
Abstract
Epigenetic drift is a key feature of aging and is associated with age-related diseases including cancer, yet the underlying molecular mechanisms remain unclear. Here, by analyzing DNA methylation and gene expression data from healthy and cancerous human colon samples, we identify an aging and colon cancer-associated DNA methylation (DNAm) drift. We find evidence that this drift is conserved in the mouse intestinal epithelium, where we demonstrate its origin within intestinal stem cells and identify its cell-intrinsic and non-mitotic characteristics, finding that its expansion is regulated via crypt clonality and fission. Mechanistically, we find that this drift is driven by age-related inflammation and reduced Wnt signaling, which dysregulate iron metabolism and impair TET activity. Despite CpG-level heterogeneity, we find that DNAm changes are consistent at the gene level, suggesting potential functionality. Our findings shed light on the epigenetic mechanisms of aging and provide a mechanistic basis for the hypermethylation observed in cancer.
Similar content being viewed by others


Main
A time-dependent change of epigenetic pattern during organismal aging is termed as epigenetic drift1. In recent years, several studies reported changes of the DNA methylation (DNAm) landscape through aging in different human tissues2,3. Some of these DNAm drifts have been used to derive epigenetic clocks such as the blood-based Hannum epigenetic clock, the multi-tissue Horvath’s epigenetic clock, or the mitotic-based EpiTOC2 to predict the age, the biological status and/or the number of cell division of a specific tissue or organism4,5,6. These aging-associated DNAm changes seem to be acquired in a stochastic manner, and the DNAm patterns at age-associated regions were found to be very heterogeneous7,8; however, it has been reported that there are age-related DNAm changes that are conserved across species and linked to developmental genes9. Stochastic epigenetic variation may represent the very early seeds for carcinogenesis, favoring clonal expansions, which may lead to neoplastic transformation and tumor development1,10. Moreover, a stronger epigenetic drift may reflect a biologically older organism, and as cancer incidence increases with age, most of these epigenetic drifts correlate with cancer risk.
An increasing body of evidence indicates that aging is associated with an accumulation of aberrant DNAm both in human and mouse tissues11,12,13. Several studies have revealed a common set of epigenomic features shared between aging and cancer11,14. Cancers have shown to have a significant age acceleration (+36 years on average) and a lower cancer age correlates with higher mutational load, suggesting that an epigenetically older tissue may need fewer mutations to develop cancer3. It has been known for a long time that DNAm plays an important role in colorectal cancer (CRC) by epigenetically inactivating oncosuppressors like the DKK and SFRP genes15,16,17,18,19. More recent studies also highlighted a causal connection between the TET1 protein, which mediates DNA demethylation, and intestinal tumorigenesis20,21. Additionally, a subset of CRCs shows a specific CpG island methylator phenotype (CIMP) that promotes cancer development and is already present in healthy colonic mucosa of CIMP+ patients22,23. Very recently, CIMP has also been observed to spontaneously arise in wild-type colon organoids during long-term culture in vitro and to promote the BRAFV600E-driven tumorigenesis24.
These observations collectively suggest that age-associated epigenetic drifts predispose healthy cells to transform into cancer cells; however, little is known about how age-dependent DNAm changes are mechanistically originated. Here, we have identified and mechanistically characterized a tissue-specific (intestine) DNAm drift in intestinal stem cells (ISCs) during aging that is highly enriched in human colon cancer. This epigenetic signature mainly involves promoter hypermethylation of a specific group of genes. Different from the CIMP that is specific to a minor fraction of colon cancers, this DNAm drift is strongly expanded in all human colon cancer types.
Results
Colon cancer associates with epigenetically drifted cells
To understand the correlation between DNAm alterations observed during aging in healthy colon tissues and colon cancer insurgence, we analyzed whole-genome bisulfite sequencing (WGBS) DNAm profiles of human colon samples from The Cancer Genome Atlas (TCGA). Principal-component analysis (PCA) separated well the healthy colon samples from cancer colon samples (Fig. 1a). In particular, the principal component (PC)1, that represented half of the total variance between all the samples in the analyzed dataset, showed the age-correlated variance in the healthy samples (Fig. 1a). Notably, the two clusters of colon cancer samples are localized further along the young–aged axis compared to the healthy samples from aged individuals, suggesting that CpGs contributing to PC1 have increased DNAm drift in cancer compared to healthy epithelium. We observed a similar number of both hypomethylated and hypermethylated gene promoters in healthy colon epithelium during aging, whereas cancer samples showed an increase in hypermethylated promoters (Fig. 1b and Supplementary Table 1). A total of 63% of the promoters that were found to be hypermethylated in aging were also found hypermethylated in cancer, whereas only 18% of the aging-hypomethylated promoters were found to be hypomethylated in cancer (Fig. 1c). The hypermethylated promoters represented a consistent aging- and colon cancer-associated epigenetic drift, named here as ACCA drift. By performing Gene Ontology (GO) analysis, we found that the genes undergoing ACCA drift were enriched in intracellular signaling pathways, such as cAMP-mediated signaling, and in stem cell-associated pathways, including the Wnt signaling pathway, which is almost always altered in colon cancer (Extended Data Fig. 1a,b)25,26. Notably, the ACCA drift gene promoters were hypermethylated both in CIMP− and in CIMP+ tumors with respect to healthy samples indicating that the ACCA drift is targeting all the human CRCs and involves a different subset of genes than the CIMP phenomenon22,23 (Extended Data Fig. 1c). Analysis of ACCA drift in cancer samples of another dataset confirmed hypermethylation in colon cancer adenocarcinoma (COAD) and showed an enrichment also in esophageal cancer (ESCA) and head and neck squamous cell carcinoma (HNSC) suggesting that ACCA drift may involve all the sections of the digestive system (Fig. 1d). Of note, DNAm analysis of a recently generated biobank of patient-derived xenografts (PDXs)27 revealed the persistence of DNA hypermethylation, indicating that ACCA drift is a robust and cell-intrinsic feature of CRCs (Extended Data Fig. 1d). Moreover, gene expression analysis of ACCA drift genes in CRC samples showed that almost half of them (45.2%) are downregulated in cancer with respect to healthy tissue, suggesting an important functional role of this epigenetic alteration in cancer (Fig. 1e and Supplementary Table 2).

a, PCA of WGBS datasets generated in human healthy and colon cancer samples of the indicated ages (yo, years old). Data were obtained from the TCGA database. b, Bar chart indicating the number of genes with hyper- or hypomethylated promoters in the indicated conditions (total n = 100; Supplementary Table 1). Differential methylation was assessed by comparing individuals older than 60 years to those younger than 15 for aging-related changes, and by comparing all CRC samples to all healthy controls for cancer-related changes. The P value was calculated using a chi-squared test (two-sided). c, Venn diagrams of the DM promoters found in aging and cancer datasets; differential methylation was assessed as described in b. The P value was calculated by a one-sided hypergeometric test. d, Boxplot indicating the DNAm level of the promoter of ACCA drift genes in colon cancer samples and nine other cancer types from the TCGA datasets: COAD, ESCA, liver hepatocellular carcinoma (LIHC), HNSC, bladder urothelial carcinoma (BLCA), kidney renal clear cell carcinoma (KIRC), breast invasive carcinoma (BRCA), lung adenocarcinoma (LUAD), glioblastoma multiforme (GBM) and skin cutaneous melanoma (SKCM). Boxplots represent interquartile range with 5–95 percentile whiskers. P values were computed using two-sided Wilcoxon rank-sum tests between healthy and tumor samples. n = 50 samples per cancer type (50 healthy + 50 tumor). e, Proportional distribution of the ACCA drift genes classified as upregulated (15.92%), downregulated (45.21%) or not regulated (39.87%) in CRC compared to healthy controls. Differential expression was determined based on both effect size (log2 fold change) and statistical significance (false discovery rate (FDR) < 0.05) (Supplementary Table 2). f, Schematic representation of an integrative analysis of DNA methylation and gene expression datasets of human healthy colon samples. g, GO analysis of the statistically correlated genes revealed in f. Numbers indicate the number of significant genes included in the relative GO term.
Next, we analyzed the transcriptomes (RNA-seq) of healthy colon samples of patients at different ages to find genes showing a higher correlation with the DNAm level of ACCA drift genes (Fig. 1f and Supplementary Table 3). GO analysis of statistically correlated genes revealed again intracellular and stem cell-associated signaling pathways (Fig. 1g and Extended Data Fig. 1e), suggesting an association between these differentially methylated (DM) genes and specific processes in intestinal cells, such as Wnt signaling. Among the genes associated with Wnt signaling, we found that all the members of the DKK and SFRP gene families (except SFRP5) showed linear promoter hypermethylation during aging (Extended Data Fig. 1f); however, these genes are expressed at low levels in human and mouse intestinal epithelium during aging and we found a significant downregulation of only Dkk3 during mouse aging, whereas no significant differences were found for the other DKK/SFRP genes (Extended Data Fig. 1g,h).
The DKK and SFRP genes are inhibitors of the Wnt pathway, whose promoters undergo hypermethylation in colon cancer, and their de-repression inhibits cell proliferation and cancer development/progression18,19,20. Of note, their DNAm level in cancer tissue is much higher than the DNAm level in age-matched healthy samples (Extended Data Fig. 1i). As usually both alleles of a cell are either methylated or unmethylated, DNAm shows a bimodal distribution and the CpG methylation percentage in a bulk sample reflects the percentage of cells having a given CpG methylated. Therefore, these data led to the hypothesis that during aging, the intestinal epithelium accumulates cells with ACCA drift and that colon cancer may originate from one of these drifted cells.
ACCA drift is conserved in mice, is specific to the intestinal tissue and is maintained at the stem cell level
To determine the main features and the origin of ACCA drift, we first investigated whether this epigenetic drift also exists in mouse intestinal epithelium, which would be an ideal experimental model for further investigations. Intestinal epithelium is composed of villi and intestinal crypts that are invaginations of the epithelium in which ISCs reside. We performed reduced-representation bisulfite sequencing (RRBS) of intestinal crypts of female and male mice at different ages to map DNAm genome-wide. PCA of the RRBS datasets accurately clustered the samples according to age (PC1) and sex (PC2), indicating that the DNAm landscape changes with age and between the intestinal crypts of female and male mice (Fig. 2a). We found that approximately 5% and 10% of the total CpGs decreased and increased their methylation level, respectively (Extended Data Fig. 2a), therefore indicating a slight overall increase of DNAm during aging. The methylation alteration trend was continuous along the different age points (2 m versus 18 m or 26 m) and with a very similar pattern among the different sexes (Extended Data Fig. 2b). Hypermethylated CpGs were more enriched on gene promoters and CpG islands (CpGi) than intragenic, intergenic and CpG-poor genomic regions with respect to hypomethylated CpGs (Extended Data Fig. 2c,d). Indeed, more than 10% of RRBS-covered CpG islands showed a significant increase in methylation, and less than 1% was hypomethylated (Extended Data Fig. 2e). CpGi hypermethylation showed strong (~90%) overlap between different age points and sexes, suggesting that the gain of DNAm on CpGi represents a conserved and systematic alteration during aging (Extended Data Fig. 2f,g). Hypermethylation of CpGi in gene promoters is a well-known epigenetic alteration occurring in CRC initiation15,28,29.

a, PCA of the RRBS datasets generated in aging female and male intestinal crypts. m, months. n = 4 mice per group were analyzed. b, Bar charts indicating the percentage of DM gene promoters found in the RRBS analysis as in a. Hyper, hypermethylated in aged samples; Hypo, hypomethylated in aged samples. c, Venn diagram showing the overlap of the hypermethylated gene promoters found in the indicated conditions. P value was calculated by a one-sided hypergeometric test. d, Schematic representation of a drifted stem cell expansion within an intestinal crypt. e, Venn diagrams of DM promoters found in Lgr5hi cells and whole crypts. P value is calculated by one-sided hypergeometric distribution test. f, Scatter-plot of the Δ delta methylation (aged minus young) on all the promoters in Lgr5hi cells and whole crypts. Only promoters covered in both analyses are shown. Linear regression was performed (lm function in R) and statistical significance of regression coefficients was assessed using two-sided t-tests; overall model fit was evaluated using an F-test (F statistic, 1.693 × 10⁴). The P value for the slope was obtained as twice the probability of observing a t-statistic as extreme as 130.11 under a Student’s t-test distribution with 17,452 degrees of freedom. Because this value is far smaller than machine precision, the exact P value cannot be reported and is conventionally written as P < 2.2 × 10⁻¹⁶. g, The DNAm level of the Dkk2 gene promoter analyzed by bisulfite (BS) pyrosequencing in the different tissues at the indicated ages. n = 3 mice per group were analyzed. SI, small intestine. h, Representative pictures of organoids cultured in the indicated conditions. Scale bar, 200 μm. For details, see the main text. i, Organoid-forming efficiency in the indicated conditions. Dots represent individual animals. n = 3 mice per group were analyzed. P value was calculated by an unpaired t-test, two-tailed. j, Budding efficiency of organoids cultured in the indicated conditions. n = 3 mice per group were analyzed. P value was calculated by an unpaired t-test, two-tailed. k, Bar chart indicating the DNAm level of the Dkk2 gene promoter analyzed by BS pyrosequencing upon 1 month of organoid culture of crypts isolated from young and aged mice and cultured in the indicated conditions. n = 3 mice per group were analyzed. P value was calculated by an unpaired t-test, two-tailed. l, Representative pictures of organoids cultured in the indicated conditions. Scale bar, 200 μm. For details, see the main text. m, Organoid forming efficiency in the indicated conditions as in l. Dots represent individual animals. n = 3 mice per group were analyzed. P value was calculated by an unpaired t-test, two-tailed. n, Budding efficiency of organoids cultured in the indicated conditions as in l. n = 3 mice per group were analyzed. P value was calculated by an unpaired t-test, two-tailed. o, Bar chart indicating the DNAm level of the Dkk2 gene promoter analyzed by BS pyrosequencing upon 1 month of organoid culture of crypts isolated from young mice and cultured in the indicated conditions as in l. n = 3 mice per group were analyzed. P value was calculated by an unpaired t-test, two-tailed. p, Bar chart indicating the DNAm level of the Dkk2 gene promoter analyzed by BS pyrosequencing upon 1 month of organoid culture of crypts isolated from young and aged mice and cultured in the presence of CHIR99021 or recombinant Wnt3a. n = 3 mice per group were analyzed. P value was calculated by an unpaired t-test, two-tailed. Error bars in the figure bar charts represent the s.d.
To understand whether these aging-associated DNAm alterations might have an actual functional role in regulating gene transcription, we further analyzed DM promoters (Fig. 2b and Supplementary Table 4). As expected, we found that DM promoters showed stronger hypermethylation during aging (probably due to the presence of CpGi). Of note, most of the gene promoters undergoing ACCA drift in human intestinal samples were also found to be hypermethylated during mouse aging (Fig. 2c), demonstrating that ACCA drift is conserved in mice.
Gut tissue renews rapidly (3–5 days) thanks to the action of ISCs that continuously proliferate and differentiate into all the cell types of the intestinal epithelium30,31. We hypothesized that ACCA drift is initiated at the stem cell level and further propagated by differentiation into the other cell types of the crypt, where it gradually accumulates over time (Fig. 2d). To verify our hypothesis, we performed WGBS of the Lgr5hi ISCs isolated from young and aged Lgr5-GFP-IRES-CreERT2 mice30 and profiled their DNAm during aging at genome-wide level (Extended Data Fig. 2h). WGBS revealed a slight increase of global DNAm in aged Lgr5hi cells with respect to young ones (Extended Data Fig. 2i); however, the DM CpGs were equally distributed among hyper- or hypomethylated ones, indicating that both the gain and loss of DNAm take place in Lgr5hi cells during aging (Extended Data Fig. 2j). Like the DNAm landscape of whole crypts, hyper-DM CpGs were enriched in gene promoters and CpGi, whereas hypo-DM CpGs were enriched in intra-/intergenic and CpG-poor genomic regions (Extended Data Fig. 2k). Correspondingly, Lgr5hi cells showed solid CpGi hypermethylation during aging (Extended Data Fig. 2l) with a significant correlation with the whole-crypt DNA methylation profile (Extended Data Fig. 3a,b) suggesting that CpGi hypermethylation is initiated in stem cells. Notably, CpGi on the Dkk2 and Sfrp1 gene promoters also showed age-dependent hypermethylation (Extended Data Fig. 3c).
More extensive analysis of gene promoters covered both in the whole crypts and in the Lgr5hi cells revealed significant overlap especially on the hypermethylated promoters (Fig. 2e). Plotting of DNA methylation delta values (aged minus young) of all the gene promoters in whole crypts and Lgr5hi cells showed a very high correlation between ISCs and whole crypt compartments (Fig. 2f). This strongly indicates that DNA hypermethylation is established in ISCs and is maintained in these cells during aging. Several recent studies have reported that adult stem cells of other tissues undergo epigenetic alterations during aging11,12,13 and we wondered whether ACCA drift could also occur in other tissues. As genome-wide analysis of DNAm profiles is time- and money-consuming, we decided to analyze the promoter of the genes of the Dkk and Sfrp families (Dkk2 and Sfrp1 throughout the study) as marker genes of ACCA drift genes. The DNAm level of ACCA drift genes was highly correlated with the average DNAm of the two marker genes (DKK2 and SFRP1), as well as when four marker genes were considered (DKK1, DKK2, SFRP1 and SFRP2) (Extended Data Fig. 3d,e). By using bisulfite pyrosequencing, which allows a fast, reliable and sensitive quantification of the DNAm, we did not observe any age-dependent DNA hypermethylation in other tissues analyzed (blood, muscle, skin and whole-brain compared to small intestine whole tissue) (Fig. 2g and Extended Data Fig. 4a).
All together, these results indicate that ACCA drift is conserved in mice, is maintained at the stem cell level and is specific to the intestinal tissue.
DNAm drift is maintained in in vitro cultures, is not driven by cell division and is modulated by the Wnt signaling pathway
Aging is a multifactorial process driven by cell-intrinsic and extrinsic changes. To understand better what contributes to ACCA drift, we isolated intestinal crypts from young and aged mice and cultured them in vitro as organoids32 (Extended Data Fig. 4b). We found that DNAm levels observed in young and aged crypts in vivo are maintained in the respective organoids cultured in vitro, suggesting that ACCA drift is not lost when intestinal epithelial cells are extrapolated from their microenvironment (Extended Data Fig. 4c). Moreover, the long-term culture of the organoids (derived from both young and aged mice) led to a further increase in DNAm levels of Dkk2 and Sfrp1 (Extended Data Fig. 4d,e). To better characterize this process, we performed a low-coverage RRBS experiment on organoids after 1 week, 1 month and 3 months of culture and compared them to the crypts in vivo during aging. The analysis of DNAm of all ACCA drift gene promoters showed a very similar trend to that of the Dkk2 and Sfrp1 genes (Extended Data Fig. 4f,g).
These results suggested that cell division might represent a DNAm drift-promoting factor as, for example, in the case of the reported DNAm drift-causing hypomethylation within partially methylated domains during aging33. To test the potential contribution of cell division/proliferation to the origin of DNAm drift, we cultured organoids for 1 month in medium containing a standard concentration of the epithelial growth factor (EGF)/Noggin/R-spondin1 (ENR) and in medium containing a reduced concentration of these growth factors to 50% (ENR 50%) or to 25% (ENR 25%) (Fig. 2h). The organoids cultured in ENR 50% or ENR 25% were growing slower, forming fewer new organoids and fewer buds (crypts-like structures that branch out from the organoid body) with respect to the control condition ENR (Fig. 2h–j); however, they did not show a decrease but rather a further increase in DNAm drift, especially in the condition ENR 25% (Fig. 2k and Extended Data Fig. 4h). We also manipulated cell proliferation using alternative strategies. We decreased either R-spondin to 25% (EN-R25) or removed EGF from the standard ENR medium (NR) or cultured organoids with low glucose (LG) (1.5%) or without glucose (0 G) as shown in Fig. 2l. Organoids showed less growing and less budding in all the four conditions (Fig. 2m,n), but we found no difference in the DNAm of the Dkk2 or Sfrp1 gene, except a slight increase in the EN-R25 condition, suggesting that the Wnt pathway may play a role in DNAm drift (Fig. 2o and Extended Data Fig. 4i). To further confirm the potential role of Wnt signaling, we performed an opposite experiment where we supplemented the organoids’ culture medium either with CHIR99021 or with the recombinant Wnt3a protein to increase cell proliferation and organoid bud formation by stimulating the Wnt pathway34,35. Accordingly, we observed a decrease in the promoter DNAm level, especially in the organoids derived from aged mice (Fig. 2p and Extended Data Fig. 4j).
In summary, these data demonstrated that DNAm drift is maintained when intestinal epithelial cells are isolated from their environment and that it is not driven by cell division. The data also suggest an important role of the Wnt signaling pathway in modulating DNAm drift in vitro.
DNAm drift is heterogeneous at the CpG level, but consistent at the gene promoter level
Next, we wanted to know how DNAm drift accumulates during aging. Our previous experiments demonstrated that DNAm drift is acquired and maintained at the ISC level. Intestinal crypts undergo clonality over the time (Fig. 3a), and all the cells of an intestinal crypt originate from a single ISC in a period of ~3 months31,36. Based on the existence of crypt monoclonality, we decided to perform single-crypt DNAm analysis, hypothesizing that it could reflect a single ISC DNAm pattern and allow us to assess DNAm heterogeneity of the DNAm drift. We established unique protocols for isolation and DNAm profiling (using pyrosequencing) of single intestinal crypts from mouse gut epithelium. Different from a single-cell approach, where reliable and complete DNAm profiles are still technically challenging to achieve, a single-crypt approach allowed us more robust analysis. We collected 50 single crypts from young mice and 50 from aged mice and analyzed the DNAm level of the promoters of the four marker genes Dkk2, Sfrp1, Dkk1 and Sfrp2 (Fig. 3b and Extended Data Fig. 5a). We also analyzed the promoter of the Actb and Dppa3 genes as controls, as they are unmethylated and fully methylated, respectively, in adult tissues (Extended Data Fig. 5b). Because of the low amount of DNA as a starting material, for some crypts we were not able to perform DNAm analysis or obtain passing filter DNAm sequences (represented as dashes in Fig. 3b and Extended Data Fig. 5a). By analyzing the DNAm data of the Dkk2 promoter, we found that young mice showed a general hypomethylation of the regions analyzed (only some CpGs were found methylated in few crypts), but the crypts from aged mice had a trinomial DNAm distribution with around half of the CpGs being not methylated (as in the young), 54 of 210 CpGs being methylated around 50% (monoallelic methylation) and 16 of 210 CpGs being 100% methylated (biallelic methylation) (Fig. 3c). This trinomial distribution in crypt from aged mice (present also in the other genes analyzed; Extended Data Fig. 5c) confirmed our hypothesis that single-crypt DNAm reflects the DNAm profile of a single (stem) cell (of origin).

a, Schematic representation of a drifted stem cell expansion within the intestinal crypt and its subsequent monoclonal conversion. b, The DNAm level of the Dkk2 gene promoter (six CpGs analyzed by BS pyrosequencing) in 80 single crypts isolated from young and aged mice (as indicated). Each circle represents the DNAm level (in percentage). The failed DNAm analysis is represented by dashes (NA; not available). n = 3 mice per group were analyzed. c, Violin plot showing the DNAm level of the Dkk2 gene promoter in single intestinal crypts isolated from young and aged mice as in b. d, Hierarchical clustering and heatmap of the Pearson correlation of the DNAm profiles of 80 single crypts as in b. e, The DNAm profile of the Dkk2 gene promoter in the crypts from the cluster 1 (crypts isolated from young mice, ‘young’ crypts cluster) (top). The DNAm profile of the Dkk2 gene promoter in the crypts from the cluster 5 (crypts isolated from aged mice, ‘aged’ crypts cluster) (bottom). f, Bar chart showing the number of aged crypts (events observed − expected) having the indicated number of methylated genes among three different genes analyzed in each single crypt. g, Spearman correlation heatmap of DNAm patterns of the four analyzed genes in a single crypt. P value, two-tailed confidence interval at 95%. h, The DNAm level of the indicated genes in crypts with high or low DNAm on the Dkk2 gene promoter. Average Y, average DNAm in young crypts; Average A, average DNAm in aged crypts.
Mostly, aged crypts showed a pronounced level of hypermethylation but not with the same DNAm pattern (Fig. 3b and Extended Data Fig. 5a), suggesting that intestinal epithelial crypts become hypermethylated heterogeneously during aging. Pearson correlation analysis of all crypts analyzed revealed the existence of a big cluster (cluster 1), mainly composed of young crypts and other smaller clusters (clusters 2–5), principally composed of aged crypts (Fig. 3d). Of note, in cluster 1, we found three crypts from aged mice suggesting that young-like DNAm patterns may be present in aged gut epithelium; we also found some young crypts in aged clusters 2–5 (Fig. 3d,e). A similar result was observed analyzing the promoter of the Dkk1 gene (Extended Data Fig. 5d,e).
To understand the inter-gene variability, we considered only aged crypts in which three gene promoters were successfully analyzed, and we calculated the average DNAm of these three promoters for each crypt (Extended Data Fig. 5f). The average DNAm level spanned from ~15% to almost 60%, indicating that some aged crypts were methylated on more genes. We counted the number of aged crypts having none, one, two or all three genes methylated (Fig. 3f). We observed that aged crypts have higher probability to have either none or all the three genes methylated and lower probability to have only one or two genes methylated (Fig. 3f) suggesting that the hypermethylation process hits multiple genes. Finally, we observed a general positive promoter DNAm correlation of the different genes in the aged crypts, except between Dkk1 and Dkk2 genes (Fig. 3g). Indeed, crypts with low promoter DNAm on the Dkk2 gene (gray bar, Fig. 3h) show high DNAm on the Dkk1 gene, whereas crypts with high promoter DNAm on the Dkk2 gene (yellow bar, Fig. 3h) show low DNAm on the Dkk1 gene, suggesting a possible mutually exclusive mechanism on these two genes.
These results indicate that DNAm drift, while very consistent and reliable in bulk analysis of the intestinal epithelium, is composed of multiple epigenetic subdrifts with clusters of crypts sharing similar hypermethylation patterns. Additionally, multigene analysis showed a positive correlation in aged crypts, suggesting the presence of crypts with an enhanced DNAm drift as well as epigenetically young-like crypts in the intestinal epithelium of aged mice.
DNAm drift is expanded and selected by crypt fission during aging
To better understand the origin and the expansion of these DNAm patterns and crypt clusters, we decided to investigate whether they might be generated by crypt fission (Fig. 4a), a homeostatic mechanism through which intestinal epithelium expands and maintains tissue homeostasis37. Recently, it has also been observed that crypt fission contributes to the fixation and spread of somatic mutations in human colon38. We isolated neighboring crypts from intestinal sections by laser-capture microdissection (LCM) and analyzed the DNAm level at single-crypt resolution (Fig. 4b and Extended Data Fig. 6a). We analyzed the Dkk2 gene promoter in four groups of neighboring crypts (16, 8, 11 and 11 crypts) from four intestinal regions of two different aged mice (Fig. 4c and Extended Data Fig. 6b). DNAm profiles of these neighboring crypts revealed the existence of pairs of neighboring crypts with correlating DNAm patterns on the Dkk2 promoter (red arrows, Fig. 4c,d and Extended Data Fig. 6b,c). DNAm profiling of the Dkk1 gene promoter gave similar results and revealed the probable existence of triplets of epigenetically similar neighboring crypts (Extended Data Fig. 6d,e). The analysis of all the sequenced neighboring crypts showed that crypts that are neighboring to each other have a higher correlation of DNAm profiles on the analyzed gene promoter with respect to randomly selected crypts (Fig. 4e and Extended Data Fig. 6f). Moreover, highly correlating neighboring crypts on the Dkk1 promoter also showed higher correlation on Dkk2 with respect to low-correlating ones, demonstrating the epigenetic heredity during crypt fission (Fig. 4f and vice versa in Extended Data Fig. 6g).

a, Schematic representation of a monoclonal crypt expansion by fission. b, Schematic representation of LCM-coupled DNAm analysis of single crypts by BS pyrosequencing. c, DNAm profiles of the Dkk2 gene promoter of 16 (left, region A) and 8 (right, region B) neighboring crypts. The red arrows indicate neighboring crypts with very similar DNAm profiles. d, Correlation heatmap of the neighboring crypts as in c. e, Bar chart showing the Pearson correlation of the DNAm profiles of the Dkk2 gene promoter in the randomly picked (n = 45) and/or neighboring (n = 42) crypts. P value was calculated by a two-tailed Mann–Whitney U-test. f, Bar chart showing the Pearson correlation of the DNAm profiles of the Dkk2 gene promoter in the crypts with high (n = 20) and low (n = 20) correlation of the DNAm profiles of the Dkk1 gene promoter. P value was calculated by a two-tailed Welch’s t-test. g, Schematic representation of a Confetti-reporter induction experiment. h, Representative images of transversal sections of small intestine after 2 months (left) and 1 year (right) upon Confetti transgene activation. n ≥ 3 mice per group were analyzed. i, Bar chart showing the probability of a crypt to have a neighboring crypt of the same color after 2 months (n = 328) or 1 year (n = 370) upon Confetti transgene activation. P value was calculated by a two-tailed, unpaired t-test. j, The DNAm profiles of the Dkk2 gene promoter of single crypts of the same color coming from the same cluster. In total nine independent clusters were analyzed. n = 3 mice were used. k, The DNAm profiles of the Dkk2 gene promoter of single scattered crypts of the same color as the crypt clusters analyzed in j. l, Bar chart showing the Pearson correlation of the Dkk2 DNAm patterns of crypts of the same color and coming from the same cluster (n = 55) with respect to scattered crypts (n = 28) of the same color as the cluster. P value was calculated by a two-tailed Welch’s t-test. m, Bar chart showing the Dkk2 DNAm level of the crypts of the same color and coming from the same cluster (n = 34) with respect to scattered crypts (n = 8) of the same color as the cluster. P value was calculated bya two-tailed Mann–Whitney U-test. Error bars in the bar charts represent the s.d. n, Bar charts showing the Dkk2 DNAm level of the crypts of the same color (clustered; n = 4) or of different colors (not-clustered; n = 5) isolated from the same region of the cluster 2 and 4 analyzed in Fig. 4j. P value was calculated by a two-tailed Mann–Whitney U-test. Error bars in the figure bar charts represent the s.d.
To further confirm these data and to assess whether DNAm-drifted crypts could be positively selected during crypt fission, we decided to employ the Confetti-reporter mouse model31. Based on the observation that approximately 1–2 months after activation of the Confetti multifluorescent reporter the intestinal crypts become monochromatic, we decided to assess whether crypt clonality and expansion could occur and be followed in aging gut epithelium (Fig. 4g) as previously observed over a shorter time for the crypts carrying oncogenic mutations39. To verify the case, we analyzed the probability of a crypt having a neighboring crypt of the same color after 2 months or 1 year upon Confetti transgene activation. This analysis revealed that the probability after 1 year is almost twice as high as after 2 months (Fig. 4h,i; for Fig. 4h, single channels are provided as Source Data), suggesting that, on average, one event of crypt fission may take place in 1 year. We then analyzed the single-crypt DNAm level of the Dkk2 gene promoter of nine clusters of the same-color neighboring crypts in mice following 1 year of Confetti activation (Fig. 4j). For each cluster of crypts microdissected from a different slide we also captured and analyzed one scattered crypt of the same color as the cluster but located far from the cluster as a control (Fig. 4k). The crypts in the cluster showed a higher intra-cluster correlation of the DNAm pattern with respect to crypts of other clusters and to scattered crypts (Fig. 4l and Extended Data Fig. 6h,i). Remarkably, the crypts in clusters showed a higher DNAm level on the Dkk2 gene promoter than the scattered crypts, indicating that crypts with DNAm drift have been positively selected over time (Fig. 4m). To further confirm this last observation, for clusters 2 and 4, we also analyzed cluster-adjacent crypts of a different color (blue or green) and not being in a cluster of same-color crypts. DNAm analysis showed that crypts in clusters have higher DNAm on the Dkk2 gene with respect to nonclustered crypts isolated from the same region (Fig. 4n).
Taken together, these data demonstrate that DNAm drift expands through a crypt fission mechanism during aging, forming patches of epigenetically drifted crypts that are positively selected over time; however, other mechanisms are supposed to have a role in driving DNAm drift during aging, for example, the de novo establishment of DNA (hyper)methylation in unmethylated crypts.
Crypts with high DNAm drift have an impaired iron metabolism
To better characterize the origin of DNAm drift, we decided to analyze the transcriptome of the crypts with high DNAm drift. First, we optimized the isolation protocol to obtain sufficient amount of both DNA and RNA from single-isolated crypts (Fig. 5a). We processed around 150 crypts from aged mice, analyzed their DNAm level on the four marker gene promoters (Dkk1, Dkk2, Sfrp1 and Sfrp2) and calculated the average DNAm level (Fig. 5b). Then we selected the crypts with higher or lower DNAm drift (respectively, high or low 5mC crypts) and performed single-crypt RNA-seq (Fig. 5b,c). Of note, high 5mC crypts were significantly clustered apart from low 5mC crypts and showed a more scattered distribution in PC analysis (Fig. 5c). We checked the expression of the main DNAm-related genes (Dnmt and Tet gene families), but we did not find any significant differences between low and high 5mC crypts (Extended Data Fig. 7a,b). Therefore, we performed a GO analysis of the genes differently expressed in high versus low 5mC crypts, which revealed an enrichment of pathways related to inflammation and cellular transport/metabolism, including two pathways related to iron homeostasis and cellular response to iron (Extended Data Fig. 7c). Notably, transferrin receptor messenger RNA (Tfrc aka TfR1) (a key gene for cellular iron import) was downregulated in high versus low 5mC crypts, whereas ferroportin mRNA (Slc40a1 aka FPN1) (the only known exporter of non-heme iron in mammals) was upregulated (Fig. 5d). We also found the same trend as in low versus high 5mC crypts when we analyzed the transcriptome of young versus aged ISCs (Extended Data Fig. 7d). To confirm this result, we checked the protein level of these two genes in intestinal crypts isolated from young and aged mice (Fig. 5e). We found a significant decrease of TfR1 as well as a significant increase of FPN1 proteins in crypt cells of aged compared to young mice (Extended Data Fig. 7e). Finally, we assessed nuclear iron levels in crypts isolated from young and aged mice and observed a reduced amount of Fe2+ iron in aged samples (Fig. 5f).

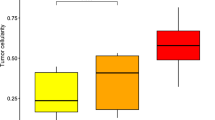
a, Schematic representation of a single-crypt DNAm analysis coupled with RNA sequencing. b, Scatter-plot showing the average DNAm level on the Dkk1, Dkk2, Sfrp1 and Sfrp2 gene promoters in single crypts isolated from aged mice. The most methylated crypts (the top 10%, high 5mC crypts) and the least-methylated crypts (the bottom 10%, low 5mC crypts) were selected and analyzed by RNA-seq. n = 4 mice were used. c, PCA of single crypt RNA-seq datasets generated in crypts of the top 10% (high 5mC crypts, red dots) and the bottom 10% (low 5mC crypts, blue dots) as in b. To assess the statistical significance of the separation between two groups, a MANOVA test using Pillai’s trace was performed, yielding a highly significant result (P = 4.235 × 10−5). d, Transferrin receptor (Tfrc) and ferroportin (Slc40a1) expression levels in high (n = 16) and low (n = 12) 5mC crypts as in b. P value was calculated by a two-tailed Welch’s t-test. Each dot represents a single crypt. n = 4 mice were used. e, Western blot analysis of the protein levels of TfR1 and ferroportin (FPN1) in small intestinal crypts isolated from young and aged mice. Actin was used as a loading control. n = 5 mice per group were analyzed. f, Ferrous (Fe2+) iron levels in small intestinal crypts isolated from young and aged mice. n = 3 mice per group were analyzed. P value was calculated by a two-tailed Welch’s t-test. g, Schematic representation of our hypothesis. h, Quantification of TET hydroxymethylase enzymatic activity in intestinal crypts isolated from young (n = 5) and aged (n = 6) mice. P value was calculated by a two-tailed Welch’s t-test. i, Quantification of DNMT enzymatic activity in the intestinal crypts isolated from young (n = 4) and aged (n = 5) mice. P value was calculated by a two-tailed Welch’s t-test. j, Bar chart showing DNAm level of the Dkk2 gene promoter analyzed by BS pyrosequencing at the indicated time points upon C35 TET inhibitor treatment of organoids derived from young intestinal crypts. n = 3 mice per group were analyzed. P value was calculated by a two-tailed Welch’s t-tests. k, Bar charts showing DNAm level of the Dkk1 and Dkk2 gene promoters in intestinal crypts isolated from wild-type (WT) and Tet2/3-dKO mice. n = 3 mice per group were analyzed. P value was calculated by a two-tailed Welch’s t-test. l, Bar chart showing DNAm level of the Dkk2 gene promoter analyzed by BS pyrosequencing in organoids treated with the iron chelator, DFO for 1 and 2 months. n = 3 mice per group were analyzed. P value was calculated by a two-tailed paired t-test. Error bars in the figure bar charts represent the s.d.
These analyses indicate that the crypts with high DNAm drift have an impaired transcriptome and metabolism, in particular the cellular iron metabolism that leads to a reduced amount of iron in the intestinal epithelial aged cells.
DNAm drift is driven by an impaired TET enzymatic activity in aged intestinal cells
To better understand the connection between iron metabolism and DNAm drift, we hypothesized that, in aged intestinal cells, the reduced level of transferrin receptor and the augmented level of ferroportin lead to a reduced level of intracellular bioavailable iron (Fe2+). Within the cell, the majority of iron is stored as ferric iron (Fe3+), while only a small portion exists as a transient redox-active form of ferrous iron (Fe2+) and constitutes the labile iron pool (LIP). The LIP is essential as a cofactor for numerous iron-dependent enzymes, including nuclear TET enzymes, which play a key role in the active DNA demethylation process (Fig. 5g)40. Thus, we measured TET hydroxymethylase activity in the intestinal crypts isolated from young and aged mice and observed a significant reduction of TET enzymatic activity in aged crypts (Fig. 5h), whereas no differences were observed for DNA methyltransferase (DNMT) enzymatic activity (Fig. 5i). Chromatin immunoprecipitation (ChIP) analysis revealed that TET2 and TET3, but not TET1 were bound to the promoter of the Dkk2 and Sfrp1 genes (Extended Data Fig. 7f,g). Previous studies reported that TET enzymes can prevent the promoter hypermethylation of oncosuppressor genes such as the DKK or SFRP gene families20 by converting 5mC into 5-hydroxymethylcytosine (5hmC). By using a 5hmC DNA immunoprecipitation (hMeDIP) technique, we found an enrichment of 5hmC on the promoter of the Dkk2 and Sfrp1 genes in intestinal epithelial crypts isolated from young mice, which was significantly reduced in crypts isolated from aged mice (Extended Data Fig. 8a,b). Therefore, we decided to employ organoid culture for functional experiments. We derived intestinal organoids from crypts isolated from young mice and treated them with the C35 TET inhibitor (Extended Data Fig. 8c). C35 inhibitor treatment led to a reduction of 5hmC on the Dkk2/Sfrp1 gene promoters (Extended Data Fig. 8d) and to a dose- and time-dependent increase of DNAm on these promoters (Fig. 5j and Extended Data Fig. 8e). To further demonstrate the involvement of TET2/3 enzymes in promoter hypermethylation of Dkk/Sfrp genes, we analyzed DNAm in double knockout Tet2/3 (Tet2/3-dKO) mice41. We found that Tet2/3-dKO mice have a higher DNAm level on these gene promoters (Fig. 5k and Extended Data Fig. 8f). To demonstrate that iron deficiency drives promoter hypermethylation of these genes, we treated the organoids with the iron chelator, deferoxamine (DFO) for 1 and 2 months and found that DFO-treated organoids have higher DNAm on the Dkk2 and Sfrp1 gene promoters with respect to control conditions (Fig. 5l and Extended Data Fig. 8g).
Our data indicated that TET activity (in particular, TET2 and TET3) is necessary to keep the promoters of Dkk2 and Sfrp1 genes hypomethylated and that this DNA demethylation mechanism is impaired in aged gut epithelial cells because of a deficiency of Fe2+ iron, which serves as a cofactor for the TET enzymes.
DNAm drift is prevented by transferrin receptor re-expression in aged intestinal cells and Wnt signaling can restore its expression upon proinflammatory challenges
To assess the importance of the aging-reduced transferrin receptor, we re-expressed it by transducing Tfrc in intestinal organoids derived from aged mice (Fig. 6a). We found that Tfrc-expressing organoids have increased TET activity and lower DNAm on the Dkk2 and Sfrp1 gene promoters with respect to the mock condition after 40 days of culturing (Fig. 6b,c and Extended Data Fig. 8h).

a, Western blot analysis of the protein levels of the transferrin receptor (TfR1) in intestinal organoids derived from aged mice and analyzed 40 days after transduction with a 3xFlag-Tfrc- expressing lentivirus. Mock (parental vector) was used as a negative control. Actin was used as a loading control. n = 3 mice per group were analyzed. b, Quantification of TET hydroxymethylase enzymatic activity in intestinal organoids as in a. n = 3 mice per group were analyzed. P value was calculated by a two-tailed Welch’s t-test. c, Bar chart showing the DNAm level of the Dkk2 gene promoter analyzed by BS pyrosequencing in intestinal organoids as in a. Parental organoids, untransduced organoids at day 0. n = 3 mice per group were analyzed. P value was calculated by a two-tailed Welch’s t-test. d, Representative images of the western blot analysis of the protein levels of TfR1 and FPN1 in intestinal organoids cultured in different media as indicated. ENRW (EGF, Noggin, R-spondin1, Wnt3a); ENRC (EGF, Noggin, R-spondin1, CHIR99021). GAPDH was used as a loading control. n = 3 mice per group were analyzed. For details, see the main text. e, Ferrous (Fe2+) iron levels in intestinal organoids as in d. n = 3 mice per group were analyzed. P value was calculated by one-way analysis of variance (ANOVA). f, Quantification of TET hydroxymethylase enzymatic activity in intestinal organoids as in d. n = 3 mice per group were analyzed. P value was calculated by one-way ANOVA. g, Representative images of the western blot analysis of the protein levels of the TfR1 and FPN1 in intestinal organoids treated with IFNγ (0.2 ng ml−1) or with both IFNγ (0.2 ng ml−1) and Wnt3a (100 ng ml−1) for 5 days as indicated. GAPDH was used as a loading control. n = 3 mice per group were analyzed. For details, see the main text. h, Ferrous (Fe2+) iron levels in intestinal organoids as in g. n = 3 mice per group were analyzed. P value was calculated by one-way ANOVA. i, Quantification of TET hydroxymethylase enzymatic activity in intestinal organoids as in g. n = 3 mice per group were analyzed. P value was calculated by ordinary one-way ANOVA. Error bars in bar charts represent the s.d.
As our data showed an inverse correlation between Wnt signaling and ACCA drift (Fig. 2) and Wnt signaling is reduced in aging42,43, we wondered whether modulation of Wnt signaling could regulate expression of Tfrc. We cultured small intestinal organoids in a medium with reduced R-spondin (EN-R25 medium) or in standard medium supplemented with Wnt3a (ENRW) or CHIR99021 (ENRC). We observed a slight downregulation of the TfR1 protein level and a reduced (though not significant, P = 0.11 and P = 0.17) iron level and TET activity in cells cultured in EN-R25 medium (Fig. 6d–f). An opposite effect has been observed in organoids cultured in ENRW or ENRC medium (Fig. 6d–f). Previous studies have shown that an interferon (IFN)γ-induced inflammaging impaired the homeostasis, the epigenome and the functionality of the intestinal epithelium44,45,46,47. Transcriptional analysis of crypts with high DNAm drift revealed an inflammatory signature, in particular, IFN signaling (Extended Data Fig. 7c). Therefore, we tested whether IFNγ could lead to impaired iron homeostasis and whether overactivation of Wnt signaling could rescue it. Of note, we found that IFNγ treatment did not modulate TfR1 protein levels, but it increased the level of FPN1 and impaired iron homeostasis and TET activity (Fig. 6g–i). Treatment of the organoids with either IFNγ and Wnt3a led to upregulation of TfR1 protein and could restore iron levels and TET activity (Fig. 6g–i).
These data indicated aging-associated proinflammatory stress and impaired Wnt signaling as driving factors promoting altered iron homeostasis and DNAm drift. Our data also showed that transferrin receptor restoration, as well as Wnt factor supplementation can rescue iron homeostasis and protect from DNAm drift.
Discussion
Epigenetic alterations represent one of the most conserved hallmarks of aging in different species48 and recently, the loss of epigenetic information has been proposed as a cause of mammalian aging49. In recent years, DNAm drifts have been increasingly explored and identified in several tissues of different species3,50,51 and are often used to build epigenetic clocks6,52. The main identified DNAm drifts are a cell-intrinsic and tissue-type independent drift, which allows highly accurate quantification of chronological age (Horvath’s epigenetic clock3); a mitotic drift that may be connected to the DNAm machinery inefficiency during DNA replication5,33; and a nonmitotic drift, which is tissue specific and affected by cell-extrinsic factors (such as systemic factors, inflammation and lifestyle) that can be used to build predictors of the organismal biological age8,53. In our study, we identified an intestinal-specific, DNAm drift not driven by cell proliferation that may contribute to the generation of precancerous cells, which represent the cells of origin of colorectal cancers. The ACCA drift consists of the hypermethylation of hundreds of gene promoters, some of them found epigenetically silenced in human cancers16,19. The assumption that tumors derive from epigenetically drifted epithelial cells may mechanistically explain the DNAm alterations found in CRC. Our data confirmed the occurrence of the ACCA drift also in mouse aging where we found a predominant increase in promoter DNAm, consistent with a previous study that found that 21% of genes are hypermethylated, whereas 13% of genes are hypomethylated in the aged mouse intestine54. To better characterize ACCA drift, we employed intestinal organoid cultures that have been recently validated to be a viable model to study DNAm and aging55. By in vitro analysis, we demonstrated that ACCA drift is not driven by cell division, as cells cultured in a poor medium that have a lower proliferation rate, showed no difference or even higher ACCA drift. These experiments may indicate that ACCA drift favors cell resistance and survival to hostile environments similarly to specific DNAm events that drive cancer cell survival56.
Previous studies reported that the DNAm patterns at age-associated regions are very heterogeneous at the CpGs level and showed low correlation at individual CpGs, reflecting a strong stochastic component7,8; however, recent analysis revealed that biological age is better predicted by nonstochastic processes53. We found that DNAm drift shows crypt-specific DNAm patterns and, even though it is heterogeneous at the single CpG level, it remains consistent at the gene level, supporting the idea that it is functional to biological processes.
We often link ACCA drift to cancer development, but we cannot exclude that it may also impair intestinal tissue functionality as DNAm has been demonstrated to play a key role in the ISC self-renewal and differentiation into all the epithelial cells of the crypt–villus axis57,58,59; however, further studies should be performed to assess the influence of ACCA drift on the homeostasis of the gut epithelium.
Our study confirms previous observations that DNAm patterns can be used to identify and analyze clonality of the human colon epithelium60,61. Indeed, we found that DNAm drift is maintained and expanded through homeostatic mechanisms of the intestinal epithelium, such as crypt clonal dominance and crypt fission. We observed that epigenetically drifted crypts bear a selective advantage and can spread and fix faster than epigenetically young crypts, reflecting somehow the behavior of crypts carrying oncogenic mutations38,39,62.
All together, our data explain the origin of an intestinal DNAm drift that initiates in healthy stem cells bearing dysfunctional iron metabolism and then propagates to the surrounding cells via crypt clonality and crypt fission mechanisms. The origin and the evolution of human epigenetic drift are long-standing questions in the field of the epigenetics of aging. It is tempting to speculate that the observed epigenetic drifts in each tissue are the sum of multiple epigenetic subdrifts, each of which has specific driving mechanisms and functional consequences. Understanding the global picture may allow a better comprehension of human aging and possible areas where interventions can be developed to prevent, diagnose and treat age-related diseases.
For further discussion, please see the Supplementary Information file.
Limitations of the study
Our study identifies an intestinal DNAm drift and characterizes it in human and mouse organisms during aging; however, functional experiments to demonstrate that loss of iron homeostasis and cell clonality drives the insurgence and the amplification of this drift have been conducted in mouse models. Further experiments should be performed to validate these mechanisms also in human organisms. We have shown that the ACCA drift is associated with human colon cancers, but functional experiments should be performed to demonstrate that it is a driver alteration in cell transformation.
Methods
Mice
All animal experiments were conducted according to protocols approved by the state government of Thuringia (license numbers: TG/J-0002858/A; TG/J-0003616/A; TG/J-0003681/A; FLI-18–016; O_FN_18–20 and FLI-18–005). We used young (2-4 months old) and aged (18–26 months old) male and female wild-type C57BL6/J and Lgr5-ki-e-GFP-creER mice. The Rosa26-lsl-Confettiki/+; Villin-creERT2tg/+ mice have been described previously63. All the mice were group housed and maintained in a specific opportunist pathogen-free animal facility in Fritz Lipmann Institute (FLI) with a 12-h light–dark cycle and fed with a standard mouse chow. The Tet2/3fl/fl and Tet2/3fl/fl VillinCre mice have been described previously41.
Small intestinal and colon crypts isolation
Small intestinal crypts were isolated using the established protocol64 with minor modifications. In brief, mouse small intestine was dissected, washed in cold PBS and opened longitudinally. Villi were removed by scraping with a coverslip. The villi-free intestinal pieces (2 cm) were washed four times in cold PBS and transferred to 5 mM EDTA–PBS, followed by two 30-min incubations at 4 °C on a rotator. The tissue was transferred to fresh cold PBS and manually shaken for 30 s. The crypt solution was filtered using a 70-µm cell strainer and centrifuged at 120g for 5 min at 4 °C. The crypts pellet was resuspended in 1 ml of cold PBS and evaluated for purity microscopically. Isolated crypts were aliquoted, snap frozen in liquid nitrogen and stored at −80 °C for further experiments. For RNA isolation, the crypts were immediately resuspended in QIAzol Lysis Reagent (QIAGEN) and stored at −80 °C. Colon crypts were isolated as previously reported65.
Intestinal stem cells isolation and sorting
For Lgr5–eGFP ISC isolation, freshly isolated crypts were dissociated in complete TrypLE medium (10 mM Tris-HCl, pH 7.5, 5 mM CaCl2, 2.5 mM MgCl2, 20 µM Y27632 and 1 mg ml−1 DNase I) for 30 min in a water-bath at 37 °C with brief vortexing every 10 min. The single-cell suspension was then passed through a 40-µm cell strainer and centrifuged at 800g for 5 min at 4 °C. The cell pellet was resuspended in 3 ml FACS staining medium containing PBS supplemented with 2% fetal bovine serum, 2.5 mM EDTA, 10 µM Y27632 and 4,6-diamidino-2-phenylindole (DAPI; 1:1,000 dilution). The single-cell suspension was applied to a FACS LSRII (BD Biosciences) and Lgr5-eGFPhi ISCs were sorted for downstream analysis as previously carried out44.
Organoid culture and treatment
Small intestinal organoids were cultured according to the established protocol44. In brief, freshly isolated crypts (roughly 250) were mixed with 20 µl of Matrigel and plated in a 24-well plate. After polymerization of Matrigel for 30 min at 37 °C, the ENR crypt culture medium (Advanced DMEM/F12, 1× GlutaMAX, 10 mM HEPES, N2 supplement (1:100 dilution), B27 supplement (1:50 dilution), 0.5 U ml−1 penicillin–streptomycin supplemented with 50 ng ml−1 mouse recombinant EGF, 100 ng ml−1 mouse recombinant Noggin and 500 ng ml−1 human recombinant R-spondin1) was added and refreshed every 2 days.
For the proliferation experiment, the organoids were cultured in the standard ENR medium (a control condition) or in medium containing different concentrations of the growths factors as follows: the 50% ENR medium (containing 25 ng ml−1 mouse recombinant EGF, 50 ng ml−1 mouse recombinant Noggin and 250 ng ml−1 human recombinant R-spondin1), the 25% ENR medium (containing 12.5 ng ml−1 mouse recombinant EGF, 25 ng ml−1 mouse recombinant Noggin and 125 ng ml−1 human recombinant R-spondin1), the EN-R25 medium (containing 50 ng ml−1 mouse recombinant EGF, 100 ng ml−1 mouse recombinant Noggin and 125 ng ml−1 human recombinant R-spondin1), the NR medium (containing 100 ng ml−1 mouse recombinant Noggin and 500 ng ml−1 human recombinant R-spondin1), the LG medium (standard ENR medium containing 1.5 g l−1 glucose) and the 0 G medium (standard ENR medium without glucose).
For the IFNγ treatment, 1-week-grown organoids were passaged and treated with IFNγ (0.2 ng ml−1) for 5 days. Wnt3a (100 ng ml−1 final concentration) and CHIR99021 (3 µM final concentration) were added to the medium where indicated.
The TET inhibitor C35 (Aobious) was added to the standard ENR medium at a final concentration of 5 µM or 7.5 µM, as indicated and refreshed every 24 h. DFO was added to the standard ENR medium at a final concentration of 5 µM as indicated.
The images of intestinal organoids in culture were acquired using a Primovert 12V DC microscope (Zeiss).
DNA constructs
The complementary DNA of murine transferrin receptor (Tfrc) was purchased from Origene (plasmid MC221416) and cloned into the CSII-EF Venus-P2A-3F-MCS vector64 using the BamHI and NotI restriction sites.
Lentivirus production and organoids transduction
Lentivirus was produced in the Lenti-X 293T cell line by transfection with the lentiviral vectors CSII-EF Venus-P2A-3F-Tfrc, CSII-EF Venus-P2A-3F-MCS (mock), and the packaging vectors pCMV-VSV-G (Addgene #8454), pRSV-Rev (Addgene #12253), and pMDLg/pRRE (Addgene #12251). Supernatants containing lentivirus were collected 24 h and 48 h after transfection, pooled together and passed through a 0.45-μm filter. The lentiviruses were concentrated by using the Lenti-X concentrator (Clontech) following the manufacturer’s instructions. Organoids were transduced as previously described64.
Induction of the Confetti transgene
The Rosa26-lsl-Confettiki/+; Villin-creERT2tg/+ mice were injected intraperitoneally with tamoxifen (100 mg kg−1 body weight) for 5 consecutive days to induce the Cre-mediated recombination and euthanized after 21 days or 1 year after tamoxifen injection as previously reported63.
Whole-mount preparation
Intestinal whole mounts were prepared as previously described63. In brief, the small intestine was flushed with ice-cold PBS and the tissue was fixed in 4% paraformaldehyde (PFA) overnight at 4 °C on a rotator. The next day, the tissue was washed in PBS (3 ×5 min), opened longitudinally and villi were scraped off with a coverslip. After three washes in ice-cold PBS, the tissue was directly mounted between two coverslips using DePeX (Serva 18243.01) and imaged on an Axiovert 200 ApoTome (Zeiss).
Laser-capture microdissection
For LCM of neighboring crypts, the proximal intestine was briefly washed in cold PBS, a small piece (2 cm) was directly embedded and slowly frozen (using liquid nitrogen) in optimal cutting temperature compound without fixation and stored at −80 °C. Subsequently, 8-µm sections were cut from the frozen block on a cryostat, mounted on a PET membrane-covered glass slide (Zeiss, 415190-9051-000), stained with crystal violet for 20 s at room temperature and washed with ddH2O. Longitudinal crypt sections were microdissected on a PALM laser-capture microdissection and optical tweezer microscope (Zeiss). Single crypt fragments were catapulted and collected in 0.2-ml adhesive cap tubes (Zeiss, 415190-9201-000) and the capturing success was visually confirmed after the collection session.
For LCM of crypts in clusters isolated from tamoxifen-induced Rosa26-lsl-Confettiki/+; Villin-creERT2tg/+ mice, intestinal tissue was prepared according to ref. 66 to preserve the fluorescence signal. In brief, the proximal intestine was washed in cold PBS, a small piece (2 cm) was immersed for 15–20 s in 2-methylbutane prechilled with liquid nitrogen and 8-µm transversal sections were immediately cut on a cryostat. The crypt clusters were visualized by fluorescent channels and the microdissection procedure was the same as described above.
RNA and DNA isolation
Genomic DNA from bulk crypts and tissues was isolated using a DNeasy Blood and Tissue kit (QIAGEN) following the manufacturer’s instructions. Total RNA from bulk crypts was isolated using QIAzol Lysis Reagent (QIAGEN) followed by isopropanol precipitation. Genomic DNA and total RNA from Lgr5-eGFPhi ISCs sorted by FACS were isolated using a ZR-Duet DNA/RNA MiniPrep Plus kit (Zymo Research) following the manufacturer’s instructions.
Genomic DNA and RNA from single crypts were isolated using a Quick-DNA/RNA Microprep Plus kit (Zymo Research) following the manufacturer’s instructions.
Isolated DNA and/or RNA were quantified on a Nanodrop 8000 (Thermo Fisher Scientific) and on a Qubit 3.0 (Thermo Fisher Scientific). The quality of isolated genomic DNA was analyzed by agarose gel electrophoresis. The quality of isolated RNA was analyzed by a Fragment Analyzer (Agilent).
Reduced-representation bisulfite sequencing library preparation
For RRBS library preparation, 100–250 ng of RNase-treated genomic DNA was digested with MspI enzyme (NEB) at 37 °C for 12 h and purified using AMPure XP beads (Beckman Coulter) following the manufacturer’s instructions. The MspI-digested genomic DNA was used as input for RRBS library preparation using a NEXTflex Bisulfite-Seq kit (PerkinElmer) following the manufacturer’s instructions. Bisulfite conversion was performed using a Methylation-Lightning kit (Zymo Research) following the manufacturer’s instructions.
Whole-genome bisulfite sequencing library preparation
For WGBS library preparation, 10 ng of RNase-treated genomic DNA was used as input and the WGBS library was prepared following an established protocol67.
Bisulfite pyrosequencing
Bisulfite conversion of genomic DNA was performed using a Methylation-Lightning kit (Zymo Research) following the manufacturer’s instructions. Bisulfite-treated DNA was amplified using PyroMark PCR kit (QIAGEN). PCR products were sequenced on a PyroMark Q48 Autoprep (QIAGEN) using PyroMark Q48 Advanced CpG Reagents (QIAGEN) and analyzed by PyroMark Q48 Autoprep Software (QIAGEN, v.2.4.2). Bisulfite conversion-specific PCR primers were designed using PyroMark Assay Design Software v.2.0 (QIAGEN). The primers’ sequences are listed in Supplementary Table 5.
Single-crypt bisulfite pyrosequencing
For the experiments where only DNAm was analyzed, each single crypt was first digested with Proteinase K in 20 µl of M-Digestion Buffer 1× (Zymo Research) at 50 °C for 20 min and subsequently bisulfite-converted in the presence of an RNA carrier (20 ng) using a Methylation-Lightning kit (Zymo Research). Bisulfite-converted DNA was purified in the presence of 1 µg ml−1 RNA carrier added to the M-binding buffer, following the manufacturer’s instructions. Pyrosequencing was performed as described above.
LCM-coupled bisulfite pyrosequencing
The bisulfite pyrosequencing of LCM neighboring crypts was performed as for single crypts with the modification that Proteinase K digestion was performed in the tube cap for the first 2 min, then the tube was spun for 10 s and again incubated at 50 °C for 18 min to complete the digestion reaction. Bisulfite pyrosequencing of LCM crypt clusters was performed as above but the Proteinase K digestion was incubated at 50 °C for 4 h.
Single-crypt RNA-seq library preparation
RNA-seq library preparation of single crypts was performed using a NEB Next Single Cell/Low-Input RNA Library Prep kit (NEB) following the manufacturer’s instructions.
Targeted bisulfite DNA methylation analysis
For deep DNA bisulfite sequencing, 100 ng of genomic DNA from each mouse sample was treated with bisulfite using the EpiTect Bisulfite kit (QIAGEN) according to the manufacturer’s instructions. The bisulfite-treated DNA was then amplified with sequence-specific primers containing Illumina Nextera handle sequences. The forward overhang sequence was 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-(locus-specific sequence) and the reverse overhang sequence was 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-(locus-specific sequence). Primers were designed to amplify a sequence shorter than 200 bp containing 7–16 CpG sites. PCR products ranging from 150 to 200 bp were gel-extracted using the QIAquick Gel Extraction kit (QIAGEN). Equimolar amounts of all amplicons for each sample were pooled in a single tube. To incorporate the index sequences, the pooled PCR products were further amplified by limited cycle-number PCR using Nextera complementary primers. Each indexed pool was then gel-extracted again using the QIAquick Gel Extraction kit. Equimolar amounts of all indexed pools were pooled into a single tube and processed for sequencing. Sequencing was performed using the MiSeq Reagent kit v2 on a MiSeq platform (Illumina). Sequenced reads were separated by barcode, aligned to the target sequence, and methylation ratios were determined using a Python script (methratio.py). The primers’ sequences are listed in Supplementary Table 5.
High-throughput sequencing
All the samples for genome-wide experiments were sequenced on the HiSeq2500, and NextSeq500 or MiSeq platform (Illumina).
ChIP–qRT–PCR analysis
ChIP was performed as described previously68. In brief, freshly isolated intestinal crypts (from whole small intestines) were crosslinked by addition of formaldehyde to 1% for 10 min at room temperature on a rotator, quenched with 0.125 M glycine for 5 min at room temperature on a rotator, then washed twice with cold PBS. The crosslinked crypts were resuspended in SDS ChIP Buffer (50 mM Tris-HCl, pH 8.0, 10 mM EDTA, 1% SDS and protease inhibitors), incubated on a rotator for 30 min at 4 °C, sonicated for 18 cycles on a high power setting (30 s ON, 30 s OFF) using the Bioruptor Next Gen (Diagenode) and centrifuged at 12,000g for 15 min at 4 °C. The isolated chromatin was diluted tenfold with ChIP dilution buffer (16.7 mM Tris-HCl, pH 8.0, 0.01% SDS, 1.1 % Triton X-100, 1.2 mM EDTA and 167 mM NaCl) and incubated with 5 μg of antibody overnight at 4 °C on a rotator. Protein G-conjugated magnetic beads (Dynal, Thermo Fisher Scientific) were saturated with PBS/1% BSA and sonicated salmon sperm overnight at 4 °C. Next day, samples were incubated with saturated beads for 2 h at 4 °C on a rotator, and subsequently washed with 1 ml of cold low-salt buffer (20 mM Tris-HCl, pH 8.0, 0.1 % SDS, 1% Triton X-100, 2 mM EDTA and 150 mM NaCl), 1 ml of cold high-salt buffer (20 mM Tris-HCl, pH 8.0, 0.1 % SDS, 1% Triton X-100, 2 mM EDTA and 500 mM NaCl), 1 ml of cold LiCl buffer (10 mM Tris-HCl, pH 8.0, 1% DOC, 250 mM LiCl, 1 mM EDTA and 1% NP-40) and twice with 1 ml of cold TE buffer (10 mM Tris-HCl, pH 8.0 and 1 mM EDTA). The immunoprecipitated chromatin was eluted with 200 μl of elution buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% SDS, 150 mM NaCl and 5 mM dithiothreitol (DTT)) for 30 min at room temperature on a rotator and de-crosslinked at 65 °C overnight. The de-crosslinked DNA was purified using a QiaQuick PCR Purification kit (QIAGEN) according to the manufacturer’s instructions. The antibodies used for ChIP were rabbit polyclonal TET1 antibody (N1) (cat. no. GTX125888, GeneTex), rabbit monoclonal TET2 (D9K3E) antibody (cat. no. 92529, Cell Signaling), rabbit polyclonal anti-TET3 antibody (cat. no. ABE290, Merck) and normal rabbit IgG polyclonal antibody (cat. no. 12-370, Millipore). The immunoprecipitated DNA was analyzed by quantitative real-time PCR using the SYBR GreenERkit (Invitrogen) as previously reported44. The primers’ sequences are listed in Supplementary Table 5.
5hmC DNA immunoprecipitation
For hMeDIP, genomic DNA isolated from crypts or organoids was sheared by sonication (Bioruptor Pico, Diagenode) to obtain 200–500-bp fragments and purified with Agencourt AMPure XP beads (Beckman Coulter) following the manufacturer’s instructions. Approximately 1 μg of sheared DNA was denatured for 10 min at 95 °C, immediately cooled on ice for 10 min and then immunoprecipitated using a hMeDIP kit (Active Motif) following the manufacturer’s instructions. The immunoprecipitated DNA was treated with Proteinase K for 2 h and purified using the MinElute PCR purification kit (QIAGEN). The purified DNA was analyzed by quantitative real-time PCR by using a SYBR GreenER kit (Invitrogen) as previously reported44. The primers’ sequences are listed in the Supplementary Table 5.
DNA methyltransferase activity
Nuclear extracts were prepared from freshly isolated intestinal crypts using the EpiQuik Nuclear Extraction Kit I (Epigentek) following the manufacturer’s instructions. DNMT activity was measured by using the EpiQuik DNA Methyltransferase Activity/Inhibition Assay kit (Epigentek) following the manufacturer’s instructions.
TET enzymatic activity
Nuclear extracts were prepared from freshly isolated intestinal crypts using the EpiQuik Nuclear Extraction Kit I (Epigentek) following the manufacturer’s instruction. The TET enzymatic activity was measured by using Epigenase 5mC Hydroxylase TET Activity/Inhibition Assay kit (Epigentek) following the manufacturer’s instructions and omitting cofactor 3 in the assay buffer.
Protein extraction and western blotting
For total protein extraction, crypts or organoids were resuspended in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% DOC and 0.1% SDS) supplemented with antiproteases, incubated on ice for 15 min and sonicated using the Bioruptor Pico (Diagenode) for five cycles (30 s ‘ON’, 30 s ‘OFF’). After centrifugation at 20,000g for 15 min at 4 °C, the supernatants were recovered and quantified using Pierce BCA Protein Assay kit (Thermo Fisher Scientific) following the manufacturer’s instructions. To analyze the protein levels of transferrin receptor (TfR1) and ferroportin (FPN1), the protein samples were mixed with SDS loading dye, incubated for 5 min at 37 °C (ref. 69) and immediately run on 4–20% Mini-PROTEAN TGX Stain-Free Gels (Bio-Rad). To analyze the protein levels of actin, the samples were prepared as described above but incubated with SDS loading dye for 5 min at 95 °C. After SDS–PAGE, the proteins were transferred to nitrocellulose membranes (iBlot 2 Transfer Stacks; Thermo Fisher Scientific). The membranes were blocked in T-PBS (0.1% Tween 20 in PBS) supplemented with 5% milk for 1 h at room temperature and then incubated with specific primary antibodies in T-PBS + 5% BSA or 5% milk overnight at 4 °C. After incubation with primary antibody, the membranes were washed five times with T-PBS for 25 min at room temperature and then incubated with secondary antibody (1:3,000 dilution) in T-PBS + 5% milk for 1 h at room temperature. After incubation with secondary antibody, the membranes were washed five times with T-PBS for 25 min at room temperature and then developed on a ChemiDoc Imaging System (Bio-Rad).
The primary antibodies used for western blotting were rabbit polyclonal ferroportin/SLC40A1 antibody (cat. no. NBP1-21502 from Novus Biologicals, 1:250 dilution); mouse monoclonal CD71/TFRC/transferrin receptor antibody (cat. no. sc-65882 from Santa Cruz, 1:2,000 dilution), mouse monoclonal anti-β-actin antibody (cat. no. A5316 from Sigma-Aldrich, 1:20,000 dilution) and rabbit polyclonal anti-GAPDH antibody (cat. no. sc-25778 from Santa Cruz, 1:3,000 dilution).
Measurement of iron contents
The iron contents were measured using an Iron Assay kit (Abcam). In brief, the isolated crypts or organoids were resuspended in Iron Assay Buffer on ice and sonicated using the Bioruptor Pico (Diagenode) for three cycles (30 s ‘ON’, 30 s ‘OFF’). After centrifugation at 16,000g for 10 min at 4 °C, the supernatants were recovered and immediately used for the assay following the manufacturer’s instructions.
Analysis of TCGA data on human colon samples
In brief, DNA methylation data from TCGA (Illumina 450K array) for CRC and multiple other cancer types was normalized and analyzed at promoter regions to identify age- and cancer-associated methylation changes. Differential methylation was tested using t-tests with multiple correction, PCA was applied for sample variability, correlations with Wnt antagonist genes were computed with permutation tests and functional/pathway analyses were performed using IPA alongside coexpression analysis from matched expression data. Additional methods regarding quantification and statistical analyses are available in the Supplementary Information file. Informed consent was obtained according to TCGA policies and the data are anonymized and publicly available. Source data can be found in Supplementary Table 6.
Statistics and reproducibility
Sample size was not predetermined. The experiments were conducted with n > 3 biological replicates. Sample sizes were selected on research-based common standards and on the minimum number of samples allowing statistics70. No data were excluded from the analysis. All the experiments were successfully replicated at least three times also for experiments where only one representative image or result is shown. Biological samples were randomly selected for the experiments using simple randomization methods. Data collection and analysis were not performed blind to the conditions of the experiments. Data distribution was assumed to be normal but this was not formally tested.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw sequencing data reported in this paper are deposited in the Gene Expression Omnibus database under the following accession numbers: RRBS Crypts, GSE129712; WGBS LGR5, GSE129767; and RNA-seq Single Crypts, GSE271646. DNA methylation data of PDX are deposited in GSE208713. Other data are available from the corresponding author upon request. Source data are provided with the paper.
Code availability
All the codes used in this study are publicly available and described in the Methods.
References
Zheng, S. C., Widschwendter, M. & Teschendorff, A. E. Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics 8, 705–719 (2016).
Horvath, S. et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 13, R97 (2012).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013).
Teschendorff, A. E. A comparison of epigenetic mitotic-like clocks for cancer risk prediction. Genome Med. 12, 56–17 (2020).
Horvath, S. & Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 23, 223 (2018).
Han, Y. et al. New targeted approaches for epigenetic age predictions. BMC Biol. 18, 71 (2020).
Meyer, D. H. & Schumacher, B. Aging clocks based on accumulating stochastic variation. Nat. Aging https://doi.org/10.1038/s43587-024-00619-x (2024).
Lu, A. T. et al. Universal DNA methylation age across mammalian tissues. Nat. Aging 3, 1144–1166 (2023).
Ermolaeva, M., Neri, F., Ori, A. & Rudolph, K. L. Cellular and epigenetic drivers of stem cell ageing. Nat. Rev. Mol. Cell Biol. 19, 594–610 (2018).
Rakyan, V. K. et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 20, 434–439 (2010).
Beerman, I. et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12, 413–425 (2013).
Maegawa, S. et al. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 20, 332–340 (2010).
Teschendorff, A. E. et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 20, 440–446 (2010).
Rhee, I. et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416, 552–556 (2002).
Sato, H. et al. Frequent epigenetic inactivation of DICKKOPF family genes in human gastrointestinal tumors. Carcinogenesis 28, 2459–2466 (2007).
Linhart, H. G. et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 21, 3110–3122 (2007).
Aguilera, O. et al. Epigenetic inactivation of the Wnt antagonist DICKKOPF-1 (DKK-1) gene in human colorectal cancer. Oncogene 25, 4116–4121 (2006).
Suzuki, H. et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat. Genet. 36, 417–422 (2004).
Neri, F. et al. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene 34, 4168–4176 (2015).
Tricarico, R. et al. TET1 and TDG suppress inflammatory response in intestinal tumorigenesis: implications for colorectal tumors with the CpG island methylator phenotype. Gastroenterology 164, 921–936.e1 (2023).
Toyota, M. et al. CpG island methylator phenotype in colorectal cancer. Proc. Natl Acad. Sci. USA 96, 8681–8686 (1999).
Kawakami, K. et al. DNA hypermethylation in the normal colonic mucosa of patients with colorectal cancer. Br. J. Cancer 94, 593–598 (2006).
Tao, Y. et al. Aging-like spontaneous epigenetic silencing facilitates Wnt activation, stemness, and BrafV600E-induced tumorigenesis. Cancer Cell 35, 315–328.e6 (2019).
Network, C. G. A. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012).
Fearon, E. R. & Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990).
Leto, S. M. et al. XENTURION is a population-level multidimensional resource of xenografts and tumoroids from metastatic colorectal cancer patients. Nat. Commun. 15, 7495 (2024).
Suzuki, H., Maruyama, R., Yamamoto, E. & Kai, M. Epigenetic alteration and microRNA dysregulation in cancer. Front. Genet 4, 258 (2013).
Jair, K.-W. et al. De novo CpG island methylation in human cancer cells. Cancer Res. 66, 682–692 (2006).
Barker, N. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007 (2007).
Snippert, H. J. et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144 (2010).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Endicott, J. L., Nolte, P. A., Shen, H. & Laird, P. W. Cell division drives DNA methylation loss in late-replicating domains in primary human cells. Nat. Commun. 13, 6659 (2022).
Yin, X. et al. Niche-independent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat. Methods 11, 106–112 (2014).
Sato, T. et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772 (2011).
Lopez-Garcia, C., Klein, A. M., Simons, B. D. & Winton, D. J. Intestinal stem cell replacement follows a pattern of neutral drift. Science 330, 822–825 (2010).
Bruens, L., Ellenbroek, S. I. J., Rheenen, J. van & Snippert, H. J. In vivo imaging reveals existence of crypt fission and fusion in adult mouse intestine. Gastroenterology https://doi.org/10.1053/j.gastro.2017.05.019 (2017).
Nicholson, A. M. et al. Fixation and spread of somatic mutations in adult human colonic epithelium. Cell Stem Cell 22, 909–918.e8 (2018).
Snippert, H. J., Schepers, A. G., Es, J. H. van, Simons, B. D. & Clevers, H. Biased competition between Lgr5 intestinal stem cells driven by oncogenic mutation induces clonal expansion. EMBO Rep. 15, 62–69 (2014).
Tahiliani, M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009).
Ansari, I. et al. TET2 and TET3 loss disrupts small intestine differentiation and homeostasis. Nat. Commun. 14, 4005 (2023).
Flanagan, D. J. et al. NOTUM from Apc-mutant cells biases clonal competition to initiate cancer. Nature 594, 430–435 (2021).
Nalapareddy, K. et al. Canonical Wnt signaling ameliorates aging of intestinal stem cells. Cell Rep. 18, 2608–2621 (2017).
Omrani, O. et al. IFNγ-Stat1 axis drives aging-associated loss of intestinal tissue homeostasis and regeneration. Nat. Commun. 14, 6109 (2023).
Funk, M. C. et al. Aged intestinal stem cells propagate cell-intrinsic sources of inflammaging in mice. Dev. Cell 58, 2914–2929.e7 (2023).
Patrick, R. et al. The activity of early-life gene regulatory elements is hijacked in aging through pervasive AP-1-linked chromatin opening. Cell Metab. 36, 1858–1881.e23 (2024).
Zhao, D. et al. Inflammation-induced epigenetic imprinting regulates intestinal stem cells. Cell Stem Cell 31, 1447–1464.e6 (2024).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Yang, J.-H. et al. Loss of epigenetic information as a cause of mammalian aging. Cell 186, 305–326.e27 (2023).
Jones, M. J., Goodman, S. J. & Kobor, M. S. DNA methylation and healthy human aging. Aging Cell 14, 924–932 (2015).
Hernando-Herraez, I. et al. Ageing affects DNA methylation drift and transcriptional cell-to-cell variability in mouse muscle stem cells. Nat. Commun. 10, 4361–11 (2019).
Bell, C. G. et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 20, 249–24 (2019).
Tong, H. et al. Quantifying the stochastic component of epigenetic aging. Nat. Aging https://doi.org/10.1038/s43587-024-00600-8 (2024).
Maegawa, S. et al. Caloric restriction delays age-related methylation drift. Nat. Commun. 8, 539 (2017).
Lewis, S. et al. DNA methylation analysis validates organoids as a viable model for studying human intestinal aging. Cell Mol. Gastroenterol. Hepatol. https://doi.org/10.1016/j.jcmgh.2019.11.013 (2019).
De Carvalho, D. D. et al. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell 21, 655–667 (2012).
Sheaffer, K. L. et al. DNA methylation is required for the control of stem cell differentiation in the small intestine. Genes Dev. 28, 652–664 (2014).
Cullen, S. M. & Goodell, M. A. Rising from the crypt: decreasing DNA methylation during differentiation of the small intestine. Genome Biol. 14, 116 (2013).
Kaaij, L. T. et al. DNA methylation dynamics during intestinal stem cell differentiation reveals enhancers driving gene expression in the villus. Genome Biol. 14, R50 (2013).
Graham, T. A. et al. Use of methylation patterns to determine expansion of stem cell clones in human colon tissue. Gastroenterology 140, 1241–1250.e9 (2011).
Fearon, E. R. & Bommer, G. T. Ancestries hidden in plain sight: methylation patterns for clonal analysis. Gastroenterology 140, 1139–1143 (2011).
Sirvinskas, D., Annunziata, F. & Neri, F. Intestinal stem cells heterogeneity and clonal dominance during aging: two faces of the same coin?. Mech. Ageing Dev. 189, 111247 (2020).
Annunziata, F. et al. Paneth cells drive intestinal stem cell competition and clonality in aging and calorie restriction. Eur. J. Cell Biol. https://doi.org/10.1016/j.ejcb.2022.151282 (2022).
Freter, R. et al. Establishment of a fluorescent reporter of RNA-polymerase II activity to identify dormant cells. Nat. Commun. 12, 3318–16 (2021).
Širvinskas, D. et al. Single-cell atlas of the aging mouse colon. iScience 25, 104202 (2022).
Meng, H. et al. Tissue triage and freezing for models of skeletal muscle disease. J. Vis. Exp. https://doi.org/10.3791/51586 (2014).
Krepelova, A. & Neri, F. Low-input whole-genome bisulfite sequencing. Methods Mol. Biol. 2351, 353–368 (2021).
Krepelova, A. et al. Myc and max genome-wide binding sites analysis links the Myc regulatory network with the polycomb and the core pluripotency networks in mouse embryonic stem cells. PLoS ONE 9, e88933 (2014).
Tsuji, Y. Transmembrane protein western blotting: Impact of sample preparation on detection of SLC11A2 (DMT1) and SLC40A1 (ferroportin). PLoS ONE 15, e0235563 (2020).
Bland, J. M. & Altman, D. G. Analysis of continuous data from small samples. Brit. Med. J. 338, a3166 (2009).
Acknowledgements
We thank the staff members of the FLI FACS, Imaging, Sequencing and Animal facilities. We also thank all the members of the Neri research group for their support and helpful discussions. This work was supported by funding from Sofja Kovalevskaja Award starting grant from the von Humboldt Foundation (ITA1164767SKP), the Leibniz Institute on Aging (FLI), the Associazione Italiana per la Ricerca sul Cancro (MFAG 2021 ID 26038) and the Fondazione Ricerca Molinette.
Author information
Authors and Affiliations
Contributions
A.K. and F.N. conceived and designed the study. A.K. performed most of the experiments, including organoid treatments, RRBS, WGBS, RNA-seq library preparation, bisulfite pyrosequencing, ChIP, hMeDIP and enzymatic activity assays, western blot analysis and iron content measurements. F.A. and O.O. contributed to biological sample collection. F.A. performed induction of the Confetti transgene and LCM. E.W. and P.E.P. helped with experiments related to iron metabolism and conceptually contributed to the study design. I.A., D.B. and Y.B. performed the targeted bisulfite DNA methylation analysis in Tet2/3-dKO mice. M.R., J.L., C.G. and F.N. performed the computational analyses. A.K., M.R., C.G. and F.N. made the figures. A.K. and F.N. wrote the manuscript with the contribution from all the authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Aging thanks the anonymous, reviewer(s) for their contribution to the peer review of this work
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Colon cancer associates with epigenetically drifted cells.
a, Correlation heatmap of the categories of the gene ontology analysis of the hypermethylated genes or the genes undergoing the Aging- and Colon Cancer-Associated epigenetic drift (the ACCA drift). b, Gene ontology analysis of the statistically correlated genes revealed in Fig. 1c. Canonical pathway enrichment was performed using Ingenuity Pathway Analysis (Qiagen). Statistical significance of pathway enrichment was assessed using a right-tailed Fisher’s exact test. Reported values represent –log₁₀ of the adjusted (Benjamini–Hochberg) p-value for each pathway. c, Boxplot indicating the DNAm level of the promoter of the ACCA drift genes in healthy colon and in colon cancer samples subdivided in CIMP negative, CIMP low and CIMP positive cancers. Number of samples: healthy (9-15 y) = 19; healthy (40-60 y) = 11; healthy (>60 y) = 34; (CIMP-) = 30; (CIMPlow) = 16; (CIMP + ) = 9. Box plots represent the median (center line), the 25th and 75th percentiles (bounds of the box), and whiskers extending to the most extreme data points within 1.5× the interquartile range. d, Boxplot indicating the DNAm level of the promoter of the ACCA drift genes in healthy colon samples, CRC samples and in patient-derived xenografts (PDX) (both from primary colorectal cancers (CRCs) and CRC liver metastasis samples). Number of samples: 50 healthy, 50 tumor, 76 PDX. e, Correlation heatmap of the categories of the gene ontology analysis performed by using the correlated genes revealed in co-expression analysis in Fig. 1g. Boxplots represent interquartile range with min to max whiskers. f, Dotplot indicating the DNAm level of the promoter of the DKK and SFRP family genes in healthy human colon samples at the indicated ages. Data are presented as mean values ± SEM (samples as in (a)). p-value was calculated by Welch’s t-test, one-tail. Number of samples: (9-15 y) = 19; (40-60 y) = 11; (>60 y) = 34. g, Boxplot indicating normalized expression counts of DKK and SFRP family genes in healthy human colon samples. For each gene, expression is shown separately for each age group. Box plots represent interquartile range with 5-95 percentile whiskers. P-values were computed using two-sided Wilcoxon rank-sum tests between (40-60 y) and (>60 y) samples. Number of samples: (40-60 y) = 12; (>60 y) = 39. h, Boxplot indicating normalized expression counts of the Dkk and Sfrp family genes in mouse intestinal crypts. For each gene, expression is shown separately for age group and sex. n = 4 mice per group were analyzed. Box plots represent interquartile range with 5-95 percentile whiskers. P-values were computed using two-sided Wilcoxon rank-sum tests between young and old or young and geriatric samples, respectively. i, Boxplot indicating the DNAm level of the promoter of the DKK2 and SFRP1 genes in healthy and colon cancer samples at the indicated ages (samples as in (a)). Number of samples: healthy (9-15 y) = 19; healthy (40-60 y) = 11; healthy (>60 y) = 34; cancer (40-60 y) = 17; cancer (>60 y) = 37. Boxplots represent the interquartile range with min to max whiskers.
Extended Data Fig. 2 The ACCA drift is conserved in the mouse intestine and originates at the stem cell level.
a, Pie charts indicating the percentage of the statistically significant differentially methylated (DM) CpGs found in the indicated conditions by using the same RRBS datasets as in Fig. 2a. m = months. b, Boxplots of the DNA methylation level of the DM CpGs that were found hypermethylated or hypomethylated during aging in female or male intestinal crypts. m = months. n = 4 mice per group were analyzed. Box plots represent the median (center line), the 25th and 75th percentiles (bounds of the box), and whiskers extending to the most extreme data points within 1.5× the interquartile range. c, Bar charts indicating the percentage of the DM CpGs found in the RRBS analysis as in Fig. 2a in different genomic features. m = months. Hyper = hypermethylated in old samples. Hypo = hypomethylated in old samples. hcp = high CpG promoters. icp = intermediate CpG promoters. lcp = low CpG promoters. CpGi = CpG island. shore = CpG island shore (±2 kb of the CpG island borders). d, Bar charts indicating the percentage of the statistically significant differentially methylated CpG (DM CpGs) found in the RRBS analysis as in Fig. 2a in different genomic features. Hyper = hypermethylated in old samples. Hypo = hypomethylated in old samples. m = months. e, Bar charts indicating the percentage of the DM CpG islands found in the RRBS analysis as in Fig. 2a. Hyper = hypermethylated in old samples. Hypo = hypomethylated in old samples. f-g, Venn diagrams of the DM CpG islands found hypermethylated in the indicated conditions in old samples as in (e). m = months. h, Hierarchical clustering and heatmap of the Pearson correlation of the whole genome bisulfite sequencing (WGBS) datasets of mouse Lgr5hi cells. Y = young; O = old; r = biological replicate. n(young) = 3 mice, n(old) = 4 mice were analyzed. i, Violin plot of the DNA methylation level on the whole genome in the indicated conditions measured by WGBS as in (h). n(young) = 3 mice, n(old) = 4 mice were analyzed. White boxes indicate the average DNAm level. j, Pie chart indicating the distribution of the DM CpGs found significantly hyper- or hypo- methylated during aging in the same datasets as in (h). k, Bar charts indicating the percentage of the DM CpGs found in the WGBS analysis of the Lgr5hi cells during aging in different genomic features. Hyper = hypermethylated in old samples. Hypo = hypomethylated in old samples. hcp = high CpG promoters. icp = intermediate CpG promoters. lcp = low CpG promoters. CpGi = CpG island. shore = CpG island shore (±2 kb of the CpG island borders). l, Bar chart showing the percentage of the DM CpG islands found in the WGBS analysis as in (h).
Extended Data Fig. 3 The ACCA drift is conserved in the mouse intestine and originates at the stem cell level.
a, Scatter plot of the delta methylation (old minus young) on all the CpG islands in the Lgr5hi cells and whole crypts. Linear regression was performed (lm function in R), and statistical significance of regression coefficients was assessed using two-sided t-tests; overall model fit was evaluated using an F-test (F-statistic: 4.27 × 10⁴). The p-value for the slope was obtained as twice the probability of observing a t statistic as extreme as 206.65 under a Student’s t distribution with 12419 degrees of freedom. Because this value is far smaller than machine precision, the exact p-value cannot be reported and is conventionally written as p < 2.2 × 10⁻¹⁶. b, Venn diagrams of the DM CpG islands found in the Lgr5hi cells and whole crypts. p-value was calculated by a one-sided hypergeometric distribution test. The computed probability of observing an overlap, given the background of 12,421 CpG islands, is far smaller than machine precision thus exact p-value cannot be reported due to numerical underflow in the calculation. c, Genomic views of the Dkk2 and Sfrp1 gene promoters showing the DNAm level in young and old Lgr5Hi ISCs. Each line is a replicate. Each rectangle is a CpG. d, Scatter plot of the average DNAm level of the ACCA drift genes and of the two marker genes DKK2 and SFRP1 (upper panel) or of the 4 marker genes DKK1, DKK2, SFRP1, SFRP2 (lower panel) in colon cancer samples. The gray line indicates linear regression; the shaded area represents the 95% confidence interval. e, Correlation coefficients between the average DNAm level of the ACCA drift genes and 100 random groups of either two (left panel) or four genes (right panel). The correlation coefficient with the average DNAm level of the two selected marker genes DKK2 & SFRP1 or four selected marker genes DKK1, DKK2, SFRP1, and SFRP2 is also shown, respectively.
Extended Data Fig. 4 The ACCA drift is cell-intrinsic and cell-cycle independent.
a, DNAm level of the Sfrp1 gene promoter in the different tissues at the indicated ages. n = 3 mice per group were analyzed. b, Schematic representation of the experimental set up. c, Bar chart indicating the DNAm level of the Dkk2 and Sfrp1 gene promoter analyzed by BS pyrosequencing in small intestinal (SI) and colon crypts and organoids at the indicated ages. n = 3 mice per group were analyzed. d, Bar chart indicating the DNAm level of the Dkk2 gene promoter analyzed by BS pyrosequencing upon long-term organoid culture of crypts isolated from young and old mice. n = 3 mice per group were analyzed. p-value was calculated by an unpaired t-test, 2-tails. e, Bar chart showing the DNAm level of the Sfrp1 gene promoter upon long-term organoid culture of crypts isolated from young and old mice. n = 3 mice per group were analyzed. p-value was calculated by an unpaired t-test, 2-tails. f, Boxplot indicating the DNAm level of the ACCA drift gene promoters in intestinal crypts isolated from young, old, and geriatric mice. For each age group, values are shown separately for female (1 F, 2 F, 3 F) and male (1 M, 2 M, 3 M) mice. n = 4 mice per group were analyzed. p-value was calculated by paired t-test, 2-tails. Box plots represent interquartile range with 5-95 percentile whiskers. g, Boxplot indicating the DNAm level of the ACCA drift gene promoters measured by RRBS upon long-term organoid culture of crypts isolated from young mice. n = 2 mice were analyzed. p-value was calculated by paired t-test, 2-tails. Box plots represent interquartile range with 5-95 percentile whiskers. h, Bar chart showing the DNAm level of the Sfrp1 gene promoter upon 1 month of organoid culture of crypts isolated from young and old mice and cultured in the indicated conditions. n = 3 mice per group were analyzed. p-value was calculated by an unpaired t-test, 2-tails. i, Bar chart indicating the DNAm level of the Sfrp1 gene promoter analyzed by BS pyrosequencing upon 1 month of organoid culture of crypts isolated from young mice and cultured in the indicated conditions as in Fig. 2l. n = 3 mice per group were analyzed. p-value was calculated by an unpaired t-test, 2-tails. j, Bar chart showing the DNAm level of the Sfrp1 gene promoter upon 1 month of organoid culture of crypts isolated from young and old mice and cultured in the presence of CHIR99021 or recombinant Wnt3a. n = 3 mice per group were analyzed. p-value was calculated by an unpaired t-test, 2-tails. Error bars in the figure bar charts represent the SD.
Extended Data Fig. 5 The DNAm drift expands via crypt clonality and shows heterogeneity at the CpG level.
a, The DNAm level of the Dkk1, Sfrp1, and Sfrp2 gene promoters analyzed by BS pyrosequencing in 59, 45, and 78 single crypts isolated from young and old mice, respectively (as indicated). Each circle represents the DNAm level (in percentage). The failed DNAm analysis is represented by dashes. n = 3 mice per group were analyzed. b, The DNAm level of the β-Actin (in 60 young and 72 old single crypts) and Dppa3 (in 25 young and 45 old single crypts) gene promoters analyzed by BS pyrosequencing in small intestinal crypts isolated from young and old mice. n = 3 mice per group were analyzed. p-value was calculated by Welch’s t-test 2-tails. Error bars represent the SD. c, Violin plot showing the DNAm level of the Dkk1, Sfrp1, and Sfrp2 gene promoters in single intestinal crypts isolated from young and old mice as in (a). d, Hierarchical clustering and heatmap of the Pearson correlation of the DNAm profiles of the Dkk1 gene promoter of 59 single crypts. e, The DNAm profile of the Dkk1 gene promoter in the crypts from the Cluster 1 (crypts isolated from young mice - ‘young’ crypts cluster) (left panel). The DNAm profile of the Dkk1 gene promoter in the crypts from the Cluster 5 (crypts isolated from old mice - ‘old’ crypts cluster) (right panel). f, Histogram plot showing the average DNAm level of three genes analyzed in single crypts as in (a) and Fig. 3b.
Extended Data Fig. 6 The DNAm drift expands via crypt fission.
a, A representative picture of laser capture microdissection (LCM) of single intestinal crypts before (upper panel) and after (lower panel) cut. b, The DNAm profiles of the Dkk2 gene promoter in two independent intestinal regions of 11 neighboring crypts (region C – left panel, region D – right panel). The red arrows indicate neighboring crypts with very similar DNAm profiles. c, Correlation heatmap of the neighboring crypts as in (b). d, The DNAm profiles of the Dkk1 gene promoter in four independent intestinal regions of 16, 8, 11, and 11 neighboring crypts (the same crypts/regions as for the Dkk2 gene). The red arrows indicate neighboring crypts with very similar DNAm profiles. e, Correlation heatmap of the neighboring crypts as in (d). f, Bar chart showing the Pearson correlation of the DNAm profiles of the Dkk1 gene promoter in the randomly picked (n = 45) and/or neighboring (n = 42) crypts. p-value was calculated by Mann-Whitney test, 2-tails. Error bars in the figure bar charts represent the SD. g, Bar chart showing the Pearson correlation of the DNAm profiles of the Dkk1 gene promoter in the crypts with high (n = 20) and low (n = 20) correlation of the DNAm profiles of the Dkk2 gene promoter. p-value was calculated by Welch’s t-test, 2-tails. Error bars in the figure bar charts represent the SD. h, Correlation heatmap of the crypts in clusters as in the Fig. 4j. i, Correlation heatmap of the scattered crypts as in the Fig. 4k.
Extended Data Fig. 7 The crypts with high DNAm drift have altered iron homeostasis and the TET enzymatic activity.
a, The expression level of the Dnmt enzymes in high (red dots; n = 16) and low (blue dots; n = 12) 5mC crypts based on RNAseq as in Fig. 5b. b, The expression level of the Tet enzymes in high (red dots; n = 16) and low (blue dots; n = 12) 5mC crypts based on RNAseq as in Fig. 5b. c, Gene ontology analysis of the differentially expressed genes (DEGs) between the crypts with high (the top 10%) and low (the bottom 10%) DNAm as in Fig. 5b. p-value was calculated by one-sided Fisher’s Exact test. d, Transferrin receptor (Tfrc) and ferroportin (Slc40a1) expression levels in the Lgr5hi cells isolated from young and old mice. n = 3 mice per group were analyzed. p-value is calculated by DESEQ2. e, Quantification of the western blot analysis as in Fig. 5e. n = 5 mice per group were analyzed. p-value was calculated by Welch’s t-test, 2-tails. f, ChIP-qRT-PCR analysis of the ChIP of TET1, TET2 and TET3 on the Dkk2 gene promoter in the intestinal crypts isolated from young and old mice. n = 3 mice per group were analyzed. IgG was used as a control. p-value was calculated by Welch’s t-test, 2-tails. g, ChIP-qRT-PCR analysis of the ChIP of TET1, TET2 and TET3 on the Sfrp1 gene promoter in the intestinal crypts isolated from young and old mice. n = 3 mice per group were analyzed. IgG was used as a control. p-value was calculated by Welch’s t-test, 2-tails. Error bars in the figure bar charts represent the SD.
Extended Data Fig. 8 The crypts with high DNAm drift have altered iron homeostasis and the TET enzymatic activity.
a, hMeDIP analysis of the Dkk2 gene promoter in the intestinal crypts isolated from young and old mice. n = 3 mice per group were analyzed. p-value was calculated by Welch’s t-test, 2-tails. b, hMeDIP analysis of the Sfrp1 gene promoter in the intestinal crypts isolated from young and old mice. n = 3 mice per group were analyzed. p-value was calculated by Welch’s t-test 2-tails. c, Schematic representation of functional experiments using organoid culture. d, hMeDIP analysis of the Dkk2 and the Sfrp1 gene promoters in the organoids treated with the C35 TET inhibitor for 1 week. n = 3 mice per group were analyzed. p-value was calculated by Welch’s t-test, 2-tails. e, Bar chart showing the DNAm level of the Sfrp1 gene promoter analyzed by BS pyrosequencing at the indicated time points upon the C35 TET inhibitor treatment of organoids derived from young intestinal crypts. n = 3 mice per group were analyzed. p-value was calculated by Welch’s t-test, 2-tails. f, Bar charts showing the DNAm level of the Sfrp1 and Sfrp2 gene promoters in intestinal crypts isolated from WT and Tet2/3-dKO mice. n = 3 mice per group were analyzed. p-value was calculated by Welch’s t-test, 2-tails. g, Bar chart showing the DNAm level of the Sfrp1 gene promoter analyzed by BS pyrosequencing in organoids treated with the iron chelator desferoxamine (DFO) for 1 and 2 months. n = 3 mice per group were analyzed. p-value was calculated by paired t-test, 2-tails. h, Bar chart showing the DNAm level of the Sfrp1 gene promoter analyzed by BS pyrosequencing in the intestinal organoids as in Fig. 6a. Parental organoids = untransduced organoids at day 0. n = 3 mice per group were analyzed. p-value was calculated by Welch’s t-test, 2-tails. Error bars in the figure bar charts represent the SD.
Supplementary information
Supplementary Information (download PDF )
Supplementary Discussion, Methods (quantification and statistical analyses) and References.
Supplementary Table 1 (download XLSX )
List of the gene promoters hypermethylated in human aging.
Supplementary Table 2 (download XLSX )
Gene expression of the ACCA drift genes in CRC.
Supplementary Table 3 (download XLSX )
Expression of the genes correlated with ACCA drift gene methylation.
Supplementary Table 4 (download XLSX )
List of DM promoters in mouse aging.
Supplementary Table 5 (download XLSX )
List of primers used in this study.
Supplementary Table 6 (download CSV )
The clinical data for the collected samples from the TCGA database.
Source data
Source Data Figs. 4 and 5 (download PDF )
Greyscale images of micrographs for Fig. 4 and unprocessed western blots for Fig. 5.
Source Data Fig. 6 (download PDF )
Unprocessed western blots for Fig. 6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Krepelova, A., Rasa, M., Annunziata, F. et al. Iron homeostasis and cell clonality drive cancer-associated intestinal DNA methylation drift in aging. Nat Aging 5, 2432–2448 (2025). https://doi.org/10.1038/s43587-025-01021-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s43587-025-01021-x
This article is cited by
-
Revealing epigenetic drift in intestinal pathogenesis with age
Nature Aging (2025)
