
Abstract
The chemical modification of natural proteins in living systems is highly desirable as cutting-edge research at the chemistry–biology interface. Recent advances in bio-orthogonal protein modification have enabled the production of chemically functionalized proteins in cultured cell systems. However, a limited number of methods are applicable in vivo because of the complexity of the three-dimensional constructs of living systems with diverse, heterogeneous cell populations and flow systems filled with tissue fluids. Here we report a genetic-engineering-free method to modify receptor proteins with various probes in the living mouse brain by combining in-brain ligand-directed chemistry with bio-orthogonal click chemistry, and propose a chemical guideline for the reaction design. The rapid and selective tethering of a set of fluorescent peptides to AMPA-type glutamate receptors allowed the synthesis of receptor-based fluorescent sensors. These probes enabled mapping of the activity of matrix metalloproteinase-9 proximal to AMPA-type glutamate receptors in the living brain to be realized with high spatial resolution.

Similar content being viewed by others

Main
Protein chemical modification is important in basic science and applied research1,2,3,4,5,6,7,8. By chemically tethering various probes to target proteins it becomes possible to expand the natural functions of these proteins for use as bioimaging tools, biosensors and biopharmaceuticals. Although the chemical modification of proteins has often been performed using purified proteins in vitro, precise modification in more natural environments, such as live cells and in vivo, is highly desirable for state-of-the-art research at the chemistry–biology interface. Compared with in vitro chemistry, achieving protein selectivity and acceptable modification yields remain challenging because live cells and living systems are spatially heterogeneous and contain multiple biomolecules under crowding conditions. Recent progress in bio-orthogonal protein chemistry, including the incorporation of non-natural amino acids by genetic code expansion coupled with a click reaction, and chemogenetic approaches using enzymes (such as Halo/SNAP)-tag-based protein engineering, has enabled the production of chemically modified proteins in cultured cell systems9,10. Although the utility of click reactions have been proved, chemistry-based tethering methods are limited for modification of endogenous proteins applicable to in vivo systems11. This is because three-dimensional (3D) constructs with diverse cell populations and a flow system of tissue fluids are very different to the two-dimensional (2D) closed systems of cells cultured on a Petri dish12,13.
The brain has one of the most complex 3D structure of all in vivo systems. In the brain, extensive networks of neurons are tightly supported by glial cells, blood vessels, cerebrospinal fluid (CSF) and other substances to maintain brain homeostasis in both structure and function. The central neural system contains many synapses of 1 µm or less in size. In these synapses, neurotransmitter receptors play crucial roles in interneuron communication and the resultant sophisticated brain functions. Neurotransmitter receptors, such as glutamate receptors, are surrounded by other proteins and enzymes, such as other receptors, synaptic organizers, and proteases and peptidases, by which their functions are tightly regulated. Although such dynamically modulated molecular networks centred on the receptors are known to be involved in brain functions and disorders, the details have not been well deciphered because of the insufficient analytical tools applicable to live brain systems. Chemistry-based methods are anticipated to uniquely contribute to the understanding of neural networks at the molecular level, but such approaches are still only in their infancy. We have recently reported in-brain ligand-directed acylimidazole (LDAI) chemistry as a method for the chemical modification of neurotransmitter receptors in the complex context of the brain14,15. This method can be used to chemically label and visualize target receptors with high selectivity in the living mouse without genetic manipulation. Although promising, there are still many challenges with this method, including the slow modification kinetics, the requirement for time-consuming individual design and synthesis of a labelling reagent for each probe14,15,16,17, and the lack of guidelines for the rational design of affinity-driven labelling reactions in the live brain. To address these issues, we sought to combine in-brain LDAI chemistry with bio-orthogonal click reactions18,19. This approach is designed to separate the anchoring step of a bio-orthogonal reaction handle to the target receptor and the step for the modification of a functional probe, to expand the flexibility for probe selection. Because of the limited examples of click reactions in the brain20 and the poor experimental data for the in-brain chemical modification of proteins, we investigated a series of LDAI labelling reagents and click reagents with varied physicochemical properties. Here we describe a design guideline for both the chemical labelling (anchoring) of target receptors with a click handle in the brain and the modification of receptors with functional probes using click reagents. Using this guideline, we achieved the rapid functional modification of a target receptor protein in a living mouse brain without the use of genetic techniques, termed ‘chemical knock-in’ (KI). The chemical KI of fluorescent peptide probes to AMPA-type glutamate receptors (AMPARs) allowed the synthesis of receptor-based fluorescent sensors in a live brain. These sensors enabled the successful mapping of the activity of matrix metalloproteinase-9 (MMP9) (a representative protease involved in the regulation of brain function) which was proximal to AMPARs, in the context of the 3D live brain.
Results
Chemical KI strategy using in-brain LDAI and click chemistry
In established bio-orthogonal protein modification using click chemistry, the incorporation of a non-natural reaction (clickable) handle, such as an azide, is required for a protein-selective reaction. A state-of-the-art genetic expansion protocol using a suppressor codon can enable the incorporation of such a handle into a target protein in live cell systems21,22,23. However, the robust application of this strategy in live animals, such as mice, is still difficult and applications in the brain have been limited to date. Although the metabolic incorporation of a clickable substrate is an alternative method, this approach generally suffers from insufficient selectivity toward the target protein24,25,26.
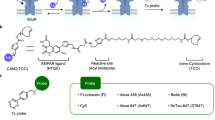
In our chemical KI approach, we used our LDAI chemistry to incorporate a clickable handle into a target receptor in the living brain. This anchoring step was followed by click chemistry to realize the functional modification of a receptor protein (labelling step). The approach is summarized in Fig. 1. Because LDAI chemistry can modify endogenous receptors, the chemical KI does not require any genetic manipulation. Chemical KI should be particularly beneficial for targeting structurally complicated oligomeric proteins in which the subunit balance is often disrupted by genetic engineering. In addition, genetic engineering techniques are still not well established in many animals. We here targeted the AMPARs, a neurotransmitter receptor involved in fast excitatory neurotransmission, for a proof-of-principle study of chemical KI in the brain. As the click handle, we selected trans-cyclooctene (TCO), which reacts with many tetrazine (Tz) derivatives with fast kinetics (1–106 M−1 s−1 in vitro)27,28,29, to enable the rapid introduction of functional molecules to AMPARs in vivo. If these two steps can be successfully performed in the animal brain, the selective functionalization of endogenous AMPARs, that is, the chemical KI of designer probes for a target protein, will be realized.

Nu, nucleophilic amino-acid residue; Lg, selective ligand for target receptor; MeTz, methyltetrazine.
However, the conditions for the chemical reactions in the live brain (in vivo) are very different from those in live cells cultured in 2D Petri dishes (in vitro). In addition to the complexity of the 3D structure with diverse and heterogeneous cell populations, the brain also has a flow system with constant irrigation and drainage of CSF30. Under such conditions, chemical reagents are gradually removed from the brain, which can hinder the distribution of the reagents over a wide area of the brain. In our recent studies of in-brain LDAI chemistry14,15, we found that the method of administration was critical for efficient delivery of the LDAI reagents to the whole brain, and the direct injection of the reagent into the lateral ventricle (LV) gave excellent results. After injection into the LV, the labelling reagent was diluted by CSF constantly produced from the choroid plexus of the LV, and the CSF flow from the LV to various areas allowed the labelling reagent to diffuse to tissues throughout the whole brain. Because the parameters required for efficient chemical reactions in the brain are expected to be different from those in vitro, we sought to compare the chemical reactivity between the conventional live cell systems and the live brain, using identical sets of LDAI and click reagents. Furthermore, considering that there are no design guidelines for either the anchoring step using LDAI or the protein modification (or labelling) step by click reaction in the live brain, we examined the reaction profiles of these two steps separately in detail.
Selective anchoring of TCO by ligand-directed chemistry
For anchoring the clickable TCO moiety to AMPARs, different LDAI reagents for AMPARs (chemical AMPAR modification reagents, CAM2) were designed and synthesized, based on the molecular structure of CAM2-Ax647 (Ax647, Alexa Fluor 647) (1), which has previously been shown to have an excellent labelling capability in live mouse brains14. The three labelling reagents (2–4) contained Ax647 and TCO connected with different spacer units, namely, an N-branched structure, Glu or Lys, respectively (Fig. 2a). We also used CAM2-TCO (5) lacking Ax647 and CAM2-Ax647 (1) lacking TCO as control compounds31,32. These five compounds had distinct physicochemical properties, which could be indexed by the values for the calculated logP (ClogP), a parameter of hydrophobicity, which were −4.4 (1), −4.2 (2), −3.2 (3), −1.8 (4) and 2.9 (5).

a, LDAI reagents for anchoring TCO to AMPARs. b, MeTz-based labelling reagents for the click reaction with a TCO group attached to AMPAR. The structures of Ax647 and Ax555 shown are the putative structures.
We first evaluated the selectivity and efficiency of the TCO-anchoring reaction in HEK293T cells expressing GluA2 (a subunit of the AMPAR) (Fig. 3a,b). CAM2 reagents (1 µM) were added to live HEK293T cells at 37 °C, followed by incubation for 3 h and cell lysis. The lysates were analysed by SDS–PAGE in-gel fluorescence. Because reagents 1–4 contain the fluorescent Ax647 moiety, we directly detected Ax647 signals. In the case of CAM2-TCO (5), the lysate was mixed with Tz-Ax647 prior to SDS–PAGE analysis. We previously reported that Tz-Ax647 reacts efficiently and rapidly with TCO-anchored AMPARs in lysate31. As shown in Fig. 3b, all the CAM2 reagents gave a single band corresponding to GluA2, indicating the high AMPAR selectivity in the TCO-anchoring step. In addition, the intensities of fluorescence bands derived from the labelled Alexa647 were almost comparable to each other. The anchoring efficiency was not dramatically different between these CAM2 reagents (1–5) within 30% variation relative to CAM2 reagent 1.

a, Experimental workflow for the results shown in this figure and in Fig. 4. b,c, In-gel fluorescence and western blotting (WB) analysis of the labelling reaction with AMPARs expressed with GluA2 under in-dish conditions (b, biological replicates, n = 4) and with endogenous AMPARs in the live brain (c, biological replicates, n = 3). d, Comparison of the relative labelling efficiency between in-dish and in-brain conditions. Data are presented as means ± s.e.m. (biological replicates, n = 3–4). e, Fluorescence images of labelled brains with 2 (Ax647) and colocalization analysis with anti-GluA2. Imaging was performed using a CLSM equipped with a 10× objective. Scale bar, 2 mm. f, High-resolution confocal image of the cerebral cortex (100×). Scale bar, 5 µm.
The selectivity and efficiency of the TCO-anchoring step using these reagents were also evaluated in living mouse brains (Fig. 3a,c). Mice were injected with 4.5 µl of each labelling reagent (100 µM) into the LV, and 24 h after injection, the cerebellum, which is relatively distant from the LV and rich in endogenous AMPARs, was isolated. The homogenates were subjected to SDS–PAGE analysis. The homogenates of the mouse brains injected with CAM2-TCO (5) were treated with MeTz-Ax647 prior to SDS–PAGE. A single band corresponding to a subunit of the AMPAR was detected for the CAM2 reagents (1–4) in the in-gel fluorescence analysis, indicating high AMPAR selectivity. In contrast, negligible bands were observed for CAM2-TCO (5), which suggested that the anchoring reaction had scarcely occurred. The anchoring efficiency was compared from the band intensities, CAM2-Ax647 (1) lacking TCO had the highest intensity, and for the TCO-bearing CAM2 derivatives, the efficiency declined gradually from the derivatives with an N-branch (2) to Glu (3), Lys (4) and CAM2-TCO (5). Figure 3d shows a plot of the anchoring efficiency against the ClogP values of the five CAM2 reagents. The efficiency of the reaction in the live brain corresponded to the ClogP value of the CAM2 reagent; that is, compounds with more negative ClogP values resulted in more efficient TCO anchoring to the AMPARs. In sharp contrast, the anchoring efficiency in HEK293T systems (in vitro) was not strongly related to the ClogP values (Fig. 3b,d). For example, CAM2-TCO (5) failed to tether the TCO group with the AMPARs in the brain, whereas this reaction was successful in vitro.
The spatial distribution of the anchoring reaction was subsequently examined using confocal laser scanning microscopy (CLSM) of the brain sections. In brain slices injected with the CAM2 reagent 2, fluorescence from Ax647 was observed from the hippocampus, cerebellum, cortex and striatum, where endogenous AMPARs are highly expressed (Fig. 3e). Although the cerebellum was distant from the administration site of the reagents, a fluorescent signal was clearly observed, revealing the excellent in-brain diffusivity and permeability of 2. Importantly, the fluorescence derived from the anchored Ax647 showed a pattern similar to that found with co-immunostaining using anti-GluA2 antibodies (Fig. 3e) and the high-resolution images of the cortex area showed numerous bright spots ∼1 µm in size. These bright spots also overlapped well with the anti-GluA2 fluorescence, implying that these spots indicated neuronal spines where endogenous AMPARs were accumulated (Fig. 3f).
Considering that the LDAI reagents need to reach target AMPARs in several distinct regions of the brain, the diffusion and permeability of the CAM2 reagents through heterogeneous brain tissues could be crucial factors for efficient ligand-directed TCO anchoring in the live brain. These factors may be partly expressed by the ClogP value and this value was found to be a helpful indicator in the design of chemical reagents for in-brain ligand-directed receptor labelling. Compared with the live brain, the cultured cell system was rather simple and thus the anchoring step might be less sensitive to the parameters reflected by the ClogP value in this system.
Highly selective and fast chemical KI using click chemistry
We next investigated the click reaction (probe labelling) step. According to previous reports of the fast reactivity with the TCO moiety and good stability in vivo, methyltetrazine (MeTz) was selected as the clickable moiety and a set of fluorescent (Cy5 core) derivatives with various ClogP values was prepared as follows: MeTz-Ax647 (ClogP = −6.0), MeTz-SulfoCy5 (−1.4), MeTz-Cya-Cy5 (1.6), MeTz-Asp-Cy5 (3.2) and MeTz-Cy5 (4.1) (Fig. 2b). For the TCO anchoring to AMPARs, CAM2 (2) bearing Ax555 instead of Ax647 was used in both live cells and the live brain study to enable the use of two distinct wavelengths to evaluate the click reaction and the TCO anchoring separately. Because fluorescent dyes with longer wavelengths are more suitable in terms of detection sensitivity and accuracy, we decided to analyse the intensity of the Ax647-fluorescent band by in-gel fluorescence analysis in the click reaction step.
We conducted the first anchoring step of AMPARs transiently expressed in HEK293T cells according to the protocol shown in Fig. 3a, (2 (Ax555), 1 µM, 37 °C, 3 h). Subsequently, the cells were incubated with each of the five MeTz derivatives (1 µM, 1 h), followed by washing and cell lysis and then the lysates were subjected to in-gel fluorescence analysis. The reaction of MeTz with the TCO-tethered AMPARs could be monitored by the Cy5 emission. As shown in Fig. 4a, single bands exhibiting almost identical emission intensity were detected for MeTz-Ax647, MeTz-SulfoCy5, MeTz-Cya-Cy5 and MeTz-Asp-Cy5. The result shows the selective labelling of AMPARs using 2 (Ax555) and these MeTz dyes. In the case of MeTz-Cy5, the non-specific labelling of proteins other than AMPARs was also observed. Given that MeTz-Cy5 has no anion charge and can easily enter cells, it may be reasonably considered that MeTz-Cy5 caused non-specific/covalent labelling to intracellular proteins compared to other MeTz dyes (Supplementary Fig. 1). The observed intensity indicated that the click reaction in HEK cells was not dependent on the ClogP value of the MeTz derivatives, which displayed a similar trend to that for the LDAI-based TCO-anchoring step.

a,b, In-gel fluorescence and WB analysis of the click reaction with AMPARs expressed with GluA2 in the in-dish conditions (a) and with endogenous AMPARs in the brain (b) (biological replicates, n = 3). c, Comparison of the labelling efficiency between the in-dish and in-brain conditions relative to MeTz-Ax647. Data are presented as means ± s.e.m. (biological replicates, n = 3). d, Kinetic analysis of in-brain click reaction with TCO-anchored AMPARs. Relative labelling efficiency normalized by the in-gel fluorescence intensity after 4 h of click reaction. Data are presented as means ± s.e.m. (biological replicates, n = 3). e, Fluorescence images of sagittal brain sections labelled by the click reaction with MeTz-Ax647 or MeTz-Cy5. A mouse was additionally injected with MeTz-Ax647 or MeTz-Cy5 (100 µM, 4.5 µl), 24 h after LV injection of 2 (Ax555) (100 µM, 4.5 µl). Then, 24 h after the LV injection of the MeTz derivatives, the mouse was transcardially perfused with a 4% PFA/PBS(–) solution. The brain was isolated and sectioned by cryostat (50-µm-thick sections). Imaging was performed using a CLSM equipped with a 10× objective. Scale bar, 2 mm. f, Zoomed image of the cerebral cortex shown in e. Imaging was performed using a CLSM equipped with a 100× objective. Scale bar, 5 µm. g, Fluorescence imaging of a tissue-cleared brain labelled with 2 (Ax555) (100 µM, 4.5 µl; left and right LV) and MeTz-Ax647 (100 µM, 4.5 µl; left and right LV). Fluorescence imaging of CUBIC-cleared whole brain was performed using FUSION, a chemiluminescence/fluorescence imaging system (excitation wavelength, 600–650 nm; detection wavelength, 690–720 nm). Three-dimensional imaging of the hippocampus area was performed using a CLSM equipped with 10× and 20× objectives. FRET signals between Ax647 (click labelling) and Ax555 (TCO anchoring) were observed with the laser excitation at 561 nm and emission at 655–770 nm.
In the live mouse brain system, the anchoring step and the click reaction step that followed were performed according to the protocol shown in Fig. 3a. The TCO-anchoring reagent 2 (Ax555) (100 µM, 4.5 µl) was injected into the LV of mice under anaesthesia, and after 24 h, a MeTz probe was injected into the LV (100 µM, 4.5 µl). The cerebellum was isolated after 24 h and the homogenates were subjected to SDS–PAGE in-gel fluorescence analysis. The single band resulting from the labelling of AMPARs was observed with MeTz dyes other than MeTz-Cy5, which also implies the selective TCO anchoring of AMPARs using 2 (Ax555) in the live brain. MeTz-Ax647 (ClogP = −6.0) and MeTz-SulfoCy5 (−1.4) showed greater click reaction efficiency, relative to the other compounds, and almost no bands were observed for MeTz-Cy5 (Fig. 4b). Figure 4c shows that MeTz derivatives with a negative ClogP value resulted in better modification yields than those with a positive ClogP value. Compared with the in-brain LDAI chemistry, the click reaction step was less sensitive and rather more tolerant to compounds with varying ClogP values. Overall, the reaction efficiency in the brain system was correlated with the ClogP value of the reagents in both the anchoring and modification steps, whereas the efficiency in the in-dish cultured cell system did not show any appreciable correlation with the ClogP values.
Using the efficient in-brain chemical KI tools (2 (Ax555) and MeTz-Ax647), we next determined the modification kinetics of endogenous AMPARs in the live brain. Mice bearing TCO-tethered AMPARs were injected with MeTz-Ax647 (100 µM, 4.5 µl) into the LV. The cerebellum was isolated at various time points after the injection and the reaction efficiency was assessed from the in-gel fluorescence band intensity (Fig. 4d). It is clear from Fig. 4d that the click reaction reached saturation in less than 60 min, indicating that the high reactivity of the inverse electron-demand Diels–Alder reaction between TCO and MeTz resulted in a rapid reaction in the live mouse brain, which enabled chemical KI of the fluorescent probe Ax647 into the target AMPARs within 1 h. MeTz-sulfoCy5 also shows similar fast kinetics (Supplementary Fig. 2). We also evaluated the duration of MeTz-Ax647 in the mouse brain without TCO-anchored AMPARs. The half-life of MeTz-Ax647 was ∼0.5 h (Supplementary Fig. 3), clearly indicating that the live brain had a flow system that enabled the chemical reagents to be removed gradually. We believe that consideration of this flow system is critically important in the design of in-brain chemical reactions with high efficiency.
We conducted imaging analysis of the brain after the click (modification) reaction (Fig. 4e–g). For MeTz-Cy5, strong fluorescence was observed in very limited areas close to the LV, the MeTz-Cy5 injection site, whereas very weak fluorescence was observed in the hippocampus and cerebellum regions where AMPARs are highly expressed (Fig. 4e). Combined with the in-gel fluorescence results, the strong signal close to the LV areas was believed to mainly result from the non-specific binding or adsorption of MeTz-Cy5. For MeTz-Ax647 and MeTz-SulfoCy5, a broad distribution of fluorescence signals was observed throughout the brain areas expressing endogenous AMPARs (Fig. 4e and Supplementary Fig. 4). Imaging at a higher magnification clearly revealed that the fluorescent bright spots, of 1 µm or less in size, of TCO-anchored AMPARs (Ax555) in the spines were well merged with the signals derived from the click modification (Ax647 emission, Fig. 4f). Interestingly, these labelled signals were substantially retained even after clearing of the whole brain by the CUBIC method33 and these punctate signals could be observed in 3D mode without 2D section preparation (Fig. 4g). These imaging analyses demonstrated that the diffusivity and permeability of the MeTz derivatives had a critical impact on the in-brain click reaction, which was reflected in the ClogP values, as was the case for the in-brain LDAI chemistry.
In addition to the two-step labelling protocol mentioned above, chemical KI can be performed by a one-step protocol using the probe-conjugated labelling reagents which were prepared by premixing with 2 (Ax555) and MeTz-probes (Ax647 or SeTau647). The one-step labelling using premixed 2 (Ax555) and MeTz-Ax647 yielded CLSM images similar to those observed in the two-step protocol (Supplementary Fig. 5a). However, in the case of the one-step labelling, 6 h were needed for the reaction saturation (Supplementary Fig. 6), which was slower than the two-step protocol for the Ax647 modification (Fig. 4d). Moreover, in the case of MeTz-SeTau647, a photostable dye suitable to two-photon imaging, the one-step (premix) labelling protocol did not work well, whereas the two-step protocol did (see imaging data in Supplementary Fig. 5b). These results suggest that the two-step labelling protocol is more flexible and appropriate in some cases.
In-brain synthesis of AMPAR-based fluorescent MMP9 sensors
Having an efficient in-brain chemical KI method in hand, we applied this method to the synthesis of receptor-based protease sensors. In the brain, proteases such as MMP9 play crucial roles in the regulation of synaptic function by reorganizing the extracellular matrix, degrading synaptic organizers and activating cytokines34,35. However, the molecular details of the activity of particular proteases are not fully understood, mainly because there are no valid methods capable of directly analysing protease activity with high spatial resolution in a live brain. Fluorescent protease biosensors that can monitor the protease activity in situ are expected to be useful for such analysis, and thus we constructed AMPAR-based sensors via chemical KI of the corresponding protease substrates (Fig. 5a). In detail, AMPARs anchored with Ax555 and TCO were modified with a variety of Ax647-appended peptides (protease substrates) via the in-brain click reaction. When a protease cleaves the peptide tethered to the AMPARs, the Ax647 emission will decrease, resulting in an increase in the ratio of Ax555/Ax647. Given that the cleavage site is <10 nm from the surface of the AMPAR, this sensor is able to detect protease activity that is proximal to the receptor. The fluorescent peptide substrates for MMP9 (MMP9 probes) were designed based on previous reports36,37, as shown in Fig. 5b. MeTz and the fluorescent Ax647 were attached at the C-, and N-termini of the peptides, respectively. Two different sequences with high and low reactivity for MMP9 were prepared (MMP9-Leu, MMP9-Abu), together with MMP9-DLeu where the l-Leu of the cleavage site was replaced with the d-isomer, as a non-cleavable control. Furthermore, an Ax647-appended peptide substrate for cathepsin B (CatB), which is an intracellular protease mainly localized in endosomes, was designed in the same manner (CatB probe, Fig. 5b)38. We separately confirmed the enzymatic cleavage of these probes by HPLC analysis (Supplementary Figs. 7 and 8).

a, Schematic illustration of the analysis of MMP9 activity proximal to AMPARs. The image of the AMPAR was rendered using Pymol with data from PDB: 5L1F. b, Chemical structure of fluorescent peptide probes for the detection of MMP9 and CatB activity. c,d, Time-course analysis of MMP9 activity using an AMPAR-based fluorescent MMP9 sensor. CLSM observations were conducted at different times after the addition of collagenase. Fluorescence imaging was performed using a Leica CLSM equipped with a 40× objective. Scale bar, 10 µm. e, Plot of Ax555/Ax647 values on the cell surface versus time. Region of interest (ROI) (n = 5). Data are presented as means ± s.e.m.
First, we prepared a fluorescent MMP9 sensor for the AMPAR scaffold transiently expressed in HEK293T cells and investigated whether the sensor could detect MMP9 protease activity. Live cell CLSM imaging showed that Ax647 fluorescence derived from the MMP9-Leu peptide was observed on the cell surface, together with Ax555 emission (Fig. 5c, 0 min), indicating that the AMPARs successfully underwent modification with the MMP9-peptide by click chemistry. When a solution of collagenase containing MMP9 (gelatinase B, type IV collagenase) was added to the HEK293T cells, the emission intensity of Ax647 decreased without a change in the Ax555 emission (Fig. 5c, 30 and 60 min). Such fluorescence changes did not occur with MMP9-DLeu modification (Fig. 5d). Ratiometric analysis of the fluorescence intensity of Ax555/Ax647 at the cell surface enabled the evaluation of the MMP9 activity more quantitatively (Fig. 5e). These results indicated that an AMPAR-based MMP9 fluorescent sensor was successfully constructed by chemical KI in a cultured cell system. We also prepared an AMPAR-based CatB fluorescent sensor on the cell surface (Supplementary Fig. 9a). The AMPAR-based CatB(L) sensor exhibits FRET signals from Ax555 to Ax647 on the plasma membrane. After endocytosis (12 h), the substrate peptide was cleaved in endosome rich in CatB, resulting in a decrease of the FRET intensity there by releasing the peptide fragment containing Ax647 (Supplementary Fig. 9b,c). In contrast, the AMPAR-based CatB(D) sensor preserved the FRET signal in endosome (Supplementary Fig. 9d). Comparing the FRET intensity between the CatB(L) sensor and CatB(D) sensor, it is clear that the AMPAR-based CatB(L) probe can indeed detect the activity of CatB (Supplementary Fig. 9e,f).
Finally, we attempted to construct a fluorescent MMP9 sensor in a live brain. After anchoring AMPARs with TCO and Ax555 by LV injection of 2 (Ax555) in a mouse brain, an Ax647-appended peptide (MMP9 or CatB probe) was subsequently injected to the LV site. The hippocampus was isolated 6 h after the second injection, and the homogenates were analysed by SDS–PAGE in-gel fluorescence. A single Ax647 fluorescence band was observed at ∼100 kDa corresponding to the endogenous AMPAR subunit, indicating the selective modification of endogenous AMPARs with these peptide probes (Fig. 6a). The hippocampal samples labelled with MMP9-Leu or MMP9-Abu showed weak fluorescence bands (lanes 1 and 4), relative to the band of the sample labelled with MMP9-DLeu (lane 3). Importantly, the fluorescence band of the sample after co-injection with MMP9-Leu and ilomastat, an MMP inhibitor, retained intensity that was almost comparable to that of the MMP9-DLeu sample (lane 2). These results were in good agreement with the results for the cleavage reactivity of the MMP9 probes in vitro using hippocampal homogenates (Supplementary Fig. 10). It was concluded that the relative intensity of the in-gel fluorescence band reflected the MMP activity; that is, the decrease in the fluorescent bands for MMP9-Leu and MMP9-Abu indicated high MMP9 activity in the hippocampal region. In contrast, the differences in the signals between the samples labelled with the CatB(L) and CatB(D) probes were small (Fig. 6a, lanes 5 and 6), which suggested that the CatB activity was low in the extracellular region of the hippocampus.

a, Comparison of in-gel fluorescence band intensity at 6 h after the injection of peptide probes (100 μM, 4.2 μl). The hippocampus homogenates from three or four mice (C57BL6/N, 5 weeks, male) were employed for the in-gel fluorescence assay. Data are presented as mean values ± s.e.m. (n = 3–4). b, Fluorescence images of sagittal brain sections labelled by the click reaction with MMP9-Leu or MMP9-DLeu. A mouse was additionally injected with MMP9-Leu or MMP9-DLeu (100 μM, 4.2 μl), 12 h after LV injection of 2 (Ax555) (100 μM, 4.2 μl). Then, 6 h after LV injection of MeTz derivatives, the mouse was transcardially perfused with a 4% PFA/PBS(–) solution. The mouse brain was isolated and sectioned by cryostat (50-μm-thick sections). Imaging was performed using a CLSM equipped with a 5× objective (numerical aperture, 0.25). Scale bar, 2 mm. c, High-resolution image of the cortical section labelled with MMP9-Abu. After co-immunostaining with pre- and postsynaptic markers, anti-Bassoon and anti-Homer1a, imaging was performed with a 100× objective and Leica Lightning deconvolution.
The fluorescence images of cryosections were expected to provide spatial information on the protease activity in the whole brain, and we observed fluorescence in sagittal brain sections 6 h after injection of MMP9-Leu using a 5× objective lens. Figure 6b shows that there was a negligible signal from Ax647 in the hippocampus and substantial fluorescence in the cerebellum, relative to the signals of the corresponding regions after MMP9-DLeu injection, suggesting distinct activity of MMP9 depending on the region. The high-resolution images indicated that the MMP9 probe was indeed tethered to AMPARs located in the dendritic spines (Fig. 6c). In these experimental set-ups, however, the decrease in the Ax647 signal may have been caused by two possible scenarios; that is, the peptide probe was cleaved before reaching the AMPARs and/or the probe was cleaved after tethering to the AMPARs. To explicitly evaluate the cleavage of peptides attached to the AMPARs, we performed snapshot CLSM imaging of brain slices after different incubation times. Perfusion fixation was conducted 3, 6 and 9 h after the probe injection. In the hippocampal regions modified with MMP9-Leu, almost no Ax647 fluorescence was observed even after incubation for 3 h (Fig. 7a, left panels). The samples modified with MMP9-Abu, in contrast, showed definite Ax647 fluorescence at 3 h and a gradual decrease over incubation for 6 and 9 h, resulting in an increase in the Ax555/Ax647 ratio (Fig. 7a, middle panels). In contrast, such changes were not observed for the samples injected with MMP9-DLeu (Fig. 7a, right panels). These results clearly indicated that with MMP9-Abu, a substantial amount of the MMP9 probe was cleaved after being tethered to AMPARs. This result may reflect the lower reactivity of MMP9-Abu for MMP9 (compared with MMP9-Leu), and the slow kinetics allowed MMP9-Abu to reach AMPARs prior to the cleavage. This MMP9-Abu probe with moderate cleavage properties is more appropriate for the investigation of the MMP9 activity proximal to a receptor of interest and we used this probe in the following studies. Note that, as expected from the results shown in Fig. 6a, there was little change in the fluorescence images of samples labelled with the CatB(L) probe and those labelled with the CatB(D) probe (Supplementary Fig. 11).

a, Hippocampus area, 5× objective. Scale bar, 0.5 mm. b, Hilus area of the hippocampal dentate gyrus, 63× objective (numerical aperture, 1.4). Scale bar, 5 μm. Similar images were observed in at least three independent experiments. c, Boxplot of the Ax555/Ax647 ratio values of the fluorescent bright spots in the hilus area of the hippocampal dentate gyrus. The number of ROIs was 240 for each of the images (n = 240). The horizontal line and × within each box indicate the median and mean, respectively. Boxes show interquartile range (IQR). Whiskers show 1.5 × IQR. d, Cerebellum area, 5× objective. Scale bar, 1 mm. e, Area near Purkinje cells, 63× objective. Scale bar, 5 μm. Similar images were observed in at least three independent experiments. f, Boxplot of the Ax555/Ax647 ratio values of the fluorescent bright spots in the posterior lobe IX of the cerebellum. The number of ROIs was 240 for each of the images (n = 240). The horizontal line and × within each box indicate the median and mean, respectively. Boxes show IQR. Whiskers show 1.5 × IQR.
The magnified CLSM images showed the MMP9 activity at a higher spatial resolution. In the hilus of the hippocampal dentate gyrus, the signals from Ax555 and Ax647 fluorescence derived from the AMPARs and the MMP9-Abu probes, respectively, were both observed as bright puncta of 1 µm or less in size and the images were well merged (Fig. 7b). Importantly, longer incubation times (from 3 to 9 h) decreased the Ax647 fluorescence and increased the value of the Ax555/Ax647 ratio in many of the spines in the samples labelled with MMP9-Abu (Fig. 7b,c), strongly suggesting that MMP9 had penetrated into the synaptic cleft, closely approached the AMPARs (within 10 nm) and then cleaved the MMP9-substrate peptide. This result may be consistent with a previous report suggesting that MMP9 was responsible for the cleavage of synaptic organizers (such as neuroligins)39. This is an important report of images that capture the MMP9 activity proximal to AMPARs in the hippocampal excitatory synapses of a live brain, which cannot be achieved using conventional methods. CLSM observations were also performed in the cerebellum, where AMPARs are highly expressed. In the fluorescence imaging of the whole cerebellum, the Ax647 fluorescence derived from AMPAR-based MMP9-Abu sensors remained considerably high, 9 h after injection of the probe (Fig. 7d). The high-magnification images of the molecular layer region also showed that many of the Ax647 bright spots remained intact over 9 h of incubation (Fig. 7e). When we compared the time-profile of the Ax555/Ax647 ratio between the hippocampus and cerebellum, at puncta-level resolution, the MMP9 activity proximal to AMPARs in the cerebellum was observed to be approximately 6-fold lower than that in the hippocampus area (Fig. 7c,f). This is important experimental evidence showing that MMP9 activity in the live brain is greatly dependent on the region.
Discussion
Over the decades, a variety of chemical methods for protein bioconjugation have been developed1,2,3,4,5,6,7,8. However, most of them cannot be used for living systems, partly because of the poor protein selectivity and inefficient reactions under the complex conditions encountered in vivo. In addition, proteins targetable by methods currently used in living animals are mainly limited to proteins that are pathologically overexpressed40,41. Modification strategies for natural proteins that are not related to pathology are still in their infancy. In the present study, we have developed a strategy for the chemical KI of functional synthetic molecules into a target receptor in a live mouse brain with no genetic manipulation. The power of the chemical KI method was clearly demonstrated by the synthesis of AMPAR-based MMP9 protease sensors, which enabled, for the first time, the mapping of MMP9 activity in proximity (~10 nm) to AMPARs in the postsynapses with high spatial resolution in a live brain context.
This entirely chemistry-based method was realized on the basis of our interesting finding of a design guideline for efficient chemical reactions in the live brain. In both steps of the method, employing LDAI and click reagents in the brain, we discovered an unprecedented correlation between the ClogP values of the reagents and the reaction efficiency that was not observed in reactions in cell-culture systems. This result implied that the ClogP value may be a useful indicator for the design of chemical reactions in the brain (and probably elsewhere in in vivo systems). Our study also indicated that the diffusivity and tissue-penetration properties of chemical reagents may be related to the target selectivity and reaction efficiency in complex biological tissues such as the brain. We believe that our findings will contribute to the progress in the emerging field of in vivo chemistry/chemical biology.
The vital roles of proteases in brain functions have not been fully characterized in molecular detail to date. Many issues remain to be elucidated, such as the existence/localization not only of proteases released inside the brain but also of proteases transported from outside the brain42. The functional analysis of proteases using genetic techniques can be hindered in complex situations in which there are a large number of genes for a target protease, and when the expression and substrate patterns of related proteases are heavily overlapped. Our chemical KI method is expected to be complimentary to these genetic methods, and can enable the installation of a set of peptides as substrates for a target receptor, which will provide the ability to monitor diverse protease/peptidase activities in a live brain, potentially leading to the elucidation of the detailed functions of extracellular proteolysis in the brain. In situ zymography, a method for detecting protease activity in tissue sections, has been used to analyse the activity of MMPs43,44. However, this method uses isolated brain slices instead of the whole live brain and has several limitations, including artefacts because of the postmortem brains used, the rather poor protease selectivity and the low spatial resolution of the images43. The method developed in the present study can be used to monitor protease activity in a living animal, which should provide results more indicative of the natural physiological state. The developed method also has high protease species selectivity because of the simplicity of synthesizing the desired peptide sequences by solid-phase peptide synthesis, and the ability to monitor protease activity in proximity to a target receptor. In addition to the recording of the activity of a target protease in the brain as demonstrated herein, we envisage that our chemical KI strategy may be extended to the high-resolution mapping of important biological environmental changes (for example, pH, metal ions and bioactive small molecules) proximal to target receptors in live brains using various supramolecular and/or chemical biology-based sensors that have been developed to date.
Methods
Synthesis
A general procedure for the synthesis of TCO-anchoring reagents
The TCO-anchoring reagents 2–4 were synthesized by condensing Boc-deprotected CAM2-Boc or S8 with S2, S3 or S7. The detailed procedures are described in the Supplementary Information.
A general procedure for the synthesis of MeTz dyes
The MeTz unit was reacted with a commercially available fluorescent dye by a condensation reaction. The detailed procedures are described in the Supplementary Information.
A general procedure for the synthesis of MeTz-peptides
The peptides were synthesized by the stepwise elongation of Fmoc-amino acids on 2-Cl-Trt(2-Cl) resin (0.2 mmol g−1, 200 mg, Watanabe Chemical) according to a reported procedure with Fmoc-AA-OH (5 equiv.). The protecting groups and the resin were removed by stirring the dried resin for 2 h at room temperature in TFA/H2O/TIPS/thioanisole (85/5/5/5). After removing the resin by filtration, the crude peptides were solidified by adding ice-cold diethyl ether and dried under reduced pressure. The crude peptides were purified by reverse-phase HPLC. MeTz-group and Ax647 were introduced into purified peptides by condensation reactions. The detailed procedures are described in the Supplementary Information.
Biochemical/biological, fluorescence imaging and animal experiments
The detailed experimental procedures are described in the Supplementary Information.
Construction of AMPAR-based MMP9 sensor on HEK293T cells
See Fig. 5. HEK293T cells transfected with GluA2 were treated with 1 μM anchoring reagents 2 (Ax555) in DMEM Glutamax including 10 mM HEPES at 17 °C for 4 h. After removal of the medium, the cells were washed with HBSS (+) (Nacalai Tesque) twice and treated with 1 μM MMP9-Leu or MMP9-DLeu in DMEM Glutamax including 10 mM HEPES at 17 °C for 30 min. After removal of the reaction medium, the cells were washed with HBSS (+) twice and replaced with DMEM Glutamax including 10 mM HEPES. Collagenase (Collagenase Type A, Fujifilm Wako, 034-24563, 1 mg ml−1 HBSS (+), 100 µl) was added to labelled cells on a microscope stage for CLSM observation. Confocal live imaging of labelled AMPARs was performed using a CLSM (TCS SP-8, Leica Microsystems) equipped with a 40× objective and a GaAsP detector. Fluorescence images were acquired using 561-nm excitation for Ax555 and 633-nm excitation for Ax647 derived from a white light laser.
Construction of AMPAR-based MMP9 sensor in the live mouse brain
See Figs. 6 and 7. At 12 h after the LV injection of anchoring reagent 2 (Ax555) (100 µM, 4.2 µl), MeTz-peptide probe (100 µM, 4.2 µl) was injected into the LV. At 3, 6 and 9 h after the injection, mice were killed under deep isoflurane anaesthesia or by transcardially perfusion with ice-cold 4% PFA/PBS (−) (pH 7.4). Brain samples were processed as described above and subjected to in-gel WB and CLSM analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The authors declare that the data supporting the findings of this study are available with the paper and its Supplementary Information files.
References
Boutureira, O. & Bernardes, G. J. L. Advances in chemical protein modification. Chem. Rev. 115, 2174–2195 (2015).
Tamura, T. & Hamachi, I. Chemistry for covalent modification of endogenous/native proteins: from test tubes to complex biological systems. J. Am. Chem. Soc. 141, 2782–2799 (2019).
Lin, S. et al. Redox-based reagents for chemoselective methionine bioconjugation. Science 355, 597–602 (2017).
Josephson, B. et al. Light-driven post-translational installation of reactive protein side chains. Nature 585, 530–537 (2020).
Xie, X. et al. Oxidative cyclization reagents reveal tryptophan cation–π interactions. Nature 627, 680–687 (2024).
Fiala, T. et al. Chemical targeting of voltage sensitive dyes to specific cells and molecules in the brain. J. Am. Chem. Soc. 142, 9285–9301 (2020).
Kosar, M. et al. Platform reagents enable synthesis of ligand-directed covalent probes: study of cannabinoid receptor 2 in live cells. J. Am. Chem. Soc. 145, 15094–15108 (2023).
Yamaura, K., Kiyonaka, S., Numata, T., Inoue, R. & Hamachi, I. Discovery of allosteric modulators for GABAA receptors by ligand-directed chemistry. Nat. Chem. Biol. 12, 822–830 (2016).
Los, G. V. et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382 (2008).
Keppler, A. et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat. Biotechnol. 21, 86–89 (2003).
Adhikari, K. et al. Trans-cyclooctene—a Swiss army knife for bioorthogonal chemistry: exploring the synthesis, reactivity, and applications in biomedical breakthroughs. EJNMMI Radiopharm. Chem. 9, 47 (2024).
Huppertz, M.-C. et al. Recording physiological history of cells with chemical labeling. Science 383, 890–897 (2024).
Mohar, B. et al. DELTA: a method for brain-wide measurement of synaptic protein turnover reveals localized plasticity during learning. Nat. Neurosci. 28, 1089–1098 (2025).
Nonaka, H. et al. Bioorthogonal chemical labeling of endogenous neurotransmitter receptors in living mouse brains. Proc. Natl Acad. Sci. USA 121, 2313887121 (2024).
Takato, M., Sakamoto, S., Nonaka, H., Tamura, T. & Hamachi, I. Photoproximity labeling of endogenous receptors in the live mouse brain in minutes. Nat. Chem. Biol. 21, 109–119 (2025).
Fujishima, S., Yasui, R., Miki, T., Ojida, A. & Hamachi, I. Ligand-directed acyl imidazole chemistry for labeling of membrane-bound protein on live cells. J. Am. Chem. Soc. 134, 3961–3964 (2012).
Arttamangkul, S. et al. Visualizing endogenous opioid receptors in living neurons using ligand-directed chemistry. eLife 8, e49319 (2019).
Chang, P. V. et al. Copper-free click chemistry in living animals. Proc. Natl. Acad. Sci. USA 107, 1821–1826 (2010).
Prescher, J. A., Dube, D. H. & Bertozzi, C. R. Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877 (2004).
Kim, E. & Koo, H. Biomedical applications of copper-free click chemistry: in vitro, in vivo, and ex vivo. Chem. Sci. 10, 7835–7851 (2019).
Lang, K. & Chin, J. W. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev. 114, 4764–4806 (2014).
Ernst, R. J. et al. Genetic code expansion in the mouse brain. Nat. Chem. Biol. 12, 776–778 (2016).
Shandell, M. A., Tan, Z. & Cornish, V. W. Genetic code expansion: a brief history and perspective. Biochemistry 60, 3455–3469 (2021).
Dieterich, D. C., Link, A. J., Graumann, J., Tirrell, D. A. & Schuman, E. M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl Acad. Sci. USA 103, 9482–9487 (2006).
Dieterich, D. C. et al. Cell-type-specific metabolic labeling of nascent proteomes in vivo. Nat. Biotechnol. 13, 897–905 (2010).
Bob, J. et al. THRONCAT: metabolic labeling of newly synthesized proteins using a bioorthogonal threonine analog. Nat. Commun. 14, 3367 (2023).
Oliveira, B. L., Guo, Z. & Bernardes, G. J. L. Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev. 46, 4895–4950 (2017).
Blackman, M. L., Royzen, M. & Fox, J. M. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels–Alder reactivity. J. Am. Chem. Soc. 130, 13518–13519 (2008).
Devaraj, N. K., Weissleder, R. & Hilderbrand, S. A. Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjugate Chem. 19, 2297–2299 (2008).
Simon, M. J. & Iliff, J. J. Regulation of cerebrospinal fluid (CSF) flow in neurodegenerative, neurovascular and neuroinflammatory disease. Biochim. Biophys. Acta 1862, 442–451 (2016).
Ojima, K. et al. Ligand-directed two-step labeling to quantify neuronal glutamate receptor trafficking. Nat. Commun. 12, 831 (2021).
Wakayama, S. et al. Chemical labelling for visualizing native AMPA receptors in live neurons. Nat. Commun. 8, 14850 (2017).
Susaki, E. A. et al. Versatile whole-organ/body staining and imaging based on electrolyte-gel properties of biological tissues. Nat. Commun. 11, 1982 (2020).
Vafadari, B., Salamian, A. & Kaczmareck, L. MMP-9 in translation: from molecule to brain physiology, pathology, and therapy. J. Neurochem. 139, 91–114 (2016).
Beroun, A. et al. MMPs in learning and memory and neuropsychiatric disorders. Cell. Mol. Life Sci. 76, 3207–3228 (2019).
Kridel, S. J. et al. Substrate hydrolysis by matrix metalloproteinase-9. J. Biol. Chem. 276, 20572–20578 (2001).
Fudala, R. et al. Fluorescence detection of MMP-9. I. MMP-9 selectively cleaves Lys-Gly-Pro-Arg-Ser-Leu-Ser-Gly-Lys peptide. Curr. Pharm. Biotechnol. 12, 834–838 (2011).
Dubowchik, G. M. et al. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjugate Chem. 13, 855–869 (2022).
Peixoto, R. T. et al. Transsynaptic signaling by activity-dependent cleavage of neuroligin-1. Neuron 76, 396–409 (2012).
Dubiella, C. et al. Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat. Chem. Biol. 17, 954–963 (2021).
Lou, J. et al. Discovery of a covalent inhibitor that overcame resistance to venetoclax in AML cells overexpressing BFL-1. J. Med. Chem. 67, 10795–10830 (2024).
Moon, H. Y. et al. Running-induced systemic cathepsin B secretion is associated with memory function. Cell Metab. 24, 332–340 (2016).
Galis, Z. S., Sukhova, G. K., Lark, M. W. & Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Invest. 94, 2493–2503 (1994).
Gawlak, M. et al. High resolution in situ zymography reveals matrix metalloproteinase activity at glutamatergic synapses. Neuroscience 158, 167–176 (2009).
Acknowledgements
The authors thank T. Gonda, Y. Nabeta and K. Nishizawa for technical support of biological experiments. The authors also thank V. Muir for editing a draft of this manuscript. This work was supported by the Japan Science and Technology Agency (JST) ERATO (grant number JPMJER1802 to I.H.) and MEXT/JSPS KAKENHI Grant-in-Aid for Specially Promoted Research (grant number 23H05405 to I.H.), MEXT/JSPS KAKENHI Grant-in-Aid for Scientific Research (B) (grant number 24K01627 to H.N.), MEXT/JSPS KAKENHI Grant-in-Aid for Scientific Research (C) (grant number 23K04960 to S.S.), Grants-in-Aid from the Takeda Science Foundation (to H.N.) and JSPS Fellows (grant number 21J15773 to K.S.).
Author information
Authors and Affiliations
Contributions
H.N. and I.H. initiated and designed the project. S.S., K.S., M.W., M.I. and H.N. performed synthesis and chemical labelling experiments in HEK293T cells and brains. H.N., S.S, K.S. and I.H. prepared the manuscript with contributions from the other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Thomas West, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Experimental details and Supplementary Figs. 1-11.
Source data
Source Data Fig. 3
Unprocessed western Blots and/or gels, Statistical Source Data.
Source Data Fig. 4
Unprocessed western Blots and/or gels, Statistical Source Data.
Source Data Fig. 5
Statistical Source Data.
Source Data Fig. 6
Unprocessed western Blots and/or gels, Statistical Source Data.
Source Data Fig. 7
Unprocessed western Blots and/or gels, Statistical Source Data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sakamoto, S., Shiraiwa, K., Wang, M. et al. In-brain synthesis of receptor-based protease sensors by coupling ligand-directed chemistry and click chemistry. Nat. Synth 4, 1128–1140 (2025). https://doi.org/10.1038/s44160-025-00815-6
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44160-025-00815-6
