent Degradation of RCHY1. PLoS One 10, e0141347 (2015).
Liu, Z. et al. Abnormally high expression of HOXA2 as an independent factor for poor prognosis in glioma patients. Cell Cycle 19, 1632–1640 (2020).
Zong, X., Fu, J., Wang, Z. & Wang, Q. The Diagnostic and Prognostic Values of HOXA Gene Family in Kidney Clear Cell Renal Cell Carcinoma. J. Oncol. 2022, 1762637 (2022).
Jiang, M. et al. LncRNA-LINC00472 suppresses the malignant progression of non-small cell lung cancer via modulation of the miRNA-1275/Homeobox A2 axis. Adv. Clin. Exp. Med 33, 283–297 (2024).
Samarelli, A. V. et al. Expression of HOXB7 in the Lung of Patients with Idiopathic Pulmonary Fibrosis: A Proof-of-Concept Study. Biomedicines 12, https://doi.org/10.3390/biomedicines12061321 (2024).
Zhang, N., Guo, F. & Song, Y. HOXC8/TGF-β1 positive feedback loop promotes liver fibrosis and hepatic stellate cell activation via activating Smad2/Smad3 signaling. Biochem Biophys. Res Commun. 662, 39–46 (2023).
Peng, L. et al. HOXA13 exerts a beneficial effect in albumin-induced epithelial-mesenchymal transition via the glucocorticoid receptor signaling pathway in human renal tubular epithelial cells. Mol. Med Rep. 14, 271–276 (2016).
Li, X. et al. HOXD10 attenuates renal fibrosis by inhibiting NOX4-induced ferroptosis. Cell Death Dis. 15, 398 (2024).
Xiao, X. et al. Hypermethylation leads to the loss of HOXA5, resulting in JAG1 expression and NOTCH signaling contributing to kidney fibrosis. Kidney Int 106, 98–114 (2024).
Zeybel, M. et al. DNA methylation profiling identifies novel markers of progression in hepatitis B-related chronic liver disease. Clin. Epigenetics 8, 48 (2016).
Wu, H., Qiu, Z., Wang, L. & Li, W. Renal Fibrosis: SIRT1 Still of Value. Biomedicines 12, https://doi.org/10.3390/biomedicines12091942 (2024).
Hu, H. et al. SIRT1 regulates endoplasmic reticulum stress-related organ damage. Acta Histochem 126, 152134 (2024).
Kim, H. et al. Anti-Fibrotic Effect of Losartan, an Angiotensin II Receptor Blocker, Is Mediated through Inhibition of ER Stress via Up-Regulation of SIRT1, Followed by Induction of HO-1 and Thioredoxin. Int. J. Mol. Sci. 18, https://doi.org/10.3390/ijms18020305 (2017).
Chang, J. W. et al. Up-Regulation of SIRT1 Reduces Endoplasmic Reticulum Stress and Renal Fibrosis. Nephron 133, 116–128 (2016).
Kim, H., Baek, C. H., Chang, J. W., Yang, W. S. & Lee, S. K. Febuxostat, a novel inhibitor of xanthine oxidase, reduces ER stress through upregulation of SIRT1-AMPK-HO-1/thioredoxin expression. Clin. Exp. Nephrol. 24, 205–215 (2020).
Zhou, W. et al. Dihydroartemisinin suppresses renal fibrosis in mice by inhibiting DNA-methyltransferase 1 and increasing Klotho. Acta Pharm. Sin. 43, 2609–2623 (2022).
Chen, Z. et al. lncRNA HOTAIRM1 promotes osteogenesis of hDFSCs by epigenetically regulating HOXA2 via DNMT1 in vitro. J. Cell Physiol. 235, 8507–8519 (2020).
Kim, H. et al. Activation of AMP-activated protein kinase inhibits ER stress and renal fibrosis. Am. J. Physiol. Ren. Physiol. 308, F226–236 (2015).
Guo, R. et al. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: An insight into endoplasmic reticulum stress response mechanism. Int J. Cardiol. 191, 36–45 (2015).
Benedetti, R. et al. ATF6 supports lysosomal function in tumor cells to enable ER stress-activated macroautophagy and CMA: impact on mutant TP53 expression. Autophagy 20, 1854–1867 (2024).
Wu, D. et al. Unfolded protein response factor ATF6 augments T helper cell responses and promotes mixed granulocytic airway inflammation. Mucosal Immunol. 16, 499–512 (2023).
Zhou, R. et al. Resveratrol Ameliorates Lipid Droplet Accumulation in Liver Through a SIRT1/ ATF6-Dependent Mechanism. Cell Physiol. Biochem 51, 2397–2420 (2018).
Wu, Q. et al. Polystyrene nanoplastics-induced lung apoptosis and ferroptosis via ROS-dependent endoplasmic reticulum stress. Sci. Total Environ. 912, 169260 (2024).
Zhou, Y. et al. TGF-β1 induces ROS to activate ferroptosis via the ERK1/2-WISP1 pathway to promote the progression of renal tubular epithelial cell fibrosis. Cytotechnology 77, 61 (2025).
Makhammajanov, Z. et al. Tubular toxicity of proteinuria and the progression of chronic kidney disease. Nephrol. Dial. Transpl. 39, 589–599 (2024).
Tsai, L. T. et al. Inhibition of Indoxyl Sulfate-Induced Reactive Oxygen Species-Related Ferroptosis Alleviates Renal Cell Injury In Vitro and Chronic Kidney Disease Progression In Vivo. Antioxidants (Basel) 12, https://doi.org/10.3390/antiox12111931 (2023).
Cao, Y. et al. Danshen injection ameliorates unilateral ureteral obstruction-induced renal fibrosis by inhibiting ferroptosis via activating SIRT1/GPX4 pathway. Front Pharm. 15, 1503628 (2024).
Tian, J. et al. SIRT1 slows the progression of lupus nephritis by regulating the NLRP3 inflammasome through ROS/TRPM2/Ca(2+) channel. Clin. Exp. Med 23, 3465–3478 (2023).
Sun, H. J. et al. Polysulfide-mediated sulfhydration of SIRT1 prevents diabetic nephropathy by suppressing phosphorylation and acetylation of p65 NF-κB and STAT3. Redox Biol. 38, 101813 (2021).
Yuan, Q. et al. MiR-185-5p ameliorates endoplasmic reticulum stress and renal fibrosis by downregulation of ATF6. Lab Invest 100, 1436–1446 (2020).
Khan, I., Amin, M. A., Eklund, E. A. & Gartel, A. L. Regulation of HOX gene expression in AML. Blood Cancer J. 14, 42 (2024).
Song, Y. P. et al. Comprehensive Landscape of HOXA2, HOXA9, and HOXA10 as Potential Biomarkers for Predicting Progression and Prognosis in Prostate Cancer. J. Immunol. Res 2022, 5740971 (2022).
De Palma, F. D. E. et al. Epigenetic regulation of HOXA2 expression affects tumor progression and predicts breast cancer patient survival. Cell Death Differ. 32, 730–744 (2025).
Mohan, K. N. DNMT1: catalytic and non-catalytic roles in different biological processes. Epigenomics 14, 629–643 (2022).
Chen, Q. Q. et al. Neuraminidase 1 promotes renal fibrosis development in male mice. Nat. Commun. 14, 1713 (2023).
Adachi, E. et al. Progranulin deficiency attenuates tubulointerstitial injury in a mouse unilateral ureteral obstruction model. Exp. Anim. 73, 293–301 (2024).
Rouschop, K. M. et al. CD44 deficiency increases tubular damage but reduces renal fibrosis in obstructive nephropathy. J. Am. Soc. Nephrol. 15, 674–686 (2004).
Badid, C. et al. Interstitial expression of alpha-SMA: an early marker of chronic renal allograft dysfunction. Nephrol. Dial. Transpl. 17, 1993–1998 (2002).
Li, X. et al. MYCT1 attenuates renal fibrosis and tubular injury in diabetic kidney disease. iScience 26, 107609 (2023).
Loos, B. et al. TGFβ signaling sensitizes MEKi-resistant human melanoma to targeted therapy-induced apoptosis. Cell Death Dis. 15, 925 (2024).
Li, X. et al. YY1-induced upregulation of LncRNA-ARAP1-AS2 and ARAP1 promotes diabetic kidney fibrosis via aberrant glycolysis associated with EGFR/PKM2/HIF-1α pathway. Front Pharm. 14, 1069348 (2023).
Yang, J. W. et al. Didymin alleviates metabolic dysfunction-associated fatty liver disease (MAFLD) via the stimulation of Sirt1-mediated lipophagy and mitochondrial biogenesis. J. Transl. Med. 21, 921 (2023).
Neveu, W. A., Mills, S. T., Staitieh, B. S. & Sueblinvong, V. TGF-β1 epigenetically modifies Thy-1 expression in primary lung fibroblasts. Am. J. Physiol. Cell Physiol. 309, C616–626 (2015).
Li, P. et al. An improved cell nuclear isolation method. Biol. Methods Protoc. 10, bpaf007 (2025).
Acknowledgements
We thank for the support from Department of Nephrology, Neurology and Anesthesiology of Fourth Affiliated Hospital of China Medical University, Department of Biological Therapy of First Affiliated Hospital of China Medical University and Department of Pathology of Shengjing Hospital of China Medical University. The study was supported by the National Natural Science Foundation of China (82402721), Liaoning Province Postdoctoral Science Fund (3110211225) and Science and Technology Project of Liaoning Province (2025080736-JH3/101).
Author information
Authors and Affiliations
Contributions
All authors approved final version of manuscript. X.L., T.M., X.D., H.L., Y.L. and H.D. conceived and designed research and performed experiments. S.H., F.M., Y.Z., Q.Y. X.D. and X.L. analyzed data. X.L., T.M., H.L. and Y.L. prepared figures and edited text. X.L., T.M., and X.D. drafted manuscript. H.L., Y.L., and H.D. edited and revised manuscript. H.L., Y.L., and H.D. given final approval of the version to be published.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Dr Toshiro Moroishi and Dr Ophelia Bu. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, X., Ma, TK., Deng, XX. et al. HOXA2 exerts anti-renal fibrosis effects through reducing endoplasmic reticulum stress via the upregulation of SIRT1. Commun Biol 9, 174 (2026). https://doi.org/10.1038/s42003-025-09453-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-09453-2
