
Abstract
The adenosine A2A receptor (A2AR) is a Class A G protein-coupled receptor (GPCR) that regulates inflammation, glucose metabolism, and energy homeostasis in metabolically active tissues. While the effects of small-molecule ligands and protein interactions with A2AR have been extensively studied, the regulatory influence of endogenous metabolites remains unexplored. To address this gap, we employed the Mass spectrometry Integrated with equilibrium Dialysis for the discovery of Allostery Systematically (MIDAS) platform to screen a library of human metabolites for interactions with A2AR. This approach identified 180 metabolites that interact with A2AR, including allosteric and orthosteric modulators. We characterized the mechanisms of three metabolites previously unreported to interact with A2AR: prostaglandin D2, an allosteric antagonist that fully inhibits receptor signaling, and two orthosteric agonists, S-adenosyl-L-homocysteine and 2′-deoxyadenosine, that fully activate A2AR. Overall, these findings highlight the potential of the MIDAS platform to uncover previously unrecognized metabolite-GPCR interactions for research and therapeutic applications.
Similar content being viewed by others


Introduction
Adenosine receptors are a subfamily of G protein-coupled receptors (GPCRs) that mediate the actions of adenosine, an important metabolite in the pathway of energy metabolism. Among them, the adenosine A2A receptor (A2AR) is the most abundant, exerting a critical regulatory influence on energy homeostasis and glucose metabolism1,2,3,4,5. Notably, under certain conditions of metabolic stress, A2AR signaling is exploited to initiate immunosuppressive responses6,7,8 and to modulate inflammation9,10.
In certain cancers, such as melanomas, non-small cell lung cancer, and breast cancers, hypoxia-induced ectonucleotidase expression elevates extracellular adenosine levels in the tumor microenvironment. This results in excessive A2AR signaling in T-cells and NK effector cells, leading to suppression of anti-tumor immune responses triggering enhanced tumor growth. Consequently, inhibiting A2AR has emerged as a promising strategy for restoring anti-tumor immunity and enhancing the effectiveness of immunotherapies8,11. In contrast, under inflammatory conditions, A2AR signaling serves a protective role by dampening the release of pro-inflammatory cytokines, thereby mitigating tissue injury. Thus, developing A2AR agonists highlights the therapeutic potential to alleviate pathological inflammation in conditions such as rheumatoid arthritis and inflammatory bowel disease10,11. While there is a long history of drug discovery studies focused on A2AR with a well-established and rich pharmacology of small molecules and antibodies, to our knowledge, the influence of endogenous metabolites on the functional regulation of A2AR has yet to be studied.
Many metabolites serve as intracellular signaling molecules by regulating enzyme activity, such as histone deacetylases, modulating metabolic pathway enzymes in feedforward or feedback loops, or binding directly to nuclear hormone receptors like PPAR12 and FXR13. However, over the past two decades, it has become evident that an expanding array of metabolites directly engage GPCRs to elicit signal transduction events. For example, a classical intracellular metabolite such as succinate that is generated as part of the TCA cycle can act as an extracellular signaling molecule for GPR9114. Other metabolites, including short-chain fatty acids, secondary bile acids, and amino acids have been shown to regulate various GPCRs15,16,17. Furthermore, evidence is emerging that GPCRs can be activated by multiple endogenous ligands. The glucagon receptor, GCGR can be activated by multiple hormones, including glucagon and oxyntomodulin18; the galanin receptor type 2, GALR2 can be activated by two distinct peptides galanin and spexin19; and the purinergic receptor, P2Y2 can be activated by adenine and uridine nucleotides20.
Given that adenosine has multiple metabolic derivatives and A2AR signaling is implicated in critical metabolic processes, both under basal conditions and in certain pathological states, we reasoned that other existing metabolites may be capable of regulating A2AR. Thus far, only a limited subset of studies has systematically screened metabolites for interaction with GPCRs, with most of these studies focused on deorphanizing GPCRs21,22,23,24,25. Moreover, these studies can be biased towards targeting the orthosteric ligand binding site and thus overlook allosteric modulators. Most GPCRs have allosteric sites that are spatially distinct from the orthosteric site which do not directly compete with the native ligand, offering advantages to improve drug selectivity and fine-tune pharmacological responses26. This is particularly relevant to A2AR, as the high sequence conservation of the orthosteric site across adenosine receptor family members presents a significant obstacle with achieving pharmacological selectivity. Historically, discovering allosteric modulators is challenging, as only a handful of molecules have successfully advanced through clinical development27. Therefore, identifying potential allosteric metabolites that interact with A2AR could lead to the development of new classes of modulators that harness the body’s own regulatory systems or offer advantages in terms of biocompatibility and reduced immunogenicity for disease treatment.
To this end, we optimized the Mass spectrometry Integrated with equilibrium Dialysis for the discovery of Allostery Systematically (MIDAS) platform28 to allow for the discovery of metabolite ligand interactions with membrane proteins, a capability previously not possible in the platform, providing a means to target more of the proteome for protein-metabolite ligand discovery. Using this advanced MIDAS platform, we assayed A2AR to identify metabolite ligand interactions. MIDAS leverages the sensitivity and specificity of mass spectrometry metabolomics combined with equilibrium dialysis to enable the detection of direct interactions for both orthosteric and allosteric ligands from a metabolite library representative of the human metabolome. We used MIDAS to identify 180 metabolites that putatively interact with A2AR, and by integrating differential scanning fluorimetry (DSF), fluorescence polarization assays, and functional activity assays—we identified prostaglandin D2 (PGD2), S-adenosyl-L-homocysteine (SAH), and 2’-deoxyadenosine (DA) as functional modulators of A2AR. We demonstrated that PGD2, an anionic lipid, acts as a negative allosteric inhibitor that achieves complete inhibition of A2AR, while SAH and DA are orthosteric agonists that achieve maximal activation of A2AR. Collectively, our systematic and robust screening approach characterized allosteric and orthosteric modulators of A2AR, paving the way for the discovery of therapeutics to regulate GPCRs.
Results
The MIDAS platform identifies metabolite interactions with A2AR
Implementing MIDAS for membrane proteins such as A2AR posed significant challenges due to the instability of A2AR over a prolonged dialysis procedure and the incompatibility of common protein buffers and detergents with flow injection analysis mass spectrometry (FIA-MS). Therefore, we purified a thermostabilized construct of A2AR (Supplementary Fig. 1), which enabled purification of the receptor at milligram-scale quantities suitable for analysis by the MIDAS platform. Next, we developed a hydrophilic interaction liquid chromatography-mass spectrometry (LC-MS) approach that ensured the compatibility of our metabolite library with common buffer components (HEPES and NaCl) and detergents commonly used for membrane protein solvation (lauryl maltose neopentyl glycol, glyco-diosgenin, and cholesteryl hemisuccinate) and optimized sample conditions to minimize the contribution of phospholipid and detergent interference with our LC-MS methods. Using MIDAS, we screened a library of 401 unique human metabolites, which was reduced to 331 quantifiable metabolites after grouping non-separable mass isomers with indistinguishable retention times. These encompassed a sizable fraction of the core human metabolome, including carbohydrates, amino acids, nucleotides, lipids, coenzymes, vitamins, and their intermediates and derivatives28 (Fig. 1a). We identified 180 unique metabolites that putatively interacted with A2AR using cutoffs of z-score-transformed normalized log2(fold-change) < −0.25 or > 0.25, broadly capturing low and high affinity ligand interactions (Fig. 1b). Among these, 118 metabolites had positive log2(fold change) and 62 metabolites had negative log2(fold change) with A2AR. Positive log2(fold change) can indicate a non-reactive protein-metabolite interaction (e.g., orthostery or allostery) or the product of an enzymatic or non-enzymatic reaction. Negative log2(fold change) can indicate a non-reactive protein-metabolite interaction or the substrate of an enzymatic or non-enzymatic reaction. Therefore, the magnitude of log2(corrected fold change) serves as a proxy for the affinity or reaction rate of the putative protein-metabolite ligand interaction. This MIDAS metabolomic analysis revealed an enrichment of lipids, phosphatidylethanolamine (PE), phosphatidic acid (PA), and phosphatidylserine (PS), with cholic acid ranked as the most enriched metabolite (Fig. 1b), consistent with the majority of currently published structures that visualize cholesterol binding29. This enrichment of lipids was anticipated, considering that A2AR functions within lipid membrane bilayers and is stabilized by amphipathic detergent micelles in our experimental conditions. We therefore used this enrichment ranking as the basis for selecting metabolites for further testing.

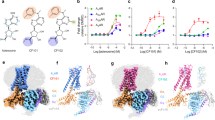
a Schematic of the MIDAS platform. b MIDAS analysis of A2AR. Each data point represents a unique metabolite from the MIDAS metabolite library. Log2(fold change) A2AR-metabolite interactions were normalized to a no protein control dialysis. Dotted lines indicate normalized z-score log2(fold change) > 0.25 and <−0.25, respectively. The MIDAS equilibrium dialysis was performed in triplicate. Statistical significance was calculated using multiple unpaired two-sided t-tests with FDR correction as shown in Supplementary Fig. 2a. c Chemical structures of the prostaglandins. d Dose-response differential scanning fluorimetry of A2AR with the enriched prostaglandins. Data points show mean ± SD of the melting temperature (Tm) at different concentrations. For data points of DMSO and 0.1 mM PGD2, n = 4, for 0.25 mM and 0.5 mM PGD2, n = 5 technical replicates. For all data points for PGE2 and PGI2, n = 3 technical replicates. Data was analyzed using multiple unpaired two-sided t-tests. DMSO vs. 0.25 mM PGD2, p = 0.0365, DMSO vs. 0.5 mM PGD2, p = 0.0018. *p < 0.05, **p < 0.01, ns not significant.
From this ranking analysis, endogenous Class A GPCR ligand isomers prostaglandin D2, E2, and I2 (PGD2, PGE2, and PGI2) collapsed into a single signal, appeared in the top 25 and were significantly enriched (p < 0.05) (Fig. 1b, Supplementary Fig. 2a). We also identified several other endogenous ligands or precursors of ligands for Class A GPCRs ranked in the top 50 which included 5-methoxytryptamine (5-MT), N-acetylserotonin, and melatonin. Interestingly, while some adenosine derivatives were also significantly enriched, such as dimethylaminopurine (DMAP) and adenine, the endogenous ligand for A2AR – adenosine, was not significantly enriched (Supplementary Fig. 2a). We also identified SAH and DA, two adenosine analogs with a similar chemical scaffold as adenosine which met the z-score cutoff criterion for enrichment but did not meet the statistical significance threshold (Fig. 1b, Supplementary Fig. 2a). We attribute this to the incorporation of thermostabilizing mutations at two key residues within the orthosteric site of A2AR – T88A and S277A. Both residues directly interact with the ribose moiety of adenosine and are key determinants of agonist binding, as demonstrated by mutagenesis studies that show mutation of T88 or S277 to alanine, results in orders of magnitude reduction of adenosine affinity30,31. Because adenosine, SAH, and DA all have this ribose moiety, binding to A2AR during the MIDAS screen is likely disrupted by these mutations. In contrast, adenine and DMAP lack a ribose group and therefore are unlikely to rely on these interactions for binding, consistent with their statistically significant enrichment (Supplementary Fig. 2b).
To orthogonally validate direct interaction of this subset of metabolites with A2AR, we performed competition fluorescence polarization (FP) assays and observed that none of the three prostaglandins – PGI2, PGE2, and PGD2 were able to displace a fluorescently-labeled orthosteric ligand – NECA (Supplementary Fig. 3a), suggesting an allosteric mode of binding. Conversely, SAH and DA reduced the affinity of NECA, indicating that these metabolites bind to A2AR at the orthosteric site (Supplementary Fig. 3b). Remarkably, of the three prostaglandins, only PGD2 exhibited a concentration-dependent increase in the melting temperature (Tm) of A2AR (Fig. 1c, d), indicating that PGD2 binds to and stabilizes the receptor. Next, we explored the possibility for functional regulation of A2AR, given our complementary results from MIDAS and our DSF assays, along with its known role as an endogenous agonist for the Class A GPCRs DP1 and DP232.
Prostaglandin-D2 is a non-competitive inhibitor of A2AR
To broaden the scope of this functional analysis, we systematically screened a representative subset of the top 75 ranked metabolites identified by MIDAS, selecting a wide range of structurally diverse molecules, while excluding those with conserved chemical architectures. Under physiological conditions, A2AR activation catalyzes the exchange of GDP for GTP of the trimeric Gs protein, resulting in its decoupling from A2AR, separation into the Gα and Gβγ subunits, and initiation of cAMP signaling. This signaling cycle is then reset when the Gα subunit hydrolyzes GTP and reforms with Gβγ to form the inactive trimeric complex33,34,35. We therefore purified functionally active A2AR and trimeric Gs (Supplementary Fig. 4), then measured Gs-mediated hydrolysis as a readout of A2AR activation.
Our initial screening approach was conducted at a single metabolite concentration to enable detection of metabolites with potentially low potency (Fig. 2a, b). Among the identified adenosine derivatives, both DA and SAH activated A2AR, whereas none of the other adenosine analogs exhibited any significant activity modulation. Notably, among the enriched endogenous ligands and precursors of ligands for Class A GPCRs identified by MIDAS, only PGD2 demonstrated any functional impact by completely inhibiting A2AR activity, whereas PGE2 and PGI2 had no effect (Fig. 2b).

a, b Metabolites were screened for (a) activation at 10 μM or (b) inhibition at 100 μM. Data was analyzed by one-way ANOVA and adjusted using Dunnett’s multiple comparisons test. For activation, (F(10,22) = 40.74, p < 0.0001), For SAH, p < 0.0001. For inhibition, F(10,22) = 151.9, p < 0.0001). For ZM and PGD2, p < 0.0001 (c) GTP hydrolysis assay demonstrating sustained inhibition of A2AR in the presence of 500 μM PGD2. (d) PGD2 inhibition in HEK293 cells stably expressing A2AR. a–d All data points shown as mean ± SD, n = 3 technical replicates, ****p < 0.0001.
To further characterize PGD2-mediated inhibition of A2AR, we performed a dose-response assay and determined the IC50 to be 180 µM (Supplementary Fig. 5a). Given that our previous FP assay indicated PGD2 binds allosterically, we carried out a competition activity assay to validate this mechanism of binding. While maintaining a fixed concentration of PGD2, we observed that even with saturating levels of adenosine up to 100 µM, PGD2 sustained near-complete inhibition of A2AR (Fig. 2c). This result indicates that PGD2 does not compete with adenosine at the orthosteric site, consistent with an allosteric mechanism of engagement.
To determine whether our in vitro findings translated to a cellular model, we evaluated PGD2-mediated inhibition in HEK293 cells stably expressing A2AR. We carried out functional assays using a homogenous time-resolved fluorescence (HTRF) cAMP detection assay to monitor cAMP responses as a readout for A2AR signaling activity. We first validated these cells expressed functional A2AR by demonstrating an increase in cAMP accumulation upon adenosine stimulation. We then performed a concentration-dependent assay and observed near-complete inhibition of adenosine-induced cAMP production in the presence of PGD2, whereas PGE2 had no significant effect (Fig. 2d), in line with our protein-based in vitro assay results (Supplementary Fig. 5a). For comparison, we observed an approximately 8-fold increase in the IC50 of PGD2 in our cell-based assay relative to our protein-based assay, which we attribute to the reduced capacity of PGD2 to partition into a heterogeneous lipid bilayer, given that PGD2 is a lipid and our previous assays demonstrate an allosteric mechanism of inhibition for PGD2. We also performed a genetic knock out of the prostaglandin transporter SLCO2A1, which is known to facilitate the cellular influx of prostaglandins36, to exclude the possibility of PGD2-mediated inhibition occurring through an indirect mechanism involving Gs (Supplementary Fig. 5b, c).
Inhibition of A2AR is driven by the head group of PGD2
Given the established role of lipids in modulating membrane properties and GPCR function37, we hypothesized that PGD2 could alter the membrane environment surrounding A2AR or engage lipid-binding sites on the receptor. To investigate potential PGD2 binding sites and elucidate the molecular basis of its inhibition, we determined cryo-EM structures of A2AR with and without PGD2 using the A2AR-BRIL construct used for MIDAS screening (Table 1 and Supplementary Figs. 6–8). Because PGD2 binds allosterically, we included the high-affinity antagonist ZM241385 to stabilize the receptor for structure determination and elucidation of the PGD2 binding pocket(s). The resulting maps with and without PGD2 were solved at 3.3 Å and 3.1 Å, respectively, which enabled reconstructions of near-complete models of A2AR and revealed four peripheral lipid-like features (Supplementary Fig. 9). While these features were present in both maps, nevertheless, we used their positions to narrow down the search space for potential sites of action. Interestingly, mutating one such site resulted in a minor reduction of inhibition for the Q163E/H75A double mutant (Supplementary Figs. 9i, j and 10). Given that the H75A point mutation had comparatively no effect (Supplementary Fig. 10), this suggests that Q163, located at the periphery of extracellular loop 2 (ECL2) is involved in PGD2-mediated inhibition, although additional sites of interaction are necessary to fully account for overall inhibitory activity. Therefore, while a single, discrete binding site cannot be conclusively defined from our cryo-EM analysis, our data show that PGD2 engages peripheral sites on A2AR, thereby exerting a cumulative inhibitory effect.
The observation that PGD2 selectively inhibits A2AR despite its structural similarity to PGE2 and PGI2, prompted us to investigate its underlying mechanism. Given that PGD2 is an anionic lipid metabolite and the peripheral surface of A2AR is net positively charged, we hypothesized that the main driving force of binding and inhibition is through electrostatic interactions. To test this hypothesis, we compared the inhibition potency of PGD2 and PGD2-methylester, an analog of PGD2 that is modified by methylating the ionic oxygen of the carboxylate group of PGD2, thereby converting the ionic fragment into a neutral moiety (Fig. 3a). We then utilized the Gs hydrolysis assay to compare the impact of this modification with the inhibition potency of PGD2. Surprisingly, our findings reveal that the IC50 values for both prostaglandins were nearly identical, indicating that the inhibitory effect is not primarily mediated by an electrostatic interaction (Fig. 3b, c). This is in stark contrast to anionic phospholipids, where electrostatic interactions between the receptor side chains and the lipid headgroups is the primary driving force for positive allosteric modulation. To identify the key fragment of PGD2 responsible for inhibition, we examined its chemical structure in greater detail. Although PGD2 and PGE2 possess nearly identical cyclopentane ring headgroups and hydrocarbon tails, they are regioisomers that differ only in the arrangement of the oxygen atoms attached to their headgroups (Fig. 3a). We hypothesized that this divergence critically influences their inhibitory potency. To interrogate the role of these functional groups, we compared additional analogs alongside PGD2 - PGK2 and PGF2α, which have either two carbonyls or two hydroxyls, respectively, enabling us to systematically evaluate how the carbonyl and hydroxyl groups impact inhibition of A2AR. Intriguingly, we discovered that substitution of the carbonyl group on the headgroup of PGD2 with a hydroxyl group reduced inhibition potency by approximately 2 orders of magnitude (Fig. 3b, c). To understand in more detail the physicochemical differences between these hydroxyl and carbonyl groups, we employed MOE (Molecular Operating Environment) to generate electrostatic maps around these moieties to visualize the surface interactions likely to bind each group. The results reveal a significantly stronger region of positively charged density surrounding the carbonyl group relative to the hydroxyl group, indicating that the carbonyl group could preferentially engage the receptor, due to A2AR’s net positive charge (Supplementary Fig. 11). In contrast, PGI2 was excluded from this fragment-based analysis because its bicyclic pentane headgroup attached to an extended carboxylate deviates substantially from the structures of PGD2, PGE2, and typical phospholipids. We also did not consider the fatty acid tails of prostaglandins in this analysis, as these hydrophobic segments are presumed to mediate non-specific interactions to hydrophobic surfaces of A2AR. Overall, our results underscore the pivotal role of the carbonyl moiety attached to the headgroup of PGD2 as a key determinant of A2AR inhibition.

a Chemical structures of selected PGD2 analogs. b Dose-response GTP hydrolysis assay comparing inhibition potency of selected PGD2 analogs. Data points shown as mean ± SD, n = 3 technical replicates. c Quantitative comparison of prostaglandin inhibition potency.
Functional and structural characterization of orthosteric agonists – SAH and DA
During our metabolite activity screen, we identified two molecules, SAH and DA, that elicited sub-maximal activation of A2AR at a concentration of 10 µM. Inspection of their chemical structures revealed both molecules share the canonical adenine moiety and a ribose scaffold similar to adenosine, yet harbor distinct chemical structure differences (Fig. 4a, Supplementary Fig. 12a). SAH contains an extended sulfur-linked homocysteine substituent on the ribose ring, whereas DA does not have the 2’ hydroxyl group attached to the ribose moiety. To clarify their agonist properties, we utilized our GTP hydrolysis assay to determine that both SAH and DA were full agonists of A2AR, albeit with markedly lower potencies compared to adenosine. Whereas adenosine exhibits an EC50 of 18 nM under our assay conditions, SAH and DA exhibited EC50 values of 5 µM and 15 µM, respectively (Fig. 4b, Supplementary Fig. 12b). We observed A2AR activation by SAH with an EC50 of ~5 µM from our cell-based cAMP detection assays, in agreement with our GTP hydrolysis results (Fig. 4c), whereas DA required millimolar concentrations for activation (Supplementary Fig. 12c).

a Chemical structures of adenosine and SAH. b Activation comparison of adenosine and SAH (EC50 = 18 nM and 5 µM, respectively). c SAH activation in HEK293 cells stably expressing A2AR. d Molecular docking of SAH into the active site of A2AR (PDB: 2YDO). All data points shown as mean ± SD, n = 3 technical replicates.
Prompted by these findings, we investigated the structural basis for A2AR activation by docking SAH into the adenosine-bound crystal structure38 (PDB: 2YDO) (Fig. 4d). We determined that SAH binds in the canonical orthosteric site, preserving many of the key interactions with A2AR as adenosine. SAH shares a core adenine moiety which rests in a hydrophobic pocket defined by V843.32, Leu2496.51, and S2777.3930. (TM3, TM6, and TM7). Importantly, SAH retained critical hydrogen bonds between the exocyclic adenine nitrogen atom to Glu169 in extracellular loop 2 (ECL2) and Asn2536.55 in TM6 (Fig. 4d). The ribose moiety aligns closely with that of adenosine, positioning the 5′-uronamide groups in proximity to S2777.42 and Ile2747.39 (TM7), consistent with established agonist-binding contacts30 (Fig. 4d).
Despite these similarities, SAH displayed distinct variations in its geometry and residue engagement within the orthosteric site. The homocysteine substituent of SAH projects toward the cytoplasmic portion of the receptor, where the amide moiety establishes a hydrogen bond with T883.36 (Fig. 4d) - a key residue for agonist-induced activation observed for all high-affinity synthetic agonists NECA, CGS21680, and UK-432097 that have been co-crystallized with A2AR30. Interestingly, this polar interaction is notably different compared to the weaker van der Waals contact formed by adenosine. SAH’s homocysteine substituent also creates an additional hydrogen bond between its terminal carboxyl group and Asn1815.42 (Fig. 4d), a key residue for ligand binding39.
Most notably, the absence of a hydrogen bond with H2506.52 is observed with SAH compared to all other structures in complex with agonists30. Instead, SAH forms a weaker van der Waals contact with H2506.52(Fig. 4d), and this lack of a polar interaction is potentially the primary determinant of the >250-fold decrease in potency. Mutating H2506.52 to alanine abolishes agonist binding entirely, whereas substituting phenylalanine – despite its limited hydrogen-bonding capacity – merely reduces agonist affinity but does not preclude binding40. Furthermore, the W2466.48 side chain, known as the ‘toggle switch41,’ is positioned closely to H2506.52. This close proximity suggests that H2506.52 is not only important for agonist binding, but also contributes to receptor activation through a hydrophobic interaction with W2466.48. Collectively, the simultaneous engagement of key residues known for agonist binding T883.36, Asn1815.42, combined with the diminished coordination with H2506.52 results in the marked reduction in activation potency.
In contrast, the primary structural difference between DA and adenosine lies in the absence of the 2’-hydroxyl group on the ribose scaffold, resulting in >800-fold reduction in potency. The absence of this moiety eliminates hydrogen bonding with His2787.43 and a hydrophobic contact with V84 (Supplementary Fig. 12d). Both H278 and V84 play key roles in high-affinity binding of agonists to A2AR, as previous studies observe >98% reduction in binding of the synthetic agonist CGS21680 when mutating either V84 or H278 to alanine39,40. Notably, the H278A mutant expressed in heterologous cells exhibited a > 30-fold loss in agonist potency, despite being able to achieve full activation. Thus, we attribute the reduction in potency for DA primarily to the loss of critical interactions with H278 and V84.
Discussion
The potential for endogenous metabolites other than adenosine to modulate A2AR signaling has not been investigated. Beginning from an unbiased screening approach using our optimized MIDAS platform, we identified several naturally occurring metabolites that bind to A2AR and subsequently characterized the functional role of a selected subset. We established that the adenosine derivatives - SAH and DA bind at the orthosteric site with an activation efficacy weaker than adenosine, despite nearly identical chemical structures. Additionally, we identified PGD2 as an allosteric antagonist of A2AR. Of interest, it is known that PGD2 is an endogenous agonist for the DP1/2 Class A GPCR, yet according to our GTPase hydrolysis and FP assays, PGD2 does not bind at the active site of A2AR. Instead, PGD2 acts as an allosteric antagonist, unique among anionic lipids by its ability to fully inhibit A2AR signaling, which was previously described to positively modulate A2AR.
While our results indicate PGD2 binds allosterically, it is important to highlight that a limitation of the current study is the lack of a defined binding site for PGD2. Our biochemical and structural analysis indicates that interactions on the surface of A2AR could cumulatively result in PGD2’s inhibitory mechanism. Intriguingly, this pattern of multiple binding sites closely resembles previous observations for cholesterol. Our MIDAS screen revealed cholic acid, a structural analogue of cholesterol, as the most enriched metabolite. This is not surprising given that previous studies using X-ray crystallography and molecular dynamics (MD) simulations clearly define 8 different cholesterol binding sites which surround the extracellular leaflet of A2AR29. Cholesterol acts as a weak positive allosteric modulator (PAM), moderately increasing basal activity and weakly enhancing agonist potency. We noticed from our cryo-EM data that at least one of the known cholesterol binding sites from prior X-ray structures aligns with a peripheral density feature into which we docked PGD2 (Supplementary Figs. 9 i, j and 13). It is tempting to speculate that PGD2 may compete with cholesterol at overlapping binding sites, invoking a possible switch in modulatory behavior based on the difference between a reactive hydroxyl group and carbonyl group. Such a regulatory switch in hydrogen bonding could conceivably alter the receptors’ conformational dynamics or the membrane fluidity properties due to the change in headgroup moieties.
We also cannot exclude the possibility that PGD2 engages a binding site located near the intracellular leaflet for PGD2. Several studies have demonstrated that anionic phospholipids function as PAMs by stabilizing receptors in active conformations and promoting G-protein coupling through discrete allosteric pockets. Examination of several experimentally determined structures of Class A GPCRs have identified lipids or sterols occupying a small, polar groove formed by TM3,4, and 5 situated just above intracellular loop 2 (ICL2)42. In each instance, these molecules act as PAMs and suggest a conserved lipid-binding site where GPCR activation is regulated by lipids or lipid-like molecules in that region. This observation aligns with earlier studies demonstrating that phosphatidylserine (PS) stabilizes agonist-bound intermediate states of rhodopsin43, while PS, phosphatidylglycerol (PG), and phosphatidylinositol (PI) shift conformations of β₂-adrenergic receptors toward activated states44. Furthermore, PG and PS contribute to β₂AR signaling by enhancing Gs coupling through the generation of a net negative local membrane charge45. Consistent with these findings, 19F-NMR and MD simulations have proposed an anionic lipid binding site between TM6 and TM7 of A2AR46. This site, characterized by a cluster of positively charged residues (H230, K233, and R291) forms an intracellular cavity that appears to prime A2AR for Gs coupling. However, we introduced a K233A mutation (Supplementary Fig. 10) and observed no change in PGD2 inhibition, suggesting PGD2 does not inhibit A2AR at this site. Therefore, our discovery that PGD2 acts as a non-competitive inhibitor of A2AR points to a fundamentally distinct mechanism of anionic lipid modulation.
Although PGD2 inhibits A2AR signaling in vitro, the physiological concentrations of PGD2 are too low47 to achieve comparable effects in vivo. PGD2’s role as a sleep hormone and its action on DP1 receptors to activate A2AR-expressing neurons suggest potential in vivo relevance, but further studies are needed to determine if it regulates A2AR signaling at physiologically relevant concentrations. On the other hand, there is emerging evidence suggesting that SAH and DA may modulate A2AR activity. While their serum concentrations are typically too low to activate A2AR under basal conditions, levels can significantly rise in pathological states such as dementia48, coronary artery disease (CAD)49, and adenosine deaminase deficiency (ADA)50,51. For instance, patients with CAD can exhibit elevated plasma homocysteine levels, which correlate with reduced A2AR signaling52. As SAH is a key precursor for homocysteine biosynthesis, it may exist at comparable concentrations in these patients, warranting further exploration of its potential to modulate A2AR in such conditions.
Finally, the unexpected identification of SAH and DA underscores the potential broader utility of MIDAS for deorphanizing GPCRs. Conventionally, “reverse pharmacology” methods match orphan GPCRs to known transmitters, with high-throughput screening methods frequently employed to address this gap53,54. However, these methods typically rely on “universal” cell-based functional assays that depend on the native or forced coupling of G-proteins or arrestin recruitment55,56, which can overlook non-canonical signaling mechanisms. This is exemplified by cases of atypical G-protein coupling, such as LGR557, GPR2758, and CXCR759, for beta-arrestin-mediated signaling with B3AR60 and GIP61, or the absence of both pathways for AT2R62. In contrast, directly measuring ligand binding makes MIDAS particularly advantageous when only the tissue localization of an orphan GPCR is known, as one can perform focused screening of tissue-specific metabolomes to identify putative endogenous ligands. MIDAS can also screen a broad chemical space and detect both orthosteric and allosteric molecules, as evidenced by our discovery of SAH and PGD2 (Fig. 5). Nonetheless, MIDAS is limited by the requirement for purified, homogenous protein and the compatibility of candidate ligands with mass spectrometry. We expect that future studies on the basis of the results presented here will not only uncover unique GPCR ligands, but also a deeper mechanistic understanding of how PGD2 inhibits A2AR.

Illustration of orthosteric activation and allosteric inhibition of A2AR by SAH and PGD2, respectively.
Methods
MIDAS metabolite library
The MIDAS metabolite library was constructed as previously described28. Briefly, all metabolites used in this study were purchased from Sigma-Aldrich, Cayman Chemicals, Avanti Polar Lipids, Enamine, Combi-Blocks, Inc., or custom-sourced from Molport. Metabolites were solvated to 10 mM in molecular grade water (Millipore Sigma W4502) or DMSO (Millipore Sigma D1435) and, where necessary to increase solubility, titrated with acid or base. The MIDAS metabolite library was arrayed 1 mL per well in 96-deep well storage plates (Greiner 780280), sealed with aluminum foil seals (VWR 60941-112), and stored at −80 °C. When working stocks were needed, metabolites were moved from the deep well storage plates and arrayed, 50 µL per well, across multiple, single-use 384-well small volume storage plates (Greiner 781280), sealed with aluminum foil seals (VWR 60941-112), and stored at −80 °C. Metabolite library management, manipulation, and pooling was conducted on a Beckman Coulter Biomek NXp SPAN-8 liquid handling robot.
MIDAS equilibrium dialysis
The day of MIDAS analysis, 384-well small volume working stock plates of the MIDAS metabolite library were defrosted at 30 °C for 5 min and metabolites were combined to generate four predetermined MIDAS metabolite pools28. The four MIDAS metabolite pools were prepared to 5 µM of each metabolite in 25 mM HEPES pH 7.4 (Fisher Scientific BP310), 100 mM NaCl (Fisher Scientific BP358), 0.00075% lauryl maltose neopentyl glycol (LMNG, Anatrace NG310), 0.00025% glyco-diosgenin (GDN, Anatrace GDN101), and 0.0001% cholesteryl hemisuccinate tris salt (CHS, Anatrace CH210) and pH-adjusted with ammonium hydroxide (Fisher Scientific 424330025). 10 µL of 368 µM detergent-solubilized A2AR protein in a buffer containing 25 mM HEPES pH 7.4 (Fisher Scientific BP310), 100 mM NaCl (Fisher Scientific BP358), 0.00075% LMNG (Anatrace NG310), 0.00025% GDN (Anatrace GDN101), and 0.0001% CHS (Anatrace CH210) or no protein control (buffer and detergents only) was arrayed in triplicate per metabolite pool across the shallow wells of a 10 kDa MWCO 96-well microdialysis plate (SWISSCI Diaplate) and sealed with aluminum foil seals (Beckman Coulter 538619) to create the protein chambers. To the paired, large volume wells on the reverse side of the plate, 300 µL of metabolite pool was aliquoted per A2AR protein replicate and sealed with aluminum foil seals (Beckman Coulter 538619) to create the metabolite chambers. The loaded microdialysis plate was placed in the dark at 4 °C on a rotating shaker (120 rpm) and incubated for 40 h. Post-dialysis, protein and metabolite chamber dialysates were retrieved, sample volume normalized and diluted 1:4 in 100% methanol (Millipore Sigma 1.15333 and 1060351000) to denature protein, incubated 30 min on ice, and centrifuged at 3200 x g for 15 min to remove precipitated protein. Processed protein and metabolite chamber dialysates were retrieved and arrayed across a 384-well microvolume plate (Thermo Scientific AB-1056), sealed with aluminum foil seals (Beckman Coulter 538619), and placed at 4 °C for metabolomic analysis.
Development of a mass spectrometry method for MIDAS analysis of membrane proteins
The previous MIDAS platform used electrospray ionization flow injection analysis mass spectrometry (FIA-MS) to quantify metabolites to identify putative protein-metabolite interactions28. FIA-MS is a high-throughput, direct injection approach that does not chromatographically separate analytes and matrix. Thus, FIA-MS does not permit common, non-volatile buffers, salts, or detergents due to deteriorated signal performance and artefacts from ion suppression. Thus, we leveraged hydrophilic interaction liquid chromatography mass spectrometry (LC-MS) for the development of a MIDAS platform that could identify metabolite ligand interactions with membrane proteins that require non-volatile buffer, salt, and detergents. Briefly, we tested the performance of 2.5 pmol injections of our four metabolite pools in the presence and absence of 25 mM HEPES pH 7.4 (Fisher Scientific BP310), 100 mM NaCl (Fisher Scientific BP358), 0.00075% LMNG (Anatrace NG310), 0.00025% GDN (Anatrace GDN101), and 0.0001% CHS (Anatrace CH210). These detergent concentrations represent the minimum required to solvate A2AR, thus mitigating ion suppression artefacts. Samples were assayed using a gradient of LC-MS grade ACN (Honeywell 34967) and 25 mM formate pH 2.7 (Millipore Sigma F0507) for positive mode LC-MS analyses or 25 mM ammonium carbonate pH 8.5 (Millipore Sigma 11204) for negative mode LC-MS analyses in water (Millipore Sigma 1.15333). The following LC method was used: 0–1.00 min, 0.15 mL/min 95% ACN; 1.00 – 10.0 min, 0.15 mL/min 95% to 1.0% ACN; 10.0–11.5 min, 0.30 mL/min 1% to 95% ACN; 11.5–12.5 min, 0.30 mL/min 95% ACN; 12.5–13.5 min, 0.40 mL/min 95% ACN; 13.5–14.5 min, 1.5 mL/min 95% ACN. Metabolite pools 1 and 2 were analyzed by positive mode LC-MS, and pools 3 and 4 were analyzed by negative mode LC-MS. Positive mode LC-MS was performed using a SeQuant ZIC-cHILIC 3 µm, 100 Å, 100 × 2.1 mm, PEEK-lined stainless-steel column (Millipore Sigma 1.50657). Negative mode LC-MS was performed using a SeQuant ZIC-pHILIC 5 µm polymer, 100 × 2.1 mm, PEEK-lined stainless-steel column (Millipore Sigma 1.50462). Columns were maintained at 40 °C during LC-MS operation using the column oven. Source conditions consisted of 40 psi ion source gas 1 and 2, 30 psi curtain gas, 250 °C source temperature, and +5500 V or −4500 V spray voltage. LC-MS method duration was 14.5 min. LC-MS auto-calibration of positive or negative mode was performed at the start of each mode. Under these conditions, we observed a reproducible reduction in signal for metabolites coeluting with HEPES and NaCl, but not below the limit of analyte quantification. The indicated concentrations of LMNG, GDN, and CHS detergent components did not perturb coeluting metabolite signal.
MIDAS liquid chromatography mass spectrometry data collection
MIDAS sample data collection was performed by hydrophilic interaction LC-MS on a Shimadzu Nexera HPLC system equipped with binary LC-20ADXR pumps, SIL-20ACXR autosampler, and CTO-20AC column oven coupled to a SCIEX X500R ESI-QTOF MS. Briefly, 2 µL of each processed protein and metabolite chamber dialysate (~2.5 pmoles per metabolite) was injected for LC-MS analysis in technical triplicate. Between injections, the autosampler injection needle was washed in 95% LC-MS grade acetontrile (ACN, Honeywell 34967) with a rinse speed of 35 µL/s in 500 µL. A gradient of LC-MS grade ACN (Honeywell 34967) and 25 mM formate pH 2.7 (Millipore Sigma F0507) for positive mode LC-MS analyses or 25 mM ammonium carbonate pH 8.5 (Millipore Sigma 11204) for negative mode LC-MS analyses in water (Millipore Sigma 1.15333). The following LC method was used: 0–1.00 min, 0.15 mL/min 95% ACN; 1.00–10.0 min, 0.15 mL/min 95% to 1.0% ACN; 10.0–11.5 min, 0.30 mL/min 1% to 95% ACN; 11.5–12.5 min, 0.30 mL/min 95% ACN; 12.5–13.5 min, 0.40 mL/min 95% ACN; 13.5–14.5 min, 1.5 mL/min 95% ACN. Metabolite pools 1 and 2 were analyzed in positive mode LC-MS, and pools 3 and 4 were analyzed in negative mode LC-MS. Positive mode LC-MS was performed using a SeQuant ZIC-cHILIC 3 µm, 100 Å, 100 × 2.1 mm, PEEK-lined stainless-steel column (Millipore Sigma 1.50657). Negative mode LC-MS was performed using a SeQuant ZIC-pHILIC 5 µm polymer, 100 × 2.1 mm, PEEK-lined stainless-steel column (Millipore Sigma 1.50462). Columns were maintained at 40 °C during LC-MS operation using the column oven. Source conditions consisted of 40 psi ion source gas 1 and 2, 30 psi curtain gas, 250 °C source temperature, and +5500 V or −4500 V spray voltage. LC-MS method duration was 14.5 min. LC-MS auto-calibration of positive or negative mode was performed at the start of each mode and between each protein and metabolite chamber sample.
MIDAS LC-MS data processing and analysis
MIDAS LC-MS spectra were processed in SCIEX OS software using a targeted method to determine metabolite abundances in the protein and metabolite chambers. Briefly, raw MS spectra were quantitatively processed in SCIEX OS Analytics using the MQ4 peak integration algorithm to quantify metabolite abundance. Metabolite extracted ion chromatograms (XICs) were identified by the intact mass determined from the chemical formula, adduct, charge, predetermined retention time, and precursor mass with 0.01 Da XIC width, 2-point Gaussian smooth width, 1000-point peak splitting, and 1 min baseline subtraction window. Metabolite abundance was quantified as the integrated area under the XIC counts per second peak trace. Following metabolite quantification, the mean of the technical triplicate injections was used to generate a metabolite abundance for each protein and metabolite chamber per dialysis replicate. For each dialysis replicate, log2(fold change) for each metabolite was calculated as the difference between the log2 metabolite abundance in the paired protein and metabolite chambers. The A2AR-metabolite log2(fold change) values were normalized to the no protein control log2(fold change) values to remove off-target metabolite interactions and z-score transformed to identify enriched or depleted metabolites in the protein chamber, indicative of putative protein-metabolite ligand interactions.
A2AR expression
For MIDAS screening and cryo-EM, the human ADORA2A cDNA was modified with an N-terminal cleavable leader sequence (gp67) signal peptide followed by a FLAG-tag. Nine point mutations were introduced (A54L, T88A, R107A, K122A, N154A, L202A, L235A, V239A, and S277A) along with replacing ICL3 with apocytochrome b562RIL (BRIL). Finally, the C-terminal region was truncated after A317, followed by the introduction of a 10x-His tag. For biophysical assay characterization, the human ADORA2A cDNA was cloned into a pFastbac vector with the following modifications for expression in SF9 cells. An N-terminal cleavable leader sequence (gp67), His10 tag, and TEV protease cleavage site, followed by thioredoxin, was used to connect to A2AR (residues V8–A316) through an EAAAKA linker. A N154A mutation was introduced to remove a potential N-linked glycosylation site, along with replacing the C-terminal region with a 3C protease cleavage site followed by a Twin Strep II tag. Both constructs were cloned into pFastbac vectors and expressed using the baculovirus expression system. SF9 cells were harvested by centrifugation 72 h post infection, resuspended in hypotonic buffer (20 mM HEPES pH 7.5, 10 mM MgCl2, 20 mM KCl, SMNE and cOmplete protease inhibitor tablets (Roche), flash-frozen in liquid nitrogen and stored at –80 °C until use.
A2AR purification
SF9 cells expressing A2AR (N194A, ΔCTD) for biophysical assays were dounce homogenized 50 times and solubilized with 0.5% (w/v) Lauryl maltose neopentyl glycol (LMNG) and nutated for 2 h. The sample was clarified by ultracentrifugation and loaded onto a 5 ml Ni-NTA column (Qiagen). The column was washed twice first with wash buffer (20 mM HEPES pH 7.5, 500 mM NaCl, 25 mM imidazole, 0.05% (w/v) LMNG, 0.005% CHS (w/v)) followed by (20 mM HEPES pH 7.5, 500 mM NaCl, 25 mM imidazole, 0.05% LMNG, 0.005% CHS, 25 mM imidazole). A2AR was eluted with elution buffer (20 mM HEPES pH 7.5, 500 mM NaCl, 300 mM imidazole, 0.05% LMNG, 0.005% CHS). The eluate was concentrated using a 50 kDa cut-off Amicon centrifugal ultrafiltration unit (Millipore) and loaded onto a Superdex 200 10/300 Increase column (GE Healthcare). Peak fractions were pooled and concentrated to approximately 1 mg/ml in a final buffer composition of 20 mM HEPES pH 7.5, 150 mM NaCl, 0.00075% LMNG, 0.0001% CHS.
To purify A2AR-BRIL for MIDAS and cryo-EM studies, SF9 cells were lysed with hypotonic buffer (20 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.0025 mg/mL Leupeptin). The membrane fraction was solubilized with 25 mM Tris-HCl pH 8.5, 300 mM NaCl, 0.5% LMNG, 0.03% CHS, 0.2% sodium cholate, 0.0025 mg/mL Leupeptin for 1 h at 4 °C. For cryo-EM studies, 1 µM ZM241385 was included throughout all purification steps. After high-speed centrifugation, the supernatant was loaded onto nickel-nitrilotriacetic acid (Ni-NTA) resin and washed to gradually decrease detergent and salt concentration into buffer C (25 mM Tris-HCl pH 8.5, 100 mM NaCl, 0.02% LMNG, 0.003% CHS, 0.02% sodium cholate, 0.0025 mg/mL Leupeptin). After affinity purification exploiting the C-terminal 10x-His tag, the eluate was then incubated with M1 anti-FLAG resin, and A2AR-BRIL bound to M1-beads was eluted with 0.13 mg/mL FLAG peptide. Eluted A2AR-BRIL was concentrated and purified to homogeneity with size-exclusion chromatography using a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in 25 mM Tris-HCl pH 8.5, 100 mM NaCl, 0.00075% LMNG, 0.00025% GDN, 0.0001% CHS. For cryo-EM analysis, fractions containing monodisperse A2AR-BRIL were pooled, mixed with excess anti-BRIL Fab and anti-Fab Nb with overnight incubation at 4 °C. The following day, unbound antibody was removed via size-exclusion chromatography, using a Superdex 200 Increase 10/300 GL column (GE Healthcare) and concentrated for cryo-EM grid preparation.
Trimeric Gs expression and purification
Wild-type trimeric Gα, Gβ, and Gγ subunits were co-expressed in Sf9 insect cells using baculoviruses generated by the Bac-to-Bac method (Expression Systems). Infected cells were harvested 72 h post-infection and lysed in a cold lysis buffer containing 20 mM HEPES pH 7.5, 500 mM NaCl, 5 mM MgCl₂, and 50 μM GDP to maintain complex stability. The lysate was clarified by centrifugation at 20,000 × g for 30 min and solubilized using the supernatant subjected to affinity purification using a nickel-nitrilotriacetic acid (Ni-NTA) resin, exploiting a His-tag on the Gα subunit. After binding, the resin was washed with wash buffer (same composition as lysis buffer supplemented with 20 mM imidazole) to remove non-specifically bound proteins. The trimeric Gs complex was eluted with lysis buffer containing 250 mM imidazole. Eluted fractions were concentrated and further purified by size-exclusion chromatography using a Superdex 200 column equilibrated in the storage buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 0.05% LMNG, 0.1 mM TCEP, 5 mM MgCl₂, and 50 μM GDP). The purified trimeric Gs complex was concentrated to 4.86 mg/mL as measured by the Nanodrop protein assay and stored at −80 °C until further use.
Trimeric Gs activity assays
The GTP hydrolysis assay was performed using a modified protocol of the GTPase-Glo assay (Promega). The reaction was started by mixing 500 nM Gαsβγ with 160 nM purified A2AR in buffer containing 20 mM HEPES, pH 7.4, 100 mM NaCl, 0.002% CHS, 0.02% LMNG, 1 mM DTT, 5 mM MgCl2. The concentration of adenosine and metabolites varied depending on the experimental setup, with a final volume of 12.5 μL. Ligands were incubated with A2AR and Gs to measure activation, or with A2AR, Gs, and 80 μM adenosine (EC₈₀) to measure inhibition. After incubation at room temperature for 1 h, 12.5 μL GTP-GTPase solution containing 10 μM GTP, and the cognate 500x reagent was added to each well, followed by a 30 min incubation at room temperature. Then, 25 μL reconstituted GTPase-Glo reagent containing 5 μM ADP was added to each sample and incubated for another 10 min at room temperature with shaking. Luminescence was measured using a Clariostar plate reader (BMG LABTECH) at 528 ± 20 nm. Analysis of data was performed by Excel and GraphPad Prism.
Fluorescence polarization assay
Fluorescence polarization (FP) assays were performed to assess the binding interactions between the A2AR and the fluorescently-labeled agonist NECA (CA200623, HelloBio). A2AR-ΔCTD (A2AR with N154A and truncated C-terminal region), NECA, and metabolites were diluted in assay buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 0.00075% LMNG, 0.0001% CHS in a 384-well plate. NECA was prepared as a 1 mM stock in DMSO and diluted to 20 nM in assay buffer. A2AR was incubated with metabolites in a total volume of 10 µL for 10 min. Next, this A2AR-metabolite mix was added to 10 µL of 20 nM NECA (10 nM final) to a final volume of 20 µL and incubated at room temperature for 10 min with gentle shaking. FP measurements were conducted using a Clariostar microplate reader (BMG LABTECH) with excitation at 633 nm and emission at 670 nm. Polarization values were analyzed using non-linear regression to determine binding affinities with GraphPad Prism software.
Differential scanning fluorimetry
The thermal stability of A2AR with association of selected metabolites was determined using the Prometheus NT.48 instrument (NanoTemper Technologies). For sample preparation, A2AR was diluted to a final concentration of 20 μM in buffer containing 20 mM HEPES pH 7.5, 800 mM NaCl, 0.00075% LMNG, 0.0001% CHS, 10% glycerol, and incubated with the respective metabolites at a concentration ranging from 1 μM to 500 µM. Before measurement, the samples were incubated for 30 min at room temperature. Melting scans were performed at a temperature range from 25–95 °C with a temperature increment of 1 °C per min. Thermal unfolding curves were recorded by measuring the ratio of the tryptophan-fluorescence at 350 nm (F350) and 330 nm (F330). The protein melting temperature (Tm) of a given sample was determined as the inflection point of the thermal unfolding curves, or the local maximum of its first derivative. The measured curves were analyzed using the manufacturer’s software (PR.ThermControl, v 2.1.2, NanoTemper Technologies). Based on the determined Tm values, ligand-induced thermal shifts (ΔTm) were determined as the difference between the melting temperature of the ligand-bound state (Tm) and the apo state.
Expression and purification of the anti-BRIL Fab and anti-Fab Nb
The anti-BRIL Fab was expressed in E. coli and purified as previously described in ref. 63. The anti-Fab Nb64 was cloned into a pET26b+ vector with an N-terminal 6x-His tag followed by a TEV protease site. The protein was expressed in E. coli BL21(DE3) cells and purified by Ni-NTA chromatography, followed by removal of the His-tag by TEV digestion. The final step in the purification was SEC using a Superdex 75 10/300 column where fractions corresponding to monodisperse Nb were obtained. A2AR was incubated with molar excess of anti-BRIL Fab and anti-Fab Nb, before the mixture was again concentrated and injected into a Superdex 200 10/300 GL column equilibrated with A2AR SEC buffer. The peak fractions were concentrated and subjected to cryo-EM analysis.
Cryo-EM data collection
Purified A2AR-BRIL-Nb35 was mixed with PGD2 (final = 0.5 mM) and without to a volume of 4.0 μl on ice before being applied to glow-discharged (30 s, 15 mA, PELCO easiGlow, Ted Pella) Quantifoil Au 1.2/1.3 300 mesh holey carbon grids. The grids were blotted using a Vitrobot Mark IV (ThermoFisher Scientific) with 3.0 s blotting time, 3 s waiting time in 100% humidity, and plunge-frozen in liquid ethane. A total of 5219 movies were collected for A2AR + ZM241385, and a total of 6857 movies were collected for the A2AR + ZM241385 + PGD2 on a Titan Krios G4 electron microscope (ThermoFisher Scientific) operating at an accelerating voltage of 300 kV at a magnification of 96,000x, corresponding to a magnified pixel size of 0.808 Å. Micrographs were recorded using a Falcon 4 direct electron camera with an exposure time of 6.2 s with a total dose of 49.92 e/Å2 and a defocus range from −1.0 μm to −2.0 μm. The total number of frames per movie was 32. Automatic data acquisition was conducted using the SerialEM software65.
Cryo-EM data processing
Dose-fractionated images in super-resolution mode were internally gain-normalized and binned by 2 in EPU during data collection. Beam-induced motion was corrected in RELION 5.066. The contrast transfer functions (CTF) were estimated for motion-corrected micrographs using CTFFIND4.167, and particles were automatically picked using the internal model using LoG-based auto-picking. Picked particles were binned 4 × 4 and subjected to two rounds of 3D classification to remove junk particles. Classes with recognizable receptor features were re-extracted and centered by applying alignment offsets. Particles were subjected to 3D auto-refinement, and a mask of A2AR-BRIL was used for local refinement to improve the resolution. Multiple rounds of CtfRefine were executed to improve resolution, and final refinement was performed using Blush regularization68.
Model building and refinement
For model building, a previously published A2AR structure (PDB: 7T32) was placed into the density using UCSF ChimeraX69 and manually adjusted in Coot70. Final models were optimized using real-space refinement in PHENIX71,72 and the quality of the final models were validated with MolProbity73 against unfiltered halfmaps and unsharpened full maps. All structural figures were prepared using ChimeraX.
Homogenous time-resolved fluorescence cAMP detection assay
HEK293-A2AR overexpressing cells were grown in RPMI (ATCC modification) + 10% FBS + 1x glutamax at 37 °C, 5% CO2. Cells were harvested at 70% confluence for the Homogenous time-resolved fluorescence (HTRF) cAMP Gs Dynamic assay (Revvity) to measure intracellular cAMP and was performed using the manufacturer’s suggested protocol. Briefly, HEK293-A2AR over-expressing cells were trypsinized with TrypLE (Invitrogen), neutralized with PBS, and pelleted. The pellet was resuspended in 1x stimulation buffer + 0.5 mM isobutylmethylxanthine (IBMX) at 2E6/mL. 5 µL of inhibitor and 5 µL of HEK293-A2AR cells were added into wells of a 384-PE OptiPlate. Cells were treated with inhibitor for 45 min at room temperature. 5 µl of agonist was added to the sample and incubated at 37 oC for 30 min. D2 and K-labeled antibodies were prepared in 1x lysis buffer and an equal volume of each was added to each sample and incubated at room temperature for 1 h. A Clariostar plate reader was used to measure time-resolved fluorescence at 620 nm and 665 nm using cisBio HTRF settings.
Molecular docking
We performed molecular docking using the MOE docking module74, employing two docking strategies: a general approach and a template-based approach guided by structural similarity to adenosine. As a template, we used the crystal structure of A2AR bound to adenosine (PDB ID: 2YDO). Prior to docking, we prepared the A2AR-adenosine complex using the MOE QuickPrep module, which involved optimizing protonation states and performing energy minimization of the crystallographic structure with the Amber10:EHT force field integrated into MOE-2024.0601. During minimization, we applied MOE default tether restraints on protein atoms and monitored the root-mean-square (RMS) gradient of the potential energy, ensuring it remained below 0.1 kcal mol−1Å−2. All molecules, except for A2AR and adenosine, were removed during the preparation process.
Subsequently, we performed molecular docking to insert SAH and DA into the adenosine binding pocket using the MOE general docking module74. This involved an initial pose generation step using the triangular matcher algorithm, which generated 1000 docking poses based on receptor shape. These poses were then screened using the London dG scoring function, and the top 100 structures were selected for further refinement. The final refinement step applied a rigid receptor replacement method combined with the GBVI/WSA dG scoring function.
In parallel, we performed template-based docking using MOE substructure docking approach74. This method involved superposing the docked molecules onto the active site based on one or more reference structures—specifically, the adenine fragment in adenosine. The scoring function accounted for ligand similarity, RMSD overlap, and protein-ligand clash terms. Given that both SAH and DA contain an adenine moiety similar to adenosine, this approach provided an additional layer of structural guidance. The resulting poses were further refined using the rigid receptor replacement method and the GBVI/WSA dG scoring function. Following docking, we visually inspected the highest-ranked poses, selecting those that closely resembled adenosine binding. These selected structures were presented in this manuscript.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The 3D cryo-EM density maps have been deposited into the Electron Microscopy Data Bank under accession codes EMD-71130 (A2AR-ZM241385) and EMD-71131 (A2AR-ZM241385-PGD2). The structure coordinates have been deposited in the PDB under accession codes 9P1T (A2AR-ZM241385) and 9P1S (A2AR-ZM241385-PGD2). All source data for the MIDAS results, and protein- and cell-based assays have been deposited in Supplementary Data 1.
References
Koupenova, M. & Ravid, K. Adenosine, Adenosine Receptors and Their Role in Glucose Homeostasis and Lipid Metabolism. J. Cell. Physiol. 228, 921–929 (2013).
Verma, N. et al. Metabolic Dysfunction in Mice with Adipocyte-Specific Ablation of the Adenosine A2A Receptor. J. Biol. Chem. 301, 108206 (2025).
Castro, C. M. et al. Adenosine A2A Receptor (A2AR) Stimulation Enhances Mitochondrial Metabolism and Mitigates Reactive Oxygen Species-Mediated Mitochondrial Injury. FASEB J. 34, 5027–5045 (2020).
González-Benı́tez, E., Guinzberg, R., Dı́az-Cruz, A. & Piña, E. Regulation of Glycogen Metabolism in Hepatocytes through Adenosine Receptors. Role of Ca2+ and cAMP. Eur. J. Pharmacol. 437, 105–111 (2002).
Jang, M.-H. & Song, J. Adenosine and Adenosine Receptors in Metabolic Imbalance-Related Neurological Issues. Biomed. Pharmacother. 177, 116996 (2024).
Helms, R. S. & Powell, J. D. Rethinking the Adenosine-A2AR Checkpoint: Implications for Enhancing Anti-Tumor Immunotherapy. Curr. Opin. Pharmacol. 53, 77–83 (2020).
Zarek, P. E. et al. A2A Receptor Signaling Promotes Peripheral Tolerance by Inducing T-Cell Anergy and the Generation of Adaptive Regulatory T Cells. Blood 111, 251–259 (2008).
Leone, R. D., Lo, Y.-C. & Powell, J. D. A2aR Antagonists: Next Generation Checkpoint Blockade for Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 13, 265–272 (2015).
Ohta, A. & Sitkovsky, M. Role of G-Protein-Coupled Adenosine Receptors in Downregulation of Inflammation and Protection from Tissue Damage. Nature 414, 916–920 (2001).
Lappas, C. M., Sullivan, G. W. & Linden, J. Adenosine A2A Agonists in Development for the Treatment of Inflammation. Expert Opin. Investig. Drugs 14, 797–806 (2005).
de Lera Ruiz, M., Lim, Y.-H. & Zheng, J. Adenosine A2A Receptor as a Drug Discovery Target. J. Med. Chem. 57, 3623–3650 (2014).
Tao, L. J., Seo, D. E., Jackson, B., Ivanova, N. B. & Santori, F. R. Nuclear Hormone Receptors and Their Ligands: Metabolites in Control of Transcription. Cells 9, 2606 (2020).
Wang, Y.-D., Chen, W.-D., Moore, D. D. & Huang, W. FXR: A Metabolic Regulator and Cell Protector. Cell Res. 18, 1087–1095 (2008).
Littlewood-Evans, A. et al. GPR91 Senses Extracellular Succinate Released from Inflammatory Macrophages and Exacerbates Rheumatoid Arthritis. J. Exp. Med. 213, 1655–1662 (2016).
Sun, M., Wu, W., Liu, Z. & Cong, Y. Microbiota Metabolite Short Chain Fatty Acids, GCPR, and Inflammatory Bowel Diseases. J. Gastroenterol. 52, 1–8 (2017).
Fiorucci, S., Biagioli, M., Zampella, A. & Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 9, 1853 (2018).
Melhem, H. et al. Metabolite-Sensing G Protein-Coupled Receptors Connect the Diet-Microbiota-Metabolites Axis to Inflammatory Bowel Disease. Cells 8, 450 (2019).
Pocai, A. Unraveling Oxyntomodulin, GLP1’s Enigmatic Brother. J. Endocrinol. 215, 223–231 (2012).
Gallagher, D. M., O’Harte, F. P. M. & Irwin, N. An Update on Galanin and Spexin and Their Potential for the Treatment of Type 2 Diabetes and Related Metabolic Disorders. Peptides 171, 171096 (2024).
Soltoff, S. P., Avraham, H., Avraham, S. & Cantley, L. C. Activation of P2Y2 Receptors by UTP and ATP Stimulates Mitogen-Activated Kinase Activity through a Pathway That Involves Related Adhesion Focal Tyrosine Kinase and Protein Kinase C*. J. Biol. Chem. 273, 2653–2660 (1998).
Colosimo, D. A. et al. Mapping Interactions of Microbial Metabolites with Human G-Protein-Coupled Receptors. Cell Host Microbe 26, 273–282.e7 (2019).
Chen, H. et al. A Forward Chemical Genetic Screen Reveals Gut Microbiota Metabolites That Modulate Host Physiology. Cell 177, 1217–1231.e18 (2019).
Aleti, G., Troyer, E. A. & Hong, S. uzi G protein-coupled receptors: A Target for Microbial Metabolites and a Mechanistic Link to Microbiome-Immune-Brain Interactions. Brain Behav. Immun. Health 32, 100671 (2023).
Jenkins, L. et al. Identification of Novel Species-Selective Agonists of the G-Protein-Coupled Receptor GPR35 That Promote Recruitment of β-Arrestin-2 and Activate Gα13. Biochem. J. 432, 451–459 (2010).
Oka, S., Ota, R., Shima, M., Yamashita, A. & Sugiura, T. GPR35 Is a Novel Lysophosphatidic Acid Receptor. Biochem. Biophys. Res. Commun. 395, 232–237 (2010).
Wold, E. A. & Zhou, J. GPCR Allosteric Modulators: Mechanistic Advantages and Therapeutic Applications. Curr. Top. Med. Chem. 18, 2002–2006 (2018).
Shen, S. et al. Allosteric Modulation of G protein-coupled Receptor Signaling. Front. Endocrinol. 14, 1137604 (2023).
Hicks, K. G. et al. Protein-Metabolite Interactomics of Carbohydrate Metabolism Reveal Regulation of Lactate Dehydrogenase. Science 379, 996–1003 (2023).
Huang, S. K. et al. Allosteric Modulation of the Adenosine A2A Receptor by Cholesterol. eLife 11, e73901 (2022).
Carpenter, B. & Lebon, G. Human Adenosine A2A Receptor: Molecular Mechanism of Ligand Binding and Activation. Front. Pharmacol. 8, 868 (2017).
Zhukov, A. et al. Biophysical Mapping of the Adenosine A2A Receptor. J. Med. Chem. 54, 4312–4323 (2011).
Pettipher, R., Hansel, T. T. & Armer, R. Antagonism of the Prostaglandin D2 Receptors DP1 and CRTH2 as an Approach to Treat Allergic Diseases. Nat. Rev. Drug Discov. 6, 313–325 (2007).
Weis, W. I. & Kobilka, B. K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 87, 897–919 (2018).
Mafi, A., Kim, S.-K. & Goddard, W. A. The Mechanism for Ligand Activation of the GPCR–G Protein Complex. Proc. Natl Acad. Sci. 119, e2110085119 (2022).
Mafi, A., Kim, S.-K. & Goddard, W. A. The Dynamics of Agonist-Β2-Adrenergic Receptor Activation Induced by Binding of GDP-Bound Gs Protein. Nat. Chem. 15, 1127–1137 (2023).
Kamo, S., Nakanishi, T., Aotani, R., Nakamura, Y. & Tamai, I. Impact of FDA-Approved Drugs on the Prostaglandin Transporter OATP2A1/SLCO2A1. J. Pharm. Sci. 106, 2483–2490 (2017).
Baccouch, R., Rascol, E., Stoklosa, K. & Alves, I. D. The Role of the Lipid Environment in the Activity of G Protein Coupled Receptors. Biophys. Chem. 285, 106794 (2022).
Lebon, G. et al. Agonist-Bound Adenosine A2A Receptor Structures Reveal Common Features of GPCR Activation. Nature 474, 521–525 (2011).
Kim, J. et al. Site-Directed Mutagenesis Identifies Residues Involved in Ligand Recognition in the Human A2a Adenosine Receptor. J. Biol. Chem. 270, 13987–13997 (1995).
Jiang, Q. et al. Mutagenesis Reveals Structure−Activity Parallels between Human A2A Adenosine Receptors and Biogenic Amine G Protein-Coupled Receptors. J. Med. Chem. 40, 2588–2595 (1997).
Katritch, V., Cherezov, V. & Stevens, R. C. Structure-Function of the G-Protein-Coupled Receptor Superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 (2013).
Powers, A. S. et al. A Non-Canonical Mechanism of GPCR Activation. Nat. Commun. 15, 9938 (2024).
Gibson, N. J. & Brown, M. F. Role of Phosphatidylserine in the MI-MIII Equilibrium of Rhodopsin. Biochem. Biophys. Res. Commun. 176, 915–921 (1991).
Dawaliby, R. et al. Allosteric Regulation of G Protein–Coupled Receptor Activity by Phospholipids. Nat. Chem. Biol. 12, 35–39 (2016).
Strohman, M. J. et al. Local Membrane Charge Regulates Β2 Adrenergic Receptor Coupling to Gi3. Nat. Commun. 10, 2234 (2019).
Thakur, N. et al. Anionic Phospholipids Control Mechanisms of GPCR-G Protein Recognition. Nat. Commun. 14, 794 (2023).
Awad, J. A., Morrow, J. D. & Roberts, L. J. Detection of the Major Urinary Metabolite of Prostaglandin D2 in the Circulation: Demonstration of Elevated Levels in Patients with Disorders of Systemic Mast Cell Activation. J. Allergy Clin. Immunol. 93, 817–824 (1994).
Mihara, A. et al. Association of Serum S-Adenosylmethionine, s-Adenosylhomocysteine, and Their Ratio with the Risk of Dementia and Death in a Community. Sci. Rep. 12, 12427 (2022).
Liu, S. et al. Association between Plasma S-Adenosylmethionine and Risk of Mortality in Patients with Coronary Artery Disease: A Cohort Study. Am. J. Clin. Nutr. 114, 1360–1370 (2021).
Borkowsky, W., Gershon, A. A., Shenkman, L. & Hirschhorn, R. Adenosine Deaminase Deficiency Without Immunodeficiency: Clinical and Metabolic Studies. Pediatr. Res. 14, 885–889 (1980).
Hershfield, M. S., Kredich, N. M. & Ownby, D. R. In Vivo Inactivation of Erythrocyte S-Adenosylhomocysteine Hydrolase by 2’-Deoxyadenosine in Adenosine Deaminase-Deficient Patients. J. Clin. Invest. 63, 807–811 (1979).
Deharo, P. et al. Homocysteine Concentration and Adenosine A2A Receptor Production by Peripheral Blood Mononuclear Cells in Coronary Artery Disease Patients. J. Cell. Mol. Med. 24, 8942–8949 (2020).
Yasi, E. A., Kruyer, N. S. & Peralta-Yahya, P. Advances in G Protein-Coupled Receptor High-Throughput Screening. Curr. Opin. Biotechnol. 64, 210–217 (2020).
Laschet, C., Dupuis, N. & Hanson, J. The G Protein-Coupled Receptors Deorphanization Landscape. Biochem. Pharmacol. 153, 62–74 (2018).
Gz Enhanced Signal Transduction assaY (GZESTY) for GPCR deorphanization | bioRxiv. https://www.biorxiv.org/content/10.1101/2024.07.26.605282v1 (accessed 2025-11-05).
Kroeze, W. K. et al. PRESTO-Tango as an Open-Source Resource for Interrogation of the Druggable Human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 (2015).
de Lau, W. et al. Lgr5 Homologues Associate with Wnt Receptors and Mediate R-Spondin Signalling. Nature 476, 293–297 (2011).
Dupuis, N. et al. Activation of the Orphan G Protein–Coupled Receptor GPR27 by Surrogate Ligands Promotes β-Arrestin 2 Recruitment. Mol. Pharmacol. 91, 595–608 (2017).
Rajagopal, S. et al. -Arrestin- but Not G Protein-Mediated Signaling by the “Decoy” Receptor CXCR7. Proc. Natl Acad. Sci. 107, 628–632 (2010).
Cao, W. et al. Direct Binding of Activated C-Src to the Β3-Adrenergic Receptor Is Required for MAP Kinase Activation*. J. Biol. Chem. 275, 38131–38134 (2000).
Al-Sabah, S. et al. The GIP Receptor Displays Higher Basal Activity than the GLP-1 Receptor but Does Not Recruit GRK2 or Arrestin3 Effectively. PLoS ONE 9, e106890 (2014).
Zhang, H. et al. Structural Basis for Selectivity and Diversity in Angiotensin II Receptors. Nature 544, 327–332 (2017).
Mukherjee, S. et al. Synthetic Antibodies against BRIL as Universal Fiducial Marks for Single−particle cryoEM Structure Determination of Membrane Proteins. Nat. Commun. 11, 1598 (2020).
Bloch, J. S. et al. Development of a Universal Nanobody-Binding Fab Module for Fiducial-Assisted Cryo-EM Studies of Membrane Proteins. Proc. Natl Acad. Sci. 118, e2115435118 (2021).
Mastronarde, D. N. Automated Electron Microscope Tomography Using Robust Prediction of Specimen Movements. J. Struct. Biol. 152, 36–51 (2005).
Scheres, S. H. W. RELION: Implementation of a Bayesian Approach to Cryo-EM Structure Determination. J. Struct. Biol. 180, 519–530 (2012).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and Accurate Defocus Estimation from Electron Micrographs. J. Struct. Biol. 192, 216–221 (2015).
Kimanius, D. et al. Data driven regularization lowers the size barrier of cryo-EM structure determination. Nat. Methods 21, 1216–1221 (2024).
Goddard, T. D. et al. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Protein Sci. 27, 14–25 (2018).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and Development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–221 (2010).
Afonine, P. V. et al. Real-Space Refinement in PHENIX for Cryo-EM and Crystallography. Acta Crystallogr. Sect. Struct. Biol. 74, 531–544 (2018).
Williams, C. J. et al. MolProbity: More and Better Reference Data for Improved All-atom Structure Validation. Protein Sci. 27, 293–315 (2018).
Molecular Operating Environment (MOE), 2024.0601 Chemical Computing Group ULC, 910-1010 Sherbrooke St. W., Montreal, QC H3A 2R7, (2025).
Acknowledgements
We thank our Calico colleagues Magdalena Lopez, Suvrajit Saha, and Sean Hackett for experimental support, as well as Vladamir Akhmetov, Faith Fowler, and Isaac Knudson for helpful comments and suggestions. We thank Rebecca Ray for her help with statistical analyses throughout the paper. We thank Jie Heng, Jing Li, and members from Shuimu Biosciences for support with protein purification of A2AR and Gs, cryo-EM sample preparation, data collection, and processing. We thank Houxia Shi and members from Biortus Biosciences for assistance with A2AR protein purification. This work was supported by Calico Life Sciences LLC.
Author information
Authors and Affiliations
Contributions
Q.H., A.M., and K.H., conceived and jointly supervised the project with additional supervision from D.E., J.P., and J.R. K.H. and A.P. performed MIDAS screening and analysis. M.R., P.R., Q.H., and A.M. designed the protein constructs and supported protein purification. P.R. and M.R. performed all protein-based binding and functional assays. M.C., B.W., and C.P. performed the cell-based functional experiments. A.M. performed molecular docking. P.R., M.R., and Q.H. designed and supported the cryo-EM work. P.R. wrote the manuscript with contributions and revisions from all authors.
Corresponding authors
Ethics declarations
Competing interests
P.R., M.R., M.C., B.W., C.P., J.P., D.E., A.M., and Q.H. are employees of Calico Life Sciences LLC and declare no other competing interests. K.G.H. and J.R. are inventors of MIDAS technology that has been licensed to Atavistik Bio, for which K.G.H. is a consultant, and J.R. is a founder. The remaining authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks Matthew J. Belousoff, Yajuan (Megan) Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rao, P., Rathinaswamy, M., Chan, M. et al. Systematic metabolite screening identifies functional regulators of the adenosine A2A receptor. Commun Chem 9, 19 (2026). https://doi.org/10.1038/s42004-025-01825-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-025-01825-3
