
Abstract
The Cd concentrations and isotope compositions of open ocean surface waters are generally thought to be governed by internal cycling, and particularly by the balance between upwelling of more Cd-rich deeper water masses and Cd uptake by phytoplankton. Here we present a new dataset of coupled Cd isotope compositions and concentrations for seawater depth profiles sampled in the western tropical Atlantic Ocean during Leg 2 of the GEOTRACES GA02 section. A box model for the Cd source and sink fluxes of the oligotrophic surface waters of the study area shows that the observed light Cd isotope compositions and low Cd concentrations are a consequence of biological Cd uptake and atmospheric deposition of isotopically light anthropogenic Cd. Aerosols enriched in anthropogenic Cd thereby contributed at least 19%, and possibly more than 45%, to the dissolved surface water Cd inventory during the sampling period. This reveals that anthropogenic emissions of volatile Cd can have a key impact on the distribution of Cd in open ocean surface waters.
Similar content being viewed by others

Introduction
Cadmium is a trace element with a marine distribution that is closely correlated with that of the nutrient PO43−, as both have low dissolved concentrations in surface waters and higher concentrations at depth1,2,3. The similar distributions suggest that Cd may be a marine micronutrient, such that the low surface water Cd concentrations are a consequence of biological uptake. The mechanism of Cd uptake remains unclear, but evidence suggests that Cd may substitute for Zn or Co in the enzyme carbonic anhydrase under Zn-limited conditions4. Some marine diatoms have a Cd-cofactored carbonic anhydrase, allowing them to substitute Zn or Co with Cd5,6. Alternatively, or in addition, non-specific uptake of Cd can occur if phytoplankton are unable to differentiate the element from other essential divalent metals5,6,7. Regardless of the exact mechanism, it is evident that in the surface ocean, there is net removal of isotopically light Cd by phytoplankton8,9, in agreement with culture experiments10,11. Closed-system Rayleigh fractionation has been used to describe the Cd isotope fractionation recorded in high nutrient low chlorophyll (HNLC) surface waters, where Cd is rapidly removed from the surface waters by biological uptake12,13,14,15. In contrast, open-system steady state conditions were employed to account for Cd isotope fractionations in regions where the removal of dissolved Cd from the surface ocean is balanced by inputs via the upwelling of deeper water masses13,16,17,18,19. Following uptake and incorporation into biomass, Cd is subsequently released into the dissolved phase through remineralization in oxyclines20. The regeneration of Cd and P is not always fully linked, leading to subsurface Cd:P maxima and distributions of particulate Cd that are not strictly coupled to PO43− or O221. Despite these complexities, biological uptake and remineralization remain key in shaping Cd and PO43− distributions in the ocean3,20.
In the deeper ocean, recent Cd isotope studies of seawater have provided new insights into the interplay between biological and physical processes in the biogeochemical cycling of Cd19,22,23. Cadmium has a long, but uncertain, oceanic residence time, estimated at between 2 \(\times\) 104 years24 and 3 \(\times\) 105 years25. This results in relatively uniform Cd isotope compositions for waters from depths >2000 m, with a mean δ114Cd value of 0.25 ± 0.04‰ (1 SD, n = 538; δ114Cd is the relative deviation of the 114Cd/110Cd ratio determined for a sample from the NIST SRM 3108 Cd isotope reference material in parts per 1000) for a global deep water dataset that excludes the North Atlantic13. The deep North Atlantic, which is dominated by North Atlantic Deep Water (NADW), has a slightly but significantly elevated δ114Cd of 0.43 ± 0.09‰ (1 SD, n = 215)8,22,26,27. This higher δ114Cd value most likely stems primarily from the shallow NADW source waters of Labrador Sea Water, Iceland-Scotland Outflow Water, and Denmark Strait Overflow Water, which are expected to be isotopically heavy from biological uptake of Cd near the surface8,19,28,29. In addition, the δ114Cd signature of NADW may reflect inputs from Antarctic Intermediate Water and Subtropical Mode Water, as the latter have relatively high δ114Cd values, which they can imprint on NADW as they move northward and mix8,9,19.
While processes within the ocean’s interior influence the marine Cd distribution, external inputs, such as atmospheric deposition and riverine inputs, are unlikely to have a clear impact on basin-wide or global scale deep waters due to the long residence time of Cd in the ocean interior26,29,30. However, such inputs could affect Cd distributions in surface waters, where Cd concentrations are generally low and Cd has a much shorter residence time of months to years31,32. Sieber et al. 202313 recently inferred that the relatively light Cd isotope compositions (with δ114Cd ≈ 0‰) observed in low-Cd surface seawater samples from the equatorial Pacific Ocean along the GEOTRACES GP15 section reflect the combined effects of biological uptake, which lowers Cd concentrations, and atmospheric deposition, which supplies isotopically light Cd. This conclusion is supported by Cd data for leachates of 10 aerosol samples collected along the section, which are strongly enriched in Cd and characterized by δ114Cd values of −1.91 ± 0.39‰ to −0.07 ± 0.25‰. Such characteristics do not appear to be unusual, as aerosol samples collected along the GA03 section in the North Atlantic and the GA06 section in the Eastern Tropical Atlantic also featured high Cd contents, most likely from anthropogenic emissions, with δ114Cd values of between −0.54 ± 0.13‰ and 0.19 ± 0.21‰33,34. Notably, biological uptake of Cd in surface waters typically leaves the remaining dissolved Cd pool isotopically heavier, while the input and solubilization of anthropogenic Cd, which is often isotopically light, could balance this effect. This interplay between biological fractionation and external inputs may therefore provide an explanation for low Cd concentrations coupled with light Cd isotope signatures of surface waters.
As for riverine Cd, major rivers have variable Cd concentrations and, based on limited data, Cd isotope compositions that are influenced by local geological and biogeochemical settings, and anthropogenic inputs35. For instance, relatively pristine Siberian rivers exhibit Cd concentrations ranging from 0.02 to 0.06 nmol kg−1, with a mean δ114Cd of 0.2 ± 0.1‰, the boreal Kalix River in Sweden has a higher [Cd] of 0.24 nmol kg−1and a lower δ114Cd of −0.38 ± 0.10‰, whilst the acid mine drainage-affected Odiel and Tinto Rivers in southern Spain revealed variable Cd concentrations of between 230 and <1 nmol kg−1, depending on proximity to the estuary, but uniform δ114Cd values of about 0.02‰35,36. These findings highlight the heterogeneity of riverine Cd inputs, with variations in riverine Cd concentrations significantly exceeding those observed in surface seawater (1 \(\times\) 10−4 to 0.8 nmol kg−1)35. Such variable riverine Cd could locally influence Cd distributions and isotope compositions in coastal and adjacent surface waters, even if their impacts on the open ocean remain limited35.
Here, new coupled Cd isotope and concentration data are presented for seawater from four stations in the western tropical North Atlantic Ocean that were sampled during Leg 2 of GEOTRACES section GA02 (cruise PE321, 13 June to 4 July 2010) to investigate the processes that impact their distributions. The new results show that horizontal mixing plays a dominant role in shaping the distribution of Cd in deep waters within this hydrologically complex region, where water masses from the Northern and Southern Hemispheres converge. At thermocline and intermediate depths, the relative contributions of remineralization and mixing processes are evaluated to explain the variations in Cd concentrations and isotope compositions. Whilst the upwelling of Cd-rich deeper waters is generally thought to be the main source of Cd to the surface waters9,19, the light Cd isotope compositions found in the study area suggest that atmospheric sources, highly influenced by anthropogenic activities, strongly impacted the Cd inventory of the surface waters in this oligotrophic open ocean region.
Results
Hydrography
The hydrography of the GA02 Leg 2 section in the western North Atlantic Ocean is extensively described in previous publications3,37,38,39. In this study, we applied an adapted version of the extended optimum multiparameter (eOMP) model originally developed by Middag et al.3, which builds on the framework of Tomczak et al. (1981)40. Our adaptation incorporates five tracers—potential temperature, salinity, NO3−, Si and O2 (see Supplementary Note 1), and the effects of remineralization. The model results of the eOMP analysis indicate that the main water masses present in the region are North Atlantic Central Water (NACW), South Atlantic Central Water (SACW), AAIW, Upper Circumpolar Deep Water (UCDW), NADW, and Antarctic Bottom Water (AABW) (Figs.1 and 2, Supplementary Fig. 1 and Supplementary Note 1).

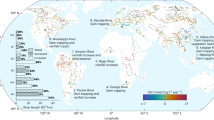
The map was produced using Ocean Data View74. The arrows indicate the predominant wind directions in the area.

Distributions of a Cd concentrations, b δ114Cd, c phosphate concentrations, and d oxygen concentrations, along the GA02 Leg 2 transect. The white contours represent different values of the variables. AABW Antarctic Bottom Water, AAIW Antarctic Intermediate Water, NACW North Atlantic Central Water, NADW North Atlantic Deep Water, UCDW Upper Circumpolar Deep Water. The map was produced using Ocean Data View74. The [Cd] and δ114Cd data are from this study, whilst the phosphate and oxygen concentrations are from the GEOTRACES Intermediate Data Product (IDP2021v2)48.
Cadmium concentrations and isotope compositions
The Cd concentration and isotope depth profiles for Stations 21, 30, 36, and 40 along the GA02 Leg 2 section display consistent patterns for both Cd concentrations and δ114Cd (Figs. 2 and 3; Table 1). In the top ~100 m, all profiles are characterized by low Cd levels and particularly low δ114Cd, with a subsurface δ114Cd maximum observed at between 101 and 200 m depth. In detail, the Cd concentrations of the surface waters in this study (with depths of about 25 to 100 m) range from 0.4 × 10–3 to 3.0 × 10−3 nmol kg−1, with isotopically lighter Cd (δ114Cd between −0.42 ± 0.37‰ and 0.37 ± 0.17‰, n = 8) compared to deeper waters (δ114Cd between 0.25 ± 0.02 and 0.63 ± 0.05, n = 38). Subsurface waters, particularly within the main thermocline and intermediate waters of NACW, SACW and AAIW/UCDW at depths of about 100 to 1000 m, exhibit Cd concentrations that generally increase with depth from 0.012 to 0.63 nmol kg−1 and show higher δ114Cd values of between 0.45 and 0.61‰. At depths exceeding ~1000 m, both Cd concentrations and δ114Cd align closely with water masses distribution. Northern-sourced water of NADW is characterized by Cd concentrations of 0.30 ± 0.05 nmol kg−1 and δ114Cd values of 0.41 ± 0.08‰ (2 SD, n = 14). In contrast, higher Cd concentrations of 0.61 ± 0.02 nmol kg−1 and lower δ114Cd of 0.27 ± 0.04‰ (2 SD, n = 2) are found at depth at the southernmost Station 40 within the AABW layer. The impact of the AABW layer is seen to a lesser extent at the northernmost stations (Figs. 2 and 3; Table 1).

Top row: Cd concentrations; middle row: δ114Cd, and bottom row: O2-def (see text for definition). Diamonds represent Station 21, squares represent Station 30, triangles represent Station 36, and inverted triangles represent Station 40. The black and blue symbols in the top and middle panels represent the measured data and the modeled [Cd]cal(mix) and δ114Cdcal(mix) values from eOMP-Monte Carlo model, respectively (see text for details). The gray shaded regions highlight the high oxygen deficiency zone associated with remineralization. The error bars for [Cd] and δ114Cd denote the 1sd and 2sd precision, respectively.
Discussion
As shown above, the GA02 seawater samples from Stations 21, 30, 36, and 40 along the western boundary of the North Atlantic encompass water masses from both the Northern and Southern Hemispheres (Figs. 1 and 2). The control of water mass mixing on deep water Cd concentrations and isotope compositions was described previously for the South Atlantic Ocean. For this ocean basin, the observed Cd isotope variations were shown to reflect the mixing proportions of NADW, AABW, and AAIW, highlighting the dominant role of large-scale ocean circulation in shaping Cd isotope distributions in the deep ocean19. The current study provides a particularly clear illustration of the systematic Cd isotope variations with latitude for the deep-water flow along the western boundary of the tropical North Atlantic, where water from the Northern Hemisphere moves southward and water from the Southern Hemisphere moves northward (Fig. 2). In addition to horizontal mixing of water masses, remineralization acts to release Cd from biological particles back into the dissolved form in the oxygen minimum layers of the western Atlantic Ocean3.
To quantify the impact of horizontal mixing and remineralization in the study area, the eOMP model was coupled with Monte Carlo simulations (Supplementary Note 1)3,41. The relative proportion of each water mass present was obtained from the eOMP model (Supplementary Figs. 1 and 2)3. Each water mass was thereby assigned an endmember Cd concentration and δ114Cd value, based on the best available data (Supplementary Table 1). For water masses formed in the North Atlantic, datasets from GA02 Leg 2 are used. For NACW, formed through winter subduction at the southern flank of the Gulf Stream42, the closest available Cd values are from GA02 Leg 2 Station 21 (31.7°N, 64.2°W). Formation of NADW is initiated by subduction of surface waters at high latitudes in the North Atlantic Ocean43. However, due to the lack of direct δ114Cd data for the formation region, the closest available Cd data are also from GA02 Leg 2 Station 21, which are used as endmember NADW Cd values. Datasets from the western South Atlantic cruise GA02 Leg 3 are employed to define the Cd endmember values of the South Atlantic-sourced water masses SACW, AAIW, UCDW, and AABW19. South Atlantic Central Water is formed in the region near the South American coast between 30°S and 45°S44, and its endmember composition is based on data for samples from GA02 Leg 3 Station 6 (40.0°S, 42.4°W)19. The formation of AAIW and AABW involves the subduction of HNLC surface waters in the Polar Frontal Zone and near the Antarctic coast, respectively. In contrast, UCDW originates from the subduction and northward advection of Circumpolar Deep Water, which upwells around Antarctica. For AAIW, UCDW, and AABW, the closest available endmember Cd values are from GA02 Leg 3 Station 2 (48.9°S, 48.7°W)19.
The Cd endmember values and the proportions of each water mass present at each location, as derived with the refined eOMP—Monte Carlo approach41 (Supplementary Note 1), were used to predict the Cd concentrations and isotope compositions based on water mass mixing alone for all samples below the surface water. The calculated results from the eOMP-Monte Carlo model, denoted as [Cd]cal(mix) and δ114Cdcal(mix) are compared with the measured data (denoted by the subscript “meas”) in Fig. 3 (see details in Supplementary Table 2). Overall, the model reproduces the observed Cd concentrations and δ114Cd values very well at depths exceeding 1002 m, confirming that conservative mixing dominates the Cd distribution at these depths as shown in Fig. 3. Similar to the results obtained using the original eOMP approach for the full GA02 section3, the modeled [Cd]cal(mix) values are significantly lower than the measured concentrations [Cd]meas for samples collected at depths that record a strong deficiency in dissolved oxygen concentrations, O2-def, with O2-def = [O2]cal(mix) – [O2]meas. Notably, at these depths the δ114Cdmeas and δ114Cdcal(mix) values are either identical within error or very similar (Fig. 3). The clear discrepancy between calculated Cd concentrations from water mass mixing and measured Cd concentrations only at depths with low dissolved oxygen concentrations, is indicative of remineralization of organic particles, which release dissolved Cd back into the ocean3,20.
The Cd concentration that is contributed by remineralization, [Cd]remin, can be determined from [Cd]meas and [Cd]cal(mix):
Calculations that apply Eq. (1) indicate that the concentration of remineralized Cd ranges from 0.03 to 0.31 nmol kg−1, contributing 22–86% ([Cd]remin/[Cd]meas) of the total measured Cd inventories for samples where mixing calculations indicate an oxygen deficiency (i.e., high O2-def; Supplementary Table 2). This demonstrates that remineralization is an important source of dissolved Cd in the study area at depths with O2-def values higher than 100 μmol kg−1 (Fig. 3). The observation that conservative water mass mixing alone appears sufficient to account for the observed δ114Cdmeas values suggests that the remineralized Cd had a similar isotope composition to that contributed by conservative mixing. This inference is explored below.
As δ114Cd data for biological particles are not available for the GA02 section, the remineralized δ114Cd, denoted as δ114Cdremin, is best estimated based on an open-system steady state model for the upper Atlantic Ocean. The data available for North Atlantic thermocline and intermediate waters of this and previous studies are thereby in excellent accord with a steady state model, if this applies an initial Cd concentration of 0.6 nmol kg−1, an initial δ114Cd 0.45‰, coupled with a biomass-seawater Cd isotope fractionation factor αbio/sw = Rbiomass/Rseawater of 0.9998, for R = 114Cd/110Cd (Supplementary Note 2 and Supplementary Fig. 3)17,19,22. The model results in a δ114Cd value of 0.45‰ for biological particles when more than 99% of the dissolved Cd is removed from seawater by biological uptake. Whilst the estimate of δ114Cdremin ≈ 0.45‰ exceeds the only directly measured particulate δ114Cd values for the North Atlantic (0.11 ± 0.31‰, 2 SD, n = 5), the difference is minor considering the measurement uncertainties8.
Following the addition of remineralized Cd with δ114Cdremin = 0.45‰, the Cd isotope composition that is expected for a water mass mixture that was also affected by subsequent remineralization, δ114Cdcal(mix + remin), can be calculated as:
After accounting for remineralization, the calculated δ114Cdcal(mix + remin) values deviate only slightly from the measured δ114Cdmeas results, typically by less than –0.11‰ (Fig. 4, Supplementary Table 2). This shows that due to the limited extent of biological isotope fractionation, remineralization had only a minor impact on the Cd isotope signatures of the sampled subsurface waters in the western tropical Atlantic. This conclusion is in accord with the results of Guinoiseau et al.17, who used a similar approach to estimate a δ114Cdremin of 0.36‰ to 0.56‰ for the eastern South Atlantic Ocean, and this altered the δ114Cd signatures of remineralization-affected waters by less than 0.08‰ when remineralization contributed 30% to the Cd inventory.
![Fig. 4: Measured δ114Cdmeas, modeled δ114Cdcal(mix), and calculated δ114Cdcal(mix+remin)plotted against measured [Cd].](http://media.springernature.com/lw685/springer-static/image/art%3A10.1038%2Fs43247-026-03211-w/MediaObjects/43247_2026_3211_Fig4_HTML.png?as=webp)
Measured δ114Cdmeas data are represented by full colored circles with black borders, modeled δ114Cdcal(mix) values based on water mass mixing from the eOMP-Monte Carlo model are shown as open circles with colored borders, whilst the calculated δ114Cdcal(mix + remin) values from water mass mixing + remineralization (see Eq. 2 in main text) are denoted by crosses with colored borders. The errorbars for δ114Cd are 2sd. The symbol colors represent the dissolved oxygen deficiency, O2-def (see text for definition). Only data for samples from depths between 101 and 1002 m are plotted in the figure. The data sources are provided in Supplementary Table 2. The errorbars for δ114Cd denote the 2sd precision.
Overall, the new results confirm earlier findings that remineralization had a major impact on Cd concentrations of intermediate depth waters that record a strong deficiency in dissolved oxygen concentrations3. However, the new δ114Cd data reveal that this process has only a minor influence on δ114Cd, as the dissolved and particulate Cd inventories had similar Cd isotope compositions. For deep waters, water mass mixing alone is sufficient to explain both Cd concentrations and isotope compositions, in accord with the results of previous studies of the Atlantic and other ocean basins15,16,28,45.
Along the GA02 Leg 2 section, the surface waters from depths of about 20 to 100 m have low Cd concentrations of 0.4 × 10–3 to 3.0 × 10–3 nmol kg−1, coupled with relatively low δ114Cd values of −0.42 to 0.37‰ (n = 8) compared to the deeper waters, which have a δ114Cd of between 0.40 and 0.60‰ (Fig. 5 and Table 1). Notably, both the Cd concentrations and δ114Cd values of the surface seawater samples are at the lower end of the ranges reported for Atlantic Ocean surface waters (Fig. 5). The surface water δ114Cd values of our samples are, furthermore, at the lower end of the global range, and the lowest values coincide with the lowest Cd concentrations, although the relationship is not statistically significant given the analytical uncertainties8,9,14. Literature observations, in contrast, suggest that preferential uptake of lighter Cd isotopes by phytoplankton at open-system steady state or closed-system Rayleigh conditions should generate low dissolved Cd concentrations coupled with δ114Cd values that are either nearly unfractionated (0.50–0.60‰) or clearly higher (>1‰), respectively, compared to upwelling deeper waters with typical δ114Cd of 0.40–0.50‰ (Supplementary Note 2; Supplementary Figs. 3 and 4)9,12,14. In the following, possible origins for the unusual surface water trend of low δ114Cd at low Cd concentrations (Fig. 5) are explored and discussed.

Four results for North Atlantic samples with δ114Cd > 2.0‰ at low [Cd] are cut off but shown in Supplementary Fig. 4 with an expanded y-axis. Orange, blue and purple symbols represent samples of this study from surface waters (<101 m), thermocline and intermediate water masses (102 – 1002 m), and deep water masses (>1002 m), respectively; these samples were collected at Stations 21 (diamonds), 30 (squares), 36 (triangles), and 40 (inverted triangles). The error bars for δ114Cd denote the 2sd precision. The gray symbols denote literature data for different regions of the Atlantic Ocean: Tropical Atlantic22, North Atlantic8, western South Atlantic19, and eastern South Atlantic17.
First, it is conceivable that analytical artefacts are responsible for the low observed δ114Cd of the surface waters, given the very low Cd masses (of ≤1 ng per sample) that were available for the isotopic analyses of these samples. Previous multiple-collector inductively coupled plasma mass spectrometry (MC-ICP-MS) studies that reported high δ114Cd values of up to 5‰ for surface waters have been questioned due to potential spectral interferences18,46. In this study, a new chemical procedure effectively removed elements that can cause spectral interference (Ge, Ga, Zr, Mo, In and Sn)47, ensuring unbiased δ114Cd results, even for low Cd concentrations (0.4 \(\times\) 10–3 – 3.0 \(\times\) 10–3 nmol kg−1). Our results also align well with previous δ114Cd values determined for surface seawater samples from the Tropical Atlantic22 and eastern South Atlantic Ocean17 (range: −0.61 ± 0.11 to 0.06 ± 0.22‰) by thermal ionization mass spectrometry (TIMS), and from the equatorial Pacific Ocean by MC-ICP-MS13. This indicates that natural processes are likely responsible for δ114Cd values observed in the surface seawater samples of this study.
Internal marine processes, such as Cd uptake by phytoplankton in Zn-depleted environments and Cd complexation by organic ligands, can only explain δ114Cd values as low as about 0.40‰ for eastern and western South Atlantic Ocean17,19, but not the much lower values observed for the western tropical Atlantic Ocean in this study and for the equatorial Pacific in a previous investigation13. Since internal processes are unlikely to be responsible, external inputs from either rivers or atmospheric deposition are considered in the following.
Despite the proximity of the study area to the Amazon River, our samples were collected outside of the main Amazon plume, where salinity exceeds 35.5, indicating minimal riverine influence (<2%). Moreover, Cd concentrations within the plume are not systematically elevated (Supplementary Fig. 5)48, suggesting no clear riverine impact on Cd distribution, consistent with previous studies22,49.
Given the limited riverine contribution, aerosol deposition emerges as the key external source that could affect the surface water δ114Cd values. Although previous studies have suggested that atmospheric deposition of natural Cd is negligible compared to the Cd upwelling fluxes in the Tropical Atlantic Ocean, where strong equatorial upwelling dominates Cd supply22, aerosol-derived inputs may still play a role in regions outside the main upwelling zone. Recent evidence furthermore indicates that anthropogenic Cd emissions can readily overwhelm natural atmospheric Cd inputs, with urban and marine aerosols showing strong enrichments of Cd coupled with δ114Cd < 0.0‰33,34,50,51. Furthermore, Sieber et al.13 proposed that the low δ114Cd (~0.0‰) of oligotrophic equatorial Pacific Ocean surface waters may originate from atmospheric deposition.
As such, atmospheric emissions enriched in anthropogenic Cd are evaluated in the following as a possible cause of the unusually low δ114Cd values observed in the surface waters of the GEOTRACES GA02 Leg 2 section in the western tropical North Atlantic.
As atmospheric fluxes to the surface ocean will increase dissolved Cd concentrations, the observed surface water trend of low δ114Cd with low Cd concentrations (Fig. 5), cannot be explained by aerosol deposition alone. Instead, it is more likely a consequence of the combined effects of atmospheric deposition of isotopically light Cd, which dominates the δ114Cd fingerprint, and biological Cd uptake, which leads to an overall reduction in dissolved Cd concentrations. A box model is used to quantitatively evaluate the surface water Cd systematics of the western tropical North Atlantic Ocean (Fig. 6), to show that atmospheric Cd deposition is indeed a key factor, as discussed below.

The black arrows represent Cd input fluxes to the surface waters, from atmospheric deposition (Fatm) and upwelling (Fup). The gray arrows represent Cd output fluxes from the surface waters, encompassing downwelling (Fdown) and particle sinking (Fsbp). The values of fatm and fsbp denote the Cd mass fractions provided by atmospheric deposition to the total Cd input flux and by sinking biological particles to the total Cd output flux, respectively. The [Cd] and δ114Cd data denote the Cd concentrations and isotope compositions of the fluxes or reservoirs.
For the surface water box model, the Cd input fluxes are from atmospheric deposition and water upwelling, the Cd output fluxes are ascribed to sinking biological particles and water downwelling (Fig. 6), and it is assumed that horizontal advection is not the major process that controls Cd cycling in the surface waters52. Considering a steady-state condition for Cd cycling within the box, the mass balance of Cd fluxes and their isotope compositions can be described by the following equations:
where Fatm and Fup represent the Cd input fluxes from atmospheric deposition and upwelling; Fdown and Fsbp are the Cd output fluxes from downwelling and sinking biological particles; δ114Cdatm and δ114Cdtcl refer to the Cd isotope compositions of atmospheric deposition and upwelled thermocline waters; and δ114Cdssw and δ114Cdsbp denote the Cd isotope compositions of the surface waters and sinking biological particles. The parameters fatm and fsbp are employed below to represent the mass fractions of Cd delivered by atmospheric deposition relative to the total Cd input flux and removed by sinking biological particles relative to the total Cd output flux, respectively:
As the results of the current study suggest that biological uptake of Cd in the Atlantic mainly reflects open-system steady-state conditions (Supplementary Note 2 and Supplementary Fig. 3), the isotope fractionation factor ε114Cdbio-sw can be used to describe the biological fractionation of δ114Cd in the surface waters:
Equations (3) to (7) can furthermore be combined to arrive at the following equation (see detailed derivation in Supplementary Note 3):
The box model, based on Eq. (8), evaluates the impact of atmospheric Cd inputs on δ114Cdssw. The aim is to determine whether reasonable parameter values, and especially for δ114Cdatm, can account for the observed surface water Cd isotope compositions, with δ114Cdssw values of −0.42‰ to 0.37‰ (Table 1). Focusing first on the output fluxes, the biological isotope fractionation factor ε114Cdbio-sw has a simple relationship to the fractionation factor αbio/sw (Eq. 7)53. With an αbio/sw value of 0.9998, which is appropriate for many regions of the Atlantic Ocean (Supplementary Note 2 and Supplementary Fig. 3)8,17,19,22, this is equivalent to ε114Cdbio-sw = −0.2‰. The value of fsbp is poorly constrained and a wide range of values of between 0.1 and 0.9 are therefore considered.
Considering the Cd input fluxes, a value of 0.60‰ is applied for δ114Cdtcl, based on the results presented in this study (Fig. 3, Table 1). The fraction of atmospheric Cd deposition fatm, is constrained by the relative Cd fluxes to the surface waters from the atmospheric deposition and water mass upwelling (Eq. 5). As the western tropical North Atlantic is not a major upwelling zone54,55, an average global upwelling rate of 1 m year−1 is employed, based on a mean oceanic depth of 4000 m and an oceanic overturning time of 4000 years19. Along the GA02 Leg 2 section, the base of the thermocline has a mean [Cd] of ∼0.4 nmol kg−1; using a seawater density of 1025 kg m−3, Fup is hence 46 μg m−2 year−1 (see details in Supplementary Table 3).
The atmospheric Cd flux Fatm was estimated based on the yearly average dust deposition of 2.97 g m−2 year−1 in the study area during the GA02 Leg 2 cruise, as derived from the dissolved Al concentration of surface seawater56. For further calculations, the Cd concentration of the aerosol matter is varied from the natural geogenic “background” abundance, with up to 200-fold enrichments of the element, to explore the impact of aerosols that are significantly enriched in anthropogenic Cd. If the Cd in the dust originates only from natural sources, the average upper continental crust [Cd]ucc of 0.09 μg g−1 is a reasonable estimate for the dust, whilst anthropogenic Cd enrichments in the aerosols are quantified by an enrichment factor defined as EF = (Cd/Al)sample/(Cd/Al)ucc57. Whether atmospheric deposition can be responsible for the low observed δ114Cdssw values is explored using figures that summarize the modeling results in plots of δ114Cdssw versus δ114Cdatm for different EF values (Fig. 7). In detail, results are shown for EF values of 1, 50, 100, and 200, with Cd solubilities following aerosol deposition of 10% and 80% for Cd of natural and anthropogenic origin, respectively, in accord with published results33,58,59. This yields overall Cd solubilities of 10.0%, 78.6%, 79.3%, and 79.7% for EF values of 1, 50, 100, and 200, respectively. The corresponding Cd deposition rates range from 0.03 to 43 μg m−2 year−1 (Supplementary Table 3), with δ114Cdatm values of between −2.0 and 1.0‰13,51,60,61.

The panels show results assuming an isotope fractionation factor αbio/sw = 0.9998 (equilavent to ε114Cdbio-sw = −0.2‰) for atmospheric deposition with Cd enrichement factors of EF = 1 (a), 50 (b), 100 (c) and 200 (d), corresponding to Cd input flux mass fractions from atmospheric deposition, fatm, of between 0.06% and 48%. Results are shown for Cd output flux mass fractions to sinking biological particles, fsbp, of 0.1 to 0.9 (blue, orange, purple lines). The gray shaded boxes show the range of δ114Cdssw values measured for the surface waters in this study (between −0.42‰ and 0.37‰). The dashed lines denote the maximum (and for EF = 200 also the minimum) δ114Cdatm values of atmospheric deposition that are in accord with the δ114Cdssw data obtained for the surface water samples.
For EF = 1, corresponding to a Cd deposition rate of 0.03 μg m−2 year−1 and fatm = 0.06%, atmospheric Cd deposition is unable to account for the low observed surface water δ114Cdssw values of the tropical western Atlantic Ocean, for all considered δ114Cdatm values (Fig. 7a). When the aerosol Cd EF is increased to 50, equivalent to a Cd deposition rate of 11 μg m−2 year−1 and fatm = 19%, δ114Cdatm values of less than −0.76‰ can account for δ114Cdssw data that exceed 0.10‰ (Fig. 7b). With an aerosol Cd EF of 100, equivalent to a Cd deposition rate of 21 μg m−2 year−1 and fatm = 32%, δ114Cdatm values of less than—0.18‰ are sufficient to explain most of the δ114Cdssw results (>−0.20‰) obtained in this study (Fig. 7c). Finally, for EF = 200, corresponding to a Cd deposition rate of 43 μg m−2 year−1 and fatm = 48%, δ114Cdatm values that range from 0.09‰ to −1.58‰ can account for all δ114Cdssw results of this study (Fig. 7d).
The modeling hence shows that the low observed δ114Cdssw can only be explained if the aerosols are characterized by high EF values for Cd and low δ114Cdatm. The model was also tested with αbio/sw values of 0.9995–0.9997, in accord with previous Cd isotope studies of the Atlantic Ocean8,17,19, and such calculations produced data consistent with those obtained with αbio/sw = 0.9998 (Supplementary Fig. 6). The results of the modeling are discussed in the context of relevant available Cd EF and δ114Cdatm data for marine aerosols, to assess whether atmospheric deposition can be responsible for the low observed δ114Cdssw values in the western tropical North Atlantic.
Notably, Cd EF data determined for total digested and ammonium acetate leachates of aerosols collected during the GA02 Leg 2 section are fully in accord with the modeled results. In detail, the Cd EFs for the total aerosol digests, also calculated using [Cd]ucc = 0.09 μg g−1, range from 4 to 370, suggesting strong anthropogenic signatures (Supplementary Table 4)57. The observed variability, with some lower Cd EF values for aerosols collected in our study area, thereby does not question the modeling results. This reflects that surface seawater integrates over the Cd deposition received for several months as a consequence of the Cd residence time in the marine surface layer (0.18–0.35 years; Supplementary Note 4). As such, the δ114Cd of surface seawater is not expected to mirror the EF values of individual aerosol sampling events, but will reflect the cumulative effect of anthropogenically impacted air masses from the same broad source region (given the stable wind pattern of the area; Fig. 1) but with Cd EFs that will inevitably show some temporal variability. Furthermore, the aerosol leachates have high [Cd]/[Al] ratios of 187 to 10,513 μg g−1 (Supplementary Table 4) and which are thus approximately 150- to 10,000-fold higher than the upper continental crust [Cd]/[Al] ratio of 1.1 μg g−157. The analyses also support the conclusion that anthropogenic Cd is more soluble than mineral-bound Cd, as the aerosol leachates released at least 50%, and in many cases more than 80%, of the total Cd (Supplementary Table 4). The measured solubility is hence very similar to the maximum overall Cd solubility of ~80% assumed for aerosols in the modeling (Supplementary Tables 3 and 4). Considering samples from other Atlantic Ocean cruises, total digested aerosols from the zonal GA03 North Atlantic section showed Cd EFs of approximately 14 within the North African dust plume, but values exceeding 10,000 outside its reach34. Aerosol samples from near Bermuda in the western North Atlantic exhibited Cd EF values of 130–230 due to anthropogenic North American emissions50, whilst analyses of aerosols from the eastern tropical Atlantic (GEOTRACES cruise GA06) showed Cd EFs ranging from 4 to 107, with δ114Cd values of between –0.54 ± 0.13‰ and 0.19 ± 0.20‰33. Meanwhile, Pacific Ocean (GEOTRACES cruise GP15) aerosol samples exhibited higher EF values of approximately 200 to 10,000 for Cd, and displayed δ114Cd of −1.91 ± 0.39 to −0.07 ± 0.25‰13. The Cd EF values of 1–200 that are used in our box model calculations are therefore within the range of previously reported marine aerosol compositions.
The Cd present in aerosols is either of natural or anthropogenic origin. As Cd has a low boiling point (of 1040 K), it is readily emitted to the atmosphere by high-temperature anthropogenic activities, akin to Pb. Processes such as combustion of fossil fuels, metal smelting, and waste incineration hence contribute to the large anthropogenic emissions of Cd to the atmosphere61,62,63,64,65,66,67. Such anthropogenic emissions, and particularly emissions from waste incineration and lead smelters, exhibit a relatively large range of δ114Cd values, with published data ranging from −0.70 ± 0.13‰ to 0.40 ± 0.19‰66,68,69. In contrast, natural Cd in aerosols is expected to be less variable in δ114Cd, as the upper continental crust has a well-defined mean δ114Cd value of 0.03 ± 0.10‰60. The above findings on the δ114Cd values of natural and anthropogenic aerosols, hence, suggest that the latter commonly feature lower δ114Cd compared to natural dust, but further work is needed to ascertain whether this is indeed a general characteristic of Atlantic Ocean aerosols. Whilst the GA02 aerosol data of this study show no clear correlation between Cd EF and Cd solubility, further studies should also investigate previous findings, which suggest that anthropogenic Cd in atmospheric deposition may be up to 10 times more soluble than natural Cd, implying preferential dissolution of the former in marine surface waters33,58,59.
Taken together, the presented evidence hence suggests that atmospheric deposition of Cd from anthropogenic sources likely plays an important role in shaping the Cd concentrations and isotope compositions of the highly Cd-depleted North Atlantic surface waters. The box model developed in this study indicates that atmospheric inputs contributed at least 19% and possibly up to 48% of the surface water Cd inventory, for atmospheric deposition with Cd EF values of 50 to 200 (Fig. 7). Such atmospheric inputs have a clear impact on the Cd cycling in this open ocean setting, with a surface water Cd residence time of less than 0.35 years determined from the results of this study (Supplementary Note 4). However, further analyses of marine aerosols from this and other regions for both Cd concentrations and isotope compositions are needed to better constrain the impact of anthropogenic emissions on the surface water Cd systematics of the western tropical North Atlantic and to investigate whether such processes are also observable in other ocean basins.
In summary, this study provides a comprehensive analysis of variations in Cd concentrations and δ114Cd values determined for seawater samples from four depth profiles in the western tropical North Atlantic. For the deep waters of the study area, the mixing of water masses derived from the Northern and Southern Hemispheres determines the observed Cd concentrations and δ114Cd values. At intermediate depths, the latter parameters are controlled by both internal biogeochemical cycling and ocean circulation, whereby in-situ remineralization accounts for about 20–85% of the dissolved seawater Cd inventory in the low oxygen thermocline and intermediate waters. Viewed in the context of surface water data for the Atlantic and other ocean basins, surface water of the study area is characterized by low Cd concentrations of 0.4 \(\times\) 10−3 to 3.0 \(\times\) 10−3 nmol kg−1 (n = 8), as typically found in oligotrophic settings, as a consequence of dissolved Cd uptake by phytoplankton. However, these surface waters also have unusually low δ114Cd values of −0.42‰ to 0.37‰. A box model approach shows that the low observed δ114Cd values are most likely a consequence of atmospheric Cd inputs that are enriched in isotopically light Cd from anthropogenic emissions, and which overprint any isotope fractionations from biological uptake. Such emissions appear to contribute more than ~20% of the surface water Cd inventory in the tropical western Atlantic Ocean. Our results thus highlight that anthropogenic Cd emissions are sufficient to clearly impact Cd cycling in the surface Atlantic Ocean, particularly in oligotrophic open ocean settings with low Cd.
Methods
Sample collection and preparation
The seawater samples were collected from the western tropical North Atlantic at four stations between 13 June and 4 July 2010, during RV Pelagia cruise PE321 for GEOTRACES section GA02 Leg 2, from Bermuda to Fortaleza (Brazil). Seawater collection utilized an ultraclean all-titanium frame CTD trace metal sampling system with novel PVDF samplers70. Following collection, the samples were filtered through 0.2 μm membranes and acidified on board with distilled HCl to a pH of about 23.
Further sample processing took place in ISO Class 4 laminar flow hoods within an ISO Class 6 clean room in the MAGIC Laboratories at Imperial College London, UK. All water used for processing was of 18.2 M Ω cm grade from a Millipore purification system, whilst mineral acids were purified in-house by sub-boiling distillation in Teflon or quartz glass stills.
The separation of Cd from the seawater matrix followed previously described methods for seawater volumes of ≥1 L47. Briefly, 1 to 11 L seawater samples were spiked with a 111Cd–113Cd double spike solution, with detailed information on seawater sample volumes and the respective ratios of spike-derived to natural Cd provided in Supplementary Table 5. After the samples were left to stand for at least 7 days with daily shaking to ensure full equilibration of the spike-derived and seawater Cd, 1 ml of 0.05 M H2O2 was added to per liter of seawater to break down any remaining organic compounds. Following addition of 1 ml of 5 M NH4OAc per liter of seawater, a variable volume of 5 M aqueous NH3 solution was added to adjust the pH to 4.8 ± 0.2, depending on the initial seawater volume and pH. The samples were then equilibrated with Nobias Chelate PA-1 resin which adsorbs Cd and other trace metals from seawater at high yields at these conditions71. The resin was separated from the seawater in a filtration rig, whereupon the adsorbed trace metals, including Cd, were eluted from the resin using the same procedure that is used in the laboratory for the separation of seawater Pb72. The metal fraction was evaporated, refluxed in a mixture of 16 M HNO3 and ~10 M H2O2 to break down organic compounds released from the resin, dried again, and then redissolved in 2 M HCl for further purification with a 2-stage anion exchange column chromatography procedure (Supplementary Table 6). The purified Cd fractions were evaporated to dryness, dried down twice with a few drops of 16 M HNO3 and then redissolved in 0.1 M HNO3 for the mass spectrometric analyses.
Determination of Cd isotope compositions and concentrations
The Cd isotope measurements were performed following established procedures27,47,73 with a Nu Instruments Nu Plasma II MC-ICP-MS instrument, using a Cetac autosampler and either a Nu Instruments DSN-100 or a Cetac Aridus II desolvation system for sample introduction. Four cadmium isotopes, 111Cd, 112Cd, 113Cd, and 114Cd, were measured to determine the Cd isotope compositions. To correct for potential isobaric interferences from 112Sn, 113In, and 114Sn, the 115In and 117Sn ion beams were measured during the Cd isotope analysis. All ion beams were monitored with Faraday collectors equipped with 1011Ω resistors. The sensitivities for Cd in the measurements were 400–900 V μg−1 ml−1 using the DSN-100 and 600–1000 V μg−1 ml−1 with the Aridus II system.
The isotope measurements employed solutions with Cd concentrations of 10–60 ng ml−1, with lower concentrations of 2–5 ng ml-1 employed for the surface water samples with low [Cd] of 0.4 \(\times\) 10–3 to 3.0 \(\times\) 10–3 nmol kg−1 (Supplementary Table 5). The sample analyses were carried out interspersed between and relative to bracketing standard solutions that were prepared as mixtures of the NIST SRM 3108 Cd isotope reference material with the Cd double spike, with closely matched ratios of spike-derived to natural Cd (deviations less than 5%) and Cd concentrations (deviations less than 10%). The secondary isotope reference material BAM I012 Cd, as well as the in-house seawater quality control material SwQC, were analyzed using the same procedures to monitor data quality. Following the measurements, the reduction of the double spike data was carried out offline using an iterative approach, which corrects for both the monitored isobaric interferences and any laboratory- or instrument-induced mass discrimination27,47,73. The obtained Cd isotope compositions are expressed as δ114Cd values relative to NIST SRM 3108 Cd:
The δ114Cd values obtained in repeated analyses of BAM I012 Cd and the SwQC seawater sample are provided in Supplementary Table 7. The Cd concentrations were determined using the isotope dilution method, from the measured and mass bias-corrected 114Cd/111Cd ratios of the spiked sample solutions. The average Cd yield from the seawater samples was 80%. Total Cd blanks for the complete analytical procedure were 10 to 20 pg. As a result, the blanks typically increased the natural Cd concentrations of the samples by only 0.02–0.07%; the maximum blank contribution, for a surface water sample, was 0.42%. The blank contributions were subtracted from the measured Cd concentrations but not corrected for the Cd isotope data, as the Cd isotope composition of the blank is not well-defined, and estimated blank corrections were smaller than the analytical uncertainties of the data.
Given the challenging nature of the Cd isotope analyses, especially for the low Cd concentration samples from the surface waters, substantial analytical efforts were expended on quality control. These efforts, documented in detail in Supplementary Note 5 and Supplementary Figs. 7–9, encompass (i) a detailed investigation and evaluation of spectral interferences, (ii) multiple analyses of Cd standard solutions and the SwQC seawater sample using run solutions with Cd concentrations of between 2 ng ml−1 and 60 ng ml−1, and (iii) intercalibration of our [Cd] and δ114Cd depth profiles with published results for samples from the same locations.
Data availability
All source data supporting the findings of this study are provided in the tables in the main manuscript and Supplementary Information. In addition, these data are publicly available via figshare (https://doi.org/10.6084/m9.figshare.30958382).
Code availability
Code and results of the eOMP analysis are currently available upon request.
References
Boyle, E. A., Sclater, F. & Edmond, J. M. On the marine geochemistry of cadmium. Nature 263, 42–44 (1976).
Bruland, K. W., Knauer, G. A. & Martin, J. H. Cadmium in northeast Pacific waters 1. Limnol. Oceanogr. 23, 618–625 (2003).
Middag, R., van Heuven, S. M. A. C., Bruland, K. W. & de Baar, H. J. W. The relationship between cadmium and phosphate in the Atlantic Ocean unravelled. Earth Planet. Sci. Lett. 492, 79–88 (2018).
Price, N. M. & Morel, F. M. M. Cadmium and cobalt substitution for zinc in a marine diatom. Nature 344, 658–660 (1990).
Cullen, J. T., Lane, T. W., Morel, F. M. M. & Sherrell, R. M. Modulation of cadmium uptake in phytoplankton by seawater CO2 concentration. Nature 402, 165–167 (1999).
Lane, T. W. & Morel, F. M. A biological function for cadmium in marine diatoms. Proc. Natl. Acad. Sci. USA 97, 4627–4631 (2000).
Horner, T. J., Lee, R. B., Henderson, G. M. & Rickaby, R. E. Nonspecific uptake and homeostasis drive the oceanic cadmium cycle. Proc. Natl. Acad. Sci. USA 110, 2500–2505 (2013).
Conway, T. M. & John, S. G. Biogeochemical cycling of cadmium isotopes along a high-resolution section through the North Atlantic Ocean. Geochim.Cosmochim. Acta 148, 269–283 (2015).
Ripperger, S., Rehkämper, M., Porcelli, D. & Halliday, A. N. Cadmium isotope fractionation in seawater — A signature of biological activity. Earth Planet. Sci. Lett. 261, 670–684 (2007).
John, S. G. & Conway, T. M. A role for scavenging in the marine biogeochemical cycling of zinc and zinc isotopes. Earth Planet. Sci. Lett. 394, 159–167 (2014).
Lacan, F., Francois, R., Ji, Y. & Sherrell, R. M. Cadmium isotopic composition in the ocean. Geochim. Cosmochim. Acta 70, 5104–5118 (2006).
Abouchami, W. et al. Modulation of the Southern Ocean cadmium isotope signature by ocean circulation and primary productivity. Earth Planet. Sci. Lett. 305, 83–91 (2011).
Sieber, M. et al. Biological, physical, and atmospheric controls on the distribution of cadmium and its isotopes in the Pacific Ocean. Glob. Biogeochem. Cycles 37, e2022GB007441 (2023).
Xue, Z. et al. Cadmium isotope variations in the Southern Ocean. Earth Planet. Sci. Lett. 382, 161–172 (2013).
Zhang, R., Jensen, L. T., Fitzsimmons, J. N., Sherrell, R. M. & John, S. Dissolved cadmium and cadmium stable isotopes in the western Arctic Ocean. Geochim. Cosmochim. Acta 258, 258–273 (2019).
George, E., Stirling, C. H., Gault-Ringold, M., Ellwood, M. J. & Middag, R. Marine biogeochemical cycling of cadmium and cadmium isotopes in the extreme nutrient-depleted subtropical gyre of the South West Pacific Ocean. Earth Planet. Sci. Lett. 514, 84–95 (2019).
Guinoiseau, D. et al. Importance of cadmium sulfides for biogeochemical cycling of Cd and its isotopes in oxygen deficient zones—A case study of the Angola Basin. Glob. Biogeochem. Cycles 33, 1746–1763 (2019).
Janssen, D. J., Abouchami, W., Galer, S. J. G. & Cullen, J. T. Fine-scale spatial and interannual cadmium isotope variability in the subarctic northeast Pacific. Earth Planet. Sci. Lett. 472, 241–252 (2017).
Xie, R. C. et al. Non-Rayleigh control of upper-ocean Cd isotope fractionation in the western South Atlantic. Earth Planet. Sci. Lett. 471, 94–103 (2017).
de Souza, G. F., Vance, D., Sieber, M., Conway, T. M. & Little, S. H. Re-assessing the influence of particle-hosted sulphide precipitation on the marine cadmium cycle. Geochim. Cosmochim. Acta 322, 274–296 (2022).
Laubach, A. et al. Particulate cadmium accumulation in the Mesopelagic Ocean. Glob. Biogeochem. Cycles 39, e2024GB008281 (2025).
Xie, R. C., Galer, S. J. G., Abouchami, W. & Frank, M. Limited impact of eolian and riverine sources on the biogeochemical cycling of Cd in the tropical Atlantic. Chem. Geol. 511, 371–379 (2019).
Zheng, L., Minami, T., Takano, S., Ho, T. Y. & Sohrin, Y. Sectional distribution patterns of Cd, Ni, Zn, and Cu in the North Pacific Ocean: relationships to nutrients and importance of scavenging. Glob. Biogeochem. Cycles 35, e2020GB006558 (2021).
Martin J-M, Whitfield M. The significance of the river input of chemical elements to the ocean. in: (eds Wong CS, Boyle E, Bruland KW, Burton JD, Goldberg ED) Trace Metals in Sea Water (Springer, 1983).
Li, Y.-H. A brief discussion on the mean oceanic residence time of elements. Geochim. Cosmochim. Acta 46, 2671–2675 (1982).
Boyle, E. A. et al. GEOTRACES IC1 (BATS) contamination-prone trace element isotopes Cd, Fe, Pb, Zn, Cu, and Mo intercalibration. Limnol. Oceanogr. Methods 10, 653–665 (2012).
Xue, Z., Rehkamper, M., Schonbachler, M., Statham, P. J. & Coles, B. J. A new methodology for precise cadmium isotope analyses of seawater. Anal. Bioanal. Chem. 402, 883–893 (2012).
Abouchami, W. et al. Biogeochemical cycling of cadmium isotopes in the Southern Ocean along the Zero Meridian. Geochim. Cosmochim. Acta 127, 348–367 (2014).
Bruland, K. W. Oceanographic distributions of cadmium, zinc, nickel, and copper in the North Pacific. Earth Planet. Sci. Lett. 47, 176–198 (1980).
Duce, R. A. et al. The atmospheric input of trace species to the world ocean. Glob. Biogeochem. Cycles 5, 193–259 (2012).
Yang, S.-C., Lee, D.-C. & Ho, T.-Y. The isotopic composition of Cadmium in the water column of the South China Sea. Geochim. Cosmochim. Acta 98, 66–77 (2012).
Hayes, C. T. et al. Replacement times of a spectrum of elements in the North Atlantic based on thorium supply. Glob. Biogeochem. Cycles 32, 1294–1311 (2018).
Bridgestock, L. et al. The Cd isotope composition of atmospheric aerosols from the Tropical Atlantic Ocean. Geophys. Res. Lett. 44, 2932–2940 (2017).
Shelley, R. U., Morton, P. L. & Landing, W. M. Elemental ratios and enrichment factors in aerosols from the US-GEOTRACES North Atlantic transects. Deep Sea Res. Part II Top. Stud. Oceanogr. 116, 262–272 (2015).
Packman, H. et al. Tracing acid mine drainage and estuarine Zn attenuation using Cd and Zn isotopes. Geochim. Cosmochim. Acta 360, 36–56 (2023).
Lambelet, M. et al. Isotopic analysis of Cd in the mixing zone of Siberian rivers with the Arctic Ocean—New constraints on marine Cd cycling and the isotope composition of riverine Cd. Earth Planet. Sci. Lett. 361, 64–73 (2013).
Lambelet, M. et al. Neodymium isotopic composition and concentration in the western North Atlantic Ocean: Results from the GEOTRACES GA02 section. Geochim. Cosmochim. Acta 177, 1–29 (2016).
Middag, R. et al. Dissolved aluminium in the ocean conveyor of the West Atlantic Ocean: Effects of the biological cycle, scavenging, sediment resuspension and hydrography. Mar. Chem. 177, 69–86 (2015).
Rijkenberg, M. J. et al. The distribution of dissolved iron in the West Atlantic Ocean. PLoS ONE 9, e101323 (2014).
Tomczak, M. A multi-parameter extension of temperature/salinity diagram techniques for the analysis of non-isopycnal mixing. Prog. Oceanogr. 10, 147–171 (1981).
Olivelli, A. et al. Vertical transport of anthropogenic lead by reversible scavenging in the South Atlantic Ocean. Earth Planet. Sci. Lett. 646, 118980 (2024).
Klein, B. & Hogg, N. On the variability of 18 Degree Water formation as observed from moored instruments at 55°W. Deep Sea Res. Part I Oceanogr. Res. Pap. 43, 1777–1806 (1996).
Stramma, L. et al. Deep water changes at the western boundary of the subpolar North Atlantic during 1996 to 2001. Deep Sea Res. Part I Oceanogr. Res. Pap. 51, 1033–1056 (2004).
Kuhlbrodt, T. et al. On the driving processes of the Atlantic meridional overturning circulation. Rev. Geophy. 45, RG2001 (2007).
Gault-Ringold, M., Adu, T., Stirling, C. H., Frew, R. D. & Hunter, K. A. Anomalous biogeochemical behavior of cadmium in subantarctic surface waters: mechanistic constraints from cadmium isotopes. Earth Planet. Sci. Lett. 341-344, 94–103 (2012).
Sieber, M. et al. Physical and biogeochemical controls on the distribution of dissolved cadmium and its isotopes in the Southwest Pacific Ocean. Chem. Geol. 511, 494–509 (2019).
Huang, Y. et al. Efficient Cd separation protocols for high-precision cadmium isotope analyses of diverse samples by double spike MC-ICP-MS. Talanta 285, 127296 (2025).
Group GIDP. The GEOTRACES Intermediate Data Product 2021v2 (IDP2021v2) (NERC EDS British Oceanographic Data Centre NOC, 2023).
Boyle, E. A., Huested, S. S. & Grant, B. The chemical mass balance of the amazon plume—II. Copper, nickel, and cadmium. Deep Sea Res. Part A Oceanogr. Res. Pap. 29, 1355–1364 (1982).
Fishwick, M. P. et al. Impact of surface ocean conditions and aerosol provenance on the dissolution of aerosol manganese, cobalt, nickel and lead in seawater. Mar. Chem. 198, 28–43 (2018).
Rehkämper, M., Wombacher, F., Horner, T.J. & Xue, Z. Natural and anthropogenic Cd isotope variations. In Handbook of Environmental Isotope Geochemistry 125–154 (Springer, 2012).
Yang, S.-C., Zhang, J., Sohrin, Y. & Ho, T.-Y. Cadmium cycling in the water column of the Kuroshio-Oyashio Extension region: insights from dissolved and particulate isotopic composition. Geochim. Cosmochim. Acta 233, 66–80 (2018).
Hayes, J.M. An introduction to isotopic calculations, 2543 (Woods Hole Oceanographic Institution, 2004).
Liao, F., Liang, X., Li, Y. & Spall, M. Hidden upwelling systems associated with major western boundary currents. J. Geophys. Res. Oceans 127, e2021JC017649 (2022).
McClain, C. R. & Firestone, J. An investigation of Ekman upwelling in the North Atlantic. J. Geophys. Res. Oceans 98, 12327–12339 (2012).
Dulaquais, G. et al. Contrasting biogeochemical cycles of cobalt in the surface western Atlantic Ocean. Glob. Biogeochem. Cycles 28, 1387–1412 (2014).
Rudnick RL, Gao S. Composition of the continental crust. in: Treatise on Geochemistry (Elsevier, 2014).
Hsieh C-C, You C-F, Ho T-Y. The solubility and deposition flux of East Asian aerosol metals in the East China Sea: the effects of aeolian transport processes. Mar. Chem. 253, 104268 (2023).
Annibaldi, A., Truzzi, C., Illuminati, S., Bassotti, E. & Scarponi, G. Determination of water-soluble and insoluble (dilute-HCl-extractable) fractions of Cd, Pb and Cu in Antarctic aerosol by square wave anodic stripping voltammetry: distribution and summer seasonal evolution at Terra Nova Bay (Victoria Land). Anal. Bioanal. Chem. 387, 977–998 (2007).
Pickard, H. et al. The cadmium and zinc isotope compositions of the silicate Earth – Implications for terrestrial volatile accretion. Geochim. Cosmochim. Acta 338, 165–180 (2022).
Shiel, A. E., Weis, D. & Orians, K. J. Evaluation of zinc, cadmium and lead isotope fractionation during smelting and refining. Sci. Total Environ. 408, 2357–2368 (2010).
Cheng, K. et al. Atmospheric emission inventory of cadmium from anthropogenic sources. Int. J. Environ. Sci. Technol. 11, 605–616 (2013).
Nriagu, J. O. & Pacyna, J. M. Quantitative assessment of worldwide contamination of air, water and soils by trace metals. Nature 333, 134–139 (1988).
Pacyna, E. G. et al. Current and future emissions of selected heavy metals to the atmosphere from anthropogenic sources in Europe. Atmos. Environ. 41, 8557–8566 (2007).
Pacyna, J. M. & Pacyna, E. G. An assessment of global and regional emissions of trace metals to the atmosphere from anthropogenic sources worldwide. Environ. Rev. 9, 269–298 (2001).
Cloquet, C., Carignan, J., Libourel, G., Sterckeman, T. & Perdrix, E. Tracing source pollution in soils using cadmium and lead isotopes. Environ. Sci. Technol. 40, 2525–2530 (2006).
Gao, B. et al. Precise determination of cadmium and lead isotopic compositions in river sediments. Anal. Chim. Acta 612, 114–120 (2008).
Wang, P. et al. Apportionment of sources of heavy metals to agricultural soils using isotope fingerprints and multivariate statistical analyses. Environ. Pollut. 249, 208–216 (2019).
Wen, H. et al. Tracing sources of pollution in soils from the Jinding Pb–Zn mining district in China using cadmium and lead isotopes. Appl. Geochem. 52, 147–154 (2015).
Rijkenberg, M. J. et al. PRISTINE”, a new high volume sampler for ultraclean sampling of trace metals and isotopes. Mar. Chem. 177, 501–509 (2015).
Biller, D. V. & Bruland, K. W. Analysis of Mn, Fe, Co, Ni, Cu, Zn, Cd, and Pb in seawater using the Nobias-chelate PA1 resin and magnetic sector inductively coupled plasma mass spectrometry (ICP-MS). Mar. Chem. 130-131, 12–20 (2012).
Griffiths, A. et al. Evaluation of optimized procedures for high-precision lead isotope analyses of seawater by multiple collector inductively coupled plasma mass spectrometry. Anal. Chem. 92, 11232–11241 (2020).
Ripperger, S. & Rehkämper, M. Precise determination of cadmium isotope fractionation in seawater by double spike MC-ICPMS. Geochim. Cosmochim. Acta 71, 631–642 (2007).
Schlitzer, R. Ocean data view. https://odv.awi.de/ (2022).
Acknowledgements
The first author is supported by an Imperial-CSC PhD scholarship. We are grateful to the captain, crew and cruise participants of the PE321 cruise on RV Pelagia (GEOTRACES section GA02, Leg 2) for the collection of the seawater samples. Steven van Heuven is thanked for sharing his MATLAB code for the eOMP analysis. Greg de Souza and Dylan Rood are thanked for the informal reviews of the manuscript, which led to improvements. The GEOTRACES 2021 Intermediate Data Product version 2 (IDP2021v2) represents an international collaboration and is endorsed by the Scientific Committee on Oceanic Research (SCOR). The many researchers and funding agencies responsible for the collection of data and quality control are thanked for their contributions to the IDP2021v2.
Author information
Authors and Affiliations
Contributions
H.X. and M.R. conceptualized and designed the study. H.X. conducted the experiments, performed the data analyses, and prepared the initial manuscript draft. H.X., M.R., Y.H., and Z.J. contributed to the interpretation of the results. K.K. and B.J. supported laboratory and instrument maintenance. R.E.T.M. facilitated data collection, A.O. contributed to the eOMP-Monte Carlo analysis, and R.M. oversaw sample collection. A.B. and R.S. analyzed the aerosols from the GEOTRACES GA02 Leg 2 cruise. H.X., M.R., and T.v.d.F contributed most to later versions of the manuscript, with all authors supplying comments and suggestions.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Ruifang Xie and the other anonymous reviewer(s) for their contribution to the peer review of this work. Primary handling editors: Sophia Johannessen and Joe Aslin. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
43247_2026_3211_MOESM2_ESM.pdf (download PDF )
Supplementary Information for Anthropogenic emissions of volatile Cd detected in western tropical North Atlantic surface seawater
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, H., Rehkämper, M., Huang, Y. et al. Anthropogenic emissions of volatile Cd detected in western tropical North Atlantic surface seawater. Commun Earth Environ 7, 181 (2026). https://doi.org/10.1038/s43247-026-03211-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43247-026-03211-w
