
Abstract
Background
Most women experience hot flashes (hot flushes) during the menopause transition. Menopausal hot flashes typically persist for years, and for a sizeable minority of women, are substantially impairing. Genetic investigations can improve understanding of hot flash etiology.
Methods
We conducted a trans-ancestry genome-wide association study (GWAS) of hot flashes (N = 149,560) among post-menopausal women age 35-88. The outcome variable was self-reported hot flashes in four samples (total n = 42,489) and menopausal hormone therapy as a proxy in one sample (n = 107,071). We estimated the heritability of hot flashes and genetic correlations with psychiatric phenotypes using linkage disequilibrium score regression.
Results
In our trans-ancestry meta-analysis, the top locus lies on chromosome 4 in the neurokinin 3 receptor gene (TACR3, p = 7.2×10-41). We also identify another locus on chromosome 4 with top SNP rs13107507 (p = 3.5×10-8). Gene results implicate TACR3, GRID1, NUDT4, and PHF21B. SNP heritability is estimated to be 8% (h2liab = .08, h2SNP = .04, se = .02). Genetic correlations are statistically significant between hot flashes and PTSD (rg = 0.25, p = 0.01), schizophrenia (rg = 0.17, p = 0.02), depression (rg = 0.21, p = 0.01), and ADHD (rg = .22, p = 0.03).
Conclusions
These genomic findings are consistent with independent, robust basic science research which led to a recently developed treatment for hot flashes, namely, a neurokinin 3 receptor antagonist. This non-hormonal class of hot flash drugs blocks the receptor coded for by the top locus reported here (TACR3, the neurokinin 3 receptor gene). Hot flash GWAS results provide an example of how GWAS findings can point to potent treatment targets for complex brain phenotypes.
Plain Language Summary
Hot flashes are sudden sensations of heat that are often accompanied by sweating, flushing, and chills. Most women experience hot flashes during the menopause transition, and for some, they occur for many years, are severe, impact sleep patterns and impair functioning in everyday life. This study aimed to investigate genetic influences on hot flashes to gain insight into their causal mechanisms and potential treatment targets. Using data from a large sample of post-menopausal women, we identified a genetic risk factor on chromosome 4 in the TACR3 gene that is significantly associated with hot flashes. This gene encodes a receptor, which is the target of a recent FDA approved drug that effectively treats hot flashes (fezolinetan/Veozah). These results provide an example of how genetic analyses that scan the human genome can point to mechanisms and clinically useful treatment targets.
Similar content being viewed by others


Introduction
The experience of a hot flash includes the sudden onset of heat, typically in the upper body, which may be accompanied by sweating, flushing, and chills1,2. Hot flashes, also called hot flushes, are commonly experienced by ~70% of women after menopause, but over half of women also experience hot flashes in the years leading up to menopause (i.e., during the late menstrual transition)1,3,4. Menopause is defined as the point at which a woman has not experienced a menstrual period for 12 consecutive months, and the average age of menopause is 51 years. Though commonly referred to separately, it has been noted that night sweats are likely hot flashes experienced during sleep2. Collectively, hot flashes and night sweats are known as vasomotor symptoms. Throughout this manuscript, we will use the term hot flashes for simplicity of phrasing and given that the term “vasomotor symptoms” is less well known.
Along with hot flashes, women often experience changes in mood and cognition during menopause. Though more research is needed, a recent review of psychiatric disorders and menopause highlighted that women with a previous diagnosis of major depressive disorder (MDD) may be at an increased risk for depressive symptoms and recurrence of MDD during menopause5. In addition, symptoms of depression, post-traumatic stress disorder (PTSD), and somatic anxiety have been found to increase risk for menopausal hot flashes6,7,8.
One of the most misunderstood aspects of menopause is the severity and duration of its symptoms, which can persist for many years for a large fraction of women. Accordingly, many women leave the workforce because of menopausal symptoms including hot flashes9,10,11. The economic impact of menopausal symptoms is substantial, with an estimated annual cost of $1.8 billion attributed solely to missed workdays9. Historically, menopausal symptoms have been downplayed, ignored, and inadequately treated. Recognizing the importance of addressing these issues, a 2024 executive order allocated $12 billion for Women’s Health Research and Innovation, with a focus on advancing menopause research as a critical component. Given that menopause is a core life stage for women, it is critical to further elucidate the genetic underpinnings of symptoms commonly experienced during the menopausal transition9.
In the first genome-wide association study (GWAS) conducted for hot flashes, Crandall et al.12 found a significant locus in the TACR3 gene in a trans-ancestry meta-analysis of 17,695 post-menopausal women from the Women’s Health Initiative (WHI). The TACR3 locus was replicated in a sample from the Study of Women Across the Nation13 and in women from the UK Biobank with linked primary health care records14. Because many samples fail to gather data about hot flashes, some studies have used proxy variables for hot flashes. In previous work using the UK Biobank, we performed a GWAS of estrogen medication usage and also identified TACR3 as the only significant locus15.
Given somewhat limited research about the genetics of hot flashes to date, it is particularly noteworthy that prior results are consistent with the known biology of hot flashes. Specifically, the TACR3 gene encodes the NK3R receptor that binds neurokinin B (NKB, also known as neurokinin 3), and NKB signaling is a causal component in a neural circuit that is sufficient for generating hot flashes16. Evidence supporting the critical role of NKB signaling in hot flashes comes from multiple complementary sources: human post-mortem tissue studies17,18, animal studies16,19, and mechanistic studies in humans20. This body of work led to the development of a non-hormonal medication treatment for hot flashes that blocks the NK3R receptor, which was approved by the Food and Drug Administration (FDA) in 2024 (fezolinetant)21,22,23,24,25.
Here, we report a GWAS of hot flashes, incorporating five cohorts and 149,560 women in a trans-ancestry meta-analysis. In addition to Crandall et al.’s prior GWAS in the WHI12, we conduct GWAS in the Nurses’ Health Study (NHS), the Nurses’ Health Study II (NHSII), and in a sample from the Queensland Institute of Medical Research (QIMR). We also conduct a GWAS of menopausal hormone therapy (MHT), formerly known as hormone replacement therapy, use in the UK Biobank as a proxy for hot flashes; this builds upon our prior work using a less specific medication variable as a proxy for hot flashes15. Here we seek to (1) identify loci and genes for hot flashes, (2) estimate SNP heritability for hot flashes, and (3) quantify genetic correlations between hot flashes and psychiatric phenotypes.
Firstly, in component analyses and the trans-ancestry meta-analysis, the top locus lies on chromosome 4 in the TACR3 gene (trans-ancestry p = 7.2 × 10−41). We identify another locus on chromosome 4 with top SNP rs13107507 (p = 3.5 × 10−8), and gene results implicate TACR3, GRID1, NUDT4, and PHF21B. Secondly, we estimate SNP heritability to be 8% (h2liab = 0.08, h2SNP = 0.04, se = 0.02). Thirdly, genetic correlations are statistically significant between hot flashes and PTSD (rg = 0.25, p = 0.01), schizophrenia (rg = 0.17, p = 0.02), depression (rg = 0.21, p = 0.01), and ADHD (rg = 0.22, p = 0.03). Taken together, these findings are consistent with separate lines of basic science research that led to the successful development of a non-hormonal treatment for hot flashes (via blockade of the neurokinin 3 receptor) and with epidemiological evidence about the co-occurrence of menopause and psychiatric symptoms.
Methods
This GWAS meta-analysis includes GWAS analyses that we conducted (in NHS, NHSII, QIMR, and UK Biobank) and summary statistics from the published GWAS by Crandall et al. (for WHI)12. Major methodological elements are described below and additional details are provided in the Supplementary Materials.
Samples
Data from 149,560 post-menopausal women were included in this analysis. To be included, women needed to be post-menopausal (per self-report, as is the standard for defining menopause). Women also needed to have answered questions about the experience of having hot flashes and/or night sweats affirmatively or negatively (i.e., women with ambiguous responses were excluded). In the UK Biobank, affirmative or negative responses for the use of MHT were required. Women also needed genome-wide genotype data passing quality control procedures to be included (see below and see Supplementary Materials for further details). Figure 1 presents an overview of sample sizes and ancestries for each cohort; Table 1 presents sample characteristics. The authors applied for access to each dataset used in this study (see Supplementary Materials for information on how to apply). Ethical approval and participant consent were obtained according to each cohort’s institutional protocols. This work was deemed by Stanford University’s Institutional Review Board to not be human subjects research. The relevant institutional review boards (IRBs) and protocols (where applicable) are as follows: The UK Biobank received ethics approval from the Research Ethics Committee (REC reference for UK Biobank is 11/NW/0382). The NHS and NHSII study protocols were approved by the IRBs of the Brigham and Women’s Hospital and the Harvard T.H. Chan School of Public Health. Ethical clearance (P3724) for use of QIMR samples was obtained from the QIMR Human Research Ethics Committee.

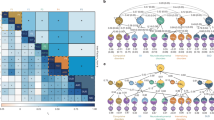
A GWAS samples by cohort with sample sizes. Solid borders denote studies that asked women about hot flashes as contrasted with dashed borders denoting the UK Biobank samples that did not ask women about hot flashes, but for whom women reported use (or not) of menopausal hormone therapy (MHT). Border colors represent major distinctions regarding ancestry, as quantified using 1000 Genomes Project groups as reference56. B Depiction of each meta-analysis conducted by component samples. NHS Nurses’ Health Study, NHSII Nurses’ Health Study II, QIMR Queensland Institute of Medical Research, WHI Women’s Health Initiative, EUR European ancestry, EAS East Asian ancestry, AFR African ancestry, SAS South Asian ancestry, AMR Hispanic/Latino ancestry.
Phenotypic measures
Across all samples, the outcome phenotype was a binary yes/no variable denoting any experience of a hot flash or night sweat, and in the UK Biobank a binary yes/no variable denoting use of MHT. In the NHS, experiences of hot flashes were queried in a 2008 questionnaire which asked women to report whether “at the beginning of menopause, did you have hot flashes or night sweats? (if you took estrogen, consider the time period before starting the treatment)”. The same question regarding hot flashes was asked in NHSII participants in 2009, 2013, and 2017. In the UK Biobank baseline assessment (2006–2010), women were asked whether they had ever used MHT (with no further specifications, e.g., about oral vs. transdermal, estrogen with or without a progestogen); women who responded “do not know” or “prefer not to say” were set to missing and therefore not included in the sample definition. In QIMR, women were drawn from two sources and queried either from 1989 to 1992 or in 1996. The questions, respectively, were: “Please tell us if you had any of the following symptoms of menopause that women sometimes experience: hot or sudden flushes or sweats”26 and “number and intensity of hot flushes and accompanying sweats, often at night?”27 Questions were answered on a 1–4 frequency scale with 1 denoting “not at all”. Responses were re-coded as a binary variable (yes/no, for experiencing hot flashes) to better match coding of data from the other cohorts.
Genotyping, imputation, quality control
Genotyping was performed using blood samples collected from 2006 to 2010 in UK Biobank, 1989–1990 in NHS, 1996–1999 in NHSII, and from 2009 onwards in QIMR12,26,27,28,29,30. In brief, data for our samples were genotyped using two arrays in the UK Biobank (UK BiLEVE, UK Biobank Axiom array), five in NHS (Illumina HumanHap Array, Illumina OncoArray, Illumina HumanCore Exome Chip, Illumina OmniExpress, Affymetrix 6.0), and three in NHSII (Illumina HumanHap Array, Illumina OncoArray, Illumina HumanCore Exome Chip). QIMR used HapMap-based (370K, 610K, 660K) or 1000-Genomes-based (Core+Exome, PsychArray) Illumina SNP arrays. Genetic data were imputed to the Haplotype Reference Consortium (HRC) reference panels31, and UK Biobank data was also imputed using the UK10K panel. Standard GWAS quality control procedures were conducted (see Supplementary Materials for details).
Statistics and reproducibility
Software summary. We conducted genome-wide association studies using logistic regression in PLINK 2.032 and in SAIGE 0.45 (QIMR only). Meta-analyses of GWAS results were conducted using METAL (version released 2011-03-25)33. Power calculations were conducted using the genpwr package34 in R (v4.0.5)35. Mendelian Randomization was conducted using MRBase36. R (v4.0.5)35 was used for other analyses and to create figures. FUMA was used for the SNP Manhattan plot37, and LocusZoom was used for the region plot38.
GWAS and meta-analyses. In NHS and NHSII cohorts, GWAS was conducted separately for each array and by batch for data from the Illumina HumanCore Exome Chip in NHSII28. In the UK Biobank, GWAS was conducted for each ancestry group (see Supplementary Fig. S1 for ancestry assignment). All GWAS were adjusted for principal components according to norms in each dataset (WHI = 10, NHS = 10, QIMR = 4, and UK Biobank = 20). GWAS in the UK Biobank were additionally adjusted for the Townsend deprivation index and the difference in years between each participant’s age at menopause and 2002 (to account for potential effects of WHI findings about MHT, released in 2002, on women’s likelihood of using MHT). Note that we found no evidence of differences in genetic effect sizes related to timing of menopause before or after 2002, when the paradigm-changing WHI findings were released39. GWAS in QIMR included covariates for imputation run (see Supplementary Materials for details). The chromosome and position of SNPs is reported here according to genome build GRCh37/hg19.
Meta-analyses. We ran multiple rounds of meta-analyses across outcome phenotypes and ancestry groups (see Fig. 1 and Supplementary Materials). Our main analysis incorporated the largest number of women (N = 149,560), thus including all ancestries available (trans-ancestry) and both phenotypic outcomes (hot flashes and the MHT proxy). For all meta-analyses, we used METAL (version released 2011-03-25)33. A fixed-effects meta-analysis with inverse variance weighting was conducted. Note that no additional loci were identified when we conducted a variable effects meta-analysis.
Post-hoc power calculations for component samples. We sought to determine whether we had the power to detect the known TACR3 locus in component samples. Power calculations were conducted using the genpwr package34 in R (v4.0.5)35 with the corresponding sample size, case rate, and minor allele frequency for the component samples. The estimated effect size for the TACR3 locus was OR = 1.63 for hot flashes and OR = 1.18 for the MHT proxy. Alpha was set at p < 5 × 10−8.
Gene-level analyses. Gene p-values were obtained with MAGMA (v1.10)40 and plots were generated using R (v.4.0.5)35.
SNP heritability and genetic correlations. We used linkage disequilibrium score regression (LDSC)41 to estimate SNP heritability and to examine genetic correlations with psychiatric phenotypes. We used the meta-analysis of hot flashes in European ancestry samples (n = 32,979; see Fig. 1) for these analyses because LDSC requires non-admixed ancestry for samples, and because this was the best powered analysis (see power calculations). We analyzed genetic correlations with psychiatric phenotypes that have well powered GWAS, using publicly available GWAS data for PTSD42, depression43, bipolar disorder44, schizophrenia45, and attention-deficit/hyperactivity disorder (ADHD)46. We also analyzed genetic correlations with age at menopause47 and testosterone levels in a female-only sample48 to determine whether these phenotypes had genetic overlap with hot flashes. We further included a sensitivity analysis in which h2SNP and genetic correlations were estimated after removing the top locus (chromosome 4:104,424,934-104,801,645).
Mendelian randomization. We used Mendelian Randomization (MR) to gain insight into possible causal relationships between hot flashes and psychiatric phenotypes. In MR, SNPs that are strongly associated with a phenotype of interest serve as instruments to estimate the putative causal effect of one phenotype on other phenotypes. For a SNP to be considered a valid instrument in MR, it must at least satisfy three main conditions: (i) the SNP is associated with the exposure phenotype of interest; (ii) it affects the outcome solely through that exposure; and (iii) it is not linked to any third factor that influences both the exposure and outcome, thereby avoiding horizontal pleiotropy. MR was performed using data from our European ancestry hot flash meta-analysis (n = 32,979), as well as European ancestry data for several other phenotypes (see Supplementary Data S1 for details). Instrumental variables for hot flashes were chosen from those present across traits examined by clumping genome-wide suggestive SNPs using PLINK (p < 5 × 10−6, 10,000 Kb and r2 < 0.001). The causal relationship between trait and hot flashes was tested using two-sample MR with the MRBase package36, employing inverse-variance weighted (IVW) MR, which combines a Wald-type estimator for each instrument in a multiplicative random effects model. To ensure consistency in the results, sensitivity analyses were performed using three additional MR methods: MR Egger, Weighted Median, and Weighted Mode. In brief, MR-Egger is similar to the IVW method but adjusts for directional horizontal pleiotropy of the SNPs used as instruments; the weighted median accounts for up to half of the instrument SNPs having a pleiotropic effect; and the weighted mode is the mode of the Wald-type estimates used in the IVW, weighted by standard error-based weights for each SNP. As a sensitivity analysis, all MR analysis was repeated using the hot flash GWAS excluding the TACR3 locus.
Extended analyses
During the course of work on this project, Ruth et al.14 published a GWAS of hot flashes using UK Biobank data. This analysis included 92,028 women, with hot flashes reported in linked primary care records as a phenotype. They included only European ancestry individuals. We used the summary statistics from Ruth et al. to compare and extend our results. Specifically, we compared effect sizes of our top locus with Ruth et al., and we used the Ruth et al. dataset in place of our UK Biobank results to conduct an alternative meta-analysis and gene-level analyses.
Results
Here, we present GWAS results for our trans-ancestry hot flash + MHT proxy meta-analysis (N = 149,560). We also present gene-level results for the UK Biobank European ancestry MHT proxy sample (n = 103,370) and the European ancestry hot flash + MHT proxy meta-analysis (n = 136,349). Finally, we present genetic correlations with psychiatric phenotypes, which were calculated using the European ancestry hot flash meta-analysis (n = 32,979). The Supplementary Materials include results from Mendelian Randomization analysis, which was also performed on the European ancestry hot flash meta-analysis (n = 32,979), as well as methods and results for polygenic risk score (PRS) analysis.
GWAS and GWAS meta-analyses
Figure 2A presents the Manhattan plot for our trans-ancestry hot flash + MHT proxy meta-analysis (N = 149,560). Overall, we found the strongest association for rs74827081, which lies in the TACR3 gene on chromosome 4 (p = 7.2 × 10−41, OR = 1.27, 95% CI = 1.23–1.32, position 104,556,732). This SNP has the highest posterior probability of causality for this locus (0.517, per LocusZoom)38. All significant (p < 5 × 10−8) SNPs in this locus are in introns of the TACR3 gene or upstream of TACR3 (see Fig. 2B). Nearby GWAS Catalog hits include age at menarche, heel bone mineral density, and testosterone concentrations49.

A Manhattan plot showing statistical significance for each SNP (-log10(p)) for the trans-ancestry hot flash + MHT proxy meta-analysis (N = 149,560). The red dashed line denotes the threshold for statistical significance (p < 5 × 10−8), which has been corrected for multiple tests. B Region plot with color denoting linkage disequilibrium between the top SNP (rs74827081) and nearby SNPs. The GWAS Catalog hits bar shows previously reported associations in this region. Both plots show the results of a meta-analysis of genome-wide association studies, which is a two-sided test. LD linkage disequilibrium, Mb megabase, cM centimorgan.
Regarding other loci, we found that rs148680409 in the NLGN1 gene was genome-wide significant (p = 6.1 × 10−9). However, as the SNP was only measured in the WHI data, we could not confirm this result in any other samples, and this is not considered a high-confidence result.
The third locus had only one significant SNP (rs13107507, p = 3.5 × 10−8) which did not intersect any genes but was nearest to the LINC02428 gene. This SNP is 0.3 Mb away (located at 4:104244426) from our top SNP but may represent a distinct signal given there are two recombination peaks in between the two SNPs (see Fig. 2B). Because the rs13107507 locus and TACR3 locus are relatively close to one another (albeit on opposite sides of two recombination peaks), it is possible that the rs13107507 locus has some as-yet-unspecified regulatory role on the TACR3 gene. Nearby GWAS Catalog hits include testicular cancer and male puberty timing. One further SNP on the X chromosome, located at 23:123,080,030, was close to reaching genome-wide significance (p = 7.6 × 10−8). Note that X chromosome data was only available for the UK Biobank and QIMR datasets. See Supplementary Fig. S2 for region plots for these additional loci.
Figure 3 presents a forest plot of effect sizes for top SNP rs74827081. The effect size from the hot flash + MHT proxy meta-analysis (OR = 1.27) is in between the effect size for hot flashes (OR = 1.63, 95% CI = 1.52–1.75) and the MHT proxy variable (OR = 1.18, 95% CI = 1.13–1.23), consistent with expectation of an attenuated effect size for the proxy variable (i.e., 1.63 to 1.18). For top SNP rs74827081 (G/C), the Gallele increases the risk of hot flashes and likelihood of MHT use. It is notable that the effect size for the TACR3 locus (OR = 1.63) is much larger than effect sizes for any other variants that impact hot flashes, including rs13107507 (OR = 1.43, see Supplementary Fig. S3 for a forest plot of effect sizes for this variant). Prior to this analysis, it was not known that the TACR3 locus had an effect size considerably larger than other variants that influence hot flash liability. In other words, one might have expected other hot flash loci to have effect sizes similar to the TACR3 locus, and a corresponding Manhattan plot with peaks of more similar heights, rather than what is observed here, namely, one locus that is over 30 orders of magnitude more significant than all the others.

Odds ratios are plotted as diamonds. Large diamonds denote meta-analytic results as contrasted with small diamonds for component samples. Color denotes ancestry according to 1000 Genomes Project classifications, and grey represents trans-ancestry meta-analyses56. Each odds ratio is plotted along with its 95% confidence interval (as needed, arrows indicate that the interval extends beyond the range of the plot). Samples are grouped by outcome variable. The first grouping includes all hot flash samples. The second grouping includes all samples with the MHT proxy variable. The third is the combined meta-analysis of hot flash and proxy samples. Note that the final sample size (n = 148,929) is smaller than the full sample size (N = 149,560) because this locus was not available in the East Asian subsample (n = 631).
The QIMR and UK Biobank South Asian ancestry samples, which did not yield significant results (p = 0.82 and p = 0.41, respectively), have odds ratios less than 1 for the TACR3 locus (0.92 and 0.8, respectively). Given that these results are insignificant, this is very likely due to low power: these samples have 0.01% and 0.00% power to detect the TACR3 locus, respectively (see Table 2). With such low power for these analyses and correspondingly wide confidence intervals, low statistical power is an entirely sufficient explanation for odds ratios that indicate the opposite direction of effect.
In gene-level analysis, we identified two other genes besides TACR3 that were significantly associated with hot flashes: the GRID1 gene on chromosome 10 and the NUDT4 gene on chromosome 12 (see Fig. 4). Full gene-level results are presented in Supplementary Data S2 and S3.

Gene Manhattan plots for A UK Biobank European ancestry MHT proxy sample (n = 103,370) and B European ancestry hot flash + MHT proxy meta-analysis (n = 136,349). The grey line denotes the threshold for statistical significance (p < 2.6 × 10−6), which has been corrected for multiple tests (19,171 genes). Both plots show the results of a gene association analysis, which is a two-sided test.
To demonstrate real-world correlates of the effect of TACR3 genotypes on women’s likelihood of developing hot flashes, we obtained the prevalence of hot flashes by rs74827081 genotype in the NHS and NHSII datasets, to which we had access to individual level data (see Fig. 5). In this sample, 57% of women with two protective alleles experienced hot flashes (CC), as contrasted with 72% of women with one risk allele (CG), and 80% of women with two risk alleles (GG). Using population allele frequencies from gnomAD50, we note that the “high-risk” genotype (GG) is the most common globally; this genotype ranges in frequency from ~89% in European ancestry women to ~100% in East Asian women.

A Percentage of women who experienced hot flashes (shaded in red) as a function of rs74827081 genotype in NHS and NHSII. B The frequency of each genotype across ancestries as calculated using frequencies from the gnomAD database.
Post-hoc power calculations
Power calculations for component datasets demonstrate that significant effects were identified as expected according to statistical power. As shown in Table 2, at alpha = 5 × 10−8, power was very low (less than 5%) for all datasets in which we failed to detect the TACR3 locus (including all non-European ancestry samples), showing that this is a sufficient explanation for the samples that yielded non-significant results. However, using alpha = 0.05 for the WHI African ancestry sample and the WHI Hispanic/Latino ancestry sample, power was 69% and 91% respectively, and indeed, we found nominally significant associations for rs74827081 in these samples (p = 0.0012 and p = 0.00075, respectively).
SNP Heritability, genetic correlations, and Mendelian Randomization. Using LDSC, the estimated SNP heritability for hot flashes was h2SNP.liability = 0.08 on the liability scale (h2SNP.observed = 0.04, se = 0.02). Removing the TACR3 locus, the heritability estimate was very similar h2SNP.liability = 0.08 (h2SNP.observed = 0.04, se = 0.02). This value means that 8% of phenotypic variance in the likelihood of developing hot flashes is explained by variance in the SNPs analyzed in this study. Note that across complex trait genetics, it is typically the case that SNP heritability estimates are approximately ¼ the size of twin heritability estimates. Here, we used the liability scale because our phenotype is binary (yes/no hot flashes). To our knowledge, this is the first SNP heritability estimate for hot flashes, and we are not aware of any twin study heritability estimates for hot flashes.
Using LDSC, we found significant (p < 0.05) genetic correlations between hot flashes and PTSD (rg = 0.25, se = 0.09, p = 0.01), depression (rg = 0.21, se = 0.08, p = 0.01), schizophrenia (rg = 0.17, se = 0.07, p = 0.02), and ADHD (rg = 0.22, se = 0.10, p = 0.03); see Fig. 6. The genetic correlations changed very little when the TACR3 locus was removed (solid vs. dashed bars; all SNPs in this window removed for the latter: chromosome 4:104,424,934-104,801,645). This suggests that the shared genetic etiology between hot flashes and these other phenotypes is largely independent of the TACR3 locus and is instead attributable to more diffuse polygenic contributions shared between hot flashes and these other phenotypes. Neither the genetic correlation with age at menopause (rg = 0.04, se = 0.09, p = 0.67) nor the genetic correlation with testosterone levels (rg = 0.10, se = 0.09, p = 0.31) was significant, suggesting minimal shared genetic factors. After correcting for multiple testing, none of the Mendelian Randomization results were significant (see Supplementary Data S4). Similarly, none of the PRS results were significant (see Supplementary Data S5).

Genetic correlations were calculated between the European ancestry hot flash meta-analysis (n = 32,979 post-menopausal women) and the listed phenotypes. Each genetic correlation is plotted as a bar along with its 95% confidence interval; the measure of center for the error bars is the estimate of the genetic correlation. Each phenotype has two bars: the solid bar is the genetic correlation with the hot flash meta-analysis including all loci, while the striped bar excludes the top locus (4:104556732). PTSD: posttraumatic stress disorder, ADHD: attention-deficit/hyperactivity disorder.
Extended analyses. Our top SNP (rs74827081) in the Ruth et al.14 data had an effect size of 1.28 (p = 2.8 × 10−20, 95% CI = 1.22–1.36), which is between that of our proxy meta-analysis (OR = 1.18) and our hot flash meta-analysis (OR = 1.63). This suggests that Ruth and colleagues’14 outcome variable, hot flashes reported in linked primary care records, is a better proxy for hot flashes than self-reported MHT use, but still is not equivalent to self-reported hot flashes. We performed an alternative trans-ancestry hot flash + proxy meta-analysis, with Ruth et al. replacing our UK Biobank European ancestry data (n = 138,218). As expected, the TACR3 locus became even more significant (p = 2.7 × 10−52) compared to our original trans-ancestry hot flash + MHT proxy meta-analysis (p = 7.2 × 10−41). We also performed an alternative gene-level analysis, with Ruth et al. replacing our UK Biobank data in the European ancestry hot flash + proxy meta-analysis (n = 125,007); see Supplementary Data S6 for full results and Supplementary Fig. S4. TACR3 remained significant (p = 1.8 × 10−13) and an additional gene, PHF21B, was also significant (p = 2.1 × 10−7); to our knowledge this is the first time that PHF21B has been associated with hot flashes.
Discussion
In this study, we meta-analyzed GWAS of additional cohorts with prior hot flash GWAS and confirmed a highly significant locus in the TACR3 gene. We also identified another locus on chromosome 4 (with top SNP rs13107507). We also report three putative genes for hot flashes (GRID1, NUDT4, and PHF21B) and several psychiatric phenotypes that have statistically significant genetic correlations with hot flashes. Interestingly, these genetic correlations with multiple psychiatric disorders do not depend on the TACR3 locus. Rather, it is other polygenic contributions to hot flashes that overlap with genetic risk for psychiatric disorders.
Our main finding in the TACR3 gene is notable for being so clearly consistent with basic science research about hot flashes and with clinical trials data for a recently developed non-hormonal hot flash medication. Specifically, prior research demonstrated that the neurokinin B (NKB) receptor in the hypothalamus plays a causal role in hot flash generation. In rats, optogenetic stimulation of NKB-expressing neurons in the preoptic area induced a hot flash, and this effect was abolished when NKB receptors were chemically blocked19. Furthermore, in humans, intravenous administration of NKB in pre-menopausal women induced hot flashes20. This preclinical work led to the development of an NKB receptor antagonist that is highly effective at treating hot flashes and is now FDA approved21,22,23,24,25. Given that our top locus is in the TACR3 gene, and that the TACR3 gene encodes the NKB receptor, we see that this GWAS of hot flashes serves as a strikingly clear example of how GWAS can pinpoint causal biology and potent treatment targets51.
In addition to the TACR3 locus, we identified another locus on chromosome 4 with top SNP rs13107507. Because the rs13107507 locus is near the TACR3 locus (0.3 Mb away), we hypothesize that it may have some regulatory effect on TACR3 given its proximity to this gene that is known to be causally related to hot flash physiology. Further research is needed to determine how this locus may contribute to hot flashes and whether this effect occurs via effects on TACR3. Also of note was a nearly significant SNP on the X chromosome. This locus did not intersect any genes (closest protein coding gene STAG2). Given that menopause is a sex-restricted phenomenon, including X chromosome data will be particularly important for future analyses of hot flashes and other menopausal symptoms.
We sought to identify loci for hot flashes in non-European ancestry groups. Our analysis included GWAS results from six non-European ancestry sub-samples, and we conducted the first African ancestry meta-analysis. Unfortunately, power to detect the largest known effect size for hot flashes (TACR3 locus) was very low in these samples (i.e., <2% statistical power). This was not only due to smaller sample sizes, but also to the lower minor allele frequency for the risk allele in non-European ancestry populations. As an example, though the African Ancestry meta-analysis had a sample size of 8089, which is very close to the WHI European ancestry sample size of 8185 that was sufficient for the detection of the TACR3 locus, the minor allele frequency was ~1% in African ancestry samples as compared to European ancestry samples ~6%, and this resulted in inadequate power. Continuing to collect multi-ancestry data is a priority for future work in this space.
Gene level analyses identified the GRID1, NUDT4, and PHF21B genes as significant in European ancestry meta-analyses. The GRID1 gene codes for a glutamate receptor subunit, and the top tissues for GRID1 expression include several regions of the brain (including the hypothalamus), as well as the thyroid and uterus. The NUDT4 gene is involved in intracellular trafficking, and the top tissues for NUDT4 expression include the arteries, heart, esophagus, and ovary. Given expression in this set of tissues, it is possible that this gene may play a role in the vasodilation that occurs during a hot flash. The top tissues for PHF21B include the cervix, pituitary, and vagina. The data used for describing tissue-level expression of these genes was obtained from the GTEx Portal on 8/26/2452,53,54.
We identified several psychiatric phenotypes that have significant genetic correlations with hot flashes. The directions of these genetic correlations are consistent with epidemiological findings. For example, women with depression have been found to be more likely to experience hot flashes6, and the genetic correlation findings reported here suggest that shared genetic risk factors may partially explain this above-chance comorbidity. Regarding PTSD, both trauma and PTSD symptoms have been found to be associated with higher risk of hot flashes7. Further, these shared genetic influences on hot flashes and psychiatric disorders may help to explain why antidepressant medications are helpful for some women experiencing hot flashes, and vice versa, why hormonal medications are sometimes helpful for psychiatric symptoms.
Also notable is the observation that hot flashes have a genetic architecture that is more similar to cigarette smoking or alcohol consumption, in which the top loci are tens of orders of magnitude more significant than the many additional loci contributing to polygenic influences on these phenotypes51. In contrast, psychiatric disorders have polygenic influences without such striking high-end outlier loci. While genetic drift might account for these high-end outlier effect sizes, there may prove to be evolutionary explanations for these discrepant genetic architectures. Future studies could investigate the way in which shifting environmental landscapes across human history, occurring after ancestral human migrations out of Africa, may have led to different selection pressures across time and regions of the world. For example, different human ancestral populations had different exposure to substances including nicotine, alcohol, and opioids. Similarly, human lifespans may have differed across populations; it is possible that longer lifespans in women led to “unmasking” of selection pressures on post-menopausal functioning in women. Note that, even after the childbearing years, women continue to contribute to survival and reproductive success of their offspring.
Regarding limitations, it is true that MHT is used for symptoms other than hot flashes. It is also only used by a minority of women, whereas most women experience hot flashes. For these reasons, MHT use is an imperfect proxy for hot flashes, and we expected to have false negatives in our proxy variable (women who actually had hot flashes, but were not taking MHT). Consistent with this, the effect size for the top locus for the MHT proxy was much smaller than the effect size for hot flashes (OR = 1.18 vs. 1.63, with non-overlapping 95% confidence intervals). Nevertheless, the proxy variable used here (MHT) yielded the same top locus (TACR3). Similarly, we found that the phenotype used by Ruth et al.14., hot flashes reported through linked primary care records, was better than the MHT proxy but was still not equivalent to self-reported hot flashes (OR = 1.28). Nevertheless, including Ruth et al. in our alternative trans-ancestry meta-analysis increased the significance of the TACR3 locus by ten orders of magnitude, and led to the detection of one additional gene for hot flashes.
Additionally, we were not able to include anxiety in our genetic correlation analysis because there currently is not a well-powered GWAS of anxiety for which the data is publicly available. Somatic anxiety has been associated with an increased risk for experiencing moderate to severe menopausal hot flashes8. Given this, it will be important to address potential shared genetic liability for anxiety and hot flashes in future work.
In medical genetics, there are many intronic variants statistically associated with diseases, but few strong examples of how those intronic variants influence the gene’s function. In the unusual example reported here, we have very high confidence that TACR3 is the true risk gene for hot flashes, given what is already known about how this receptor (neurokinin B), in a particular brain region (hypothalamus), causes hot flashes (as demonstrated in human and animal studies), and finally that blocking this receptor (with neurokinin B antagonists) prevents hot flashes. Thus, we can be nearly 100% certain that TACR3 is the risk gene, and therefore we have a special opportunity to investigate how intronic variation impacts TACR3 functioning. Changes caused by intronic variation in TACR3 could include increased or decreased receptor abundance, altered spatial distribution of receptors, and changes in receptor conformation, etc. These alternatives could be explored in human brain tissue using spatial transcriptomics to identify cell types of interest, combined with protein labeling to visualize neurokinin B receptors. This can be done in brain tissue samples for which TACR3 genotypes are known. Studies like this would reveal how the brains of women with the three risk genotypes (CC, CG, and GG) differ in terms of neurokinin B receptor abundance, distribution, and conformation.
In summary, to our knowledge, this is the largest trans-ancestry GWAS of hot flashes to date. We replicate a prior chromosome 4 locus and report another locus on chromosome 4 with top SNP rs13107507. Moreover, our top locus in the TACR3 gene points directly to an effective treatment target for a complex brain phenotype. Specifically, this locus is in the neurokinin 3 receptor gene, and a neurokinin 3 receptor antagonist (fezolinetant) was recently FDA approved for the treatment of moderate to severe hot flashes55. We also identified several potential genes for hot flashes (GRID1, NUDT4, and PHF21B), and found significant genetic correlations between hot flashes and psychiatric phenotypes.
Data availability
Cohort data may be made available upon reviewed requests from WHI, NHS, and UK Biobank, and further details relevant to this project are provided in the Supplementary Materials. Genome-wide summary data from the present study can be downloaded from GWAS Catalog using the following accession codes: GCST90570521 (European ancestry hot flash meta-analysis), GCST90570522 (European ancestry hot flash + MHT proxy meta-analysis), GCST90570523 (African ancestry meta-analysis), GCST90570524 (trans-ancestry hot flash meta-analysis), GCST90570525 (trans-ancestry MHT proxy meta-analysis), and GCST90570526 (trans-ancestry hot flash + MHT proxy meta-analysis). The source data for Fig. 2 can be downloaded from GWAS Catalog using the accession code GCST90570526 (trans-ancestry hot flash + MHT proxy meta-analysis). The source data for Fig. 4A is in Supplementary Data S2, and the source data for Fig. 4B is in Supplementary Data S3.
References
Bansal, R. & Aggarwal, N. Menopausal hot flashes: a concise review. J. -life Health 10, 6 (2019).
Kronenberg, F. Hot flashes: epidemiology and physiology. Ann. N. Y. Acad. Sci. 592, 52–86 (1990).
Gold, E. B. et al. Longitudinal analysis of the association between vasomotor symptoms and race/ethnicity across the menopausal transition: study of Women’s Health Across the Nation. Am. J. Public Health 96, 1226–1235 (2006).
Harlow, S. D. et al. Executive summary of the Stages of Reproductive Aging Workshop + 10: addressing the unfinished agenda of staging reproductive aging. Menopause 19, 387–395 (2012).
Brown, L. et al. Promoting good mental health over the menopause transition. Lancet 403, 969–983 (2024).
Reed, S. D. et al. Depressive symptoms and menopausal burden in the midlife. Maturitas 62, 306–310 (2009).
Gibson, C. J. et al. Associations of intimate partner violence, sexual assault, and posttraumatic stress disorder with menopause symptoms among midlife and older women. JAMA Intern. Med. 179, 80 (2019).
Freeman, E. W. & Sammel, M. D. Anxiety as a risk factor for menopausal hot flashes: evidence from the Penn Ovarian Aging cohort. Menopause 23, 942–949 (2016).
Faubion, S. S. et al. Impact of menopause symptoms on women in the workplace. Mayo Clin. Proc. 98, 833–845 (2023).
Keshishian, A., Wang, Y., Xie, L. & Baser, O. The economic impact of symptomatic menopause among low-socioeconomic women in the United States. Expert Rev. Pharmacoecon. Outcomes Res. 16, 305–313 (2016).
Evandrou, M., Falkingham, J., Qin, M. & Vlachantoni, A. Menopausal transition and change in employment: evidence from the National Child Development Study. Maturitas 143, 96–104 (2021).
Crandall, C. J. et al. Association of genetic variation in the tachykinin receptor 3 locus with hot flashes and night sweats in the Women’s Health Initiative Study. Menopause 24, 252–261 (2017).
Zhao, W. et al. Genetic variants predictive of reproductive aging are associated with vasomotor symptoms in a multiracial/ethnic cohort. Menopause 28, 883–892 (2021).
Ruth, K. S. et al. Insights into the genetics of menopausal vasomotor symptoms: genome-wide analyses of routinely-collected primary care health records. BMC Med. Genomics 16, 231 (2023).
Meijsen, J. J. et al. Shared genetic influences on depression and menopause symptoms. Psychol. Med. 53, 2241–2251 (2023).
Rance, N. E., Dacks, P. A., Mittelman-Smith, M. A., Romanovsky, A. A. & Krajewski-Hall, S. J. Modulation of body temperature and LH secretion by hypothalamic KNDy (kisspeptin, neurokinin B and dynorphin) neurons: a novel hypothesis on the mechanism of hot flushes. Front. Neuroendocrinol. 34, 211–227 (2013).
Rance, N. E., McMULLEN, N. T., Smialek, J. E., Price, D. L. & Young, W. S. Postmenopausal hypertrophy of neurons expressing the estrogen receptor gene in the human hypothalamus. J. Clin. Endocrinol. Metab. 71, 79–85 (1990).
Rance, N. E. & Young, W. S. Hypertrophy and increased gene expression of neurons containing neurokinin-B and substance-P messenger ribonucleic acids in the hypothalami of postmenopausal women. Endocrinology 128, 2239–2247 (1991).
Padilla, S. L., Johnson, C. W., Barker, F. D., Patterson, M. A. & Palmiter, R. D. A neural circuit underlying the generation of hot flushes. Cell Rep. 24, 271–277 (2018).
Jayasena, C. N. et al. Neurokinin B administration induces hot flushes in women. Sci. Rep. 5, 8466 (2015).
Depypere, H. et al. Treatment of menopausal vasomotor symptoms with fezolinetant, a neurokinin 3 receptor antagonist: a phase 2a trial. J. Clin. Endocrinol. Metab. 104, 5893–5905 (2019).
Fraser, G. L. et al. A phase 2b, randomized, placebo-controlled, double-blind, dose-ranging study of the neurokinin 3 receptor antagonist fezolinetant for vasomotor symptoms associated with menopause. Menopause 27, 382–392 (2020).
Santoro, N. et al. Effect of the neurokinin 3 receptor antagonist fezolinetant on patient-reported outcomes in postmenopausal women with vasomotor symptoms: results of a randomized, placebo-controlled, double-blind, dose-ranging study (VESTA). Menopause 27, 1350–1356 (2020).
Lederman, S. et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet 401, 1091–1102 (2023).
Johnson, K. A. et al. Efficacy and safety of fezolinetant in moderate to severe vasomotor symptoms associated with menopause: a phase 3 RCT. J. Clin. Endocrinol. Metab. 108, 1981–1997 (2023).
Medland, S. E. et al. Common variants in the trichohyalin gene are associated with straight hair in Europeans. Am. J. Hum. Genet. 85, 750–755 (2009).
Do, K. A. et al. Predictive factors of age at menopause in a large Australian twin study. Hum. Biol. 70, 1073–1091 (1998).
Lindström, S. et al. A comprehensive survey of genetic variation in 20,691 subjects from four large cohorts. PLoS ONE 12, e0173997 (2017).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Tworoger, S. S., Sluss, P. & Hankinson, S. E. Association between plasma prolactin concentrations and risk of breast cancer among predominately premenopausal women. Cancer Res. 66, 2476–2482 (2006).
Huang, J. et al. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nat. Commun. 6, 8111 (2015).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Moore, C. & Jacobson, S. genpwr: Power Calculations Under Genetic Model Misspecification. 1.0.4 https://doi.org/10.32614/CRAN.package.genpwr (2019).
R. Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2021).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 7, e34408 (2018).
Watanabe, K., Taskesen, E., Van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Boughton, A. P. et al. LocusZoom.js: interactive and embeddable visualization of genetic association study results. Bioinformatics 37, 3017–3018 (2021).
Writing Group for the Women’s Health Initiative Investigators Risks and benefits of estrogen plus progestin in healthy postmenopausal women principal results women’s health initiative randomized controlled trial. JAMA. 288, 321–333 (2002).
De Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Schizophrenia Working Group of the Psychiatric Genomics Consortium et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Nievergelt, C. M. et al. Genome-wide association analyses identify 95 risk loci and provide insights into the neurobiology of post-traumatic stress disorder. Nat. Genet. https://doi.org/10.1038/s41588-024-01707-9 (2024).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352 (2019).
Mullins, N. et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. 53, 817–829 (2021).
Trubetskoy, V. et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022).
Demontis, D. et al. Genome-wide analyses of ADHD identify 27 risk loci, refine the genetic architecture and implicate several cognitive domains. Nat. Genet. 55, 198–208 (2023).
Ruth, K. S. et al. Genetic insights into biological mechanisms governing human ovarian ageing. Nature 596, 393–397 (2021).
Sinnott-Armstrong, N. et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat. Genet. 53, 185–194 (2021).
Sollis, E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2023).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Duncan, L. & Deisseroth, K. Are novel treatments for brain disorders hiding in plain sight? Neuropsychopharmacol 49, 276–281 (2024).
Lonsdale, J. et al. The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
GTEx Consortium, T. he et al. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020).
Commissioner, O. of the. FDA Approves Novel Drug to Treat Moderate to Severe Hot Flashes Caused by Menopause. FDA https://www.fda.gov/news-events/press-announcements/fda-approves-novel-drug-treat-moderate-severe-hot-flashes-caused-menopause (2023).
The 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Acknowledgements
We thank all participants for making this work possible. This work was supported by the Jaswa Innovator Award to LD and by the National Institute of Mental Health (NIMH) to LD (R01 MH123486 & R21 MH125358). We thank the participants and staff of the NHS and NHSII for their valuable contributions and acknowledge the Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital, and Harvard Medical School for managing the cohorts. This study was supported by National Institute of Health grants UM1 CA186107 (for NHS infrastructure), R01 CA49449 (for NHS blood collection), U01 CA176726 (for NHSII infrastructure), and R01 CA67262 (for NHSII blood collection). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
L.E.D. designed the experiment, reviewed all the data and analyses, and drafted the manuscript. K.E.W. and R.B.L. analyzed the data, drafted relevant manuscript sections, and created the figures. M.T.S., T.L., B.L.M., H.S., S.D.G., and B.K. analysed the data. C.J.C. provided essential expertise about clinical characteristics of the menopause transition. K.E.W., R.B.L., M.T.S., T.L., B.L.M., H.S., S.D.G., B.K., C.S., M.V., A.R., J.M., A.H.S., C.K., K.C.K., C.J.C., N.G.M., and L.E.D. edited and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Gabriela Chavarría-Soley and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Werwath, K.E., Lawn, R.B., Salem, M.T. et al. Trans-ancestry GWAS of hot flashes reveals potent treatment target and overlap with psychiatric disorders. Commun Med 6, 51 (2026). https://doi.org/10.1038/s43856-025-01305-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-025-01305-8
