
Abstract
Liquid-liquid phase separation (LLPS) facilitates functionally-relevant proteins to gather in high concentrations at specific locations. Several interacting proteins encapsulated in the membraneless droplets tend to form abnormal protein aggregates which are linked to numerous pathological diseases. Thus direct observation of LLPS, successive protein aggregates and the potential conversion processes is significant for understanding associated cell function and pathology. In this review, we firstly introduce the techniques for imaging LLPS, including in vitro reconstitution and intracellular visualizing strategies. Then we elaborate recent advances in the development of the imaging approaches for protein aggregates, followed by highlighting the monitor of protein conformational changes during aggregation process. Furthermore, the similarities and differences are discussed along with these state-of-the-art technologies. Finally, the major challenges and potential in exploration of detection methods for visualizing LLPS and protein aggregates are also pointed out. With these advances, it is becoming possible both to ‘see’ independent state of LLPS or protein aggregates, and realize the real-time monitor of the whole process.

Similar content being viewed by others
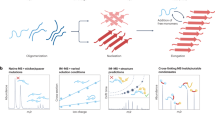
Introduction
Liquid-liquid phase separation (LLPS) and the protein aggregation that comes out of it have been widely observed and play significant roles in both biological processes and pathological diseases1,2,3,4. Cells orchestrate numerous fundamental biochemical activities through LLPS, the process by which biomacromolecules (typically protein or RNA) condense into nanoscale matter structures and separate into independent membraneless compartments5,6,7,8. Specific proteins and nucleic acids dynamically exchange biomolecules with the surrounding cytoplasm and/or nucleoplasm1,9. Such precise system facilitates to meet the biological requirement for spatiotemporal regulation of intracellular components and reactions10,11,12. Visualization of LLPS courses and resulted biomolecule products are therefore important for the understanding of normal biological functions and pathological processes that involve in the onset of the diseases.
Recent publications have verified disease-associated protein aggregates frequently nucleate through phase separation13,14,15. Remarkably, the intrinsically disordered regions (IDRs) with low sequence complexity offer these proteins enough conformational heterogeneity and flexibility to undergo LLPS16,17,18,19. Such characteristics of proteins are synergistic to generate the multivalent and weak self-attracting interactions that drive LLPS. To be specific, protein monomers of certain concentration will assemble into liquid-like droplets20,21,22,23. These condensed droplets can progressively gelate (some liquid droplets do not go through this stage) and further convert into fibrillar solid phase24,25,26,27. It has been demonstrated that plenty of human pathologies are linked to abnormal amyloid fibrils, especially neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD)28,29,30,31. These diseases are becoming more and more prevalent among a younger age group32,33,34. Thus, exploration of LLPS structures and resulted protein aggregates, especially dynamic monitor of the potential conversion processes, may reveal the general mechanism through which membraneless organelles can direct biological activities in neurodegenerative diseases.
Since growing research interest in LLPS and corresponding protein aggregates has been witnessed over the past two decades, several excellent reviews summarizing the techniques for studying liquid-phase organelles, amyloid aggregates, and the structure/function relationship of protein aggregates have been published24,35,36,37,38,39,40,41,42. However, systematic overview of imaging techniques for LLPS, generated protein aggregates and their relevance have been rarely explored, hindering the deeper understanding of how the structure of the protein molecules change in LLPS and convert to the final protein aggregates. In this review, we mainly address the recent advances in imaging strategies for LLPS and protein aggregation both in vitro and in vivo, with the main focus on the fluorescent techniques (Fig. 1). We begin by explaining the development and application of these state-of-the-art technologies, then discuss their similarities, differences and the principles behind. Finally, the major challenges of this research field are highlighted and future perspectives are also envisioned.

In the middle schematic, the pathway in which protein monomers go through LLPS and generate protein aggregation was shown.
Visualizing liquid–liquid phase separation
Conventional biochemical approaches have been applied to study the composition of many condensates in LLPS, such as nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry, antibody staining, Raman spectroscopy and fluorescent colocalization analysis43,44,45,46,47. Although these techniques were not able to achieve real-time visualization of the droplets formation and fusion during the transition, they can quantitatively detect LLPS at the nanometer-submicrometer scale. In addition, some of them may provide particular parameters which are significant for analyzing LLPS. For example, NMR can quantify the physical parameters such as tie-line, diffusion coefficient and line tension of cholesterol-driven LLPS48,49. Since the initial proposal of the phase transition phenomenon by Brangwynne et al., microscopy has been essential for gaining structural and biophysical insights into the phase separation processes50. Researchers have studied the molecular transport dynamics and associated properties through a number of imaging approaches. The details are discussed below.
In vitro reconstitution and imaging of LLPS
There are several technologies involved in the study of phase separation, among which in vitro reconstruction is proved to be a feasible system for the mechanism investigation51,52,53,54. As the key point of phase separation research is to demonstrate the target protein (or RNA) undergoing a dynamic and reversible aggregation process, it can be realized through the expression and purification of the target molecule in a test tube. Turbidity measurement is the initial way to record the macroscopic variation of LLPS55,56,57. However, the method can only detect the monomer assemblies without the information of size, shape or basic formation principle58,59. Thus turbidity measurement is suggested to be applied together with microscopic tools, which can track the droplet dynamics during the environmental perturbation.
The most frequently used techniques for imaging LLPS in vitro are bright-field and fluorescence microscopy (Fig. 2A). The solution of macromolecule monomers are recorded first before any assemblies formed. Then by incubation (from minutes to hours) of the sample with changing salt, RNA, pH or temperature as driving force, the emergence of liquid droplets could be observed by bright-field differential interference contrast (DIC) imaging60. If the components in the droplets are fluorescently labeled, the concentration of the dense phase could be evaluated with the standard curve of the fluorophore61. And for an identified driving force, LLPS would be suppressed after removing it. It is worth noting that specific RNA-binding proteins are sensitive and they will crosslink when exposing to light, thus experimentally induced gelation should be excluded62,63,64.

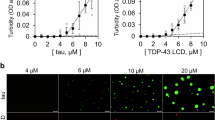
A The schematic of reconstitution process in test tubes and subsequent imaging using confocal microscopy. B Unlabeled tau and tau-GFP both undergo liquid-liquid phase separation at physiological concentrations. Scale bars are 5 µm. Images minimally modified from ref. 1 with permission from Springer Nature. C FUS converts from a liquid droplet to fibrous aggregates during in vitro “aging” experiment over 8 h. The corresponding “starbursts” structure and photobleaching test results of wild-type and G156E FUS are also shown. Images minimally modified from ref. 65 with permission from Elsevier.
Combs and coworkers studied the process of tau monomer transforming to pathological aggregates through dynamic LLPS at physiological protein levels in vitro1. They established molecular crowding conditions in buffers as physiological mimics with polyethylene glycol (PEG) as crowding reagent. In a standard HEPES buffer (10 mM HEPES, 150 mM NaCl, 0.1 mM EDTA, 2 mM DTT, pH 7.4), liquid droplets appeared for unlabeled full-length tau (2 µM) only when 10% PEG was present (Fig. 2B, left). Similarly, tau-GFP also formed spherical liquid droplets with 10% PEG (Fig. 2B, right). This work investigated the droplet dynamics systematically, providing deep insights into the mechanisms of tau toxicity with phase separation in vitro. In another rigorous research, Alberti et al. 65 first demonstrated prion-like FUS protein formed liquid compartments at DNA damage sites as well as in cytoplasm under stress in cells. Then in order to further confirm this phenomenon, an “aging” experiment was designed in test tube which showed FUS liquid droplets converting to fibrous aggregates over time (Fig. 2C). The droplets may act as centers of fiber nucleation and growth, and these “starbursts” structures were possibly the transition structures of a droplet converting into fibers. Fluorescence recovery after photobleaching (FRAP) is one of the most effective strategies to study molecular dynamics change for both in vitro and in vivo phase separation. In this research, FRAP tests for wild-type and G156E FUS were performed to quantify their biophysical properties. At time point 0 h, both the fluorescence of wild-type and G156E droplets recovered quickly after photobleaching; but after 8 h the G156E structures exhibited no fluorescence recovery, suggesting an aggregated state at that time point.
Except for the study of the dynamic process that a target protein undergoing during LLPS, a recent work has reported the visualization of multivalent protein-RNA interactions involved in liquid-liquid phase transitions. The authors demonstrated RNA interaction drove liquid-liquid phase transition in a human protein SERF2 by fluorescence imaging. And systematic investigation of how protein disorder influenced and specific contacts involved in these transitions have been illustrated. The results provided the structural insights into SERF2 binding to RNA TERRA rG4 and how it distorted the rG4 structure, laying a foundation for future work to probe other protein-RNA pairs that can be analyzed in stress granules or elsewhere66.
Spectral and lifetime phasor analysis is an unique technology characterizing the microenvironment of proteins in different phases. It was initially used in non-disease related protein applications67,68, and recently has been applied in combination with an environment-sensitive fluorescent probe ACDAN to generate a “microenvironment map” of the proteins under various states. By inspecting the phasor plot, tiny shifts in the ACDAN emission spectrum that accompaned different protein phases (native, aggregated or condensed) can be resolved69,70. These studies showed that ACDAN-spectral phasor analysis can sensitively investigate the microenvironments and droplet properties of protein phase-transition without labels, offering a helpful tool for probing their mechanisms and regulation.
In vitro reconstitution has proven indispensable for dissecting the minimal requirements underlying LLPS of proteins such as tau and FUS. By coupling turbidity test with real-time microscopy, these studies established that macroscopic condensation can be achieved at physiologically low micromolar concentrations once macromolecular crowding is introduced, highlighting the sensitivity of the phase boundary to excluded-volume effects. Nevertheless, several limitations intrinsic to test-tube assays must be noticed. Lack of ATP-dependent chaperones and cytosolic shear stress would risk PEG-induced artefacts, or suffer from tag- or photo-crosslinking, and are typically limited to <8 h tracking. Future microfluidic, label-free and energy-regenerating systems will be in demand to verify the reversibility and kinetic parameters operative in living cells.
LLPS imaging in cells
Although in vitro imaging has helped illustrate many fundamental principles of phase separation process, it still remains unclear if the mimics are physiologically relevant. Therefore visualization of LLPS within cells is particularly necessary. Nucleolus was the first membraneless compartment discovered within the neuronal nuclei71. Later other similar compartments were observed in the eukaryotic nucleus, cytoplasm as well as on the membrane72,73,74. The location, shape, dynamics and assembly manner of these structures were revealed by high-resolution microscopy imaging75,76,77,78,79,80. As mentioned in the above section, FRAP is one of the most powerful tools for intracellular investigation of LLPS81,82,83. FRAP experiments can evaluate the internal homogeneity of liquid-like droplets or condensates, obtaining high-resolution images together with dynamic fluorescence recovery curves after photobleaching in cells84,85,86,87,88.
LLPS in cells could be divided into two operational classes. The first class is the physiological ones which are short-lived, reversible droplets (e.g., nucleoli, stress granules, BRD4/MED1 transcriptional condensates) that disassemble by chaperones/ATP. In contrast, the other is the pathological/amyloid-prone class with the same physics driven beyond safety limits, yielding persistent gels or amyloid linked to disease. Young et al. have done an innovative research to demonstrate the transcriptional coactivators BRD4 and MED1 were enriched at super-enhancers (SEs) within cells, and formed nuclear puncta with the properties of phase-separated biomolecular condensates (Fig. 3A)89. MED1 and BRD4 were first visualized as discrete bodies in the nuclei of murine embryonic stem cells (mESCs) by both fixed- and live-cell imaging. Then immunofluorescence (IF) of BRD4 or MED1 was performed together with DNA-FISH for Nanog gene and its SEs. The results exhibited that the coactivators consistently overlapped with the genomic foci, indicating both BRD4 and MED1 puncta exsit at SEs (Fig. 3B). Also FRAP experiments were carried out to study the dynamics of BRD4 and MED1 puncta. After photobleaching, the fluorescence recovered within several seconds for both endogenously tagged mEGFP-BRD4 or mEGFP-MED1 cell lines, which were similar with the behaviors of previously reported liquid-like condensates (Fig. 3B). Furthermore, the researchers proved the intrinsically IDRs of the coactivators facilitated the intracellular phase separation by light-inducible droplet formation assay. During stimulation, droplet fusion events occurred for proximal droplets as liquid-like fusion properties (Fig. 3C). These results synergistically support the point of coactivators forming phase-separated condensates at SEs that compartmentalize and concentrate the transcription apparatus89.

Images minimally modified from ref. 89 with permission from AAAS. A Schematic of phase separation of coactivators at SEs. B Top: colocalization between MED1 and the Nanog locus by immunofluorescence (IF) and DNA-FISH in fixed mouse embryonic stem cells (mESCs). Bottom: FRAP experiment within mEGFP-MED1-engineered mESCs. C Top: time-lapse images of the nucleus of an NIH3T3 cell expressing MED1-optoIDR subjected to laser excitation every 2 s. Bottom: A droplet fusion event occurs in the region highlighted in the above yellow box. Scale bars, 1 µm.
Another research group developed an integrated approach of SE profiling and core regulatory circuitry (CRC) calling algorithm to figure out how CRC formed phase-separated condensates in metastatic and chemo-resistant osteosarcoma90. They initially obtained SE landscape to define metastasis- and chemoresistance-specific CRC. Then after demonstrating the formation of CRC biomolecular condensates in vitro, they also tested the properties of the liquid-like droplets in engineered 143B cells by FRAP. The large-scale analysis illustrated the regulation function of CRC on the oncogenic transcription in mammal cells. To sum up, the reversible nature brought about by physiological LLPS provided an insight that when cells failed to maintain a dynamic balance, how the same principle of phase separation may accelerate amyloid aggregation.
The abnormal aggregation of α-Synuclein (α-Syn) is the key factor for studying the pathogenic mechanism of Parkinson’s disease pathogenesis91,92,93. Researches based on NMR techniques realized the analysis of the structure and/or interaction of α-Syn aggregation, also the metabolomics of serum/CSF used for the classification of Parkinson’s disease94,95. However, it is still hard to accomplish direct imaging of α-Syn by NMR techniques. And the events involved in the early phase-separated state of α-Syn amyloid formation are still unclear. Maji et al. employed both in vitro reconstitution and cellular models to reveal how liquid-liquid phase separation of α-Syn preceded its aggregation96. Firstly, the process of α-Syn phase-separated droplets transforming into fibrillar aggregates was analyzed in test tubes. A delicate experiment of time-dependent changes of α-Syn liquid droplets was monitored by DIC imaging, Thioflavin S (ThS) staining, fluorescence microscopy and transmission electron microscopy (TEM) imaging (Fig. 4A). As time went by, ThS molecules readily co-partitioned inside the droplets, which referred to the presence of α-Syn aggregation. And after 30 days of incubation, the sample was observed as ThS-specific mesh-like fibrillar structures. The researchers further examined α-Syn liquid-liquid phase separation in mammalian cells. HeLa cells overexpressed tetracysteine-tagged α-Syn (C4-α-Syn) were cultured for 24 and 48 h and stained with fluorescein arsenical hairpin binder (Fig. 4B). The 24 h liquid-like droplets exhibited obvious mobility, and fusion events tended to happen with a rapid relaxation into spherical assemblies. Meanwhile, the droplets were largely clustered at the perinuclear region after 48 h, with the average diameter considerably increased up to 0.61 µm. In-cell FRAP recovery of droplets at 24 and 48 h of treatment were represented in thermal pseudocolour together with normalized fluorescence recovery curves (Fig. 4C). The results indicated the 24 h α-Syn molecules maintained the properties of the liquid-like state, and at 48 h the dynamics decreased due to the liquid-to-solid-like transition of α-Syn. These detailed insights of phase-separation behavior and aggregation process of α-Syn is of great concern in understanding Parkinson’s disease pathogenesis96.

Images minimally modified from ref. 96 with permission from Springer Nature. A α-Syn phase-separated droplets transform into fibrillar aggregates in test tubes. B Confocal images of HeLa cells overexpressing C4-α-Syn stained with FlAsH-EDT2, with quantification of the diameter of the droplets. C In-cell FRAP recovery of droplets at 24 and 48 h of treatment. The images of the pre-bleach and post-bleach droplets are shown with normalized fluorescence recovery curves.
Optogenetic approaches offered precise and controllable methods to monitor the dynamic LLPS process in real time. Researchers constructed light-controlled protein systems as inducible LLPS models by fusing tau with light-reactive Crye2. By exposing to specific wavelengths of light, the target proteins were induced to undergo phase separation and form aggregates, thus simulating the LLPS process within cells. Through this method, tau oligomerization and cytoplasmic granules in primary cortical neurons were driven, but rarely observed when transduced in SH-SY5Y, N2a or HEK cells, highlighting the importance of specific cellular environments on tau LLPS97. A similar strategy was used to explore the roles of different tau functional domains in LLPS process98.
As far as we know, light-inducible systems would generate local heating and ROS in cells, risking photo-crosslinking that converts liquid to artefactual gel. And most of the work in intracellular LLPS imaging has relied on the overexpression of target proteins in living cells to evaluate the probability and dynamics of phase-separated condensates formation. Moreover, there is no simultaneous measurement of condensate viscosity, ATP/chaperone activity or endogenous RNA contribution, leaving the reversibility and kinetic parameters unverified under native conditions. A growing interest of techniques for exploring the biophysical principles underlying phase separation in natural physiological conditions is advancing. These principles not only include the role of the spectrum of condensate states (from liquid phase to viscoelastic hydrogel), but also their biological function and dysfunction35.
Interrogating protein aggregates by microscopy
As described above, protein molecules of certain concentrations going through LLPS will possibly mature and aggregate into solid state. The visualization of these protein aggregates can elucidate the relationship between aggregation and corresponding diseases99,100,101,102. Numerous techniques have been developed to study the precise composition and transition of protein monomers into higher order aggregates (mainly with amyloid structure), among which cryo-electron microscopy (cryo-EM), TEM and fluorescence spectroscopy are the most commonly used techniques103,104,105,106. The details are discussed below.
Visualizing the structure of protein aggregates in vitro
Protein aggregates exhibit different properties in many biological activities compared with the unassembled protein monomers. Therefore, it is significant to observe the molecular-level structures of the protein aggregates which determine their functions, especially the amyloid fibrils36,107,108,109.
Cryo-EM imaging is capable to clearly investigate amyloid fibrils at atomic level. Eisenberg’s group demonstrated that the flanking sequence of hnRNPA2 with a well-folded structure was intensively aligned on the surface of the rigid fibril core by Cryo-EM (Fig. 5A)110. This construction ensured the outer layer of hnRNPA2 performed a relatively high flexibility compared with the fibril core. In addition, negative-staining transmission electron microscopy (NS-TEM) was also applied to visualize the morphology of α-Syn fibrils, as well as the binding conditions between α-Syn and vRAGE (Fig. 5B)111. Immunogold labeling of vRAGE with anti-His gold nanoparticles was incubated with α-Syn fibrils. The resulting TEM images showed labeled vRAGE binding on the surface of α-Syn fibrils obviously instead of α-Syn monomers. Further combined with the binding affinity measurement between vRAGE and α-Syn, the data all indicated RAGE had strong binding to α-Syn, especially the fibril form111,112.

A The cryo-EM images of hnRNPA2 fibril with a well-folded domain. Images minimally modified from ref. 110 with permission from Springer Nature. B Immunogold TEM images of a-Syn fibrils alone (left) and a-Syn fibrils incubated with vRAGE (right). White arrows indicate the nanogolds on a-Syn fibrils. Images minimally modified from ref. 111 with permission from Elsevier. C Phase transition of hnRNPA1 upon time with schematic illustration of different states of protein. Images minimally modified from refs. 36,116 with permission from Wiley-VCH and Springer Nature.
As we know, the amyloid fibrils formed by several RNA-binding proteins are highly reversible113,114. The dynamic association and disassociation processes could be influenced by temperature or phosphorylation115. The phase separation of reversible hnRNPA1 amyloid fibrils was observed by cooling down the protein solution from room temperature to 4 °C. As shown in Fig. 5C, the solution became non-transparent after several minutes, with spherical droplets of hnRNPA1 formed in the liquid phase. Then the authors utilized NS-TEM to check the phase-separated hnRNPA1 sample and found clear amyloid fibrils inside the droplets. ThT experiments were also carried out to monitor the amyloid fibril formation in order to validate these fibrils were not artifact derived from the drying duration. Moreover, the schematic of these impressive states of hnRNPA1 protein was concisely summarized in a previous review (Fig. 5C)36. Such visualization of reversible protein fibrils formation and structure change helped to understand how they were involved in the adjustment of both material properties and liquid-droplet dynamics116.
Investigating the co-aggregation of disease-associated proteins reveals synergistic mechanisms by which pathological proteins interact to form toxic aggregates in neurodegenerative disorders. In a recent study, correlation/cross-correlation spectroscopy (FCS/FCCS) and two-color coincidence detection (TCCD) burst analysis were used to characterize tau and α-Syn co-aggregates at the single-molecule level117. The results have clarified that tau and α-Syn proteins did not cause the disease independently, but can synergistically form harmful aggregates through direct electrostatic interactions. This work not only deepened our understanding of aberrant protein aggregation in neurodegenerative diseases, but also identified phase separation processes themselves as novel therapeutic targets.
The above researches showed that Cryo-EM revealed amyloid cores whose surface-exposed domains remained accessible, while immuno-electron microscopy confirmed the ligand binding was conformation-selective for the fibrillar state. Cooling-recovery assays further demonstrated these fibrils can reversibly convert into liquid-like droplets, indicating their stability was temperature-tunable. However, the structures were imaged under dehydrated or cryogenic conditions, and reversibility was probed solely in purified-protein, temperature-jump experiments; molecular crowding, chaperones or post-translational modifications in the cellular milieu might stabilize or trap fibrils in vivo. In-cell cryo-ET and temperature-jump spectroscopy are now expected to verify whether the observed conformations and rapid phase transitions persist under physiological conditions.
Intracelluar imaging of protein aggregates
The need for reliable imaging of protein aggregates in cells has stimulated the development of numerous fluorescence-based strategies. Initially, the concept for in situ amyloid oligomers and fibrils monitoring was proposed by Faller et al. in 2014118. They tried to accomplish simultaneous and specific detection of Aβ oligomers and fibrils using Förster resonance energy transfer (FRET) between two extrinsic fluorophores. The mixture of bis-ANS and a styrylquinoxalin derivative was capable to detect the presence of oligomers and fibrils of amyloidogenic peptides in the same sample. However, this work was a proof of concept with no intracellular experimental data.
Intracellular proteins might expose the hydrophobic residues in the process of misfolding or aggregation, which will cause physical changes within local environment including obvious viscosity alterations. Thus the detection of soluble protein oligomers and insoluble aggregates can be transformed into the monitor of different viscosity. Zhang’s group has exploited a series of viscosity-sensitive fluorogenic molecules to realize direct visualization of protein aggregation in live cells. They rationally designed and synthesized 18 probes with a wide range of viscosity sensitivities, which was based on the control of their excited state rotational energy barrier. These probes formed a toolbox called AggFluor, and were applied in differentiating intracellular misfolded oligomers and insoluble aggregates (Fig. 6A)119. The probes exhibited differential turn-on and turn-off fluorescence for the two conformations of protein, resulting in an effective dual-color live-cell imaging strategy. In another work of the group, the authors proposed AggTag method by chemically installing extended π-rich alternating carbon-carbon linkages between the rotational electron donors and acceptors of molecular rotor-based fluorophores120. This method not only can examine amyloid fibrils of protein-of-interest (POI), but also exhibited a similar trend for amorphous protein aggregates (Fig. 6B). Later on, the above methods were extended to study protein aggregates using both fluorescence intensity and lifetime121. Although these viscosity-dependent strategies are nonspecific, the effective distinction between misfolded oligomers and insoluble aggregates is still very helpful due to their unique functions in live cells. There were also many other chemical probes developed for visualizing phase separation, transition and protein aggregation, empowering researchers to track these processes in cells with high spatiotemporal resolution122,123. By harnessing the physicochemical property (e.g., viscosity) as a biomarker for protein aggregation, these studies enabled real-time, non-invasive imaging in live cells and created a robust platform for early diagnosis and drug discovery. Future work would merge aggregation-sensitive cores with protein-specific antibodies or aptamers to engineer “dual-recognition” probes with superior specificity.

A AggFluor was a series of viscosity-responsive molecular rotor fluorophores. They allowed for differentiation of complex protein aggregates in live cells. Images minimally modified from ref. 119 with permission from American Chemical Society. B Scheme and representitive images of the AggTag method to monitor live-cell protein aggregation. Images minimally modified from ref. 120 with permission from Wiley-VCH. C Representative images of the hTau seeding assay. Hyperphosphorylated tau (green) was stained with AT8 antibody to show the insoluble aggregates of tau (white arrowheads). Images minimally modified from ref. 124 with permission from Springer Nature. D The intracellular and extracellular α-Syn-derived fluorescence was semi-quantitatively analyzed. Images minimally modified from ref. 125 with permission from Springer Nature.
In addition, Hyman’s group observed insoluble tau aggregates by hTau seeding assay in neurons124. When the brain extract of AD was incubated with neurons, tau aggregates were present in cells (Fig. 6C). And the quantification analysis showed similar trend compared with the intensities in the FRET-biosensor assay. In another research, the imaging of SH-SY5Y cells treated with α-Syn species was carried out using confocal scanning microscopy125. Cell membranes were labeled with red fluorescence, meanwhile α-Syn species were revealed with green fluorescence by polyclonal antibodies. The semi-quantitative statistical analysis of the intracellular and extracellular α-Syn-derived fluorescence indicated α-Syn fibrils were distributed mostly on the cellular membrane surface (Fig. 6D).
Monitor of protein conformational changes during aggregation
In recent years, the development of AlphaFold series has brought significant advancements in protein structure prediction126,127,128. However, it is worth noting that AlphaFold tools are based on existing protein structures in the Protein Data Bank (PDB) together with an optimization algorithm. Therefore, the predicted structures may have certain limitations to show single, static conformations and lack the dynamic features of proteins129. On the other hand, imaging is one of the most useful techniques to monitor the conformational evolution of protein aggregates during many physiological processes, including LLPS.
As protein misfolding diseases are highly linked to abnormal LLPS processes, the understanding of pathological protein phase transitions at molecular level may provide important insights. Perrett and Wu et al. studied the conformational changes during aberrant aggregation of neurodegenerative diseases-associated protein tau17. Single-molecule FRET and fluorescence correlation spectroscopy were performed to investigate tau conformational changes during LLPS and subsequent irreversible aggregation (Fig. 7A). To be specific, above the critical phase separation concentrations, the native protein monomers underwent LLPS to form condensed liquid-like droplet. The intra- and intermolecular changes of tau revealed the N- and C-terminal regions becoming extended and the microtubule-binding region was exposed. These changes facilitated intermolecular interactions which can promote subsequent irreversible conversion of tau into the amyloid fibrils. The results highlighted the deep links between the molecular changes of LLPS and tau aggregation, providing important insights into the mechanisms of pathological aggregates formation in cells.

A Left: schematic of tau conformational changes from native soluble monomers to condensed liquid droplets and finally amyloid fibrils. Right: corresponding characterizations of different structure changes of tau. Images minimally modified from ref. 17 with permission from American Chemical Society. B Characterization of human islet amyloid polypeptide fibrils over time. The time course involved the lag, growth, and plateau phases and representative ns-EM images at 3, 6, and 22 weeks were shown (scale bars, 80 nm). Images minimally modified from ref. 130 with permission from Elsevier.
As previously discussed, cryo-EM imaging is a powerful tool in the field of amyloid fibril structure evaluation at atomic level, including the disease-associated ones. However, most researches have focused on the structures at a single time point of long assembly processes, and the conformational evolution during fibrils formation remains unknown. A recent publication has accomplished the analysis of diabetes-related variant of human islet amyloid polypeptide (IAPP-S20G) over time to study the dynamic properties of amyloid polymorphism130. The structures of IAPP-S20G at three distinct phases of fibril assembly were observed in vitro using Cryo-EM. As shown in Fig. 7B, the fibrils formed during the lag, growth, and plateau phases obtained totally different structures with multiple IAPP-20G fibril polymorphs. And it was intriguing that as the fibrils assembly proceeded, new structure appeared while the other forms disappeared. Similarly, the time-course fibrils of wild-type hIAPP also exhibited this general property during fibrillation. Although it is still hard to determine whether different polymorphs interconvert directly, this work presented a high-resolution result of fibril maturation, which may enlighten the understanding of amyloid progression in diseases.
Super-resolution fluorescence methodologies have recently provided unprecedented nanoscale insights into amyloid assembly, including stimulated emission depletion (STED), photoactivated localization microscopy (PALM), stochastic optical reconstruction microscopy (STORM), single-molecule orientation localization microscopy (SMOLM) and Förster resonance energy transfer coupled to fluorescence lifetime imaging (FRET-FLIM). Transient-binding dSTORM with ThT enabled covalent and label-free imaging of Aβ fibrils with ~20 nm precision131. SMOLM revealed that Aβ42 fibril growth was driven by increasing molecular order, while decay and remodeling originated from local structural heterogeneity132. Two-color dSTORM resolved the directionality and kinetics of heterogeneous fibril growth133. And FRET-FLIM reported local molecular environment changes during aggregation134. These researches collectively underscored the transformative potential of super-resolution techniques for LLPS and protein aggregation analysis.
Physicochemical properties of biomolecular condensates revealed by imaging
Over the past five years, an expanding imaging-based toolkit has been deployed to map the key physicochemical signatures of biomolecular condensates—micropolarity, viscoelasticity, surface tension and internal pH—that govern their multiphase architecture and cellular function135,136,137.
Zhang’s group utilized fluorescence lifetime imaging with environment-sensitive probes to reveal the micropolarity differences drove the formation of multilayered membraneless organelles, directing their dynamic reorganization and function138. Micropolarity gradients affected layered architecture in phase-separated condensates, with lower-polarity cores recruited folded clients while higher-polarity shells binded disordered partners, leading to selective partitioning and tunable condensate structure. A subsequent study introduced a environment-sensitive dye to map the dielectric permittivity (ε) inside biomolecular condensates139. ε was estimated across the membraneless organelles and correlated with local hydrophobicity, revealing hydrophobic cores that preferentially adhered to DOPC membranes more often than hydrophilic shells, demonstrating hydrophobicity-driven membrane interactions. These findings highlighted the pivotal role of micropolarity in structuring nucleoli and other condensates.
Although biomolecular condensates appeared like droplets, an increasing number of studies have found that they possessed viscoelasticity, exhibiting the properties of both liquids and solids simultaneously. This mechanical property may influence their functions, stability and regulatory mechanisms140. Optical tweezers enable the probing of condensate viscoelasticity through both active and passive measurements141. Jawerth et al. used optical tweezers to systematically reveal the viscoelastic properties of FUS protein aggregates142. They reconstructed the phase separation system using aggregates formed by the low complexity domain of the FUS protein in vitro, and controlled the protein concentration, pH and salt concentration to provide a quantitative method for characterizing the mechanical properties of organelles without membranes. The results demonstrated FUS aggregates exhibited distinct viscoelastic properties, heterogeneity and aging effects, which offered a new perspective for understanding the abnormal protein aggregation caused by phase separation in neurodegenerative diseases. Micropipette aspiration (MPA) is also a powerful tool for measuring the surface tension and viscosity of biological condensates143. A calibration-free MPA model was presented that correctly described the hydrodynamics of unbounded condensates144. With validation against standard silicone oil of known material properties, it accurately predicted both viscosity and surface tension. Beyond the methods described here, surface tension and viscoelastic characteristics of the condensates can also be assessed using techniques such as atomic force microscopy (AFM)145 and flicker spectroscopy146.
Biomolecular condensates can serve as self-assembling, sequence-programmable pH imaging platforms, converting chemical composition into visible spatial pH signal across natural and synthetic systems. Native condensates (e.g., nucleoli, stress granules) acted as genetically-encoded pH sensors with their IDR composition setting a fixed buffer capacity. This would create stable, microscale pH gradients that can be imaged indirectly via pH-sensitive fluorescent protein reporters fused to condensate-resident IDRs147,148. Synthetic elastin-like polypeptide condensates might function as programmable pH imaging probes. Their sharp, reversible phase transition (ΔpH < 0.3) can be coupled with pH-sensitive fluorophores, converting droplet assembly/disassembly behavior into a binary fluorescence signal. This allowed ratiometric pH imaging inside vesicles or cells149.
These quantitative physicochemical read-outs of biomolecular condensates converted static phase-separated snapshots into sequence-encoded thermodynamic landscapes, providing the link between amino-acid sequence, mesoscale organization and biological output. And they enabled the distinguish of passive crowding from active sequence-encoded regulation within condensates.
Conclusions and future perspectives
There is growing lines of evidence that direct observation of LLPS and resulted protein aggregation can be useful in the revelation of associated disease mechanisms and progression150,151. Breakthroughs in detection and monitoring of phase-separated processes and protein aggregates both in vitro and in vivo are long-awaited results for the research and clinical field, representing efforts of numerous scientists over the past decades152,153,154,155.
In this review, recent advances in imaging strategies for LLPS, protein aggregates and the structural changes behind have been described, with a particular focus on the fluorescent techniques. Other microscopic tools, including cryo-EM, TEM and related quantification results are also discussed. The researches reviewed here are of remarkable importance and provide valuable guidance for the exploration of detection methods for in-depth and comprehensive understanding of disease mechanisms.
Despite the progress has been made, several challenges regarding the imaging of phase-separated processes and protein aggregates still need addressing. Firstly, there is a lack of effective method to monitor the LLPS processes, protein oligomers and final fibrils simultaneously and/or orderly due to the technical limitation. The ultimate goal is to clearly know the macroscopic performance and corresponding molecular alterations of the whole process of the disease-associated biomarkers. Second, as we have discussed above, the available strategies in cells are developed on the basis of overexpression of target proteins. Thus visualization of LLPS or protein fibrils under original physiological conditions in live cells is a potential direction, which may play significant roles in the early diagnosis of diseases. And further developed in vivo imaging systems will shed light on future research in both biological and medical applications. Finally, acquiring the spatial distribution and precise quantification information of the protein aggregates at single-molecule level can be very powerful to improve our understanding of LLPS and protein aggregation. We look forward to new technologies to study the structure, kinetics and functions of LLPS as well as protein aggregates.
References
Kanaan, N. M., Hamel, C., Grabinski, T. & Combs, B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 11, 2809 (2020). Disease-associated tau modifications promoted liquid-liquid phase separation that drove formation of toxic and non-filamentous oligomers, revealing a key pathogenic mechanism in tauopathies.
Xue, S. et al. Low-complexity domain of U1-70K modulates phase separation and aggregation through distinctive basic-acidic motifs. Sci. Adv. 5, eaax5349 (2019).
Shin, Y. & Brangwynne, C. P. Liquid phase condensation in cell physiology and disease. Science 357, eaaf4382 (2017).
Babinchak, W. M. & Surewicz, W. K. Liquid–liquid phase separation and its mechanistic role in pathological protein aggregation. J. Mol. Biol. 432, 1910–1925 (2020).
Molliex, A. et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133 (2015).
Strom, A. R. et al. Phase separation drives heterochromatin domain formation. Nature 547, 241–245 (2017).
Su, Q., Mehta, S. & Zhang, J. Liquid-liquid phase separation: orchestrating cell signaling through time and space. Mol. Cell 81, 4137–4146 (2021).
Mehta, S. & Zhang, J. Liquid–liquid phase separation drives cellular function and dysfunction in cancer. Nat. Rev. Cancer 22, 239–252 (2022).
Berry, J. et al. RNA transcription modulates phase transition-driven nuclear body assembly. Proc. Natl Acad. Sci. USA 112, E5237–E5245 (2015).
Nesterov, S. V., Ilyinsky, N. S. & Uversky, V. N. Liquid-liquid phase separation as a common organizing principle of intracellular space and biomembranes providing dynamic adaptive responses. Biochim. Biophys. Acta (BBA) - Mol. Cell Res. 1868, 119102 (2021).
Zhang, M.-L. et al. Interplay between intracellular transport dynamics and liquid‒liquid phase separation. Adv. Sci. 11, 2308338 (2024).
Ribeiro, S. S., Samanta, N., Ebbinghaus, S. & Marcos, J. C. The synergic effect of water and biomolecules in intracellular phase separation. Nat. Rev. Chem. 3, 552–561 (2019).
Harvey, Z. H., Chen, Y. & Jarosz, D. F. Protein-based inheritance: epigenetics beyond the chromosome. Mol. Cell 69, 195–202 (2018).
Jarosz, D. F. & Khurana, V. Specification of physiologic and disease states by distinct proteins and protein conformations. Cell 171, 1001–1014 (2017).
Hufnagel, D. A., Tükel, Ç & Chapman, M. R. Disease to dirt: the biology of microbial amyloids. PLOS Pathog. 9, e1003740 (2013).
Ash, P. E. A. et al. TIA1 potentiates tau phase separation and promotes generation of toxic oligomeric tau. Proc. Natl Acad. Sci. USA 118, e2014188118 (2021).
Wen, J. et al. Conformational expansion of tau in condensates promotes irreversible aggregation. J. Am. Chem. Soc. 143, 13056–13064 (2021). Single-molecule FRET revealed that Tau LLPS extended its termini to expose the MT-binding domain, nucleating nanoclusters that disease mutations P301L/S explosively converted into amyloid.
Chong, S. & Mir, M. Towards decoding the sequence-based grammar governing the functions of intrinsically disordered protein regions. J. Mol. Biol. 433, 166724 (2021).
Lin, Y., Currie, S. L. & Rosen, M. K. Intrinsically disordered sequences enable modulation of protein phase separation through distributed tyrosine motifs. J. Biol. Chem. 292, 19110–19120 (2017).
Julius, K. et al. Impact of macromolecular crowding and compression on protein–protein interactions and liquid–liquid phase separation phenomena. Macromolecules 52, 1772–1784 (2019).
Jo, Y. & Jung, Y. Interplay between intrinsically disordered proteins inside membraneless protein liquid droplets. Chem. Sci. 11, 1269–1275 (2020).
Cao, Y. et al. Bioinspired autocatalysis for polymer nanoparticle synthesis: monomer preassembly through liquid–liquid phase separation. Macromolecules 57, 8097–8108 (2024).
Zhang, Z. et al. Amyloid aggregation and liquid–liquid phase separation from the perspective of phase transitions. J. Phys. Chem. B 127, 6241–6250 (2023).
Alberti, S. & Hyman, A. A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 22, 196–213 (2021).
Lin, W.-J. et al. Magnetic fields reduce apoptosis by suppressing phase separation of Tau-441. Research 6, 0146 (2023).
Pan, Q. et al. Real-time study of protein phase separation with spatiotemporal analysis of single-nanoparticle trajectories. ACS Nano 15, 539–549 (2021).
Tejedor, A. R. et al. Protein structural transitions critically transform the network connectivity and viscoelasticity of RNA-binding protein condensates but RNA can prevent it. Nat. Commun. 13, 5717 (2022).
Eisenberg, D. & Jucker, M. The amyloid state of proteins in human diseases. Cell 148, 1188–1203 (2012).
Shahnawaz, M. et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 578, 273–277 (2020).
Dobson, C. M., Knowles, T. P. J. & Vendruscolo, M. The amyloid phenomenon and its significance in biology and medicine. Cold Spring Harb. Perspect. Biol. 12, a033878 (2020).
Peng, C., Trojanowski, J. Q. & Lee, V. M. Y. Protein transmission in neurodegenerative disease. Nat. Rev. Neurol. 16, 199–212 (2020).
Monzio Compagnoni, G. et al. The role of mitochondria in neurodegenerative diseases: the lesson from Alzheimer’s disease and Parkinson’s disease. Mol. Neurobiol. 57, 2959–2980 (2020).
Soleimani-Meigooni, D. N. et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer’s disease and other neurodegenerative diseases. Brain 143, 3477–3494 (2020).
Goedert, M. & Spillantini, M. G. Propagation of Tau aggregates. Mol. Brain 10, 18 (2017).
Bracha, D., Walls, M. T. & Brangwynne, C. P. Probing and engineering liquid-phase organelles. Nat. Biotechnol. 37, 1435–1445 (2019).
Xu, Q. et al. Protein amyloid aggregate: structure and function. Aggregate 4, e333 (2023).
Sawaya, M. R. et al. The expanding amyloid family: structure, stability, function, and pathogenesis. Cell 184, 4857–4873 (2021).
Fowler, D. M., Koulov, A. V., Balch, W. E. & Kelly, J. W. Functional amyloid: from bacteria to humans. Trends Biochem. Sci. 32, 217–224 (2007).
Mahler, H.-C., Friess, W., Grauschopf, U. & Kiese, S. Protein aggregation: pathways, induction factors and analysis. J. Pharm. Sci. 98, 2909–2934 (2009).
Amin, S. et al. Protein aggregation, particle formation, characterization & rheology. Curr. Opin. Colloid Interface Sci. 19, 438–449 (2014).
Housmans, J. A. J., Wu, G., Schymkowitz, J. & Rousseau, F. A guide to studying protein aggregation. FEBS J. 290, 554–583 (2023).
Kitamura, A. & Kinjo, M. State-of-the-art fluorescence fluctuation-based spectroscopic techniques for the study of protein aggregation. Int. J. Mol. Sci. 19, 964 (2018).
Fei, J. et al. Quantitative analysis of multilayer organization of proteins and RNA in nuclear speckles at super resolution. J. Cell Sci. 130, 4180–4192 (2017).
Putnam, A., Cassani, M., Smith, J. & Seydoux, G. A gel phase promotes condensation of liquid P granules in Caenorhabditis elegans embryos. Nat. Struct. Mol. Biol. 26, 220–226 (2019).
Tang, Y. et al. Prediction and characterization of liquid-liquid phase separation of minimalistic peptides. Cell Rep. Phys. Sci. https://doi.org/10.1016/j.xcrp.2021.100579 (2021).
Samuel, A. Z., Sugiyama, K., Ando, M. & Takeyama, H. Direct imaging of intracellular RNA, DNA, and liquid–liquid phase separated membraneless organelles with Raman microspectroscopy. Commun. Biol. 5, 1383 (2022).
Zhang, X. et al. Study liquid–liquid phase separation with optical microscopy: a methodology review. APL Bioeng. 7, 021502 (2023).
Veatch, S. L. et al. Liquid domains in vesicles investigated by NMR and fluorescence microscopy. Biophys. J. 86, 2910–2922 (2004).
Lau, S. ophie & Middleton, D. avidA. Analysis of the orientation of cholesterol in high-density lipoprotein nanodiscs using solid-state NMR. Phys. Chem. Chem. Phys. 24, 23651–23660 (2022).
Brangwynne, C. P. et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732 (2009).
Le Ferrand, H. et al. Time-resolved observations of liquid–liquid phase separation at the nanoscale using in situ liquid transmission electron microscopy. J. Am. Chem. Soc. 141, 7202–7210 (2019).
Kang, W. et al. Liquid-liquid phase separation (LLPS) in synthetic biosystems. Mater. Sci. Eng.: R: Rep. 157, 100762 (2024).
Zhang, M. et al. Molecular organization of the early stages of nucleosome phase separation visualized by cryo-electron tomography. Mol. Cell 82, 3000–3014.e3009 (2022).
Currie, S. L. et al. Quantitative reconstitution of yeast RNA processing bodies. Proc. Natl Acad. Sci. USA 120, e2214064120 (2023).
Babinchak, W. M. et al. Small molecules as potent biphasic modulators of protein liquid-liquid phase separation. Nat. Commun. 11, 5574 (2020).
Ambadipudi, S. et al. Liquid–liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat. Commun. 8, 275 (2017).
Van Lindt, J. et al. A generic approach to study the kinetics of liquid–liquid phase separation under near-native conditions. Commun. Biol. 4, 77 (2021).
Alberti, S., Gladfelter, A. & Mittag, T. Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176, 419–434 (2019).
Huang, Y. et al. Common pitfalls and recommendations for using a turbidity assay to study protein phase separation. Biochemistry 60, 2447–2456 (2021).
Alberti, S. et al. A user’s guide for phase separation assays with purified proteins. J. Mol. Biol. 430, 4806–4820 (2018).
Girelli, A. et al. Molecular flexibility of antibodies preserved even in the dense phase after macroscopic phase separation. Mol. Pharm. 18, 4162–4169 (2021).
Kramer, K. et al. Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods 11, 1064–1070 (2014).
Feng, H. et al. Structure-based prediction and characterization of photo-crosslinking in native protein–RNA complexes. Nat. Commun. 15, 2279 (2024).
Kristofich, J. & Nicchitta, C. V. High-throughput quantitation of protein–RNA UV-crosslinking efficiencies as a predictive tool for high-confidence identification of RNA-binding proteins. RNA 30, 644–661 (2024).
Patel, A. et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 (2015). ALS-linked FUS protein phase-separated into functional liquid droplets that spontaneously matured into pathological solids, and that disease-linked mutations accelerated the transition, establishing deranged liquid-solid partitioning as a core mechanism of neurodegeneration.
Sahoo, B. R. et al. Visualization of liquid-liquid phase transitions using a tiny G-quadruplex binding protein. bioRxiv 09, 561572 (2023).
Mangiarotti, A. et al. Biomolecular condensates modulate membrane lipid packing and hydration. Nat. Commun. 14, 6081 (2023).
Luca, G. D. et al. Surface-catalyzed liquid-liquid phase separation and amyloid-like assembly in microscale compartments. J. Colloid Interface Sci. 676, 569–581 (2023).
Rodríguez, L. C., Foressi, N. N. & Celej, M. S. Tracking protein transitions through fluorescence spectral phasor analysis with ACDAN. Biophys. Rep. 5, 100209 (2025).
Foressi, N. N. et al. Cation-driven modulation of tau condensates: insights into liquid–liquid phase separation and rheological properties. Biomacromolecules 26, 3605–3616 (2025).
Pederson, T. The nucleolus. Cold Spring Harb. Perspect. Biol. 3, a000638 (2011).
Liang, J. & Cai, D. Membrane-less compartments in the nucleus: separated or connected phases?. Curr. Opin. Cell Biol. 84, 102215 (2023).
Ivanov, I. et al. Directed growth of biomimetic microcompartments. Adv. Biosyst. 3, 1800314 (2019).
Gomes, E. & Shorter, J. The molecular language of membraneless organelles. J. Biol. Chem. 294, 7115–7127 (2019).
Banani, S. F., Lee, H. O., Hyman, A. A. & Rosen, M. K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017).
Feric, M. et al. Coexisting liquid phases underlie nucleolar subcompartments. Cell 165, 1686–1697 (2016).
Woodruff, J. B. et al. The centrosome is a selective condensate that nucleates microtubules by concentrating tubulin. Cell 169, 1066–1077.e1010 (2017).
Boke, E. et al. Amyloid-like self-assembly of a cellular compartment. Cell 166, 637–650 (2016).
Brangwynne, C. P., Mitchison, T. J. & Hyman, A. A. Active liquid-like behavior of nucleoli determines their size and shape in Xenopus laevis oocytes. Proc. Natl Acad. Sci. USA 108, 4334–4339 (2011).
Case, L. B., Zhang, X., Ditlev, J. A. & Rosen, M. K. Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science 363, 1093–1097 (2019).
Muzzopappa, F. et al. Detecting and quantifying liquid–liquid phase separation in living cells by model-free calibrated half-bleaching. Nat. Commun. 13, 7787 (2022).
Wang, X. et al. SMART FRAP: a robust and quantitative FRAP analysis method for phase separation. Chem. Commun. 59, 2307–2310 (2023).
Huang, X. et al. ROS regulated reversible protein phase separation synchronizes plant flowering. Nat. Chem. Biol. 17, 549–557 (2021).
Santamaria, A. et al. Quantifying surface tension and viscosity in biomolecular condensates by FRAP-ID. Biophys. J. 123, 3366–3374 (2024).
Chen, Y. et al. Enzyme-active liquid coacervate microdroplets as artificial membraneless organelles for intracellular ROS scavenging. Biomater. Sci. 10, 4588–4595 (2022).
Gibson, B. A. et al. In diverse conditions, intrinsic chromatin condensates have liquid-like material properties. Proc. Natl Acad. Sci. USA 120, e2218085120 (2023).
Gong, Q. et al. Coassembly of a new insect cuticular protein and chitosan via liquid–liquid phase separation. Biomacromolecules 23, 2562–2571 (2022).
Zhou, S. et al. DNA nanotubes in coacervate microdroplets as biomimetic cytoskeletons modulate the liquid fluidic properties of protocells. J. Mater. Chem. B 10, 8322–8329 (2022).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018). Super-enhancers induced BRD4 and MED1 to condense into liquid-like droplets that locally concentrated the transcription machinery, revealing that phase separation of co-activator IDRs was a key mechanism for robust activation of cell-identity genes.
Lu, B. et al. Pharmacological inhibition of core regulatory circuitry liquid-liquid phase separation suppresses metastasis and chemoresistance in osteosarcoma. Adv. Sci. 8, 2101895 (2021).
Winner, B. et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl Acad. Sci. 108, 4194–4199 (2011).
Singleton, A. B. et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841–841 (2003).
Maries, E. et al. The role of α-synuclein in Parkinson’s disease: insights from animal models. Nat. Rev. Neurosci. 4, 727–738 (2003).
Oliveira, G. & Silva, J. L. Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun. Biol. 2, 374 (2019).
Zhang, C. et al. C-terminal truncation modulates α-Synuclein’s cytotoxicity and aggregation by promoting the interactions with membrane and chaperone. Commun. Biol. 5, 798 (2022).
Ray, S. et al. α-Synuclein aggregation nucleates through liquid–liquid phase separation. Nat. Chem. 12, 705–716 (2020). Liquid-liquid phase separation of α-Synuclein was the initiating step that triggered its subsequent amyloid aggregation and toxicity in Parkinson’s disease, which revealed a new phase-separation-driven mechanism for neurodegeneration.
Jiang, L. et al. Interaction of tau with HNRNPA2B1 and N6-methyladenosine RNA mediates the progression of tauopathy. Mol. Cell 20, 4209–4227 (2021).
Zhang, X. et al. The proline-rich domain promotes Tau liquid–liquid phase separation in cells. J. Cell Biol. 219, e202006054 (2020).
Munishkina, L. A. & Fink, A. L. Fluorescence as a method to reveal structures and membrane-interactions of amyloidogenic proteins. Biochim. Biophys. Acta (BBA) - Biomembr. 1768, 1862–1885 (2007).
Ross, C. A. & Poirier, M. A. Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17 (2004).
Stefani, M. & Dobson, C. M. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. 81, 678–699 (2003).
Eisele, Y. S. et al. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 14, 759–780 (2015).
Ge, W.-Y. et al. Amyloid protein cross-seeding provides a new perspective on multiple diseases in vivo. Biomacromolecules 24, 1–18 (2023).
Petkova, A. T. et al. Self-propagating, molecular-level polymorphism in Alzheimer’s β-amyloid fibrils. Science 307, 262–265 (2005).
Fitzpatrick, A. W. P. et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190 (2017).
Li, D. & Liu, C. Structural diversity of amyloid fibrils and advances in their structure determination. Biochemistry 59, 639–646 (2020).
Ban, T., Yamaguchi, K. & Goto, Y. Direct observation of amyloid fibril growth, propagation, and adaptation. Acc. Chem. Res. 39, 663–670 (2006).
Fändrich, M. et al. Amyloid fibril polymorphism: a challenge for molecular imaging and therapy. J. Intern. Med. 283, 218–237 (2018).
Kaminski Schierle, G. S. et al. In situ measurements of the formation and morphology of intracellular β-amyloid fibrils by super-resolution fluorescence imaging. J. Am. Chem. Soc. 133, 12902–12905 (2011).
Lu, J. et al. CryoEM structure of the low-complexity domain of hnRNPA2 and its conversion to pathogenic amyloid. Nat. Commun. 11, 4090 (2020). Cryo-EM revealed stress-responsive hnRNPA2 low-complexity domain fibrils as kinked, reversible β-sheets whose D290V mutation collapsed them into stable and pathogenic amyloid.
Long, H. et al. Interaction of RAGE with α-synuclein fibrils mediates inflammatory response of microglia. Cell Rep. https://doi.org/10.1016/j.celrep.2022.111401 (2022). RAGE’s basic V-domain binded α-syn fibrils via their acidic tail, sparking microglial inflammation that drove PD pathology and was quenched by RAGE deletion or FPS-ZM1.
Zhang, S. et al. Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc. Natl Acad. Sci. USA 118, e2011196118 (2021).
Ramaswami, M., Taylor, J. P. & Parker, R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 154, 727–736 (2013).
Hughes, M. P. et al. Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science 359, 698–701 (2018).
Luo, F. et al. Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol. 25, 341–346 (2018).
Gui, X. et al. Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat. Commun. 10, 2006 (2019). hnRNPA1 formed reversible Asp-stacked “hnRAC” amyloid cores that tuned liquid-droplet fluidity and stress-granule recruitment, whereas ALS-linked Asp mutations locked these fibrils into irreversible pathological assemblies.
Gracia, P. et al. Molecular mechanism for the synchronized electrostatic coacervation and co-aggregation of alpha-synuclein and tau. Nat. Commun. 13, 4586 (2022).
Alies, B. et al. Concept for simultaneous and specific in situ monitoring of amyloid oligomers and fibrils via Förster resonance energy transfer. Anal. Chem. 86, 11877–11882 (2014).
Wolstenholme, C. H. et al. AggFluor: fluorogenic toolbox enables direct visualization of the multi-step protein aggregation process in live cells. J. Am. Chem. Soc. 142, 17515–17523 (2020). AggFluor, a tunable GFP-derived molecular-rotor toolkit, lighted up soluble oligomers versus insoluble aggregates in two colors in live cells, enabling real-time tracking of their interconversion by proteostasis regulators.
Ye, S. et al. A general strategy to control viscosity sensitivity of molecular rotor-based fluorophores. Angew. Chem. Int. Ed. 60, 1339–1346 (2021). Conjugated π-linker length tuned the viscosity sensitivity of molecular-rotor fluorophores, yielding RBFs that imaged oligomers and insoluble aggregates in live cells and offered a general strategy to modulate related photoisomerizable and AIE probes.
Shen, B. et al. A dual-functional BODIPY-based molecular rotor probe reveals different viscosity of protein aggregates in live cells. Aggregate 4, e301 (2023).
Owyong, T. C., Zhao, J. & Hong, Y. Small molecule fluorescent probes for the study of protein phase separation. Curr. Opin. Chem. Biol. 76, 102354 (2023).
Sun, R. et al. Chemical probes for investigating protein liquid-liquid phase separation and aggregation. Curr. Opin. Chem. Biol. 74, 102291 (2023).
Dujardin, S. et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 26, 1256–1263 (2020). Patient-specific post-translational signatures of soluble tau seeds dictated their spreading kinetics and forecastd the pace of Alzheimer’s cognitive decline, arguing for personalized anti-tau therapy.
Cascella, R. et al. The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells. Nat. Commun. 12, 1814 (2021). α-Synuclein fibrils continuously shed membrane-perforating prefibrillar oligomers from their ends, making short fibrils the fastest toxin source and linking fibril growth directly to acute neuronal damage.
Senior, A. W. et al. Improved protein structure prediction using potentials from deep learning. Nature 577, 706–710 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Tunyasuvunakool, K. et al. Highly accurate protein structure prediction for the human proteome. Nature 596, 590–596 (2021).
Zhang, B. et al. Decoding protein dynamics in cells using chemical cross-linking and hierarchical analysis. Angew. Chem. Int. Ed. 62, e202301345 (2023).
Wilkinson, M. et al. Structural evolution of fibril polymorphs during amyloid assembly. Cell 186, 5798–5811.e5726 (2023). Time-resolved cryo-EM revealed that IAPP fibril architectures were transient and evolved from lag to plateau, implying early-stage structures may seed pathology instead of the classic endpoint fibrils.
Spehar, K. et al. Super-resolution imaging of amyloid structures over extended times by using transient binding of single thioflavin T molecules. ChemBioChem 19, 1944 (2018).
Sun, B. et al. Single-molecule orientation imaging reveals the nano-architecture of amyloid fibrils undergoing growth and decay. Nano Lett. 24, 7276–7283 (2024).
Pinotsi, D. et al. Direct observation of heterogeneous amyloid fibril growth kinetics via two-color super-resolution microscopy. Nano Lett. 14, 339–345 (2014).
Algar, W. R. et al. FRET as a biomolecular research tool—understanding its potential while avoiding pitfalls. Nat. Methods 16, 815–829 (2019).
Saurabh, S. Probing the physicochemical properties of biomolecular condensates using optical nanoscopy and fluorescence spectroscopy. Biophys. J. 124, 412 (2025).
Alberti, S. et al. Current practices in the study of biomolecular condensates: a community comment. Nat. Commun. 16, 7730 (2025).
Ibrahim, K. A. et al. Label-free techniques for probing biomolecular condensates. ACS Nano 18, 10738–10757 (2024).
Ye, S. et al. Micropolarity governs the structural organization of biomolecular condensates. Nat. Chem. Biol. 20, 443–451 (2023).
Sabri, E., Mangiarotti, A. & Dimova, R. Fluorescence-based mapping of condensate dielectric permittivity uncovers hydrophobicity-driven membrane interactions. bioRxiv 09, 642144 (2025).
Català-Castro, F. et al. Measuring age-dependent viscoelasticity of organelles, cells and organisms with time-shared optical tweezer microrheology. Nat. Nanotechnol. 20, 411–420 (2025).
Alshareedah, I. et al. Programmable viscoelasticity in protein-RNA condensates with disordered sticker-spacer polypeptides. Nat. Commun. 12, 6620 (2021).
Jawerth, L. et al. Protein condensates as aging Maxwell fluids. Science 370, 1317–1323 (2020).
Wang, H. et al. Surface tension and viscosity of protein condensates quantified by micropipette aspiration. Biophys. Rep. 1, 100011 (2021).
Roggeveen, J. V. et al. calibration-free model of micropipette aspiration for measuring properties of protein condensates. Biophys. J. 123, 1393–1403 (2024).
Costa, L. et al. Liquid-liquid interfacial imaging using atomic force microscopy. Adv. Mater. Interfaces 4, 1700203 (2017).
Law, J. O. et al. A bending rigidity parameter for stress granule condensates. Sci. Adv. 9, eadg0432 (2023).
Dai, Y. et al. Biomolecular condensates regulate cellular electrochemical equilibria. Cell 187, 5951–5966 (2024).
King, M. R. et al. Macromolecular condensation organizes nucleolar sub-phases to set up a pH gradient. Cell 187, 1889–1906 (2024).
Haas, R. J. et al. pH-Responsive elastin-like polypeptide designer condensates. ACS Appl. Mater. Interfaces 15, 45336–45344 (2023).
Ye, S., Hsiung, C.-H., Tang, Y. & Zhang, X. Visualizing the multistep process of protein aggregation in live cells. Acc. Chem. Res. 55, 381–390 (2022).
Mekonnen, G., Djaja, N., Yuan, X. & Myong, S. Advanced imaging techniques for studying protein phase separation in living cells and at single-molecule level. Curr. Opin. Chem. Biol. 76, 102371 (2023).
Visser, B. S., Lipiński, W. P. & Spruijt, E. The role of biomolecular condensates in protein aggregation. Nat. Rev. Chem. 8, 686–700 (2024).
Silva, J. L. et al. Targeting biomolecular condensation and protein aggregation against cancer. Chem. Rev. 123, 9094–9138 (2023).
Bai, Y. et al. Advanced techniques for detecting protein misfolding and aggregation in cellular environments. Chem. Rev. 123, 12254–12311 (2023).
Spruijt, E. Open questions on liquid-liquid phase separation. Commun. Chem. 6, 23 (2023).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (NSFC) (grant number 22204126, 82172063, 32471230), the Natural Science Basic Research Program of Shaanxi (grant number 2022JQ-092).
Author information
Authors and Affiliations
Contributions
Jing Xue: Conceptualization, writing—original draft, writing—review & editing, resources, methodology, investigation, funding acquisition. Xiao-Wen Cao: Writing—original draft, writing—review & editing, methodology, investigation. Xiu-Lan Jia: Data curation, visualization. Xi Chen: Visualization. Wei-Hong Guo: Validation. Da-Chuan Yin: Conceptualization, supervision, writing—review & editing, resources, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xue, J., Cao, XW., Jia, XL. et al. Visualizing liquid-liquid phase separation and protein aggregates. Commun Chem 8, 370 (2025). https://doi.org/10.1038/s42004-025-01756-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01756-z
