
Abstract
The emergence of molecular oxygen on early Earth is conventionally attributed to the evolution of oxygenic photosynthesis. A persistent challenge for early life, however, was the management of reactive oxygen species such as hydrogen peroxide (H2O2), which could arise through a variety of abiotic processes. Here we report that some RNA molecules, when coordinated with ferrous iron (Fe2+), catalyze the oxidation of H2O2 into O2 and H2O under anoxic conditions that mimic the early Earth environment. This previously unrecognized RNA-based redox activity suggests that ancient RNA-metal complexes may have contributed to the detoxification of H2O2 and the management of oxidative stress prior to the evolution of protein enzymes. Such RNA–Fe complexes provide a plausible molecular mechanism linking early geochemical oxidants to primitive biological redox chemistry.
Similar content being viewed by others


Introduction
The Last Universal Common Ancestor (LUCA) represents an inferred ancestral population, with recent estimates placing its emergence at approximately 4.2 billion years ago (Ga)1. LUCA inhabited an anoxic environment rich in soluble iron2,3,4. During this period, Earth’s atmosphere lacked free molecular oxygen (O₂) until the emergence of oxygenic photosynthesis, which initiated the Great Oxidation Event around 2.4 Ga5,6. Notably, by ~3.5 Ga, robust geological evidence indicates the presence of stromatolite-forming phototrophs and sulfate-reducing bacteria7,8, underscoring that LUCA predates the emergence of these more complex microbial ecosystems.
Although the early Earth was largely anoxic, multiple abiotic processes are thought to have generated reactive oxygen species (ROS), including hydrogen peroxide (H2O2), through photochemical reactions5,9,10, mineral-water interactions11, and geodynamic surface oxidation12. As a result, early life likely faced intermittent oxidative stress even prior to the accumulation of atmospheric oxygen.
Despite this, phylogenetic analyses of proteins and ribosomal RNA (rRNA), along with reconstructed LUCA genomes, have revealed the presence of genes involved in O2 and H2O2 cycling13,14. This suggests that trace amounts of free oxygen may have existed prior to the evolution of oxygenic photosynthesis14,15. Rather than indicating sustained oxygenation, however, such genetic signatures may reflect an early need for biological systems to tolerate, detoxify, or exploit transient ROS in otherwise anoxic environments. These findings are often interpreted as the result of later evolutionary developments or horizontal gene transfer events associated with the rise of oxygenic photosynthesis16.
Another defining feature of LUCA’s environment was its high concentration of soluble iron4. Metal ions such as Fe2+ were essential for early biochemical processes, aiding the folding, assembly, and activity of proteins and RNAs. In this prebiotic context, iron played a central role in LUCA’s molecular machinery16. For instance, Williams and colleagues demonstrated that ribosomes can catalyze peptidyl transfer in vitro when Mg2+ is replaced with Fe2+ or Mn2+17. Similarly, 23S rRNA and resurrected fragments of its ancient peptidyl transfer center (PTC) have been shown to mediate electron transfer in the presence of Fe2+18,19.
The Class Ia ribonucleotide reductase (RNR), a key enzyme in the transition from RNA to DNA-based life, contains a di-iron cluster that catalyzes the reduction of ribonucleotides to deoxyribonucleotides20,21. In addition, enzymes such as monofunctional heme catalase and catalase-peroxidase, which use heme-bound Fe as a cofactor, decompose hydrogen peroxide (H2O2) into water and oxygen22,23. The origins of catalases and RNRs are thus closely tied to evolutionary adaptation to increasing oxygen levels during the Great Oxidation Event21,22. These enzymes illustrate how iron-centered redox chemistry became central to managing oxidative stress as oxygen levels increased, but they also raise the question of whether simpler, pre-proteinaceous systems could have performed analogous ROS-detoxifying functions earlier in evolution.
In the Thermus thermophilus 70S ribosome, the X-ray crystallographic structure revealed a tetradentate Mg2+ coordination site within the 23S rRNA24. A detailed inspection of this structure reveals a chelation pattern reminiscent of the heme–Fe complex. Structural superimposition of the 23S rRNA–Mg2+ chelation and the heme-Fe center in catalase reveals remarkable similarity (Fig. 1A, B). In both cases, a central metal ion is coordinated by four donor atoms: Mg2+ by four phosphate oxygen atoms from the rRNA, and Fe2+ by four nitrogen atoms of the porphyrin ring (Fig. 1C, D).

A Superposition of the tetradentate rRNA–Mg2+ coordination structure from the Thermus thermophilus 70S ribosome (Mg²⁺ 3111; PDB ID: 4v51) with the heme-Fe center from catalase (PDB ID: 1dgb). The rRNA structure coordinates a Mg2+ ion (yellow sphere) via four phosphate oxygen atoms (red spheres), while the heme group (cyan sticks) binds an Fe2+ ion (green sphere) through four nitrogen atoms (blue spheres) of the porphyrin ring. Two ten-membered rings formed by the rRNA backbone, highlighted in magenta, define the tetradentate motif. The coordination geometry of the rRNA–Mg2+ complex closely resembles that of the heme-Fe complex. For clarity, side chains and portions of the ribose are omitted. B Orthogonal view of the same superposition, rotated 90° along the x-axis. C The complexity of the complete rRNA structure chelating the tetradentate Mg2+, superimposed on the heme-Fe complex. Residue side chains of the tetradentate motif are labeled and shown in gray. D Orthogonal view of the complete rRNA tetradentate motif superimposed on the heme structure, rotated 90° along the x-axis.
This structural resemblance raises an intriguing possibility: if the tetradentate Mg2+ coordination site identified in the 23S rRNA structure were instead occupied by Fe2+, could the resulting RNA–Fe complex participate in redox chemistry relevant to the detoxification of ROS, such as the decomposition of H2O2 into O2 and H2O, prior to the evolution of protein enzymes? We therefore hypothesize that tetradentate Fe2+-chelated RNA complexes may represent an ancient, RNA-based mechanism for mitigating oxidative stress under prebiotic conditions, rather than a primary driver of planetary oxygenation.
To test this, we examined five RNA and related molecules: the universally conserved tRNA 3′-terminal trinucleotide CCA, the dinucleotide CA, the ribosomal A-site analogs: C-puromycin (CPmn; a mononucleotide-linked aminonucleoside), and puromycin (Pmn; an aminonucleoside), and the intact T. thermophilus 23S rRNA (Fig. 2). The five RNA constructs were selected to probe how backbone chemistry and molecular complexity influence Fe2+ coordination and redox catalysis. The three natural RNAs (CA, CCA, and 23S rRNA) possess canonical phosphate-based backbones and are widely regarded as evolutionarily ancient motifs. In contrast, the aminonucleoside analogs, CPmn and Pmn, contain peptide-like linkages and were included as experimental comparators to contrast RNA-based and peptide-based scaffolds under identical catalytic conditions. Using the “blue bottle” assay25, we assessed the potential of these RNAs to catalyze the oxidation of H2O2, functioning as redox-active cofactors under simulated anoxic, iron-rich conditions mimicking early Earth.

A Native trinucleotide CCA. B C-puromycin (CPmn), a ribosomal A-site analog consisting of a cytidine monophosphate covalently linked to the aminonucleoside puromycin. C Native dinucleotide CA. D Puromycin (Pmn), an aminonucleoside mimicking the ribosomal A-site substrate. E The full-length 23S rRNA from T. thermophilus. The red highlights denote atoms potentially involved in the formation of the tetradentate chelation motif.
Results and discussion
All RNAs tested, except for CA, exhibited catalytic activity for H2O2 decomposition (Fig. 3). The 23S rRNA demonstrated the highest activity, consistent with its structural complexity and high density of metal–phosphate interactions. Unexpectedly, CPmn exhibited the second-highest catalytic efficiency, surpassing that of the CCA trinucleotide. The observed activity of CCA is attributed to its ability to form a ten-membered chelation ring via adjacent phosphate oxygen atoms26, possibly enabling tetradentate coordination of Fe2+ (Fig. 2A). As a universally conserved tRNA 3’-terminus, the CCA tail is considered to be evolutionarily ancient and likely predates the LUCA, with deep roots in the ribosome origin27,28. Considering the availability of H2O2 in Earth’s prebiotic environments from geological and geochemical sources14, these results suggest that intrinsic RNA structural features endowed early biomolecules with catalytic capacities relevant to oxidative stress management.

The 23S rRNA from T. thermophilus (purple), C-puromycin (CPmn; red), CCA trinucleotide (cyan), and puromycin (Pmn; green) catalyze the oxidative decomposition of H2O2 in the presence of Fe2+. The reaction was monitored using a modified “blue bottle” assay, which detects oxygen generation through the oxidation of methylene blue, measured by absorbance at 664 nm. The CA dinucleotide (pink) showed no catalytic activity. “Minus RNA” denotes the control reaction comprising Fe2+ and H2O2 in the complete assay mixture, but with no added RNA or RNA analog. Dashed lines represent raw experimental data; solid curves represent smoothed trends derived from the data.
The dinucleotide CA did not exhibit catalytic activity consistent with its lack of adjacent phosphate groups required for chelation ring formation (Fig. 2C). Pmn displayed weak activity at 1.6 μM, fourfold higher than the concentration used for CPmn, suggesting a minimal chelating role for its primary amine nitrogen (Figs. 2D and 3).
The dependence of catalytic activity on RNAs accentuates the importance of metal coordination geometry in catalysis. The 23S rRNA coordinating Mg2+ through multiple phosphate oxygens in support of peptidyl transferase activity24,26 may use a similar geometry in Fe2+ coordination for redox catalysis. Remarkably, the minimal CPmn molecule appears to form a functional RNA–Fe2+ complex capable of catalysis. Continuous variation analysis29,30 under anoxic conditions revealed a 2:1 RNA:Fe2+ stoichiometry for CPmn (Fig. 4A), consistent with a tetradentate coordination such that the complexation geometry, as we proposed, is similar to that of heme–Fe complexes (Fig. 1). Comparable stoichiometry was observed for CCA (Fig. 4B), while Pmn displayed a 1:2 ratio (Fig. 4C), possibly reflecting an alternative, less efficient catalytic mechanism.

Continuous variation analysis for Fe2+ complexation with A C-puromycin (CPmn), B CCA trinucleotide, and C puromycin (Pmn). The mole fraction of Fe2+ (XFe2+) and RNA (XRNA) ranges from 0 to 1 along the x-axis. UV–Vis absorbance was recorded at 260 nm (OD260), and the data were normalized to OD260 per nucleotide for plotting. The discontinuity point, marked by a dashed arrow, indicates the stoichiometric ratio of Fe2+ to RNA at maximal complex formation. D The oxidative decomposition of H2O2 catalyzed by the CPmn-Fe2+ complex follows enzyme kinetics and is saturable, reaching a plateau at high [H2O2].
Kinetic analysis of CPmn-catalyzed H2O2 decomposition revealed Michaelis–Menten behavior (Fig. 4D). Non-linear regression fitting determined the kinetic parameters for CPmn, kcat = 3.8 × 10−2 s−1, Km = 1.52 M, and kcat/Km = 2.5 × 10−2 M−1S−1.
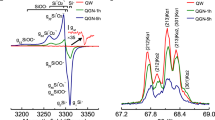
Electron paramagnetic resonance (EPR) spectroscopy was used to investigate the catalytic oxidation system involving hydrogen peroxide (H2O2), native RNA (CCA), and a mononucleotide-linked aminonucleoside (CPmn) under simulated early Earth conditions, featuring Fe2+ and anoxic environments. In the continuous wave (CW) EPR spectrum of the Fe2+/CCA/H2O2 system, CCA acting as an iron chelator for coordination of Fe2+ was observed, as compared to spectra from Fe2+ and Fe3+ alone (Fig. 5A, B). The powder spectrum of the Fe2+/CCA/H2O2 complex exhibits axial symmetry, featuring a g ≈ 4.29 signal at low magnetic field and a broad signal at high field, consistent with a low-spin Fe2+ (d6, S = 1) species (Fig. 5C). In CW EPR, the natural-abundance 57Fe isotope (~2.1%) contributes detectable hyperfine structure through electron–nuclear spin interactions. In the high-field region, hyperfine coupling is more pronounced along g⊥ ≈ 1.91 than g// ≈ 2.01 (Fig. 5A, G). Upon CCA coordination, superhyperfine interactions from 31P are observed in the g// region of 57Fe, indicating possibly the formation of a tetradentate [Fe(CCA)2]2+ complex (Fig. 5C, H).

Spectra were recorded with the x-axis plotted as magnetic field (Gauss). Control spectra collected in the absence of CCA and H2O2 are shown for A Fe2+ and B Fe3+ standards. Time-dependent spectra of the CCA/Fe2+/H2O2 reaction mixture are shown after C 2.5 h, D 3.5 h, E 4.5 h, and F 15.5 h. Expanded high-field regions for Fe2+ standard (G) and for the reaction mixture at 2.5 h (H) highlight the g-tensor features. The characteristic signals: g ≈ 4.29 at low field, g⊥ ≈ 1.91 and g// ≈ 2.01 at high field, and the transient intermediate at g ≈ 2.71, are indicated directly on the spectra.
During the RNA–Fe catalytic oxidation of H2O2, the g// ≈ 2.01 signal becomes more pronounced (Fig. 5C), suggesting initial oxidation of Fe2+ in the CCA–Fe complex. A new feature at g ≈ 2.71 appears, attributed to a ferryl cation radical species (CCA–Fe⁴⁺•), likely corresponding to a high-spin Fe4+ (d4, S = 2) center coupled to a radical via dipole–dipole interaction (Fig. 5D). As the reaction proceeds, the g ≈ 2.71 signal associated with the ferryl radical fades away (Fig. 5E), indicating the decay or transformation of the high-valent intermediate. The catalytic oxidation observed with the CPmn/Fe2+/H2O2 system followed a process comparable to that of the CCA/Fe2+/H2O2 system, except that the ferryl radical feature at g ≈ 2.71 appeared as a markedly broader signal (Supplementary Fig. 2A). This broadening may indicate that the unpaired radical in the CPmn–Fe⁴⁺• complex resides in close proximity to the ferryl cation, consistent with dipolar or exchange interactions31,32.
The prevailing view on the origin of atmospheric oxygen centers on the evolution of photosynthesis. However, some studies have proposed the existence of limited amounts of molecular oxygen on early Earth before the emergence of oxygenic photosynthesis15,33. Abiotic processes such as paleo-geochemical reactions and photochemical pathways are suggested as potential sources of free oxygen14,34,35. Recent studies have demonstrated plausible prebiotic sources of H2O2 and O2, including mineral-mediated production11, silicate surface-water vapor reactions36, geodynamic oxidation of Archean surfaces12, and photochemical pathways proposed to precede oxygenic photosynthesis5,9,10. These findings collectively indicate that early life would have been exposed to low but non-negligible fluxes of reactive oxygen species (ROS), making the management and detoxification of H2O2 a critical biochemical challenge. Among these, the oxidative decomposition of H2O2 is considered a plausible route, producing small amounts of O2 as a by-product. Accordingly, H2O2 was proposed as one of the possible transitional electron donors in the evolution of oxygenic photosynthesis5,9. Blankenship and Hartman hypothesized that H2O2 may have served as an early electron donor for primitive photosystems, releasing O2 prior to the biological oxidation of water10. They further developed models that the evolution of oxygenic photosynthesis required both the development of charge accumulation capacity and the emergence of highly oxidizing reaction center pigments.
In this study, we demonstrate that there may be biologically based free oxygen generated beyond the LUCA. Both native nucleic acids, including the full-length 23S rRNA (~2800 nucleotides) and a universally conserved tRNA 3’-terminus, CCA (a trinucleotide), and a mononucleotide-linked aminonucleoside CPmn, are capable of catalyzing the decomposition of H2O2 into molecular oxygen. Williams and coworkers have shown that the 23S rRNA can facilitate electron transfer in the presence of Fe2+ and low concentrations of H2O218, suggesting that RNA itself may function as a primitive charge-accumulating system. Here, both CCA and CPmn demonstrate the ability to bind Fe, presumably forming a tetradentate RNA–Fe complex that is structurally reminiscent of heme-Fe coordination. This RNA–Fe complex serves as an oxidizing center with sufficient redox potential to drive the oxidation of H2O2 to O2. Note that CPmn is not a natural or ancestral biomolecule; it is an aminonucleoside analog commonly used as an A-site mimic in ribosomal assays37. CPmn included here is not to imply evolutionary origin, but to provide a structural comparator for assessing whether Fe2+-mediated H2O2 oxidation depends on a phosphate-based RNA backbone.
The catalytic capability of RNA for H2O2 oxidation appears to originate from its flexible, yet rotameric backbone38. Adjacent phosphate oxygens within RNA act as Fe chelators and are proposed to form two ten-membered rings26 with a single Fe2+ ion, constituting a tetradentate geometry. This type of coordination is believed to predate LUCA and may represent a primitive metal-binding motif in early biochemical evolution19,26,39. Thus, the RNA-derived scaffolds may have contributed to prebiotic redox chemistry.
In contrast to the CCA system, which operates as a sustained catalytic cycle (Fig. 5F), the CPmn system behaves differently. Although CPmn can catalyze H2O2 oxidation, its activity appears to be consistent with a “suicide ribozyme” mechanism, in which catalysis is coupled to self-degradation (Supplementary Fig. 2A, B). This mechanistic divergence indicates that while both phosphate- and peptide-linked scaffolds of RNAs are capable of redox catalysis, only the phosphate-based RNA backbone provides the stability required to sustain oxidative function. Such chemical resilience may possibly have driven the evolutionary selection for RNA over peptide-like analogs as the primary catalytic and informational polymer40.
Under simulated anoxic early Earth conditions, RNA–Fe complexes catalyze the oxidation of H2O2, producing free oxygen as the byproduct. This O2 could, in turn, oxidize soluble Fe²⁺ to Fe³⁺, introducing the challenge of iron insolubility for life during early evolution41. In our EPR experiments, an overnight reaction of the CCA/Fe2+/H2O2 system led to the oxidation of Fe2+ to Fe3+ (Fig. 5F), likely due to O2 generated during H2O2 decomposition. Interestingly, the reaction mixtures remained transparent, with no visible Fe3+ precipitate, suggesting that the RNA–Fe complex may help stabilize Fe3+ in solution, thus mitigating the challenge of iron insolubility faced by early life.
However, the RNA-mediated production of biologically derived oxygen may not primarily be the driver for the oxygenated Earth. Rather, such chemistry could have conferred selective advantages. In particular, RNA’s ability to both generate and tolerate oxidative conditions may have contributed to its evolutionary persistence by offering protection against reactive oxygen species.
Methods
RNA and chemical preparation
The 23S rRNA from Thermus thermophilus HB8 was generated by in vitro transcription using a previously described pUC19 construct containing the full-length 23S rRNA sequence (pUC19/23STT)42. The pUC19/23STT plasmid was linearized with HindIII-HF (New England Biolabs) and purified using a commercial DNA purification kit (Zymoclean Gel DNA Recovery Kit, Zymo Research). Linearized template DNA (1.0 µg) was transcribed using the MEGAscript Transcription Kit (Thermo Fisher Scientific) in 20 µL reaction volumes at 37 °C for 4 h. The resulting RNA was recovered by ammonium acetate precipitation and resuspended in nuclease-free water. Transcript integrity and purity were verified by electrophoresis on a 4% denaturing 8 M urea PAGE and visualized using SYBR Green II (Lonza) RNA gel stain under 254-nm UV illumination.
C-puromycin (CPmn) was purchased from Abcam Ltd. (UK), and puromycin (Pmn) from Dharmacon, Inc. (USA). The RNA oligomers CCA and CA were obtained from KriSan Biotech Co., Ltd. (Taiwan); they were HPLC-purified and characterized by mass spectrometry. All chemical solutions were freshly prepared in an anaerobic chamber (Coy). Deionized, nuclease-free water was deoxygenated using argon gas.
All RNAs were pre-treated with Chelex 100 resin (Bio-RAD) to remove trace metal ions. The mixtures were incubated at 60 °C for 1 min, recovered using a 0.22 μm Ultrafree-MC Centricon (Millipore), lyophilized, and transferred into the anaerobic chamber for subsequent use.
Modified “blue bottle” assay
A modified “blue bottle” assay25 was used to monitor RNA-mediated H2O2 decomposition under anoxic conditions. In this assay, leucomethylene blue, a colorless reduced dye, is oxidized by O2 to blue methylene blue. Glucose under alkaline conditions regenerates leucomethylene, allowing continuous monitoring of O2 production via redox cycling.
The reduced dye solution was freshly prepared by mixing 250 mM NaOH, 4 mM glucose, and 0.4 mM methylene blue. Reactions were assembled in an order where RNA was first mixed with Fe2+ at 25 °C, followed by the addition of H2O2. Reactions were carried out in 50 mM HEPES-Tris buffer (pH 8.0) containing 6 μM FeSO4·7H2O and 500 mM deoxygenated H2O2, with a total volume of 500 μL. RNA concentrations per reaction were as follows: 200 pmol for CPmn and 23S rRNA, 400 pmol for CCA and CA, and 800 pmol for Pmn.
The dye was put in a 100 μL volume cuvette sealed with a cap and connected via tubing to the reaction tube (Supplementary Fig. 1). UV–Vis spectra were recorded at 664 nm or across the full spectrum (200–1000 nm) using an Ocean Optics USB 2000 + XR1-ES spectrometer inside the anaerobic chamber. Cuvettes were gently vortexed for one minute intermittently every four minutes of recording during the assay.
Continuous variation analysis
To examine RNA–Fe2+ complex formation, continuous variation experiments were performed using RNA and Fe2+ at a constant total concentration of 40 μM for CCA and Pmn, and 4 μM for CPmn. The molar ratios of RNA to Fe2+ were varied between 0 and 1. Samples were prepared in 50 mM Tris-Cl buffer (pH 8.0) and incubated at 25 °C for 2 h (CCA and Pmn) or 30 min (CPmn), followed by storage at 4 °C. Absorbance at 260 nm was measured at 25 °C using a Nanodrop spectrometer (Thermo) inside the anaerobic chamber. Each measurement was repeated five times.
Data analysis for continuous variation
Absorbance data were normalized as OD260 per nucleotide and plotted against the mole fraction of RNA or Fe2+. Outliers were excluded if they deviated by 1.5 standard deviations (1.5 σ) from the mean. The point of discontinuity, indicating complex stoichiometry, was determined from the intersection of two fitted linear regressions.
Michaelis–Menten kinetics
Chelex-treated CPmn was first resuspended in 50 mM HEPES-Tris buffer (pH 8.0) containing 6 μM FeSO4·7H2O. The initial reaction rate was determined by the addition of deoxygenated H2O2 at varying final concentrations: 56, 83, 125, 250, 278, 357, 500, 833, and 1250 mM. Oxygen production was monitored via the increase in absorbance at 664 nm, corresponding to the formation of methylene blue (ε = 7.4 × 104 M−1cm−1)25,43. All kinetic measurements were performed inside the anaerobic chamber.
Kinetic data analysis
Initial velocities were plotted against H2O2 concentrations and fitted using non-linear regression based on the Michaelis–Menten equation. Curve fitting and extraction of Vmax and Km values were conducted using JMP Student Edition 18 (SAS Institute Inc.).
EPR experiments
Sample mixtures were freshly prepared by first mixing Chelex-treated CPmn or CCA with Fe2+, followed by the addition of H2O2. Reactions were constituted in 50 mM HEPES-Tris buffer (pH 8.0) containing CPmn (8 μM) or CCA (16 μM), FeSO4·7H2O (120 μM), and H2O2 (500 mM). Samples were incubated on ice for the indicated reaction times prior to measurement.
Continuous wave (CW) EPR spectra were recorded at 77 K on a Bruker EMX X-band spectrometer operating at 9.5 GHz. Instrument settings were as follows: microwave power, 20 mW; modulation frequency, 100 kHz; and modulation amplitude, 10 G.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated and analyzed in this study are included in this published article and its Supplementary Information. Primary datasets supporting the main findings are provided as Supplementary Data files: Supplementary Data 1 (Fig. 3), Supplementary Data 2 and 3 (Fig. 4), and Supplementary Data 4 (Fig. 5).
References
Moody, E. R. R. et al. The nature of the last universal common ancestor and its impact on the early Earth system. Nat. Ecol. Evol. 8, 1654–1666 (2024).
Schopf, J. W., Kudryavtsev, A. B., Agresti, D. G., Wdowiak, T. J. & Czaja, A. D. Laser-Raman imagery of Earth’s earliest fossils. Nature 416, 73–76 (2002).
Holland, H. D. The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. B: Biol. Sci. 361, 903–915 (2006).
Anbar, A. D. Elements and evolution. Science 322, 1481–1483 (2008).
Olson, J. M. & Blankenship, R. E. Thinking about the evolution of photosynthesis. Photosynth Res. 80, 373–386 (2004).
Sessions, A. L., Doughty, D. M., Welander, P. V., Summons, R. E. & Newman, D. K. The continuing puzzle of the great oxidation event. Curr. Biol. 19, R567–R574 (2009).
Awramik, S. M. The oldest records of photosynthesis. Photosynth Res. 33, 75–89 (1992).
Ueno, Y., Ono, S., Rumble, D. & Maruyama, S. Quadruple sulfur isotope analysis of ca. 3.5Ga Dresser Formation: new evidence for microbial sulfate reduction in the early Archean. Geochim. Cosmochim. Acta 72, 5675–5691 (2008).
Olson, J. M. Evolution of photosynthesis. Science 168, 438 (1970).
Blankenship, R. E. & Hartman, H. The origin and evolution of oxygenic photosynthesis. Trends Biochem. Sci. 23, 94–97 (1998).
He, H. et al. A mineral-based origin of Earth’s initial hydrogen peroxide and molecular oxygen. Proc. Natl. Acad. Sci. USA 120, e2221984120 (2023).
Wu, X. et al. Geodynamic oxidation of Archean terrestrial surfaces. Commun. Earth Environ. 4, 132 (2023).
Slesak, I., Slesak, H. & Kruk, J. Oxygen and hydrogen peroxide in the early evolution of life on earth: in silico comparative analysis of biochemical pathways. Astrobiology. 12, 775–784 (2012).
Stone, J., Edgar, J. O., Gould, J. A. & Telling, J. Tectonically-driven oxidant production in the hot biosphere. Nat. Commun. 13, 4529 (2022).
Schwartz, R. M. & Dayhoff, M. O. Origins of prokaryotes, eukaryotes, mitochondria, and chloroplasts. Science 199, 395–403 (1978).
Weiss, M. C. et al. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 1, 16116 (2016).
Bray, M. S. et al. Multiple prebiotic metals mediate translation. Proc. Natl. Acad. Sci. USA 115, 12164 (2018).
Hsiao, C. et al. RNA with iron(II) as a cofactor catalyses electron transfer. Nat. Chem. 5, 525–528 (2013).
Lin, S.-Y., Wang, Y.-C. & Hsiao, C. Prebiotic iron originates the peptidyl transfer origin. Mol. Biol. Evol. 36, 999–1007 (2019).
Cotruvo, J. A. & Stubbe, J. Class I ribonucleotide reductases: metallocofactor assembly and repair in vitro and in vivo. Annu. Rev. Biochem. 80, 733–767 (2011).
Poole, A. M., Logan, D. T. & Sjöberg, B.-M. The evolution of the ribonucleotide reductases: much ado about oxygen. J. Mol. Evol. 55, 180–196 (2002).
López, M. B., Oterino, M. B. & González, J. M. In Macromolecular Protein Complexes V: Structure and Function (eds Robin Harris, J. & Jon Marles-Wright) 33–47 (Springer International Publishing, 2024).
Zámocký, M., Gasselhuber, B., Furtmüller, P. G. & Obinger, C. Turning points in the evolution of peroxidase-catalase superfamily: molecular phylogeny of hybrid heme peroxidases. Cell Mol. Life Sci. 71, 4681–4696 (2014).
Hsiao, C. et al. In Nucleic Acid Metal Ion Interactions (ed. Hud, N.) 1–35 (The Royal Society of Chemistry, 2008).
Limpanuparb, T., Areekul, C., Montriwat, P. & Rajchakit, U. Blue bottle experiment: learning chemistry without knowing the chemicals. J. Chem. Educ. 94, 730–737 (2017).
Hsiao, C. & Williams, L. D. A recurrent magnesium-binding motif provides a framework for the ribosomal peptidyl transferase center. Nucleic Acids Res. 37, 3134–3142 (2009).
Petrov, A. S. et al. History of the ribosome and the origin of translation. Proc. Natl. Acad. Sci. USA 112, 15396–15401 (2015).
Fox, G. E. Origin and evolution of the ribosome. Cold Spring Harb. Perspect. Biol. 2, a003483 (2010).
Cantor, C. & Schimmel, P. Biophysical Chemistry (I-III) (Academic Press, 1984).
Job, P. Studies on the formation of complex minerals in solution and on their stability. Annal. De. Chim. Fr. 9, 113–203 (1928).
Bandarian, V. & Reed, G. H. Hydrazine cation radical in the active site of ethanolamine ammonia-lyase: mechanism-based inactivation by hydroxyethylhydrazine. Biochemistry 38, 12394–12402 (1999).
Equbal, A. et al. Role of electron spin dynamics and coupling network in designing dynamic nuclear polarization. Prog. Nucl. Magn. Reson. Spectrosc. 126-127, 1–16 (2021).
Cloud, P. Working model of primitive Earth. Am. J. Sci. 272, 537–548 (1972).
He, H. et al. An abiotic source of Archean hydrogen peroxide and oxygen that pre-dates oxygenic photosynthesis. Nat. Commun. 12, 6611 (2021).
Balk, M. et al. Oxidation of water to hydrogen peroxide at the rock–water interface due to stress-activated electric currents in rocks. Earth Planet. Sci. Lett. 283, 87–92 (2009).
Xia, Y. et al. Contact between water vapor and silicate surface causes abiotic formation of reactive oxygen species in an anoxic atmosphere. Proc. Natl. Acad. Sci. USA 120, e2302014120 (2023).
Schmeing, T. M. et al. A pre-translocational intermediate in protein synthesis observed in crystals of enzymatically active 50S subunits. Nat. Struct. Biol. 9, 225–230 (2002).
Murray, L. J., Arendall, W. B. 3rd, Richardson, D. C. & Richardson, J. S. RNA backbone is rotameric. Proc. Natl. Acad. Sci. USA 100, 13904–13909 (2003).
Petrov, A. S., Bowman, J. C., Harvey, S. C. & Williams, L. D. Bidentate RNA-magnesium clamps: on the origin of the special role of magnesium in RNA folding. Rna 17, 291–297 (2011).
Fried, S. D., Fujishima, K., Makarov, M., Cherepashuk, I. & Hlouchova, K. Peptides before and during the nucleotide world: an origins story emphasizing cooperation between proteins and nucleic acids. J. R. Soc. Interface 19, 20210641 (2022).
Sánchez, M., Sabio, L., Gálvez, N., Capdevila, M. & Dominguez-Vera, J. M. Iron chemistry at the service of life. IUBMB Life 69, 382–388 (2017).
Athavale, S. S. et al. Domain III of the T. thermophilus 23S rRNA folds independently to a near-native state. Rna 18, 752–758 (2012).
Baranovskii, S. F., Bolotin, P. A. & Evstigneev, M. P. Aggregation of 1,3,7-trimethylxanthine with methylene blue in aqueous solution. J. Appl. Spectrosc. 73, 171–177 (2006).
Acknowledgements
The authors thank Drs. Steve Sheng-Fa Yu and Ivan Tsai (Institute of Chemistry, Academia Sinica, Taiwan) for training and access to the EPR facility. We also acknowledge Drs. S. Yu, I. Tsai, J. Lin, and C. Chang for helpful discussions. This work is supported by the National Science and Technology Council (NSCT-112-2311-B-002-010).
Author information
Authors and Affiliations
Contributions
Y.C.W., J.H.T., L.C.Y., and C.H. conceived and designed the study. Y.C.W. and J.H.T. performed the experiments and analyzed the data. Y.C.W. and L.C.Y. provided key materials. C.H. supervised the project. Y.C.W., J.H.T., L.C.Y., and C.H. co-wrote the manuscript. All authors discussed the results and contributed to the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, YC., Tu, JH., Yu, LC. et al. RNA−Iron complexes catalyse prebiotic oxygen generation. Commun Chem 9, 124 (2026). https://doi.org/10.1038/s42004-026-01935-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-026-01935-6
