
Abstract
Septic cardiomyopathy, one manifestation of multiple organ dysfunction syndrome, is a challenging complication in sepsis, and cytopathic hypoxia has been proposed to have a key role in the pathophysiology of multiple organ dysfunction syndrome. However, the underlying mechanisms remain unknown. Here, we show that upregulation of hypoxia-inducible factor-1α (HIF-1α) in cardiomyocytes following lipopolysaccharide (LPS) treatment suppresses mitochondrial respiration via inducible nitric oxide synthase-dependent nitric oxide, leading to cytopathic hypoxia. Cardiac-specific heterozygous deletion of HIF-1α ameliorates mitochondrial and contractile dysfunction in a mouse model of septic cardiomyopathy. Mechanistically, nuclear factor-κB (NF-κB)-mediated upregulation of cyclooxygenase 2 (COX2) and secretory phospholipases A2 (sPLA2) enhances HIF-1α expression following LPS exposure, whereas their inhibition prevents LPS-induced HIF-1α upregulation, cytopathic hypoxia and contractile dysfunction. In addition, phospholipid metabolites (prostaglandins and lysophospholipids/free fatty acids, respectively) stabilize HIF-1α via protein kinase A activation. These findings highlight a crucial role of excessive HIF-1α, driven by LPS-enhanced phospholipid metabolism, in septic cardiomyopathy through induction of cytopathic hypoxia.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others
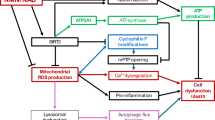

Data availability
The microarray data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE297683. Owing to concerns regarding potential misuse or unauthorized secondary use, all other raw data will be made available upon reasonable request. Requests should be directed to the corresponding author, M.I.
References
Evans, L. et al. Surviving Sepsis Campaign: international guidelines for management of sepsis and septic shock 2021. Crit. Care Med. 49, e1063–e1143 (2021).
Singer, M. et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315, 801–810 (2016).
Beesley, S. J. et al. Septic cardiomyopathy. Crit Care Med 46, 625–634 (2018).
Blanco, J. et al. Incidence, organ dysfunction and mortality in severe sepsis: a Spanish multicentre study. Crit. Care 12, R158 (2008).
Fink, M. Cytopathic hypoxia in sepsis. Acta Anaesthesiol. Scand. 110, 87–95 (1997).
Fink, M. P. Cytopathic hypoxia. Mitochondrial dysfunction as mechanism contributing to organ dysfunction in sepsis. Crit. Care Clin. 17, 219–237 (2001).
Hotchkiss, R. S. et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit. Care Med. 27, 1230–1251 (1999).
van Boxel, G. I., Doherty, W. L. & Parmar, M. Cellular oxygen utilization in health and sepsis. Contin. Ed. Anaesth. Crit. Care Pain 12, 6 (2012).
Majmundar, A. J., Wong, W. J. & Simon, M. C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309 (2010).
Semenza, G. L. Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 (2012).
Simon, M. C. Coming up for air: HIF-1 and mitochondrial oxygen consumption. Cell Metab. 3, 150–151 (2006).
Papandreou, I., Cairns, R. A., Fontana, L., Lim, A. L. & Denko, N. C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197 (2006).
Kim, J. W., Tchernyshyov, I., Semenza, G. L. & Dang, C. V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 (2006).
Deguchi, H. et al. Roxadustat markedly reduces myocardial ischemia reperfusion injury in mice. Circ. J. 84, 1028–1033 (2020).
Bishop, T. & Ratcliffe, P. J. HIF hydroxylase pathways in cardiovascular physiology and medicine. Circ. Res. 117, 65–79 (2015).
Kido, M. et al. Hypoxia-inducible factor 1-α reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J. Am. Coll. Cardiol. 46, 2116–2124 (2005).
Holscher, M. et al. Cardiomyocyte-specific prolyl-4-hydroxylase domain 2 knock out protects from acute myocardial ischemic injury. J. Biol. Chem. 286, 11185–11194 (2011).
Moslehi, J. et al. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation 122, 1004–1016 (2010).
Holscher, M. et al. Unfavourable consequences of chronic cardiac HIF-1α stabilization. Cardiovasc Res. 94, 77–86 (2012).
Bekeredjian, R. et al. Conditional HIF-1α expression produces a reversible cardiomyopathy. PLoS ONE 5, e11693 (2010).
McGettrick, A. F. & O’Neill, L. A. J. The role of HIF in immunity and inflammation. Cell Metab. 32, 524–536 (2020).
Ruan, H., Zhang, Q., Zhang, Y. P., Li, S. S. & Ran, X. Unraveling the role of HIF-1α in sepsis: from pathophysiology to potential therapeutics-a narrative review. Crit. Care 28, 100 (2024).
Rius, J. et al. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 453, 807–811 (2008).
Jung, Y. J., Isaacs, J. S., Lee, S., Trepel, J. & Neckers, L. IL-1β-mediated up-regulation of HIF-1α via an NFκB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 17, 2115–2117 (2003).
Xu, S. et al. IL-6 promotes nuclear translocation of HIF-1α to aggravate chemoresistance of ovarian cancer cells. Eur. J. Pharmacol. 894, 173817 (2021).
Zhou, J., Schmid, T. & Brune, B. Tumor necrosis factor-α causes accumulation of a ubiquitinated form of hypoxia inducible factor-1α through a nuclear factor-κB-dependent pathway. Mol. Biol. Cell 14, 2216–2225 (2003).
Salabei, J. K., Gibb, A. A. & Hill, B. G. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 9, 421–438 (2014).
Poderoso, J. J., Helfenberger, K. & Poderoso, C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 88, 61–72 (2019).
Liu, X. H. et al. Prostaglandin E2 induces hypoxia-inducible factor-1α stabilization and nuclear localization in a human prostate cancer cell line. J. Biol. Chem. 277, 50081–50086 (2002).
Chang, T. C. et al. Stabilization of hypoxia-inducible factor-1α by prostacyclin under prolonged hypoxia via reducing reactive oxygen species level in endothelial cells. J. Biol. Chem. 280, 36567–36574 (2005).
Liu, L. & Clipstone, N. A. Prostaglandin F2α induces the normoxic activation of the hypoxia-inducible factor-1 transcription factor in differentiating 3T3-L1 preadipocytes: potential role in the regulation of adipogenesis. J. Cell. Biochem. 105, 89–98 (2008).
Stasinopoulos, I., O’Brien, D. R. & Bhujwalla, Z. M. Inflammation, but not hypoxia, mediated HIF-1α activation depends on COX-2. Cancer Biol. Ther. 8, 31–35 (2009).
Lee, S. J. et al. Regulation of hypoxia-inducible factor 1α (HIF-1α) by lysophosphatidic acid is dependent on interplay between p53 and Kruppel-like factor 5. J. Biol. Chem. 288, 25244–25253 (2013).
No, Y. R., Lee, S. J., Kumar, A. & Yun, C. C. HIF1α-Induced by lysophosphatidic acid is stabilized via interaction with MIF and CSN5. PLoS ONE 10, e0137513 (2015).
Seo, J. et al. Fatty-acid-induced FABP5/HIF-1 reprograms lipid metabolism and enhances the proliferation of liver cancer cells. Commun Biol. 3, 638 (2020).
Otake-Kasamoto, Y. et al. Lysophosphatidylserines derived from microbiota in Crohn’s disease elicit pathological Th1 response. J. Exp. Med. 219, e20211291 (2022).
MacPhee, M. et al. The secretory phospholipase A2 gene is a candidate for the Mom1 locus, a major modifier of ApcMin-induced intestinal neoplasia. Cell 81, 957–966 (1995).
Takahama, M. et al. Organism-wide analysis of sepsis reveals mechanisms of systemic inflammation. Preprint at bioRxiv https://doi.org/10.1101/2023.01.30.526342 (2023).
Bullen, J. W. et al. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci. Signal 9, ra56 (2016).
Li, L. F. et al. Reduction in ventilation-induced diaphragmatic mitochondrial injury through hypoxia-inducible factor 1α in a murine endotoxemia model. Int. J. Mol. Sci. 23, 1083 (2022).
Sano, M. et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 446, 444–448 (2007).
van Hensbergen, V. P., Wu, Y., van Sorge, N. M. & Touqui, L. Type IIA secreted phospholipase A2 in host defense against bacterial infections. Trends Immunol. 41, 313–326 (2020).
Dore, E. & Boilard, E. Roles of secreted phospholipase A(2) group IIA in inflammation and host defense. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1864, 789–802 (2019).
Snider, J. M. et al. Group IIA secreted phospholipase A2 is associated with the pathobiology leading to COVID-19 mortality. J. Clin. Invest. 131, e149236 (2021).
Kano, K., Aoki, J. & Hla, T. Lysophospholipid mediators in health and disease. Annu. Rev. Pathol. 17, 459–483 (2022).
Kimura, I., Ichimura, A., Ohue-Kitano, R. & Igarashi, M. Free fatty acid receptors in health and disease. Physiol. Rev. 100, 171–210 (2020).
Lee, E. H. et al. Diagnosis and mortality prediction of sepsis via lysophosphatidylcholine 16:0 measured by MALDI-TOF MS. Sci. Rep. 10, 13833 (2020).
Cho, W. H. et al. Clinical significance of enzymatic lysophosphatidylcholine (LPC) assay data in patients with sepsis. Eur. J. Clin. Microbiol. Infect. Dis. 31, 1805–1810 (2012).
Rice, T. W. et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit. Care Med. 38, 1685–1694 (2010).
Bernard, G. R. et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis study group. N. Engl. J. Med. 336, 912–918 (1997).
Kurumbail, R. G., Kiefer, J. R. & Marnett, L. J. Cyclooxygenase enzymes: catalysis and inhibition. Curr. Opin. Struct. Biol. 11, 752–760 (2001).
Zeiher, B. G. et al. LY315920NA/S-5920, a selective inhibitor of group IIA secretory phospholipase A2, fails to improve clinical outcome for patients with severe sepsis. Crit. Care Med. 33, 1741–1748 (2005).
Levy, R. J. et al. Evidence of myocardial hibernation in the septic heart. Crit. Care Med. 33, 2752–2756 (2005).
Levy, R. J. Mitochondrial dysfunction, bioenergetic impairment, and metabolic down-regulation in sepsis. Shock 28, 24–28 (2007).
Dengler, V. L., Galbraith, M. & Espinosa, J. M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 49, 1–15 (2014).
Ryan, H. E. et al. Hypoxia-inducible factor-1α is a positive factor in solid tumor growth. Cancer Res. 60, 4010–4015 (2000).
Agah, R. et al. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Invest. 100, 169–179 (1997).
Honda, S. et al. Marginal zone B cells exacerbate endotoxic shock via interleukin-6 secretion induced by Fcα/muR-coupled TLR4 signalling. Nat. Commun. 7, 11498 (2016).
Furusawa, S. et al. Cardiac autoantibodies against cardiac troponin I in post-myocardial infarction heart failure: evaluation in a novel murine model and applications in therapeutics. Circ. Heart. Fail. 16, e010347 (2023).
Tadokoro, T. et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 5, e132747 (2020).
Ikeda, M. et al. Excessive hypoxia-inducible factor-1α expression induces cardiac rupture via p53-dependent apoptosis after myocardial infarction. J. Am. Heart Assoc. 10, e020895 (2021).
Miyamoto, H. D. et al. Iron overload via heme degradation in the endoplasmic reticulum triggers ferroptosis in myocardial ischemia-reperfusion injury. JACC Basic Transl. Sci. 7, 800–819 (2022).
Abe, K. et al. Doxorubicin causes ferroptosis and cardiotoxicity by intercalating into mitochondrial DNA and disrupting Alas1-dependent heme synthesis. Sci. Signal 15, eabn8017 (2022).
Ikeda, M. et al. The Akt–mTOR axis is a pivotal regulator of eccentric hypertrophy during volume overload. Sci. Rep. 5, 15881 (2015).
Inoue, T. et al. Twinkle overexpression prevents cardiac rupture after myocardial infarction by alleviating impaired mitochondrial biogenesis. Am. J. Physiol. Heart. Circ. Physiol. 311, ajpheart 00044 02016 (2016).
Ikeda, M. et al. Heart rate reduction with ivabradine prevents cardiac rupture after myocardial infarction in mice. Cardiovasc. Drugs Ther. 36, 257–262 (2021).
Ishimaru, K. et al. Deferasirox targeting ferroptosis synergistically ameliorates myocardial ischemia reperfusion injury in conjunction with cyclosporine A. J. Am. Heart Assoc. 13, e031219 (2024).
Arai, S. et al. Functional loss of DHRS7C induces intracellular Ca2+ overload and myotube enlargement in C2C12 cells via calpain activation. Am. J. Physiol. Cell Physiol. 312, C29–C39 (2017).
Ikeda, M. et al. Overexpression of TFAM or twinkle increases mtDNA copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS ONE 10, e0119687 (2015).
Ikeda, M. et al. Immunomodulatory cell therapy using αGalCer-pulsed dendritic cells ameliorates heart failure in a murine dilated cardiomyopathy model. Circ. Heart. Fail. 15, e009366 (2022).
Ackers-Johnson, M. et al. A simplified, langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ. Res. 119, 909–920 (2016).
Acknowledgements
The authors thank M. Sato for her technical assistance. This work was supported by the Japan Society for the Promotion of Science KAKENHI (grant numbers JP24K22274 and JP24K02449 to M.I. and JP23H05481 to K.-I.Y.), the Japan Foundation for Applied Enzymology (Vascular Biology of Innovation to M.I.), MSD Life Science Foundation, the Public Interest Incorporated Foundation (to M.I.), SENSHIN Medical Research Foundation (to M.I.), The Cardiovascular Research Fund, Tokyo, Japan (to M.I.), and The Yomiuri Telecasting Charity Fund Research Grant (to M.I.).
Author information
Authors and Affiliations
Contributions
M.W. was responsible for data curation, formal analysis, investigation, methodology, visualization, validation and writing the original draft. M.I. was responsible for conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, writing the original draft and review and editing. Ko Abe, S. Furusawa, K.I., T. Kanamura., S. Fujita, H.D.M., E.K., Y.S.I., Y.I. and T. Kai were responsible for investigation. T.H., S.M., T.I. and H.T. were responsible for review and editing. K.-I. Y., K.Y. and Kohtaro Abe were responsible for supervision and review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks Patrick Lawler, Edward Thorp, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 A murine model of septic cardiomyopathy.
(a) Left ventricular end-systolic diameter (LVESD) and left ventricular end-diastolic diameter (LVEDD) of C57BL/6 J mice 6 h post-LPS administration (n = 3, each group). (b) Echocardiographic images of the left ventricle of C57BL/6 J mice 24 h post-LPS administration. (c) Left ventricular ejection fraction (LVEF), LVESD, and LVEDD of C57BL/6 J mice 24 h post-LPS administration (n = 3 in the control [Ctrl] group and n = 4 in the LPS group). (d) Hif1a gene expression in the myocardium of C57BL/6 J mice 24 h post-LPS administration, treated with LPS (n = 3 in the Ctrl group and n = 5 in the 24 h group). (e) Western blot analysis of HIF-1α expression in the myocardium of C57BL/6 J mice 24 h post-LPS administration and its quantification (n = 6, each group). GAPDH was used as an internal control. Data are presented as the mean ± SEM and analyzed using a two-sided t-test. *P < 0.05, **P < 0.01.
Extended Data Fig. 2 Biological and histological features of the septic cardiomyopathy model.
(a) Blood pH of mice 6 h after LPS administration (n = 3 in the control [Ctrl] group and n = 4 in the LPS group). (b) Malondialdehyde (MDA) 6 and 24 h after LPS administration (n = 3 in the Ctrl group, n = 3 in the LPS 6 h group, and n = 5 in the LPS 24 h group). (c) Western blot analysis of acrolein-modified proteins in mice 6 and 24 h after LPS administration and their quantifications (n = 3 in the Ctrl group, n = 3 in the LPS 6 h group, and n = 5 in the LPS 24 h group). (d) Hematoxylin/eosin staining of the myocardium 6 and 24 h after LPS administration (n = 6 in Ctrl group, n = 6 in the LPS 6 h group, and n = 3 in the LPS 24 h group). Scale bars, 50 μm. (e) Western blots of CD3, B220, and CD107b in mice 6 and 24 h after LPS administration and their quantifications (n = 3 in the Ctrl group, n = 3 in the LPS 6 h group, and n = 5 in the LPS 24 h group). (f–h) Gene expression of inflammatory cytokines, such as Il1b, Il6, and Tnf, in the myocardium 6 and 24 h after LPS administration (n = 3 in the Ctrl group, n = 3 in the LPS 6 h group, and n = 5 in the LPS 24 h group). GAPDH was used as an internal control. Data are presented as the mean ± SEM. Data were statistically analyzed using a two-sided t-test (a) and two-sided Dunnett’s test (b–h). *P < 0.05, **P < 0.01.
Extended Data Fig. 3 HIF-1α upregulation and impaired mitochondrial respiration in a LPS dose-dependent manner.
(a) Hif1a gene expression in cultured cardiomyocytes treated with LPS (1, 10, 100 ng/mL; n = 6, each group). (b) Western blot analysis of HIF-1α expression in cultured cardiomyocytes treated with LPS (10, 100, 1000 ng/mL) and their quantifications (n = 6, each group). GAPDH was used as an internal control in western blotting experiments. (c) Overall flow of the Mitostress test using the Flux Analyzer in cultured cardiomyocytes treated with LPS (10, 100, 1000 ng/mL; n = 4, each group). (d) Summary of oxygen consumption rates (OCRs) in the Mitostress test shown in panel (c) (n = 4, each group). Mitochondrial reserve capacity (MRC) was calculated by subtracting basal OCR from maximal respiration OCR shown in panel (c). (e) Hif1a gene expression in isolated adult mouse cardiomyocytes treated with LPS (100 ng/mL, 24 h) (n = 4, each group). (f) Maximal OCR under FCCP treatment and Rotenone (Rot)/antimycin (AMA) using the Flux Analyzer in isolated adult mouse cardiomyocytes treated with LPS (n = 4, each group). Data are presented as the mean ± SEM and analyzed using a two-sided Dunnett’s test (a and b), one-way ANOVA with Tukey’s post hoc test (d), and two-sided t-test (e and f). *P < 0.05, **P < 0.01.
Extended Data Fig. 4 Roles of TLR4, inflammatory cytokines, and NO in HIF-1α upregulation and cytopathic hypoxia in cardiomyocytes under LPS treatment.
(a and b) Western blot analysis of HIF-1α expression in cultured cardiomyocytes treated with LPS (a, 100 ng/mL; n = 6, each group) or lipoteichoic acid (LTA) (b, 50 μg/mL; n = 3, each group) and TAK-242 (1 μM). (c) Overall flow of the Mitostress test using the Flux Analyzer (upper) and summary of oxygen consumption rates (OCRs) in the Mitostress test (lower) in cultured cardiomyocytes treated with LPS (100 ng/mL; n = 4, each group) and TAK-242 (1 μM). (d) Overall flow of the Mitostress test using the Flux Analyzer (upper) and summary of OCRs in the Mitostress test (lower) in cultured cardiomyocytes treated with LTA (50 μg/mL; n = 4, each group) and TAK-242 (1 μM). Mitochondrial reserve capacity (MRC) was calculated by subtracting basal OCR from maximal respiration OCR. (e) Western blot analysis of HIF-1α in cultured cardiomyocytes treated with IL-1β (10 ng/mL, 24 h) (left), IL-6 (10 ng/mL, 24 h) (middle), and TNF (3 ng/mL, 24 h) (right) (n = 6, each group). (f and g) Overall flow of the Mitostress test using the Flux Analyzer in LPS-treated cultured cardiomyocytes, in which inducible NOS (iNOS) was silenced by specific siRNA (f; n = 4, each group) or NO was scavenged by 100 μM C-PTIO (g; n = 8, each group). GAPDH was used as an internal control in western blotting experiments. Data are presented as the mean ± SEM. Data were statistically analyzed using 1-way ANOVA with Tukey’s post hoc test (a–d) and a two-sided t-test (e). *P < 0.05, **P < 0.01.
Extended Data Fig. 5 Echocardiographic data and the myocardial expression of HIF-1α in control [Ctrl] mice and cardiomyocyte-specific HIF-1α hetero knockout (caHetKO) mice 24 h after LPS treatment.
(a) Echocardiographic images of the left ventricle of Ctrl and caHetKO mice 24 h after LPS administration. (b) Left ventricular ejection fraction (LVEF, left), left ventricular end-systolic diameter (LVESD, middle), and left ventricular end-diastolic diameter (LVEDD, right) in Ctrl and caHetKO mice 24 h after LPS administration (n = 8, each group). (c) Western blot analysis of HIF-1α expression in the myocardium of Ctrl and caHetKO mice 24 h after LPS administration and its quantification (n = 8, each group). GAPDH was used as an internal control. Data are presented as the mean ± SEM and analyzed using a two-sided t-test. **P < 0.01.
Extended Data Fig. 6 Roles of the NF-κB family in cultured cardiomyocytes under LPS treatment.
(a) Western blots of p105/p50 and HIF-1α expression under LPS (100 ng/mL) treatment in cultured cardiomyocytes with or without Nfkb1 silencing and quantification of HIF-1α (n = 6, each group). (b) Hif1a gene expression in cultured cardiomyocytes treated with siRNA against Nfkb1 (n = 9, each group). (c) Western blots of RELA and HIF-1α expression under LPS treatment in cultured cardiomyocytes with or without Rela silencing and quantification of HIF-1α (n = 6, each group). (d) Hif1a gene expression in cultured cardiomyocytes treated with siRNA against Rela (n = 6, each group). (e) Western blots of c-Rel and HIF-1α expression under LPS treatment in cultured cardiomyocytes with or without Rel silencing and quantification of HIF-1α (n = 6, each group). (f) Hif1a gene expression in cultured cardiomyocytes treated with siRNA against Rel (n = 9, each group). (g) Western blots of p100/p52 and HIF-1α expression under LPS treatment in cultured cardiomyocytes with or without Nfkb2 silencing and quantification of HIF-1α (n = 3, each group). (h) Western blots of RelB and HIF-1α expression under LPS treatment in cultured cardiomyocytes with or without Relb silencing and quantification of HIF-1α (n = 3, each group). GAPDH was used as an internal control in western blotting experiments. Data are presented as the mean ± SEM and analyzed using 1-way ANOVA with Tukey’s post hoc test. *P < 0.05, **P < 0.01.
Extended Data Fig. 7 Roles of COX2 and secretory phospholipase A2 (sPLA2) in LPS-induced HIF-1α expression.
(a–b) Overall flow of the Mitostress test using the Flux Analyzer in LPS (100 ng/mL)-treated cultured cardiomyocytes, in which COX2 was silenced by specific siRNA (a) (n = 4, each group) or inhibited by flurbiprofen (b) (n = 8, each group). (c) Hif1a gene expression in cultured cardiomyocytes treated with prostaglandin E2 (10 μM, 24 h) (left; n = 4) and prostaglandin I2 (100 μM, 24 h) (right; n = 4). (d) Western blot analysis of HIF-1α in cardiomyocytes treated with U-46619 (30 μM, 24 h), an agonist for the thromboxane A2 receptor, and its quantification (n = 6, each group). GAPDH was used as an internal control in western blotting experiments. Data are presented as the mean ± SEM and analyzed using a two- sided t-test.
Extended Data Fig. 8 Roles of secretory phospholipase A2 (sPLA2) and lysophosphatidylcholine (lysoPC) in septic cardiomyopathy.
(a) Ptgs2 gene expression in cultured cardiomyocytes treated with LPS (100 ng/mL) and siRNA targeting Nfkb1, Rela, and Rel (n = 6 per group). (b) Enpp2 gene expression in cultured cardiomyocytes treated with LPS and siRNA against Nfkb1, Rela, and Rel (n = 4 in the siNfkb1 group, n = 4 in the siRela group, and n = 3 in the siRel group). (c and d) Western blots of autotaxin (ATX) and HIF-1α expression under LPS treatment in cultured cardiomyocytes with Enpp2 silencing and their quantifications (n = 3, each group). GAPDH was used as an internal control in western blotting experiments. (e) Overall flow of the Mitostress test using the Flux Analyzer in LPS-treated cultured cardiomyocytes, in which PLA2G2A was silenced by specific siRNA (n = 4, each group). (f) Experimental protocol for treatment of C57BL/6 J mice with Varespladib (an inhibitor against PLA2G2A, PLA2G5, and PLA2G10) under LPS administration. (g) Echocardiographic images of the left ventricles of C57BL/6 J mice with VPL treatment 6 h after LPS administration. (h) LVEF of C57BL/6 J mice 6 h after LPS administration with or without VPL treatment (n = 6, each group). (i) Western blot analysis of HIF-1α expression in the myocardium of C57BL/6 J mice 6 h after LPS administration with or without VPL and its quantification (n = 6, each group). GAPDH was used as an internal control in western blotting experiments. (j) The levels of lysoPCs (left, 16:0; middle, 18:0; right, 18:1) in the blood of mice treated with PLA2G5-neutralizing antibody, 6 h after LPS administration (n = 6, each group). (k) Western blot of autotaxin (ATX) expression in the plasma of C57BL/6 J mice 6 h after LPS administration and its quantification (n = 6, each group). Coomassie Brilliant Blue (CBB) was used as an internal control in western blotting experiments. Data are presented as the mean ± SEM. Data were statistically analyzed using a two-sided t test (a, c, k) and 1-way ANOVA with Tukey’s post hoc test (d, h–j). *P < 0.05, **P < 0.01.
Extended Data Fig. 9 Roles of protein kinase A (PKA) in HIF-1α upregulation in cardiomyocytes, which is induced by prostaglandin E2 (PGE2), lysophosphatidylcholine (lysoPC), and oleic acid (OA) and HIF-2α in cultured cardiomyocytes treated with LPS.
(a) Western blot analysis of HIF-2α in cultured cardiomyocytes, in which HIF-2α was silenced by specific siRNA (n = 3, each group). (b) Overall flow of the Mitostress test using the Flux Analyzer in LPS-treated cultured cardiomyocytes, in which HIF-2α was silenced by specific siRNA. (c) Summary of oxygen consumption rates (OCRs) in the Mitostress test shown in panel (b) (n = 4, each group). Mitochondrial reserve capacity (MRC) was calculated by subtracting basal OCR from maximal respiration OCR shown in panel (e). (d–f) Western blot analysis of HIF-1α in cultured cardiomyocytes treated with PGE2 (10 μM, 24 h) (d), lysoPC (200 μM [100 μg/mL], 3 h) (e), and OA (30 μM, 24 h) (f) (n = 6, each group). H-89 (30 μM) was co-treated for an inhibition of PKA. GAPDH was used as an internal control in western blotting experiments. Data are presented as the mean ± SEM and analyzed using one-way ANOVA with Tukey’s post hoc test. *P < 0.05, **P < 0.01.
Extended Data Fig. 10 Graphical abstract depicting the present findings.
COX2, cyclooxygenase 2; FFA, free fatty acid; iNOS, inducible nitric oxide synthase; NO, nitric oxide; LPS, lipopolysaccharide; LTA, lipoteichoic acid; PG, prostaglandin; PKA, protein kinase A; TLR4, toll-like receptor-4.
Supplementary information
Supplementary Table 1 (download XLSX )
Supplementary Table 1.
Source data
Source Data Fig. 1 (download PDF )
Unprocessed western blots in Fig. 1.
Source Data Fig. 3 (download PDF )
Unprocessed western blots in Fig. 3.
Source Data Fig. 4 (download PDF )
Unprocessed western blots in Fig. 4.
Source Data Fig. 5 (download PDF )
Unprocessed western blots in Fig. 5.
Source Data Fig. 6 (download PDF )
Unprocessed western blots in Fig. 6.
Source Data Fig. 7 (download PDF )
Unprocessed western blots in Fig. 7.
Source Data Fig. 8 (download PDF )
Unprocessed western blots in Fig. 8.
Source Data Extended Data Fig. (download PDF )
Unprocessed western blots in Extended Data Fig. 1.
Source Data Extended Data Fig. (download PDF )
Unprocessed western blots in Extended Data Fig. 2.
Source Data Extended Data Fig. (download PDF )
Unprocessed western blots in Extended Data Fig. 3.
Source Data Extended Data Fig. (download PDF )
Unprocessed western blots in Extended Data Fig. 4.
Source Data Extended Data Fig. (download PDF )
Unprocessed western blots in Extended Data Fig. 5.
Source Data Extended Data Fig. (download PDF )
Unprocessed western blots in Extended Data Fig. 6.
Source Data Extended Data Fig. (download PDF )
Unprocessed western blots in Extended Data Fig. 7.
Source Data Extended Data Fig. (download PDF )
Unprocessed western blots in Extended Data Fig. 8.
Source Data Extended Data Fig. (download PDF )
Unprocessed western blots in Extended Data Fig. 9.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Watanabe, M., Ikeda, M., Abe, K. et al. Excessive HIF-1α driven by phospholipid metabolism causes septic cardiomyopathy through cytopathic hypoxia. Nat Cardiovasc Res 4, 1077–1093 (2025). https://doi.org/10.1038/s44161-025-00687-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s44161-025-00687-1
This article is cited by
-
Vitamin C is associated with improved outcomes in patients with sepsis-induced myocardial injury: insights from the MIMIC-IV database
BMC Infectious Diseases (2025)
-
Linking phospholipid metabolism to septic cardiomyopathy via HIF-1α overactivation
Nature Cardiovascular Research (2025)
