
Abstract
Towards target diagnostics of low-concentrated molecules in biofluids, NMR spectroscopy faces limitations due to low sensitivity, signal overlap, and high equipment costs. Hyperpolarization methods such as photo-chemically induced dynamic nuclear polarization (photo-CIDNP) can mitigate some of these challenges. In this study, we explore the potential of steady-state photo-CIDNP to quantify target molecules in urine samples. Matrix interference poses a significant challenge to quantitative measurements, and we thus establish two counteracting methods: spiking and biofluid dilution. Experiments conducted in both high (14.1 T) and low (1.9 T) magnetic fields demonstrate the effectiveness of photo-CIDNP-based quantification at micromolar levels for the analytes sumatriptan and paracetamol. We report limits of quantification (LOQs) in complex matrices down to 3.5 μM and average errors of less than 26% with the spiking method and less than 11% using biofluid dilution. This proof-of-concept study highlights the potential of NMR supported by photo-CIDNP as a target diagnostic tool for rapid drug quantification and clinical monitoring applications, especially with low-cost benchtop NMR devices.
Similar content being viewed by others

Introduction
NMR spectroscopy is a powerful technique with plenty of applications, providing information on structure and dynamics1. In the context of metabolomics, NMR spectra identify individual sample components by their specific signal fingerprints2,3,4,5. These signals can then also be quantified to determine analyte concentrations in complex mixtures6,7,8, positioning NMR spectroscopy as a promising tool in diagnostics applications. For routine diagnostics, however, NMR has several limitations, including an inherently low sensitivity, spectra complexity, and high instrument and maintenance costs9,10. Typical metabolites or active pharmaceutical ingredients interesting for diagnostic assessments are present in biofluids at nano- to micromolar concentrations10,11, extending the acquisition time and making diagnostics with NMR inefficient and expensive. Moreover, prolonged measurements can lead to sample aging12,13. So far, NMR spectroscopy has been shown to be clinically relevant to picture the metabolome and associate the metabolomic differences to the physiological states with specific conditions, e.g., heart diseases14,15. Advanced statistical methods, such as principal component analysis or models supported by artificial intelligence, provide classification between healthy and unhealthy patients12,16,17,18,19. However, identifying unique individual signals is not a priority for these methods. In contrast, targeted diagnostics of single biomolecules in low concentration is challenging because of low analyte signal intensity and overlapping signals20. However, all the mentioned limitations can be mitigated by hyperpolarization techniques that selectively enhance specific peaks in the NMR spectrum. One versatile hyperpolarization method in NMR is photochemically-induced dynamic polarization (photo-CIDNP)21. Compared to traditional NMR, photo-CIDNP requires only two additional experimental components: sample illumination and a dye with absorption at the used wavelength. Specific interactions between the illuminated dye and certain sample components then increase signal intensities beyond the Boltzmann distribution. Steady-state photo-CIDNP experiments with prolonged laser irradiation and low laser power are easy to set up and can be adapted to low-field benchtop NMR spectrometers22. Benchtop NMR lowers costs and complexity, making them more available to diagnostics centers and hospitals.
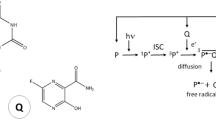
Photo-CIDNP was discovered in 1967 independently by Bargon and Fischer23 and Ward and Lawler24. The phenomenon can be observed in both liquid and solid-state NMR25. Initially it was used to prove the occurrence of transient radicals and to solve reaction mechanisms23,26 and later to study protein dynamics and structure by observing hyperpolarized amino acids, such as tryptophan or tyrosin27,28. Recently, its application was extended to drug discovery22,29, dissociation constant determination30, and magnetic resonance imaging (MRI)31. Also, technological and methodological improvements like the development of new dyes resistant to prolonged irradiation32,33, improved enzymatic systems to scavenge oxygen34, and application of LED lights as irradiation source35,36,37, or adaptation to microfluidics systems38 made photo-CIDNP more available and universal for research. Photo-CIDNP is based on the interaction between the photo-excited dye and the target molecule25,26,39,40,41. Upon irradiation, the dye goes to the excited singlet state and then, via intersystem crossing, to the long-lived triplet state. This is the most common mechanism40,42,43. The triplet state is highly reactive and prone to react with free oxygen, which therefore should be removed from the sample before measurement34. Photo-CIDNP can also originate directly from photoexcited singlet states, if they live long enough to interact with the target molecule26,43. Toward the photo-CIDNP signal, the dye in singlet or triplet state interacts with the target molecule, forming a radical pair by electron or proton transfer40. These radical pairs subsequently undergo singlet-triplet mixing and finally break into diamagnetic molecules. Because the singlet-triplet mixing is modulated by the hyperfine interactions, and these have different signs for the α and β spin states, an imbalance in the spin state population occurs43. This mechanism is known as spin-sorting and, depending on molecular parameters, can lead to a high spin state population difference, resulting in a signal enhancement that exceeds the Boltzmann population significantly. In steady-state photo-CIDNP experiments, prolonged light irradiation leads to continuous generation of radical pairs. Among these, F-pairs arise when radicals escape the solvent cage before recombination and undergo paramagnetic relaxation43. The F-pairs also originate from radical pairs that did not contribute directly to the spin sorting mechanism. They can feed the spin sorting loop again, contributing to the photo-CIDNP enhancement. Additionally, during irradiation, the nuclei can partially relax or transfer polarization between them. The sign of the enhanced signals can be explained by the Kaptein rules, depending on the radical pairs reaction mechanism44.
In samples of pure components, the photo-CIDNP effect follows quantitative relationships, such that it is linear within certain range33. Photo-CIDNP enhancement has also been qualitatively observed in complex matrices such as urine, serum45 or E.coli extract46. However, in such complex mixtures, the signal enhancement depends on the sample composition in a complicated, non-linear manner. On one hand, in such samples, the presence of multiple photo-CIDNP active molecules with different dye affinities can create competition for the dye40. Increasing the dye concentration is not usually a feasible solution to the problem, as dye-dye interactions promote self-quenching32, which reduces the population of the excited states required for photo-CIDNP enhancement. Moreover, the pool of free radicals of the target molecule, which later contributes to the F-pairs formation, can be affected by reductive electron transfer between photo-CIDNP active molecules40. As a result, the enhancement for photo-CIDNP molecules in complex mixtures is no longer quantitatively comparable with the enhancement in pure solutions. Such alteration of an analytical signal caused by matrix components is generally referred to as matrix interference effect47.
Photo-CIDNP spectra can be acquired by modifying standard NMR experiments, such as the perfect-echo W5 water suppression sequence48, by introducing a laser irradiation period following the interscan recycle delay. However, in complex mixtures, extracting photo-CIDNP signals becomes increasingly challenging, even for highly intense peaks, due to spectral overlap and background interference. This limitation can be addressed using an alternative approach, gated illumination during a pulse train experiment. This method selectively retains magnetization generated solely by photo-CIDNP hyperpolarization49. While it offers reduced sensitivity compared to conventional approaches, it significantly improves spectral clarity. In such background-free spectra, signals from naturally photo-CIDNP-active metabolites present in urine, such as tryptophan and tyrosine50, can be clearly distinguished. Previous studies, presented by Kuhn et al.45 have demonstrated this technique for qualitative identification of photo-CIDNP-active components in biofluids like urine and serum, showing reproducible enhancements for the same urine samples45. However, these studies did not address quantitative analysis in complex matrices or consider matrix interference effects, critical for diagnostic applications.
In this paper, we explored the potential of steady-state photo-CIDNP towards its use as a diagnostic tool, i.e., to detect and quantify target concentrations in complex biofluids. As a model system, we studied two photo-CIDNP active molecules as target analytes in human urine: the antimigraine compound sumatriptan and the common analgesic paracetamol51,52. As a dye, we used fluorescein due to its resistance to prolonged irradiation and the broad range of target molecules it can interact with32. To enable signal quantification on the background of the matrix interference effects, we established two approaches, one based on spiking and the other on biofluid dilution. LOQs were established in a high magnetic field of 14.1 T and on a benchtop NMR spectrometer with 1.9 T field strength.
Results and Discussion
Experiment optimization and matrix effect on photo-CIDNP
In our experiments, we considered the resonances at 6.8 ppm for paracetamol and at 7.6 and 3.2 ppm for sumatriptan. The sign of the peaks calculated by the Kaptein rules suggested that the hyperpolarization of paracetamol originates from the recombination product, while for sumatriptan it originates from the escape product (Supplementary Equation 1, Supplementary Table 1). Since the singlet excited state of fluorescein is short-lived, the photo-CIDNP signal likely comes from a triplet precursor32.
As a proof of concept in a realistic situation with variable biofluid composition, we performed experiments on a set of six different human urine samples obtained from healthy volunteers. We started with optimizing the sample conditions, including the dye concentration, pH of the solution, and number of acquired scans. We assessed four different dyes, fluorescein, 4’,5’-dibromofluoresein, riboflavin and flavin mononucleotide (FMN) for best suitability. Among these, fluorescein was most resistant to photo-bleaching, gave by far the highest enhancement and exhibited the highest absorbance at our wavelength of 450 nm (Supplementary Figs. 1,2), hence it was chosen for all further studies. The photo-CIDNP activity of the target compounds was tested at fluorescein concentrations of 25–100 μM, not showing any significant variation (Supplementary Fig. 3). Then, we tested the influence of pH in the range 5–8 (Supplementary Fig. 4). For sumatriptan and paracetamol, the photo-CIDNP enhancement dropped by 20–30% at low pH (pH 5–6) compared to physiological pH (pH 7.1–8). For the high field measurements at 14.1 T field strength, we found that 64 scans delivered the optimal total sensitivity, as the photo-CIDNP enhancement is non-linear due to radical reactions and photobleaching (Supplementary Fig. 5). For the low field measurements at 1.9 T field strength, optimal results were achieved with 32 scans. It is important to note that for samples containing 100 μM of paracetamol, conventional spectra measured at 1.9 T did not provide detectable signals at 32 scans. The signal-to-noise ratio (SN) obtained in 32 scans laser experiment is comparable with dark spectrum measured with 10,000 scans (Supplementary Fig. 6). To perform the analysis in biofluids, we used lyophilized urine samples to prevent changes over time and reconstituted them immediately before measurements in either H2O or D2O. We verified that the lyophilization did not affect the photo-CIDNP performance by comparing the photo-CIDNP-enhanced signal for a reference compound in freshly collected urine and lyophilized urine (Supplementary Fig. 7). As a reference signal for quantification, we used 5-fluoro-2-methylindole, which features a particularly high photo-CIDNP effect. This reference compound provides the additional benefit that its most enhanced signal at 6.15 ppm is well separated from the complex signal array of the urine background matrix.
To investigate the matrix interference effect on the photo-CIDNP performance, for both analytes, paracetamol and sumatriptan, we measured samples with increasing urine content from 0 to 70% (Fig. 1). The photo-CIDNP effect was analyzed as the signal-to-noise enhancement22 (SNE, Supplementary Equation 2) for the peak at 6.8 ppm for paracetamol and for the peaks at 7.6 and 3.2 ppm for sumatriptan. Compared to the buffer, we observed a drop in photo-CIDNP enhancement by 80–95% upon adding 70% of urine to the sample. Even only 5% urine caused a severe photo-CIDNP signal decrease of around 30% for all tested urines and both analytes. Importantly, the drop of the hyperpolarized signal was not uniform across different urines and for the different analytes, indicating that the particular urine composition had a specific impact on the signal enhancement. This finding thus implies a specific and strong matrix interference on the photo-CIDNP performance. However, at 5% urine content, the variation in photo-CIDNP enhancement drop between different urine samples was much less pronounced than at higher urine concentrations, reaching maximally a deviation from the mean of 20% (Fig. 1d–f). In contrast, the deviation reached over 100% at high urine content. This suggests sample dilution could be a way to overcome the matrix interference effect partially and to apply a universal calibration curve for highly diluted samples.

a 1D 1H photo-CIDNP NMR spectra of paracetamol in different dilutions of urine. The corresponding SNEs is indicated, starting from 0% (sample measured in pure buffer) to 70% of urine. The position of the resonance is indicated by pink circles on the structure. b, c Same for two sumatriptan signals. Spectra were recorded with 64-scan gated illumination photo-CIDNP at 14.1 T with a 0.2 s laser irradiation block. Samples contained 100 μM of the analyte and 100 μM of fluorescein in 100 mM sodium phosphate buffer at pH 7.1. SNEs were calculated relative to the dark spectrum measured with modified W5 and no urine addition. d–f Dependance between the urine content and the photo-CIDNP active signal integral with the percentage deviation from the mean at each urine content for each resonance, respectively.
To study the nature of the matrix interference effect further, we probed the effect of the oxygen removal system. Two samples were prepared using either the enzymatic scavenging system or conventional nitrogen degassing. We found no difference in signal enhancement between the two systems for samples containing 40% urine and 5-fluoro-2-methylindole (Supplementary Fig. 7), leading to the conclusion that the observed signal enhancement drop is not dependent on the oxygen scavenging system. Human urine contains a small fraction of large proteins53 to which small molecules could potentially bind in a specific or non-specific fashion, becoming unavailable for photo-CIDNP. We removed proteins with molecular weight larger than 3 kDa by ultra-filtration, but we observed no difference in signal intensities between filtrated and non-filtrated urine (Supplementary Fig. 7), thus ruling out dye- or compound-binding as a potential cause of the observed photo-CIDNP drop. Therefore, we concluded that the source of the photo-CIDNP drop is a competition effect between the photo-CIDNP active molecules and potential triplet state quenchers that might be a part of the biological matrix. Moreover, the photo-CIDNP active target molecule is most likely also affected by exchanging electrons in its neutral or radical state with other molecules by reductive electron transfer. Trying to resolve which are the major contributors to the photo-CIDNP drop, we investigated a series of samples containing 40% of a solution of individual urine components, including creatinine, urea, thiamine, d-mannitol, succinate, formate, pantothenic acid, glucuronate, cytosine, sodium pyruvate, nicotinamide, bilirubin, a mixture of amino acids, uric acid, lactic acid and salts mixture, in the urinary excretion range54 (Supplementary Table 2, Supplementary Fig. 8). In photo-CIDNP enhanced spectra we observed a 68% signal drop upon uric acid addition, 30% upon salts, 20% upon bilirubin and 16% upon amino acids. Notably, uric acid was buffered to pH 7.4 in 100 mM sodium phosphate buffer, and this pH value should not have affected the photo-CIDNP enhancement. The mix of all individually tested components led to a photo-CIDNP enhancement drop of 30%, which does not fully match the value observed in 40% urine. The non-additivity of the single influences implicates the presence of complicated interactions between the mixture components and confirms that the problem arises from multiple species, affecting the pool of fluorescein and analyte.
We specifically tested bilirubin due to its structural similarity to urobilin, a compound typically present in urine54. The absorption spectrum was recorded for bilirubin (Supplementary Fig. 2). As bilirubin absorbs at the excitation wavelength used in our experiments, it may act as an efficient triplet quencher. To assess whether the removal of colored compounds such as urobilin could enhance the photo-CIDNP signal, by reducing triplet quenching and minimizing competition for light absorption, we photobleached urine samples using a 600 mW laser at 450 nm for 256 s (Supplementary Fig. 9). Absorption spectra were then compared before and after photobleaching, along with the photo-CIDNP spectra of samples containing 40% urine and the reference compound 5-fluoro-2-methylindole. The results showed only a negligible improvement in photo-CIDNP enhancement following photobleaching. This indicates that although eliminating colored substances can slightly enhance the signal, they are not the primary contributors to signal loss in complex matrices. These findings support our earlier conclusion that part of the photo-CIDNP signal loss in complex matrix is due to the presence of bilirubin.
To ensure that the differences in photo-CIDNP enhancement do not stem from altered fluorescein absorbance, we spiked three samples, 40% urine I, II, or IV, with 100 μM fluorescein and measured their absorption spectra (Supplementary Fig. 10). These were then compared to a reference sample containing fluorescein in pure buffer. We observed negligible differences in absorbance at 450 nm, indicating that the urine matrix at 40% concentration does not affect the absorption properties of fluorescein.
Evaluation of NMR experiments for photo-CIDNP in urine
To develop methods for analyte quantification, we produced mock patient samples where urine was enriched with a known amount of sumatriptan or paracetamol in the concentration range of 12.5–250 μM, matching the natural urinary excretion range55,56. Samples were then diluted with buffer to 40% urine. At this urine content, we could still observe photo-CIDNP signal enhancement. We measured non-irradiated (dark) modified perfect-echo W5 experiments and gated illumination experiments (laser) for samples containing 40% urine without adding external photo-CIDNP active compounds to investigate the variability in the used urine compositions (Supplementary Fig. 11). As expected, we observed considerable spectral differences between the urine samples, both in the dark spectra and in the photo-CIDNP active molecules. Various photo-CIDNP active compounds natively occurring in urine further support the hypothesis that the competition effect affects the photo-CIDNP performance of the target molecule.
To evaluate the two NMR experiments used for detection, modified perfect-echo W5 water suppression and gated illumination, we collected spectra from samples with 100 μM paracetamol and/or 100 μM sumatriptan (Fig. 2, Supplementary Fig. 12, 13). We measured the spectra of a single analyte in buffer, and with the addition of 40% of urine I, II, or IV. We also verified that none of the urine samples contained the analytes. First, by comparing spectra of pure analytes in buffer, we noted that, although gated illumination helps to extract signals corresponding to photo-CIDNP active species and reduces spectral complexity, it results in around 2.5 times lower SNE than the standard experiment based on the modified W5 water suppression. The background removal by gated illumination might not seem crucial for samples containing 100 μM paracetamol, however, at lower analyte concentrations, the signal of interest would overlap with background signals in modified W5 water suppression experiments and can only be detected by gated illumination (Fig. 2). Furthermore, for this type of experiment, similarly like in work presented by Kuhn et al.45, we found that the reconstitution of urine in D2O provided better background nulling and was thus further used as the solvent.

a 1D 1H NMR spectra of 100 μM paracetamol in pure buffer measured in dark mode (no photo-CIDNP), laser mode (1 s of laser illumination) and with gated illumination (0.2 s of laser irradiation). b–d Same after doping samples with 40% of urine I, II, or IV, respectively. Black spectra are reference spectra of the urine background without paracetamol. All spectra were measured at 14.1 T with 64 scans. SNEs were calculated in comparison to the dark spectrum measured with modified W5 and no urine addition.
As a step towards quantification of analyte concentration, we next tested the use of a reference compound that would serve as a concentration standard (Fig. 3). We examined samples containing 40% of four different urines and the analytes of interest. As possible concentration standards, we tested two different compounds, 5-fluoro-2-methylindol and 2-(2-amino-1,3-thiazol-4-yl)propan-2-ol as internal references. Both were chosen due to their high photo-CIDNP activity, the former verified by us, the latter known in the literature22. We tested 1:1 mixtures (100 μM) of analytes with internal references, including a 1:1 mixture of paracetamol and sumatriptan. The study showed that the ratio of the integrated signals for the analyte and reference varied significantly between different urine matrices. This suggests that native urine components influence the photo-CIDNP enhancement of the target and reference molecules in distinct ways, likely by competing for the dye or by electron transfer between radicals. In some cases, the photo-CIDNP-enhanced signals for sumatriptan could not be detected anymore in the spectrum. This highlights the sample composition’s influence on the gained polarization. Unfortunately, these findings excluded the option of an internal reference application due to the massive non-linearity of the matrix interference effect. To circumvent this problem, we developed two methods that bring back linearity and thus allow the quantification of analytes by photo-CIDNP in complex mixtures. We discuss these two methods in the following paragraphs.

a Chemical structures of analytes (paracetamol, sumatriptan) and internal references (5-fluoro-2-methylindole and 2-(2-amino-1,3-thiazol-4-yl)propan-2-ol) with highlighted protons used in analysis. b–d Photo-CIDNP 1D 1H NMR gated illumination spectra of samples containing analytes. Pink spectra contain 100 μM of one analyte, black spectra contain analyte, reference compound, and 40% of urine I–IV. The integral ratios of the analyte (pink dashed box) over the reference (black dashed box) signal are given. Spectra were recorded with 64 scans and 0.2 s of laser irradiation at 14.1 T.
Method 1: quantification with spiking
For the spiking approach, a given sample with analyte concentration in the 12.5–250 μM range was split into two halves. One half of the sample remained unmodified, and the other half was spiked with a known amount of the analyte. From the signal difference of the two samples, the initial concentration can be calculated under the assumption that the signal depends linearly on the concentration in the concentration range spanned by the spiking. We tested this approach for paracetamol and sumatriptan as target analytes in samples containing 40% urine using gated illumination experiments, measuring each point in triplicates (Fig. 4 and Supplementary Fig. 14). The data show that the relation between the photo-CIDNP signals and the analyte concentration is indeed linear in the examined concentration range (Fig. 4a, Supplementary Fig. 14a). Notably, the slope had a strong variation between different urine samples, corresponding to vastly different compositions of the biofluids. For the quantification, we chose peaks with high SNE enhancements. For paracetamol, the signal at 6.8 ppm corresponding to aromatic protons in the ortho position, and for sumatriptan, the methylene signal at 3.2 ppm was selected (Fig. 4b, Supplementary Fig. 14b). Spiking was performed for four native concentrations in the range of 12.5–250 μM in three different urines, at 25, 75, 125, and 150 μM.

a Calibration curves for three sets of 40% urine I, II, III with paracetamol concentration of 12.5–250 μM (before dilution, so 5–100 μM after dilution). Each point corresponds to the average integral from triplicated experiments. The indicated LOQ corresponds to SN = 3 in native urine samples. RSDs are calculated for the points with the lowest analyte concentration (12.5 μM before dilution). b NMR signal intensities for paracetamol for an unknown concentration (light blue) and after spiking with 20 μL of 0.5 mM stock solution (dark blue). c–e Correlation of the experimentally determined with the true concentration in three different urines I–III. The difference between the true and the determined concentration is indicated next to each point with the percentage error in the brackets. All photo-CIDNP 1D 1H NMR experiments were measured at 14.1 T, using gated illumination with an irradiation time of 200 ms and 64 scans.
LOQ were estimated based on the SN of individual peaks. Instead of using the conventional SN threshold of 10, we applied a lower threshold of 357,58,59,60. This choice was justified by the high measurement precision observed, as reflected in the relative standard deviation (RSD) values58. Due to the limited volume of biological samples available, RSDs were calculated at the lowest concentration point in the calibration curves rather than directly at the LOQ. In most cases, the RSDs were below 20%, which falls within the acceptable range for analytical tests in the early stages of development58. In a few cases where the RSD exceeded 20%, we still used an SN of 3 for LOQ estimation. This was due to the current experimental setup introducing greater signal variability, as discussed in the following section. Importantly, these deviations are not fundamental limitations and can be minimized through improvements in the setup as explained in the next paragraph.
Because different urines affect photo-CIDNP to varying degrees, the LOQ depend significantly on the sample used as well as the photo-CIDNP activity of the measured analyte, with LOQs ranging from 3.5 to 12 μM for paracetamol and 13 to 42 μM for sumatriptan in native urine samples (before dilution). By measuring the experiments in triplicate, we quantified the experimental variation of the measurements, which resulted from our technical setup at 14.1 T. In this setup, illumination is applied from the top, and the inevitable slight variations in the fiberglass position then lead to a difference in illumination at the individual samples. Because the spiking method depends on the comparison of two samples rather than an internal standard, errors induced by this mechanical variation accumulate. Nonetheless, the accuracy of the spiking method was below 30% for most samples and reached errors of below 5% for some samples (Fig. 4c–e, Supplementary Fig. 14c–e). It can thus be expected that a mechanically improved setup, such as a fixed illumination source, will readily eliminate this error source and lower the RSDs. This could be achieved, e.g., by introducing laser optics from the bottom of the spectrometer, in-situ illumination by specialized NMR devices, or including LED light sources inside the probe head37,61.
Method 2: quantification with dilution method
An alternative quantification method was developed based on the observation that the variation in the photo-CIDNP enhancement between different urine samples was much smaller when the urine content was diluted to 5% compared to samples with higher urine concentrations. At 5% urine, the photo-CIDNP signal enhancement is reduced compared to pure buffer, but the variation between different urines is much less pronounced. The non-linear matrix effects, which result from bimolecular reactions, are significantly diminished compared to the overall effect, which scales with the dilution (Fig. 1d–f). The slopes and intercepts of the calibration curves for spiking at 5% urine are thus nearly identical for different urines, in contrast to those obtained at 40% urine (Fig. 5a, b). We worked on samples containing 5% of urines IV-VI, with original analyte concentrations of 20–200 μM (Fig. 5). The high dilution enabled the enhanced perfect-echo W5 water suppression experiments, offering greater sensitivity than gated illumination. Each point of the calibration curve was measured in triplicate. For paracetamol, we selected the same enhanced peak (6.8 ppm) as for the spiking method at high magnetic field (Fig. 5c). For sumatriptan, a peak at 7.6 ppm was used for quantification because of signal overlap with peaks originating from thermal equilibrium (Fig. 5d). By calculating the SN for all points in the calibration curves and then determining the concentration at SN = 3, we determined LOQ, as the RSDs are usually below 20%57,58,59,60. The LOQ for the undiluted urines was in range 6.4–7.1 μM for paracetamol and 15–19 μM for sumatriptan. We then determined the average slopes and intercepts towards a universal calibration curve for each analyte at each magnetic field. Analysis of the errors showed that almost all samples had percentage errors lower than 30%, and half of the samples had a percentage error lower than 10% (Fig. 5e, f).

a,b Calibration curves for paracetamol and sumatriptan in range 20–200 μM (before dilution, so 1–10 μM after dilution) in three different urines. The indicated LOQs correspond to SN = 3 in native urine samples. RSDs are calculated for the points with the lowest analyte concentration (20 μM before dilution). All points were measured in triplicates using W5 water suppression, 1 s of laser irradiation, and 64 scans at 14.1 T. c,d 1D 1H NMR spectra at five different analyte concentrations. e,f Correlation of the experimentally determined with the true concentration in 5% three different urines IV–VI. The difference between the true and the determined concentration is indicated next to each point with the percentage error in the brackets.
For future use in low-cost settings, we also verified this approach on a benchtop spectrometer with a low magnetic field of 1.9 T. At this field, paracetamol showed a strong signal enhancement by photo-CIDNP, whereas sumatriptan was not strongly photo-CIDNP active (Supplementary Fig. 15). We worked with concentrations of paracetamol in native urine in the 60–200 μM range and diluted to 5% (Fig. 6a). The peak at 7.3 ppm was used to quantify the signals, and intensities were used instead of integrals, as this resulted in smaller overall errors (Fig. 6b). The determined LOQ ranged from 40–47 μM for the undiluted urine samples. The percentage errors in the low magnetic field calculated for universal calibration curves were comparable to those in the high magnetic field. However, the calculated RSDs are significantly lower than 20%, confirming that the improved setup with a fixed laser position enhances the precision of the measurements compared to those performed in a high magnetic field, where laser illumination via a fiberglass cable from the top results in slight variations in illumination efficiency between measurements. The benchtop setup demonstrates that this issue can be easily avoided.

a Calibration curves for paracetamol in range 60–200 μM (before dilution, so 3–10 μM after dilution), in three different urines. The indicated LOQs correspond to SN = 3 in native urine samples. RSDs are calculated for the points with the lowest analyte concentration (60 μM before dilution). All points were measured in triplicates using W5 water suppression, 1 s of laser irradiation, and 32 scans at 1.9 T. b 1D 1H NMR spectra at five different paracetamol concentrations for samples diluted to 5% urine V with the signal used for analysis indicated by the dashed line. c Correlation of the experimentally determined with the true concentration in three different urines I–III. The difference between the true and the determined concentration is indicated next to each point with the percentage error in the brackets.
Comparing the two methods established for analyte quantification, the dilution method was more convenient than the spiking, as it requires the measurement of only one sample instead of two. At the same time, it provides smaller errors, reaching an average below 11% for dilution compared to 26% for spiking. Since the limit of quantification represents a compromise between the matrix effect, which reduces photo-CIDNP enhancement, and the amount of analyte introduced with the urine sample, the appropriate quantification method (spiking or dilution) should be chosen based on the strength of matrix interference and photo-CIDNP activity. For samples that do not exhibit strong quenching effects, spiking appears to allow for lower limits of detection compared to dilution methods (e.g., spiking for paracetamol in Urine II and III versus dilution in Urine IV–VI). For sumatriptan, both methods appear to yield comparable results. The best suitable quantification method should therefore in each case be chosen dependent on the analyte concentration, photo-CIDNP activity, and strength of the matrix interference effect.
Conclusions and outlook
This paper reports the application of photo-CIDNP NMR for the quantitative detection of analytes in complex biofluids, with potential future extension to targeted diagnostics. For the first time, we demonstrate that photo-CIDNP-enhanced signals can be successfully quantified in complex biofluids using two strategies: spiking and sample dilution. The presented quantification methods are effective at both low and high magnetic fields, achieving LOQs as low as 3.5 μM for common drug analytes in real biofluid samples. These results serve as a proof of concept and also as a basis for further development and signal improvement.
As we showed, the matrix interference effect presents, however, a significant limitation in achieving satisfactory photo-CIDNP enhancement in complex biofluids. Complex mixtures lead to a noticeable reduction in photo-CIDNP enhancement compared to pure buffer. For instance, for some samples containing 40% urine, the signal intensity is only slightly higher than that measured without laser irradiation. The intensity is thus also highly dependent on the urine composition. Overall, the photo-CIDNP performance in biofluids still needs to be optimized to get an effective sensitivity enhancement compared to conventional NMR.
In contrast, samples containing only 5% urine show a more significant enhancement, as the concentrations of the quenching components of the matrix decrease with dilution. In parallel, high dilution also reduces the contribution of the thermal signals, such that the gated illumination experiments can be replaced by standard NMR experiments with irradiation block. Standard NMR measurements are feasible in 100% urine samples, but the signal overlap caused by the matrix typically hinders accurate quantification.
A primary advantage of photo-CIDNP thus remains the reduction of spectral complexity. In conventionally measured spectra without laser irradiation, certain signals are undetectable, as they overlap with other signals. In biofluids, gated illumination experiments reduce the spectral complexity, which shown by Kuhn et al.45 and in the present study. This could be further enhanced by exploiting the high specificity of the dye–target interaction. By using specialized dyes with tuned irradiation wavelengths or by adjusting the solution’s pH to influence the ionization of the analytes62, one can steer which signals in the samples will be enhanced. Photo-CIDNP could thus be tuned into a method to obtain completely different spectra from the same biosample just by varying experimental conditions.
A further major advantage of photo-CIDNP is its utility for quantification at low magnetic fields, resulting in significant time savings, mostly because photo-CIDNP hyperpolarization becomes more pronounced as the magnetic field strength decreases22,40. This clearly indicates that while photo-CIDNP has great potential for routine diagnostic applications, further optimization is required. Identifying new dyes and target molecules that are more resistant to matrix interference is crucial, as the matrix effect does not affect all molecules equally. This has been shown in our systematic studies involving mixtures of two photo-CIDNP-active components in complex matrices.
Another important matter is the space of potential target analytes in the future diagnostics tests. One limitation of photo-CIDNP is that not all molecules are active. Active molecules typically contain aromatic structures, as these facilitate stabilizing interactions with the dye through π–π stacking and allow for effective radical stabilization. Therefore, extensive screening of photo-CIDNP-active compounds and in-depth studies of the underlying enhancement mechanisms are essential for identifying potential diagnostic targets. To support this effort, screening studies such as the one presented by Torres et al.29 or Tada et al.63, and the development of technologies that enable high-throughput photo-CIDNP measurements22,29 or connection to microfluidics systems64 are of great importance. Fortunately, many analytes and pharmaceutical compounds contain aromatic systems, making them promising candidates for photo-CIDNP-based detection. According to the studies65,66, significant proportion (80%) of the approved drugs contain aromatic rings, making them potential targets to photo-CIDNP-supported analysis. At the same time, only about 20% of metabolites contain aromatic rings, however, among these are important compounds for diagnostics, such as amino acids, microbial metabolites, and aging biomarkers67,68. As presented by Kuhn et al.45, photo-CIDNP can also be observed in serum, which may facilitate analyte quantification by universal calibration due to the more consistent composition of serum across individuals, unlike urine69.
Future photo-CIDNP-based tests will compete with state-of-the-art diagnostic methods such as LC-MS/MS, GC-MS, HPLC coupled with UV or fluorescence detection, immunoassays, enzymatic assays, and spectrophotometry70,71. The main limitation of photo-CIDNP-based diagnostics, compared to those methods, is sensitivity. While photo-CIDNP can achieve detection limits in the nanomolar range22,36 in the absence of matrix effects, it generally falls short of the sensitivity offered by other currently used techniques, which enable detection at pico- or femtomolar concentrations70. However, the key advantages of photo-CIDNP include very rapid data acquisition (from seconds to few minutes), providing high-throughput measurements, and the much lower cost of benchtop spectrometers and laser setups compared to instruments like mass spectrometers or HPLC systems22. Additionally, benchtop spectrometers are user-friendly, do not require highly qualified personnel to operate, and require barely any maintenance22. In combination with a flow system64, fast quantification of analytes that demand extensive control will become possible, e.g., in the kinetic part of clinical trials or for drugs with narrow therapeutic windows.
To conclude, photo-CIDNP offers substantial advantages, especially when measured at low magnetic fields. With appropriate strategies to mitigate matrix effects—whether through the use of more robust dyes or optimized sample preparation—the technique could become significantly more effective45. Nonetheless, a more systematic investigation of matrix effects is essential for further development of the diagnostic strategies based on the photo-CIDNP hyperpolarization. The approaches developed here have the potential to advance targeted diagnostics by combining the high stability of NMR measurements with the low cost of benchtop instruments and their versatility for various targets and samples.
Materials and methods
Sample preparation
Sumatriptan succinate was purchased from TCI, 2-(2-amino-1,3-thiazol-4-yl)propan-2-ol) from Enamine, 4’,5’-dibromofluorescein from Thermo Scientific, riboflavin from Roth, fluorescein and FMN from Sigma Aldrich, paracetamol and 5-fluoro-2-methylindol from Fluorochem. All NMR samples were prepared in 100 mM sodium phosphate buffer and contained an enzyme oxygen scavenging system (if not stated otherwise), composed of 200 nM glucose oxidase, 140 nM catalase, and 2.5 mM D-glucose. 5 mM stocks solution of 5-fluoro-2-methylindol, 2-(2-amino-1,3-thiazol-4-yl)propan-2-ol) were prepared in DMSO-d6. 5 mM, 0.5 mM, and 0.05 mM stock solutions of sumatriptan succinate and paracetamol were prepared in H2O or D2O. The urine samples were obtained from healthy volunteers. Ethical approval was not required for the current studies, as confirmed by the Ethikkommission Nordwest- und Zentralschweiz (BASEC-ID Req-2025-00689). The urine samples were lyophilized, stored at 253 K, and reconstituted in the corresponding H2O or D2O-based buffer before measurement. The sample volume for all experiments was 500 μL, except for the spiked samples, where the sample volume was 520 μL. All samples prepared in an H2O-based buffer and measured at a high magnetic field additionally contained 5% of D2O for locking. All samples were measured in H2O-based buffer pH 7.1 (if not stated otherwise). The samples measured in D2O-based buffer had pH 7.1 (pD 7.4)72.
Experiment optimization was performed for samples containing 10 μM of paracetamol or sumatriptan and 5% urine IV. The concentrations of the dye were tested for 25, 50, and 100 μM of fluorescein. The influence of pH on photo-CIDNP was tested for pH range 5–8. The optimal number of scans was determined using samples containing 50 μM of fluorescein. The impact of urine lyophilization, oxygen scavenging system, or filtration was investigated using samples containing 5 or 40% of urine I, lyophilized and reconstituted in buffer or freshly collected without lyophilization. One of the urine samples was additionally centrifuged by using a filter with a cut-off of 3 kDa. Another sample containing lyophilized and reconstituted urine was degassed for 20 min with nitrogen without the addition of an enzyme mixture for oxygen scavenging. All samples contained 100 μM 5-fluoro-2-methylindole and 25 μM of fluorescein. The influence of urine content was measured for samples containing urine I-IV at 0, 5, 10, 20, 40, and 70% of the total sample volume. All samples contained 100 μM of paracetamol or sumatriptan and 100 μM of fluorescein. The influence of individual urine components on the photo-CIDNP performance was investigated for samples containing 100 μM of 2-methyl-5-fluoroindole, 25 μM of fluorescein, and 200 μL (40% of sample volume) of individual urine component solutions with indicated concentrations and the mixture (Supplementary Table 1). The stock solutions were prepared in 10 or 100-fold concentrations in pure H2O, except for the stock solution of uric acid, which was prepared in 100 mM sodium phosphate buffer and buffered to pH 7.4. The four different dyes (fluorescein, 4’,5’-dibromofluorescein, riboflavin, FMN) were tested at 50 with 100 μM of sumatriptan and paracetamol, respectively.
The characterization of different urines was performed for samples containing 40% urines I–VI, 25 μM of fluorescein, in the D2O-based sodium phosphate buffer. To compare two photo-CIDNP 1D NMR experiments (based on W5 water suppression and gated illumination), spectra were measured for samples containing 100 μM of paracetamol or sumatriptan, 100 μM of fluorescein with or without 40% urine I, II, IV, in a D2O-based buffer. The assessment of an internal reference application for photo-CIDNP was evaluated using four samples containing 40% of urine I–IV, paracetamol or sumatriptan and 5-fluoro-2-methylindol or 2-(2-amino-1,3-thiazol-4-yl)propan-2-ol as internal references, 100 μM of fluorescein, in D2O-based buffer. The linearity of the signal integrals versus concentration was evaluated for sumatriptan and paracetamol using the following concentrations: 5, 20, 50, 80, 100 μM (after dilution), and 100 μM of fluorescein, 40% urines I-III or urines IV-VI, respectively. The spiking was performed for the following concentrations: 10, 30, 50 and 60 μM (after dilution). The spiked samples were freshly prepared with the addition of 20 μL of 0.5 mM solution of paracetamol or sumatriptan, respectively. All samples for spiking were prepared in a D2O-based buffer. Calibration curves for the quantification with the dilution method were obtained for samples containing 50 μM of fluorescein and the following paracetamol or sumatriptan concentrations: 1, 3, 5, 8, 10 μM (after dilution) for samples measured in the high magnetic field and 3, 5, 6.5, 8, 10 μM (after dilution) in the low magnetic field, respectively. For all calibration curves each point was measured in triplicates, every time with freshly-prepared sample.
NMR measurements
1D 1H NMR spectra at the high magnetic field were measured on a 14.1 T Avance III spectrometer (Bruker) equipped with an inverse triple-resonance (TXI) room temperature probe and processed in software TopSpin 3.6.2. Spectra on the low magnetic field of 1.9 T were measured on a Magritek Spinsolve 80 Ultra benchtop spectrometer and processed in MestReNova 15.1.0. All NMR experiments were performed at room temperature (298 K). For experiments with a number of scans greater than one, 4 dummy scans and 3 s of recycle delay were included. Experiments measured with a perfect-echo W5 water suppression48 with the added laser irradiation period after the d1 recycle delay were recorded with 1 s of laser irradiation, whereas the gated-illumination experiments49 were recorded with 200 ms of laser irradiation. The optimization of fluorescein concentration and buffer pH, as well as pH influence on photo-CIDNP enhancement, used 64 scans. The effect of urine treatment (lyophilization, filtration, oxygen scavenging system) on photo-CIDNP enhancement was tested with just 1 scan, while urine characterization involved 16 scans with perfect-echo W5 and 32 scans with gated illumination. Quantification experiments using spiking and dilution methods employed 64 scans at 14.1 T and dilution tested at 1.9 T 32 scans.
All the samples were irradiated using a laser diode emitting at 450 nm purchased from Thorlabs (L450P1000MM) with an output of 600 mW at the end of the optical fiber. The tip of the optical fiber was inserted directly into a 5 mm tube, touching the surface of the liquid for the experiments measured at 14.1 T. For experiments measured on the 1.9 T benchtop NMR the optical fiber tip was inserted from the bottom and placed 2 mm below the NMR tube.
All numerical data is provided in Supplementary Data 1.
Absorption spectra measurements
Absorption spectra were recorded for 200 μM solutions of fluorescein, 4’,5’-dibromofluorescein, riboflavin, FMN and bilirubin. Spectra were also measured for urine samples before and after photobleaching (at 450 nm using 600 mW of power for 256 seconds), as well as for 40% urine samples spiked with 100 μM fluorescein. All measurements were performed on a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific), using water as the blank.
DFT calculations
Density functional theory (DFT) calculations were performed to optimize the molecular geometry (Supplementary Data 1 and 2) using the B3LYP functional and def2-TZVP basis set with incorporated Conductor-like Polarizable Continuum Model (CPCM) with a water solvent. The calculations were performed in Orca 5.073. The electronic structures to determine the g factors and the isotropic hyperfine interactions were calculated for the negatively charged fluorescein radical and positively charged radicals of the target molecules.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Atta-ur-Rahman, Choudhary, M. I. Applications of NMR spectroscopy, Vol. 8. (Bentham Science Publishers, 2020).
Riswanto, F. D. O. et al. Metabolite fingerprinting based on 1H-NMR Spectroscopy and liquid chromatography for the authentication of herbal products. Molecules 27, 1198 (2022).
Yang, J. Concepts and applications of chemical fingerprint for hit and lead screening. Drug Discov. Today 27, 103356 (2022).
Romero, C. A. NMR fingerprints, an integrated approach to uncover the unique components of the drug-like natural product metabolome of termite gut-associated streptomyces species. RSC Adv. 5, 104524–104534 (2015).
Nurani, L. H. et al. Metabolite fingerprinting using 1H-NMR spectroscopy and chemometrics for classification of three curcuma species from different origins. Molecules 26, 7626 (2021).
Holzgrabe, U. Quantitative NMR spectroscopy in pharmaceutical applications. Prog. Nucl. Magn. Reson Spectrosc. 57, 229–240 (2010).
Simmler, C., Napolitano, J. G., McAlpine, J. B., Chen, S.-N. & Pauli, G. F. Universal quantitative NMR analysis of complex natural samples. Curr. Opin. Biotechnol. 25, 51–59 (2014).
Bharti, S. K. & Roy, R. Quantitative 1H NMR spectroscopy. TrAC Trends Anal. Chem. 35, 5–26 (2012).
Silva Elipe, M. V. Advantages and disadvantages of nuclear magnetic resonance spectroscopy as a hyphenated technique. Anal. Chim. Acta 497, 1–25 (2003).
Bjerrum, J. T. Ed. Metabonomics: methods and protocols. In Methods in Molecular Biology. Vol. 1277 (Springer, 2015).
Crook, A. A. & Powers, R. Quantitative NMR-based biomedical metabolomics: current status and applications. Molecules 25, 5128 (2020).
Letertre, M. P. M. Giraudeau, P. de Tullio, P. Nuclear magnetic resonance spectroscopy in clinical metabolomics and personalized medicine: current challenges and perspectives. Front. Mol. Biosci. 8, 698337 (2021).
Mulder, F. A. A. Practical considerations for rapid and quantitative NMR-based metabolomics. J. Magn. Reson. 352, 107462 (2023).
Lema, C. et al. 1H NMR serum metabolomic profiling of patients at risk of cardiovascular diseases performing stress test. Sci. Rep. 10, 17838 (2020).
Vignoli, A. et al. NMR-based metabolomics identifies patients at high risk of death within two years after acute myocardial infarction in the AMI-florence II cohort. BMC Med. 17, 3 (2019).
Puchades-Carrasco, L., Palomino-Schätzlein, M., Pérez-Rambla, C. & Pineda-Lucena, A. Bioinformatics tools for the analysis of NMR metabolomics studies focused on the identification of clinically relevant biomarkers. Brief. Bioinform. 17, 541–552 (2016).
Kuhn, S., de Jesus, R. P. & Borges, R. M. Nuclear magnetic resonance and artificial intelligence. Encyclopedia 4, 1568–1580 (2024).
Bahado-Singh, R. O. et al. Artificial intelligence and the analysis of multi-platform metabolomics data for the detection of intrauterine growth restriction. PLoS One 14, e0214121 (2019).
Speyer, C. B. & Baleja, J. D. Use of nuclear magnetic resonance spectroscopy in diagnosis of inborn errors of metabolism. Emerg. Top. Life Sci. 5, 39–48 (2021).
Emwas, A. H. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol. Biol. 1277, 161–193 (2015).
Lee, J. ungH. o, Lee, J. H., Sekhar, A. & Cavagnero, S. 1H-Detected 13C photo-CIDNP as a sensitivity enhancement tool in solution NMR. J. Am. Chem. Soc. 133, 8062–8065 (2011).
Stadler, G. R. et al. Fragment screening and fast micromolar detection on a benchtop NMR spectrometer boosted by photoinduced hyperpolarization. Angew. Chem. Int Ed. 62, e202308692 (2023).
Bargon, J., Fischer, H. & Johnsen, U. Kernresonanz-Emissionslinien während rascher Radikalreaktionen. Z. Für Naturforsch. A 20, 1551 (1967).
Ward, H. R. & Lawler, R. G. Nuclear magnetic resonance emission and enhanced absorption in rapid organometallic reactions. J. Am. Chem. Soc. 89, 5518–5519 (1967).
Matysik, J. et al. Photo-CIDNP in solid state. Appl Magn. Reson 53, 521–537 (2022).
Kuhn, L. T. Hyperpolarization methods in NMR spectroscopy. Top. Curr. Chem. 338, 1–25 (2013).
Kaptein, R., Dijkstra, K. & Nicolay, K. Laser photo-CIDNP as a surface probe for proteins in solution. Nature 274, 293–294 (1978).
Kuhn, L. T. Photo-CIDNP NMR spectroscopy of amino acids and proteins. Top. Curr. Chem. 338, 229–300 (2013).
Torres, F. et al. Ultrafast fragment screening using photo-hyperpolarized (CIDNP) NMR. J. Am. Chem. Soc. 145, 12066–12080 (2023).
Bütikofer, M. et al. Rapid protein-ligand affinity determination by photoinduced hyperpolarized NMR. J. Am. Chem. Soc. 146, 17974–17985 (2024).
Bernarding, J., Bruns, C., Prediger, I. & Mützel, M. Plau-mann, M. detection of sub-nmol amounts of the antiviral drug favipiravir in 19F MRI using photo-chemically induced dynamic nuclear polarization. Sci. Rep. 14, 1527 (2024).
Okuno, Y. & Cavagnero, S. Fluorescein: a photo-CIDNP sensitizer enabling hypersensitive NMR data collection in liquids at low micromolar concentration. J. Phys. Chem. B 120, 715–723 (2016).
Sobol, A., Torres, F., Aicher, A., Renn, A. & Riek, R. Atto Thio 12 as a promising dye for photo-CIDNP. J. Chem. Phys. 151, 234201 (2019).
Lee, J. H. & Cavagnero, S. A novel tri-enzyme system in combination with laser-driven NMR enables efficient nuclear polarization of biomolecules in solution. J. Phys. Chem. B 117, 6069–6081 (2013).
Bernarding, J. et al. Low-cost LED-based photo-CIDNP enables biocompatible hyperpolarization of 19F for NMR and MRI at 7 T and 4.7 T. Chem. Phys. Chem. 19, 2453–2456 (2018).
Yang, H., Hofstetter, H. & Cavagnero, S. Fast-pulsing LED-enhanced NMR: a convenient and inexpensive approach to increase NMR sensitivity. J. Chem. Phys. 151, 245102 (2019).
Bramham, J. E. & Golovanov, A. P. Sample illumination device facilitates in situ light-coupled NMR spectroscopy without fibre optics. Commun. Chem. 5, 90 (2022).
Mompeán, M. et al. Pushing nuclear magnetic reso-nance sensitivity limits with microfluidics and photo-chemically induced dynamic nuclear polarization. Nat. Com. 9, 108 (2018).
Köckenberger, W.; Matysik, J. Hyperpolarization Methods in NMR. Encyclopedia of Spectroscopy and Spectrometry. (Elsevier, 2017).
Okuno, Y.; Cavagnero, S. Photochemically induced dynamic nuclear polarization: basic principles and applications. eMagRes 6, 283–314 (2017).
Goez, M. An introduction to chemically induced dynamic nuclear polarization. Concepts Magn. Reson. 7, 69–86 (1995).
Morozova, O. B. & Ivanov, K. L. Time-resolved chemically induced dynamic nuclear polarization of biologically important molecules. ChemPhysChem 20, 197–215 (2019).
Hore, P. J. & Kaptein, R. Photochemically induced dynamic nuclear-polarization (photo-CIDNP) of biological molecules using continuous wave and time-resolved methods. ACS Symp. Ser. 191, 285–318 (1982).
Kaptein, R. Simple rules for chemically induced dynamic nuclear polarization. J. Chem. Soc. D Chem. Com. 14, 732 (1971).
Kuhn, L. T., Weber, S., Bargon, J., Parella, T. & Pérez-Trujillo, M. Hyperpolarization-enhanced NMR spectroscopy of unaltered biofluids using photo-CIDNP. Anal. Chem. 96, 102–109 (2024).
Yang, H. et al. Selective isotope labeling and LC-photo-CIDNP enable NMR spectroscopy at low-nanomolar concentration. JACS 6, 11608–11619 (2022).
Harris, D. C. Quantitative Chemical Analysis, 7th ed., 2nd print. (Freeman, 2007).
Adams, R. W., Holroyd, C. M., Aguilar, J. A., Nilsson, M. & Morris, G. A. Perfecting” WATERGATE: clean proton NMR spectra from aqueous solution. Chem. Commun. 49, 358–360 (2013).
Goez, M., Mok, K. & Hore, K. H. P. J. Photo-CIDNP experiments with an optimized presaturation pulse train, gated continuous illumination, and a background-nulling pulse grid. J. Magn. Reson 177, 236–246 (2005).
Anesi, A. et al. Metabolic Profiling of human plasma and urine, targeting tryptophan, tyrosine and branched chain amino acid pathways. Metabolites 9, 261 (2019).
Anderson, B. J. Paracetamol (Acetaminophen): mechanisms of action. Paediatr. Anaesth.18, 915–921 (2008).
Dahlöf, C. G. H. Sumatriptan: pharmacological basis and clinical results. Curr. Med. Res. Opin. 17, s35–s45 (2001).
Adachi, J., Kumar, C., Zhang, Y., Olsen, J. V. & Mann, M. The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome Biol. 7, R80 (2006).
Bouatra, S. et al. The human urine metabolome. PLoS ONE 8, e73076 (2013).
Lau, S. N. The estimation of paracetamol and its major metabolites in both plasma and urine by a single high-performance liquid chromatography assay. J. Pharm. Biomed. Anal. 12, 1563–1572 (1994).
Lacey, L. F., Hussey, E. K., Fowler, P. A. Single dose pharmacokinetics of sumatriptan in healthy volunteers. Eur. J. Clin. Pharmacol. 47, 543–548 (1995).
U.S. Food and Drug Administration. Bioanalytical Method Validation: Guidance for Industry. (FDA, Silver Spring, MD, USA, 2018).
The fitness for purpose of analytical methods: a laboratory guide to method validation and related topics, 2nd ed., Eurachem, Teddington (2014).
Abraham, J. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, in Handbook of Transnational Economic Governance Regimes; Tietje, C., Brouder, A., Eds.; Brill, Nijhoff, 1041–1053 (2010).
European Medicines Agency. Guideline on Bioanalytical Method Validation, EMEA/CHMP/EWP/192217/2009 Rev. 1 Corr. 2, (EMA, 2011).
Paululat, T. Rabe, M. Berdnikova, D. V. Modification of an NMR probe for monitoring of photoreactions. J. Magn. Reson. 327, 106990 (2021).
Hore, J. & Broadhurst, R. W. Photo-CIDNP of biopolymers. Prog. Nucl. Magn. Reson. Spectrosc. 25, 345–402 (1993).
Tada, T., Shimajiri, T., Nishimura, K., Matsumoto, N. & Yanai, N. Dye–Quencher pair screening for efficient photo-CIDNP: the role of molecular diffusion. J. Chem. Phys. 162, 104302 (2025).
Gomez, M. V., Baas, S. & Velders, A. H. Multinuclear 1D and 2D NMR with 19F-photo-CIDNP hyperpolarization in a microfluidic chip with untuned microcoil. Nat. Com. 14, 3885 (2023).
Mao, F. et al. Chemical structure-related drug-like criteria of global approved drugs. Molecules 21, 75 (2016).
Aldeghi, M., Malhotra, S., Selwood, D. L. & Chan, A. W. E. Two- and three-dimensional rings in drugs. Chem. Biol. Drug Des. 83, 450–461 (2014).
Pautova, A. K. Metabolic profiling of aromatic compounds. Metabolites 14, 107 (2024).
Sol, J. et al. Plasma acylcarnitines and gut-derived aromatic amino acids as sex-specific hub metabolites of the human aging metabolome. Aging Cell 22, e13821 (2023).
Ricós, C. et al. Application of biological variation – a review. Biochem. Med. 19, 250–259 (2009).
Dutta, S. et al. A comprehensive review on analytical techniques for the quantification of pharmaceutical compounds in biological matrices: recent advances and future directions. J. Cardiovasc Dis. Res. 12, 712–747 (2024).
Caroli, S. Analytical Techniques For Clinical Chemistry: Methods And Applications, 1st ed., 60-828 (Wiley, 2012).
Krȩżel, A. & Bal, W. A formula for correlating pka values determined in D2O and H2O. J. Inorg. Biochem. 98, 161–166 (2004).
Neese, F. Software update: The ORCA program system—version 5.0. WIREs Comput. Mol. Sci. 12, e1606 (2022).
Author information
Authors and Affiliations
Contributions
M.S. (Conceptualization, methodology, investigation, validation, writing—original draft, visualization), S.H. (Conceptualization, supervision, writing, and funding acquisition), T.M. (Methodology, validation, and supervision).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Nikolay E. Polyakov and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Stefańska, M., Müntener, T. & Hiller, S. Photo-CIDNP for quantification of micromolar analytes in urine. Commun Chem 8, 223 (2025). https://doi.org/10.1038/s42004-025-01626-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01626-8
