
Abstract
Imperfect attempts at organ repair after repeated injury result in aberrant formation of extracellular matrix (ECM) and loss of tissue structure. This abnormal ECM goes from being a consequence of cellular dysregulation to become the backbone of a persistently fibrotic cell niche that compromises organic function and ultimately drives systemic disease. Here, we review our current understanding of the structure of the ECM, the mechanisms behind organ-specific fibrosis, resolution, healing and regeneration, as well as the development of anti-fibrotic strategies. We also discuss the design of biomarkers to investigate fibrosis pathophysiology, track fibrosis progression, systemic damage, and fibrosis resolution.
Similar content being viewed by others
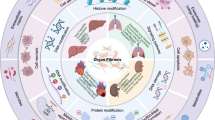
The organized complexity of the extracellular matrix
The mechano-chemical properties of the extracellular matrix (ECM) (Fig. 1) are necessary for cells to differentiate, specialize, locate themselves in relation to other cell populations, and build the functional units that characterize multicellular anatomy. There is a reciprocity between function, developmental stage and ECM structure that results in specialized ECMs, where components are connected in organ- and stage-specific patterns1. These ECMs are made of different protein combinations2 with different turnover cycles, dependent on strictly regulated building, dismantling, and remodeling cycles.

Although ECM is organ specific, there are characteristics that are common to most tissues, like the presence of a basement membrane and an interstitial matrix. Equally, disease may induce sui generis remodeling, abnormal formation and degradation are shared by several conditions (e.g., IPF, MASLD, COPD, etc.).
Pioneering work on Mass Spectrometry (MS) has cataloged ECM components (the matrisome), divided in core-matrisome (~300) and matrisome-associated proteins (~1000)2. The core-matrisome, or structural ECM proteins, includes 28 collagens, elastin, fibronectin, and laminin isoforms, as well as proteoglycans (like perlecan) and glycoproteins (like nidogens). Matrisome-associated proteins regulate ECM structure (e.g., proteolytic enzymes, matrix metalloproteinases-MMP, etc) but are also controlled by ECM, e.g., transforming growth factor beta, (TGF-β), vascular endothelial growth factor, (VEGF) superfamilies, other growth factors and cytokines2,3. The emergence of spatial proteomics and 3D ECM mapping4,5,6 is revealing the 3D structure of the ECM, showing that healthy and diseased tissue share ECM components, but their amount, distribution, density, and articulation in space differs. It is likely that functional units within an organ (e.g., nerve trunks and terminals, vessels, specialized structures like follicles, glomeruli, alveoli, or acini) have a function-specific ECM6. This seems to be the case of capillary follicles and skin7,8.
ECM structure is generally divided in two compartments (Fig. 1): Basement Membrane (BM) and Interstitial Matrix (IM). BM is a cloth-like surface, adhesive to epithelia, glandular epithelia, endothelium, myocytes, and adipocytes, among other cells5. The BM is based on a Collagen type IV backbone supporting a Laminin surface9. Glycoproteins, such as Nidogens and Perlecan, bind the Laminin and Collagen type IV layers. Others, like Netrin-4, regulate BM mechanical properties10.
IM is structured by fibrillar collagens (type I, II, III, V) and elastin. In turn, collagen fibrils are bridged by Fibril-associated Collagens with Interrupted Triple Helices (reviewed elsewhere11) and linked to the BM by non-fibrillar collagens, like Collagen type VII12. Fibronectin (reviewed here13,14), regulates ECM assembly, collagen fiber assembly, embryo development, and is also critical during wound healing, ECM maturation and cancer progression. Cell-secreted fibronectin is a mediator of scar tissue formation15. Upon fibrosis progression, IM undergoes extensive remodeling, notably, abundant cross-linking mediated by enzyme families, like transglutaminases and lysyl-oxidases16, and collagen glycation, a process associated to diabetes and aging17. The overgrowth of crosslinked collagen results in a stiff, more viscoelastic18, linearized IM with deleterious consequences for disease progression19.
ECM stores and releases growth factors, cytokines, and bioactive peptides, controlling their location, density, and activity. Fibronectin binds VEGF, hepatocyte growth factor (HGF), platelet-derived growth factors (PDGF), among others, and growth factor-binding domains are abundant in matrisomal proteins3. Injury responses can, by increasing binding sites, signaling oligopeptides, and structural domains, enhance fibrogenesis as well as mechanosignaling20. Pioneering experiments exposing radiolabeled PDGF isoforms keratinocyte growth factor and HGF to collagen chains demonstrated binding to collagens type I, III, IV, V, and VI as well as preserved growth factor bioactivity21,22,23. The affinity of collagen domains extends to inflammatory mediators and cytokines (e.g., interleukin-2, oncostatin)24,25. Isolating cell-ECM structure interactions in an ECM scaffold-based bioreactor shows that kinase activity (including growth-factor activity) and cell-driven ECM remodeling follow anatomical cues, supporting the notion of positional regulation26. Therefore, the ECM acts as a spatial regulator of cellular activity.
By-products of ECM protein synthesis add an additional layer of complexity to ECM dynamics. These by-products can be bioactive, paracrine, and endocrine regulators27. Collectively, they are called matrikines28. Notable examples of this family29 include endotrophin, a pro-peptide of collagen type VI, linked to visceral adipose tissue (VAT) dysregulation, including fibrosis, leading to metabolic disorders30; endostatin, a pro-peptide of collagen type XVIII that is an endogenous inhibitor of angiogenesis31 and potentially fibrogenesis32; and tumstatin33, a collagen type IV pro-peptide, that suppresses inflammation and angiogenesis, and therefore has been shown to play a regulatory role in multiple inflammatory and oncogenic conditions.
Balance and imbalance between tissue formation and destruction
All possible combinations of ECM proteins in presence, abundance, and density could suggest high ECM variability34, yet normal development35 and adult homeostasis follow predictable patterns. The relative pathological regularity of fibrosis serves as an indicator of disease stage36 and suggests the existence of organ- and disease-specific (rather than patient-specific) variations of ECM topography. This notion has important implications: there is considerable evidence pointing to the ECM as a key source of cellular regulation, thus, fibrotic ECM has been identified as an actor, not a bystander, of disease progression37,38. As the structure of fibrosis is largely predictable, so should be the biological effects of that structure39,40. These mechanisms seem to depend on context: an example is the function of the TGF-β superfamily41. The latent form of TGF-β1 is bound by ECM structure, however, TGF-β1 signaling is swayed by its binding substrate, e.g., binding to Fibulin 4 decreases TGF-β1 signaling42, while binding to Fibulin 2 enhances TGF-β143. Moreover, TGF-β1 is also bound and regulated by Fibrillins44, Fibronectin45, among others46,47. Similarly complex interactions are likely the norm for growth factors48.
Cumulative damage resulting from aging49, injury50,51, acute52,53, chronic disease54,55 can interact with reparative reactions, including inflammation56, metabolic dysregulation57 and immune response58, to incline the ECM balance towards fibrogenesis and overgrowth59. Some organisms respond to injury by recreating the original tissue60, mammalians however appear to have only a very partial version of this ability, most of it lost after birth61. With few exceptions, skewed ECM formation results in fibrotic scars.
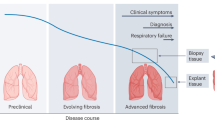
Fibrosis can be staged histologically by scoring it in biopsies62, or non-invasively by using imaging techniques63, to measure organ biomechanics64 and by probing biochemical variables (or biomarkers) that track fibrosis progression65,66. Importantly, biomarkers have helped establish a distinction between staging disease severity and assessing the dynamics of disease activity67. Staging describes the net result of fibrotic accumulation, while biomarkers of fibrogenesis, a measure of disease activity, open a window on the timing and rhythm of disease progression, revealing periods of quiescence as well as bouts of ECM remodeling or accumulation. In advanced disease, these bouts may determine the prognosis of a patient. Differences in etiology and fibrogenic activity in advanced fibrosis also reveal patient heterogeneity, leading to further advantages of using biomarkers in a personalized approach: selecting patients according to disease (and fibrosis) endotype and, it follows, determining whether a treatment affects fibrogenesis or fibrous tissue degradation during periods of ECM remodeling (Fig. 2).

Homeostatic balance can be broken by repeated injury (infection, environmental exposure, physical or chemical insult, mutations, etc.) and subsequent, insufficient, dysregulated reparative response. How this balance tilts towards determines a distinct functional disruption (endotype). Dynamic biomarkers should identify these endotypes and the activities driving disease as well as a potential regeneration.
Recent progress in drug development, in research and in clinical trials, shows that it is possible to modulate the balance between ECM formation and degradation and removal68,69,70, raising new questions about mammalian and human ability to resolve fibrosis, and then initiate a program of functional tissue repair and regeneration. This progress must be matched by biomarkers designed to detect, differentiate, and quantify disparate processes dynamically. Such biomarkers should become useful tools for development and efficacy monitoring of antifibrotic and pro-regenerative therapies.
Core mechanisms of fibrosis
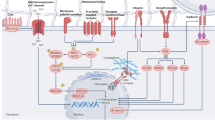
“Core” pro-fibrotic mechanisms71 are myofibroblast-associated pathways found in different fibroses. A bare bones myofibroblast definition could be that of an “activated” mesenchymal cell characterized by a dense α-smooth muscle actin (αSMA) cytoskeleton, enhanced contractility, and ECM protein overexpression. Myofibroblast progenitors can be traced to be adipocytes72, pericytes73, smooth muscle cells74, immune cells75, mesenchymal stem cells76, endothelium77, epithelia78, bone-marrow derived and organ-specific fibroblasts79. This heterogeneity suggests that more than a cell type, “myofibroblast” may denote a behavior. Myofibroblast activation largely depends on a signaling network that hovers around the TGF-β superfamily41,79, beginning with the release of TGF-β1 from the TGF-β latency associated peptide (LAP) in the ECM, prompted by multiple mechanisms, including mechanical stress sensing by the LAP-binding integrins αVβ1 on myofibroblasts and αvβ6 on activated epithelia80,81, binding to TGF-β receptors 1 and 2, canonical SMAD signaling, translocation of SMAD2, SMAD3 and SMAD4 to the nucleus and promotion of the genes encoding αSMA (ACTA2) and ECM proteins. Other pathways also lead to activation: TGF-β282,83 and TGF-β384, and non-canonical TGF-β signaling through mitogen-activated protein (MAP) kinase85.
Signaling and mechanical changes are linked. Injury attracts fibroblasts, and they contract injured tissue, increasing stiffness. An initially soft ECM is then substituted by scar tissue (discussed below) rich in fibronectin and collagen crosslinked by transglutaminases86 and lysyl oxidases87,88. Myofibroblasts transmit and perceive force through cell surface receptors, notably integrins87, discoid domain receptors (DDR)89, vanilloid receptors90, G-protein coupled receptors91, and hyaluronan receptor CD4492 Cell-ECM contact induces the synthesis of cytoskeleton proteins and cell adhesion complexes, calling for further ECM contraction. Integrins αvβ1, αvβ3, αvβ5 and αvβ6 activate latent TGF-β by mechanically pulling LAP79, thus linking both biochemical and mechanical signaling in one positive activation loop. The ADAMTS (A disintegrin-like and metalloproteinase with thrombospondin motifs) superfamily has 19 members that remodel the ECM, partly by cleaving latent TGF-β complexes, changing cell mechanics, and increasing tension as well as TGF-β release93. A subgroup of ADAMTS members bind to fibrillin and fibronectin94, belong in the fibrillin microfibril niche, a mechanosensing hub, and regulate elastic fiber assembly through TGF-β95. ADAMTS-like 2 variants produce geleophysic dysplasia, a syndrome associated to cardiac and interstitial fibrosis96, and is overexpressed in adults with chronic liver disease97. Recessive mutations in ADAMTS10 can cause Weill-Marchesani Syndrome, associated with cardiac fibrosis98.
The TGF-β superfamily also includes a subgroup of cytokines, activins, that bind to membrane receptors (activin receptors type I and II) to phosphorylate the activin-like kinase 4 (ALK4), which in turn phosphorylates Smad proteins 2 and 3 to transduce activin signaling into the nucleus99. Activin action is downregulated by follistatins, which bind to the ECM (e.g., to heparan sulfate proteoglycans)100 and trap activin, so it can be cleaved by proteolysis. Activin upregulation results in follistatin overexpression and activin attenuation. The activin-follistatin system is implicated in scarring and regeneration across several organs. In the liver, activin is overexpressed in models of liver fibrosis, activating hepatic stellate cells (HSCs)101, whilst Follistatin blocks activin and inhibits TGF-β signaling as well as collagen production102. In the kidneys, activin is overexpressed after injury and during kidney fibrosis, mimicking TGF-β signaling and hampering regeneration103, but follistatin blockade promotes epithelial proliferation and repair104. In the lungs, activin promotes myofibroblast proliferation, ECM formation, and TGF-β overexpression, which in turn induces activin, creating a persistent fibrotic niche99. Follistatin gene therapy has been trialed for Becker muscular dystrophy, resulting in reduced muscle fibrosis and muscle performance improvement105. Activin A inhibition with a monoclonal antibody (Garetosmab106) reduces new heterotopic bone lesion formation in fibrodysplasia ossificans progressive. Sotatercept, an activin inhibitor, reduced the risk of death in patients with pulmonary arterial hypertension107.
The Wnt/ β-catenin pathway can also drive fibrogenesis. Wnt is a homolog of integrase-1 and the wingless gene in Drosophila108. There are 19 Wnt proteins, essential for development and homeostasis. The canonical Wnt pathway involves binding to the Frizzled (FZD) transmembrane receptors, the translocation of β-catenin into the cell nucleus, and activation of target genes by transcription factors T-cell factor (TCF) and Lymphoid enhancer factor (Reviewed extensively in109). The Wnt/β-catenin pathway is activated in fibrogenic diseases. Wnt1, Wnt7b, Wnt10b, FZD2, FZD3, β-catenin are overexpressed during idiopathic pulmonary fibrosis (IPF), a disease marked by the overproduction of ECM. Wnt in pulmonary fibrosis promotes fibroblast proliferation, recruitment and activation110. Interestingly, chronic obstructive pulmonary disease (COPD) a disease marked by alveolar ECM destruction, has reduced Wnt/β-catenin activity111. Activation of Wnt/β-catenin seems to attenuate COPD progression112. Wnt/β-catenin can be controlled by TGF-β and thus promote myofibroblast differentiation113. Monoclonal antibodies Vantictumab and Ipafricept block Wnt to FZD receptors and decrease human tumor growth114. β-catenin inhibitors, ICG-001 and PRI-724 reduce markers of fibrogenesis and myofibroblast differentiation, collagen, and inflammation115.
Organ-specific fibrosis, fibrosis resolution, and the perspective of regeneration
Human regenerative capacity is limited, but there are examples of complete regeneration in nature that serve as experimental models and point to the mechanisms humans lack: Hofstenia miamia, an Acoel worm, can regenerate its whole body116, the sea slug Elysia cf. marginata self-decapitates to grow a new body117, the axolotl (Ambystoma mexicanum) can grow an exact replica of almost any tissue118,119, and the Zebra fish (Danio rerio) can regenerate organs upon mutilation120. Sequencing the Acoel genome uncovered Early Growth Response (egr), a master control gene induced by mutilation that epigenetically regulates other wound control response genes (vertebrates bear homologs of egr)116. Zebra fish respond to amputation with the formation of a “blastema”, a mass of undifferentiated cells that proliferate and specialize to regenerate tissue. Remarkably, epigenetic control exerted by the Kdm6b.1 demethylase over zebra fish genes, associated to embryonic patterning, switches on after injury, activating regenerative programs121.
The discovery of scarless healing122 established that regeneration exists in mammalians but vanishes as intrauterine development ends123. After injury, fetal coagulation forms porous clots that are weakly crosslinked122, followed by the deposition of an ECM with a higher collagen type III to collagen type I ratio123. Fetal wound ECM lacks oxidative stress15. TGF-β1 and TGF-β3, regulate the injury response in mammalians but seem to play opposite roles. TGF-β1 is central to the activation of fibroblasts41, while TGF-β3 can be anti-fibrotic124. Both bind to multiple ECM sites125, but only TGF-β1 becomes activated by increasing ECM stiffness126. Moreover, while TGF-β1 is only shortly activated after injury in fetuses, it persists in adults. TGF-β3 is only briefly activated at the end of adult scarring. It is notable that scarless healing disappears as the mature immune response emerges127. Mechanosignaling plays a role in switching the repair response towards scar formation, via activation of Engrailed 1 (En1) in fibroblasts by injury. High stiffness produces fibrotic scars, but low stiffness and inhibition of the Yes-associated protein favors scarless healing and recreation of specialized structures128. ECM abnormalities in adult animals (e.g., increasing stiffness) deregulate fibroblast signaling, enhancing their survival by activating RAS (derived from rat sarcoma virus) activity129, while controlling multipotency in epithelial lineages130. Regenerative mechanisms seem to be absent in post-fetal mammalians121 and are certainly lacking in humans, but our regenerative plasticity, while limited, is not non-existent (Fig. 3). Human regeneration is heterogeneous, with high variation along developmental stages and among organs.

Schematic highlighting the state-of-the-art of organ regeneration technology (left), and the “Organ death races” that are triggered by local, ECM-associated disease, emphasizing liver and adipose tissue as sources of syndrome-like and ultimately lethal events (right).
Liver
In mammalians, the liver stands out for its ability to regenerate completely from a loss of up to 75% of its mass, until it reaches at least 85% of hepatocyte function131. This process can be completed in less than 2 weeks132. In contrast, injury to the myocardium frequently results in the formation of a non-functional scar133. Livers recur to specialized repair mechanisms to maintain hepatic function after injury or hepatectomy134. Liver injury triggers the urokinase-type plasminogen (uPA) activator135 and matrix metalloproteinases to start ECM degradation and remodeling136. uPA leads to the release of HGF bound to ECM22,137,138. A subpopulation of annexin 2 (ANXA2) positive hepatocytes139 has been shown to respond to HGF and drive the regenerative response to injury. This seems to coincide with a coordinated proliferation of hepatocytes, which proceeds from the portal spaces towards the central vein140, although tracing reveals proliferating hepatocytes to be more abundant in zone 2 of the hepatic lobule141. ECM composition seems to run a parallel course to hepatocytes: pioneering data compiled in rat models134,142 suggest that ECM gene expression patterns are choreographed, successively increasing proteoglycan expression, tissue inhibitor of metalloproteinase 1 (TIMP-1), and then interstitial collagens type I and III.
A cohort of patients with MASLD and treated with bariatric surgery saw fibrosis regressing in 70% of all participants after 5 years, including resolution in 45% of patients with advanced fibrosis at baseline143. Bariatric surgery reduces the expression of pro-fibrotic genes144 as part of a vast impact on metabolic and endocrine physiology that includes decreased levels of the “hunger hormone” Ghrelin, the “satiety hormone” leptin and pro-inflammatory cytokines like Tumor Necrosis Factor Alpha (TNF-a)145. Remarkably, patients with chronic hepatitis B virus and cirrhosis at baseline regressed their histological fibrosis staging and were diagnosed as non-cirrhotic after long-term treatment with tenofovir146. Similar results seem to be mirrored in patients with hepatitis C147. Together, these results suggest liver fibrosis to be reversible (Fig. 3). Moreover, newer therapies appear on the immediate horizon148. Dual agonists of the glucagon receptor and the glucagon-like peptide-1 (GLP-1) produce fibrosis regression in up to one third of MASH patients149 and fibroblast growth factor 21 (FGF21) analogs also improve fibrosis in MASH150. New mechanisms keep being dissected, e.g., targeting claudin-1 a component of tight cell junctions, has been shown to be an anti-fibrotic strategy151.
Liver biomarkers have advanced apace. The enhanced liver fibrosis test (ELF)152, a composite biomarker synthesizing blood levels of the tissue inhibitor of metalloproteinase-1 (TIMP-1), procollagen III amino terminal peptide (PIIINP) and hyaluronic acid (HA), and the N-terminal of procollagen type III (PRO-C3)153 are routinely used in the clinical evaluation of chronic liver disease and in drug development, to diagnose, prognosticate, measure disease activity and evaluate drug effect70,148,154.
Heart
Upon insult, the adult myocardium answers with fibrogenesis, not regeneration155,156. Nonetheless, adult cardiomyocytes can re-enter the cell cycle, and conclusive evidence emerged from an elegant study that measured carbon-14 incorporation in cardiomyocytes from individuals born around the partial ban in nuclear testing of 1963157, when environmental isotope levels decreased exponentially. The key finding established that ≈0.5-1% of adult human cardiomyocytes re-enter the cell cycle per year, not enough to sustain regeneration.
The key to heart regeneration may not be in its beating cells, but in those maintaining its structure (Fig. 3). Scar-building cells in the heart come from resident fibroblasts158, recruited bone marrow progenitors (fibrocytes), and endothelial cells that complete endothelial-mesenchymal transition. They become myofibroblasts under TGF-β signaling159. These cells depose ECM after being exposed to hypoxia or inflammatory cytokines like interleukin-2 and tumor necrosis factor160, but in another example of the contextual nature of fibrosis, the persistence of myofibroblasts is not necessarily damaging. Mice engineered to produce myofibroblast-rich post-infarction tissue showed reduced scar formation161. Still, groundbreaking work162 in cardiac fibroblasts engineered to express ovoalbumin peptide to mark them as targets for CAR (chimeric antigen receptor) CD8+ T-cells (i.e., anti-fibroblast immunotherapy) led to a reduction of cardiac fibrotic injury. Tantalizingly, fibrillar collagen deposition was reduced in treated hearts that showed histologically normal myocardium, suggesting that a restitution of normal architecture could take place after eliminating the cells producing abnormal ECM. The optimal window for such a therapy needs to be determined.
Cardiac biomarkers are extensively reviewed elsewhere163,164. Benchmark biomarkers include B-type natriuretic peptide (BNP) and N-terminal BNP (NT-proBNP) (diagnostic and prognostic in heart failure patients), cardiac troponins (myocardial necrosis), Suppression of Tumorigenicity (ST2), an interleukin receptor upregulated in response to injury (prognostic for heart failure), Galectin-3, a lectin, that marks cardiac fibroblastic activity and C-terminal type VIa3 pro-collagen (PRO-C6), prognostic in heart failure with preserved ejection fraction165.
Lungs
Like the heart, the lungs are subject to constant mechanical demands fulfilled by a highly specialized ECM (Fig. 3). Unlike the heart, the lungs are directly exposed to a variety of environmental irritants that can trigger the destruction of normal ECM structure and its substitution by excessive scar tissue (e.g., interstitial lung disease- ILD) or a protracted dismantling of pulmonary airways and alveoli (emphysema in COPD).
In ILD, functional parenchyma is gradually substituted by an ECM that reduces alveolar area to dysfunctional remnants, while building a fibrotic callus. A prominent form of ILD, idiopathic pulmonary fibrosis (IPF)166 has so far evaded mechanistic dissection. Repeated micro-injury to the alveolar epithelium167 can work together with mutations in the surfactant protein gene (SFTPC) expressed by Alveolar Type 2 epithelial cells (AT2), intracellular accumulation of abnormal surfactant and cell senescence168, including telomere dysfunction, to induce fibroblast-to-myofibroblast activation. More specifically, AT2 cells lose regenerative capacity during ILD, leading to their substitution by progenitor airway cells invading the alveoli169. These airway cells have basal cell (basaloid) characteristics, including keratin 17 production (KRT5-/KRT17+ cells) and expression of ECM genes170 and locate on fibrotic lesions. Mechanochemical signaling is also associated to fibrosis development. Loss of the cell division control protein 42 homolog (Cdc42) in mice renders them unable to regenerate alveoli after pneumonectomy, which increases mechanical tension and triggers a TGF-β activation loop, driving peripheral fibrosis that advances toward the lung hilum171.
Basic knowledge is being gradually translated to the clinic. Pirfenidone inhibits TGFB signaling and collagen production172. In IPF patients, Pirfenidone slows disease progression and respiratory decline173. Nintedanib is a competitive inhibitor of non-receptor and receptor tyrosine kinases, including platelet derived growth factor receptor (PDGFR), fibroblast growth factor receptors 1, 2 and 3 and VEGF receptors 1, 2 and 3174. Nintedanib slows lung function decline but is not curative175.
Functional tests are paramount in pulmonary drug development. Forced vital capacity (FVC), forced expiratory volume in 1 s (FEV), and 6-min walk distance are used along lung imaging, arterial gases diffusion and quality of life measurements. A non-invasive biomarker, plasma fibrinogen, was qualified as a drug development tool by the food and drugs administration (FDA)153. Another biomarker, Eosinophil count, has been instrumental in the development of new drugs to treat COPD exacerbations176,177. Novel markers, sensitive to ECM remodeling during lung disease are increasingly used to evaluate drug performance66.
Kidney
At homeostasis, the potential plasticity of tubular epithelium translates into a capacity to repair the kidney parenchyma after acute injury, acting in concert with endothelium, fibroblasts, and macrophages, and through the activation of developmental pathways, like Notch, Wnt/B-catenin, SOX9 transcription factor, and the Sonic hedgehog pathway178. However, repeated aggression results in a maladaptive response to damage that subverts these pathways and ultimately results in the recruitment of pro-inflammatory cells, and the activation of myofibroblasts (including tubular epithelial cells that undergo mesenchymal-to-epithelial transition, resident mesenchymal cells, and fibroblasts)179.
Once the bridge to chronic kidney disease (CKD) has been crossed reparative ability diminishes but is not completely erased. In diabetic patients suffering from CKD, pancreas transplantation reverted ECM remodeling (more specifically, BM thickening, it is unclear if this process extends to the IM) after 10 years180 (Fig. 3). In a porcine preclinical model, a nephrectomy followed by the implantation of a segment of decellularized ECM results in the recellularization of the matrix with structures reminiscent of glomeruli, vessels, and tubules181. Unfortunately, clinical progress toward the reversal of kidney fibrosis is still partial. Blocking TGF-β signaling, the main driver of ECM deposition, has shown no beneficial effect on kidney damage182. In type 2 diabetics with CKD however, GLP-1 inhibitors reduce the risk of death, heart and kidney outcomes183.
Biomarkers of ECM remodeling and turnover assess kidney fibrosis progression184. Collagen type III turnover biomarkers PRO-C3 and C3M (a segment resulting from MMP-9 collagen type III cleavage) correlate with kidney fibrosis degree. C3M/creatinine ratio is highly discriminative for advanced kidney fibrosis185. Lysyl oxidase (LOX) crosslinks fibrillar collagen and is increased in patients with kidney fibrosis186. Dickkopf-related protein 3 (DKK-3) is a glycoprotein secreted upon tubular injury that promotes scarring that significantly predicts estimated glomerular filtration rate (eGFR) decline and identifies patients at high risk of CKD progression187. PRO-C6 is associated with kidney fibrosis and outcomes in acute kidney injury188,189.
Gut
Inflammatory Bowel Disease (IBD, including its variants Crohn’s Disease-(CD); and Ulcerative Colitis (UC) progression involves chronic inflammation and mucosal damage leading to abnormal ECM remodeling in the intestine190,191,192,193,194,195,196,197,198,199,200,201,202. This combination is called Fibro-inflammation, an emerging concept in IBD, covering processes related to immune cell activity, mucosal damage, intestinal fibrogenesis, and fibrosis resolution203. In late stages of fibro-inflammation, fibrosis progresses independently of inflammation, accompanied by visceral adipose tissue (creeping fat). Creeping fat has been associated with intestinal fibrosis progression and luminal narrowing190 Along with fibrosis, CD produces a thickening of the muscularis layer at the expense of submucosal layers, hypertrophic nerve trunks and vessels with hyperplastic muscularity also leading to strictures204. Intestinal fibrosis is a clinical feature of UC but rarely causes strictures205,206. UC and CD-associated fibrosis has been associated with an absence of clinical response to anti-inflammatory treatments such as biologics and small molecules207.
As in other organs, myofibroblasts are held responsible for ECM overproduction, and their activation depends on the interplay between fibroblasts, endothelium, epithelium, and immune cells. Single cell mRNA sequencing has begun to reveal the cellular complexity of these fibrosis/inflammation interactions: M2a macrophages are profibrotic, but regulatory M2c deactivate myofibroblasts and canonically activated M1 and M2a macrophages208. Similarly, T helper 2 cells are fibrogenic but T helper 1 cells are antifibrotic. This intricate cell niche is underpinned by a comparably complex molecular landscape, Pro-inflammatory interleukin family members IL-1, IL-6, IL-18, IL-33, and IL-36 have been associated to IBD209, while TGF-β1 may be anti-inflammatory in IBD209 but profibrotic, ushering myofibroblast activation and ECM formation210. An observational study describes TGF-β2 overexpression in human biopsies of ulcerative colitis patients211. Other factors that also stimulate myofibroblast proliferation, platelet derived growth factor subunit A (PDGFA), platelet derived growth factor subunit B (PDGFB), and insulin-like growth factor-1 (IGF-1)212. The molecular drivers of gut fibrosis are partly conditioned by the intestinal microbiota and the integrity of the gut barrier. Increased antibacterial antibodies are common in patients with CD213 and antibiotic treatment leading to reduced bacterial diversity and numbers is anti-inflammatory (e.g., downregulating the expression of NF-κB, TGFβ and αSMA in the intestinal wall)214. The nature of the immune reaction invoked also plays a role, for example by downregulating eosinophil frequency and altering their function, resulting in fibrogenesis and defective repair215. Neutrophils, and their extracellular traps (NET) have been implicated in IBD216 and in intestinal fibrogenesis. NETs enhance fibroblast differentiation into myofibroblast and increase collagen production in vitro217. Escherichia coli sp. exacerbate fibrosis and inflammation in mice, including epithelial-myofibroblast transition218. In humans, bacterial products like outer-membrane protein C, flagellin and Saccaromyces cerevisiae are associated with CD progression and surgery219,220. Conversely, Lactobacillus acidophilus decreases αSMA and collagen deposition in mice221. Genetically modified Lactococcus lactis carrying Il-10 could impair colitis activity, showing that host-microbiota interaction and a compromised gut barrier could be leveraged to tread IBD222. The nature of the microbiome makes it suitable for systems biology biomarker approaches that detect bacterial species with dynamics that could be diagnostic for CD and UC223, but they haven’t substituted established non-invasive biomarkers like calprotectin, CRP, anti-neutrophil, and anti-S cerevisiae antibodies. Janus kinase inhibitor Upadacitinib induces endoscopic remission224 and ECM remodeling225, in a clinical trial of CD patients, suggesting the gut can engage repair processes upon treatment194,226.
Adipose tissue
Fat deposits covering viscera and underlying the skin compose an endocrine organ that regulates metabolism, immunity, and homeostasis. Adipose tissue (AT) dysfunction has wide-ranging, systemic consequences, and fibrosis is both one of its sequels and drivers.
White adipose tissue (WAT) (Fig. 3) stores energy, while brown-beige (BAT) is thermogenic (i.e., it dissipates energy as heat)227,228,229. BAT sits in the paravertebral, axillary, supraclavicular, and periadrenal areas but WAT is widespread: subcutaneous fat lies beneath the dermis and represents ~80% of total body fat; visceral fat surrounds intrathoracic (e.g., pericardial, epicardial) and intraperitoneal organs (e.g., omental, mesenteric). AT secretes signaling polypeptides (adipokines) that regulate metabolism230, e.g., adiponectin promotes insulin sensitivity231, whilst resistin and lipocain promote insulin resistance230. It produces leptin, an adipokine that signals to the hypothalamus and other brain regions, promoting satiety and energy expenditure. Leptin resistance is associated to obesity232. WAT regulates immunity through pro-inflammatory cytokines TNF-a and interleukins 1B, 6, 8, and 18230. WAT deposits have different expansion-contraction patterns233 and transcriptomic profiles233, but expansion by hypertrophy is associated to hypoxia234 and hypoxia-factor 1α (HIF1α) secretion, which in WAT calls for the transcription of ECM associated genes235, fibrosis, and collagen crosslinking. This AT fibrotic response increases the synthesis of collagen type VI and collagen type VI C-terminal pro-collagen, endotrophin236. Collagen type VI correlates with insulin resistance in humans (ref), but interestingly, in COL6−/− animals, AT hypertrophy fails to invoke a fibrotic response, leading to a soft ECM, probably due to a decrease in circulating levels of endotrophin30.
ECM formation in distant organs, downstream of AT expansion, is associated to ECM formation in distant organs. Approximately one third of systemic angiotensinogen is produced by WAT237, activating angiotensin receptors (e.g., angiotensin 1b receptor) in the kidneys that are inflammatory in mice238 and humans239. Overstimulation of leptin receptors in the kidney is associated to progressing renal disease240 and overexpression of TGF-β1, collagen type IV, and fibronectin241,242.
Another example of AT driving disease is gut creeping fat, which surrounds the exterior of the intestines wrapping the intestines. Creeping fat is linked to the release of pro-inflammatory cytokines and fibrotic mediators which enhance ECM remodeling and collagen deposition of the affected intestinal tissue and is highly associated with the development of intestinal fibrosis and strictures226,243.
Skin
During homeostasis, adult epidermis regenerates continuously, turning over every 4–6 weeks244,245 under the control of epidermal stem cells in the basal layer of the skin246, however, upon injury, post-natal skin forms scar tissue, while fetal skin heals faster and regenerates completely247. Hypertrophic scars resulting from trauma (e.g., surgery, physical injury or loss of tissue integrity), are filled with parallelized collagen fibers in the upper skin, while another form of abnormal repair, keloids, proliferate beyond wound limits, accumulating disorganized collagen fibers sustained by angiogenesis248,249,250. Hypertrophic scars are amenable to surgical, laser, physical or anti-inflammatory therapies. The same treatments are less effective in keloids. Both hypertrophic scars and keloids form exaggerated ECM structures, but different collagen organization, composition and proliferation dynamics suggest different cross-linking and pro-fibrotic mechanisms. Keloids are driven by persistent VEGF and TGF- β1 signaling accompanied by dysregulated syndecan and integrin signaling along with inflammation, but inflammation and fibrosis in hypertrophic scars are self-limiting247,251,252.
Excessive collagen deposition and cross-linking are also characteristic of skin fibrosis during systemic sclerosis (SSc)253. SSc produces a thick dermis with remodeled hair follicles, sweat glands, and cutaneous blood vessels, accompanying systemic manifestations like adipose fibrosis254,255. The complex systemic progression of SSc, with multi-organ involvement and diffuse fibrosis, highlight the importance of biomarkers that predict disease evolution. SSc is an autoimmune disease, autoantibodies like Antitopoisomerase I (ATAs) and anticentromere antibodies (ACAs) are found in around 95% of all SSc patients upon diagnosis256,257,258. ATAs (Anti-Scl-70 antibodies) have been associated with poorer prognosis, increased mortality, pulmonary fibrosis, and cardiac involvement259,260,261,262,263. However, autoantibodies do not evaluate disease activity or its correlation to progressing fibrosis. Composite biomarkers like ELF152, correlated with modified Rodnan Skin Score, a measure of skin fibrosis and thickness264. Blood MMP-12 is an indicator of skin fibrosis severity and blood IL-6 has been associated with pulmonary fibrosis, FVC decline, and increased mortality265,266,267.
Biomarkers quantifying degraded collagens (C3M, C4M, C6M, and C7M), and Chemokine (C-C-motif) ligand 18 (CCL18), are lower in SSC patients treated with the autotaxin inhibitor Ziritazestat, showing impaired disease activity and fibrosis improvement268. Similarly, C3M, C4M and collagen synthesis biomarkers PRO-C4 and PRO-C3 were prognostic for worsening skin thickness in patients treated with an anti-IL-6 Ab (Tocilizumab)269.
In contrast to SSc, Stiff skin syndrome (SSS) is non-inflammatory. SSS is characterized by thickened, indurated skin, and limited joint movement in the absence of systemic symptoms (such as Raynaud’s phenomenon, periungual changes, or visceral involvement)270. SSS also suffer persistent TGF-β1 signaling, leading to increased expression of COL1A1 and COL3A1271. SSS is extremely rare and no established guidelines for patient care exist, most patients are treated with immunosuppressive agents, with a high variation in treatment results.
Designing biomarkers for fibrosis, fibrosis-driven organ death races, and fibrosis resolution
The dysfunction that produces and sustains ECM structural alterations in an organ reverberates in the body, damaging distant tissues and triggering adverse events (Figs. 3, 4). Fibrotic disease in an organ can drive end-stage disease in distant organs, characterized by simultaneous ECM remodeling, albeit at different rates, and with considerable individual variation. Consider how Metabolic dysfunction-associated steatotic liver disease (MASLD, Fig. 4), closely linked to metabolic syndrome, and characterized by excess accumulation of lipids in the liver, inflammation/hepatocyte ballooning degeneration, and hepatic fibrosis272,273 impacts multiple systems. Before developing cirrhosis, MASLD patients die of cardiovascular disease and extrahepatic cancer with more frequency than from a liver-related event274,275 (Fig. 4).

According to Sanyal AJ et al.160, patients with chronic steatohepatitis are at higher risk of cardiovascular and renal adverse events than liver-related events. This schematic illustrates the organ death race driven by MASLD progression. (Line thickness represent probability of event per 100 patients).
Liver disease is by no means unique. Fibrotic progression in WAT is correlated with higher risks of infection276, cancer (including breast, uterus, ovaries, colon, stomach, esophagus, rectum, liver, pancreas, kidney, meninges, and blood), metabolic, kidney, cardiovascular, and psychiatric disease.
Detecting, predicting, and tracking ECM formation or degradation is challenging. There are multiple molecular mechanisms and proteins involved, affecting tissues with different shape, function, resilience, and regenerative potential. Conceptually, a pharmacodynamic biomarker should either measure fibrogenic activity, i.e., determine the de novo formation of ECM proteins, or fibrolysis, i.e., ECM degradation and removal. A combination of such biomarkers could mirror the balance between fibrogenesis and fibrolysis. ECM biomarkers measure ECM component synthesis dynamically, reflecting how active fibrosis progression pathways are during measurement. They can discriminate between cumulative damage (which is the parameter assessed by a biopsy) and an actual snapshot of the biological status of the disease. Dynamic biomarkers would reflect distinct patient endotypes, characterized by different formation-degradation balances represented by different ECM parameters (Fig. 2).
There are different technological paths to build a biomarker strategy. One passes by combining large-scale biological data and data mining. Omics-based biomarker research have been gaining momentum with the establishment of national biobanks (including the UK Biobank277, and Biobank Japan278) and disease specific international patient registries (including the European MASLD Registry279). These large databases have increased the depth, quality, and availability of Omics data as the cost of large-scale data generation has decreased, creating a conducive background for biomarker discovery. Mapping the human proteome280,281 was a significant step towards assessing multiple molecular pathways simultaneously, opening a conceptual window into complex biological processes. Recently, leveraging RNA-seq and plasma proteomics resulted in organ-specific protein profiles that reveal tissue aging, thus building a proteomics-based biomarker strategy282. Plasma proteomics, used in Alcohol-related Liver Disease (ALD), detected circulating proteins associated to fibrosis and metabolic dysfunction, predictive of future liver-related events and all-cause mortality283. Complementary approaches in MASLD utilizing a proteo-transcriptomic strategy to characterize the liver-derived circulating proteome across the full disease spectrum284. However, there is evidence to suggest that different technologies (i.e., based on aptamers or antibodies) may affect protein quantification and comparability285,286. Epigenetics and metabolomics can also perform to a similar level: mapping DNA methylation in whole blood has found associations between disease, age, ancestry and all-cause mortality and specific cytosine-phosphate-guanine sequences, with substantial prognostic improvement for neoplasia-associated death287. A metabolomic platform found prognosticators all-cause mortality in a diverse population288, identifying a panel of 14 metabolic biomarkers that could perform as well as conventional risk factors of mortality.
These panoramic approaches stand in contrast to individual and composite biomarkers supported by mechanistic research. Sensing post translational changes in a fundamental disease pathway is an effective biomarker strategy, with direct clinical impact. This is made evident by the FDA list of approved companion diagnostic devices, where single genetic biomarkers underpin decisions that have reduced disease burden and mortality for millions of cancer patients (e.g., BRCA1, BRCA2, HER1, HER2, KRAS, PD-L1, etc.289). A central feature of fibrosis is the formation of ECM, therefore, detecting fibrogenesis and ECM remodeling should be an obvious goal to assess disease activity. In particular, the intracellular synthesis of fibrillar procollagens is often followed by the cleavage of pro-peptides that are then released into the bloodstream. These procollagens have been proven to be a surrogate of several complex pathophysiological events involving increased ECM synthesis and turnover. PRO-C3290, produced by fibroblasts as they deposit collagen type III, predicts fibrosis progression153, reflects fibrosis stage291, and can predict future lethal events292, disease outcome293, and importantly, monitors disease activity during and after therapeutic intervention154 across cohorts subject to different diseases or treatments. Another example of a composite biomarker centered on ECM biology is the Enhanced Liver Fibrosis (ELF) score152,294. ELF measures the tissue inhibitor of metalloproteinases 1 (TIMP-1), hyaluronic acid (HA), and the N-terminal propeptide of procollagen type III (PIIINP). ELF predicts clinical outcome and event-free survival295,296.
By-products of ECM remodeling can act as signaling messengers, driving fibrosis and metabolic dysfunction. Collagen type VI is a minor but ubiquitous, microfilamentous interstitial collagen of most organs, including the cardiovascular system and WAT, where it plays a role in the regulation of tissue expansion and WAT fibrosis297. In mice, a collagen V1(α1) knockout protects against WAT fibrosis235 and myocardial infarction298, suggesting a mechanistic role for collagen type VI remodeling in the chain of events during systemic disease. Endotrophin, a cleavage product of the C-terminal propeptide of the α) chain of procollagen type VI(α3) chain297,299, is a potent adipokine, activating fibroblasts and recruiting immune and endothelial cells to trigger and promote fibrosis progression. Increased expression of collagen type VI(α3) chain has also been demonstrated to enhance the adhesion of T-cells in tissues from UC and CD299, indicating a potential link to sustaining chronic inflammation in IBD. It also reduces energy expenditure, increases triglycerides, leads to hepatic steatosis, and ultimately metabolic disease30. Endotrophin, lying at the center of fundamental disease pathways, opens a window into organ death races driven by ECM dysregulation. An endotrophin-derived biomarker, PRO-C6, is associated to outcome in COPD300,301, chronic liver disease302, acute kidney disease189, kidney transplant303, heart failure with preserved ejection fraction165, multiple solid neoplasias304,305,306, and metabolic disease307,308,309. Although more research is required, the mechanisms of collagen type VI synthesis are a bellwether of disease activity and a potential drug target310.
The deepening knowledge about, and emergence of successful drugs against, fibrosis311,312,313,314 announce a new challenge: how to measure the dismantling of scarred, defective ECM that would determine fibrosis resolution, and healing. Collagen degradation fragments (e.g., by matrix metalloproteinases) released into the bloodstream could indicate the turning of the tide, the tipping of the ECM balance towards fibrotic scar resolution. Developing biomarkers of ECM degradation is a complex task, as the progression of fibrotic and inflammatory diseases (including rheumatoid315 and neoplastic diseases316) involves the destruction of normal ECM, and so a degradation-inclined ECM balance could rather be interpreted as high disease activity associated with enhanced ECM turnover. However, during fibrosis, fibrillar collagen is abnormally and abundantly crosslinked317, thus, fibrosis resolution would imply the degradation of crosslinked fibrillar collagen. Biomarkers of degraded, crosslinked collagen could therefore be a successful surrogate of beneficial ECM degradation and repair. The recent development of an ELISA to detect a crosslinked fragment of collagen type III cleaved by MMPs318 suggests that noninvasive measurement of release of a fragment of a “bad” collagen into the bloodstream is possible and could provide a valid surrogate for scar resolution, thus complementing the armamentarium to assess the balance between fibrogenesis, e.g., represented by PRO-C3, and fibrolysis.
The FDA biomarker qualification program (reviewed in319) sets the path for analytes to be considered drug development tools. It includes, at this point, eight biomarkers, three of them non-clinical. Apart from scientific obstacles (e.g., an insufficient knowledge of the mechanistic bases of a particular disease process), one of the main barriers for a sound validation of novel biomarkers of disease is methodological: the development of standardized, replicable measurement methods. Systems biology-based biomarkers often suffer from a lack of comparability285 that hampers the transition from being research platforms to clinical tools. Single and composite biomarkers are making inroads, and ELF was given a marketing authorization for enriching MASLD patients with advanced fibrosis by the FDA, while markers like PRO-C3 and PRO-C6 have received FDA letters of support or intent, to continue research towards full qualification.
Conclusions
More than three decades of fibrosis research have established that fibrogenesis and scar-formation are not the only possible paths towards advanced disease after a loss of tissue structure. It is increasingly clear that fibrosis resolution, and possibly regeneration, can be coaxed out of mammalian tissues by disrupting profibrotic mechanosignaling, and by eliminating, inhibiting, or manipulating myofibroblast activity directly or by several ‘upstream’ interventions311,313. The arrival of treatments like anti-myofibroblast immunotherapy, GLP-1 agonists, FGF21 analogs and integrin inhibitors, among others, may be a harbinger of a wave of anti-fibrotic therapies and the beginning of the end for the “death races” that are spurred by organ-specific fibroses. To support this progress, powerful drug development tools will be increasingly necessary, to evaluate therapeutic effect and effectivity, and more specifically to measure the balance of ECM remodeling, inflammation, and reparative response in early and late clinical developments. Two approaches, one guided by big data-driven scanning of biological products, the other based on probing critical pathways active in disease progression, are opening windows into fibrosis activity, regression, and systemic damage. In conclusion, accelerated translational medicine and advanced non-invasive diagnosis may be gradually bringing fibrosis, a condition associated with a heavy healthcare burden, poor prognosis and systemic disease, into the realm of manageable diseases.
Data availability
No datasets were generated or analyzed during the current study.
References
Reuten, R., Mayorca-Guiliani, A. E. & Erler, J. T. Matritecture: mapping the extracellular matrix architecture during health and disease. Matrix Biol. 14, 100102 (2022).
Naba, A. et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol. Cell. Proteom. 11, M111.014647 (2012).
Hynes, R. O. & Naba, A. Overview of the matrisome–an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 4, a004903 (2012).
Bhatia, H. S. et al. Spatial proteomics in three-dimensional intact specimens. Cell 185, 5040–5058.e19 (2022).
Mayorca-Guiliani, A. E. et al. ISDoT: In situ decellularization of tissues for high-resolution imaging and proteomic analysis of native extracellular matrix. Nat. Med. 23, 890–898 (2017).
Mayorca-Guiliani, A. E. et al. Decellularization and antibody staining of mouse tissues to map native extracellular matrix structures in 3D. Nat. Protoc. 14, 3395–3425 (2019).
Li, J. et al. Spatially resolved proteomic map shows that extracellular matrix regulates epidermal growth. Nat. Commun. 13, 4012 (2022).
Tsutsui, K. et al. Mapping the molecular and structural specialization of the skin basement membrane for inter-tissue interactions. Nat. Commun. 12, 2577 (2021).
Pozzi, A., Yurchenco, P. D. & Iozzo, R. V. The nature and biology of basement membranes. Matrix Biol. 57–58, 1–11 (2017).
Reuten, R. et al. Basement membrane stiffness determines metastases formation. Nat. Mater. 20, 892–903 (2021).
He, Y., Sardar, S., Bay-Jensen, A. C., Port, H. & Karsdal, M. A. Type IX collagen. In Biochemistry of Collagens, Laminins and Elastin 89–95. https://doi.org/10.1016/B978-0-443-15617-5.00034-2 (Elsevier, 2024).
Chen, M. et al. Restoration of type VII collagen expression and function in dystrophic epidermolysis bullosa. Nat. Genet. 32, 670–675 (2002).
Patten, J. & Wang, K. Fibronectin in development and wound healing. Adv. Drug Deliv. Rev. 170, 353–368 (2021).
Dalton, C. J. & Lemmon, C. A. Fibronectin: molecular structure, fibrillar structure and mechanochemical signaling. Cells 10, 2443 (2021).
Moretti, L., Stalfort, J., Barker, T. H. & Abebayehu, D. The interplay of fibroblasts, the extracellular matrix, and inflammation in scar formation. J. Biol. Chem. 298, 101530 (2022).
Barker, H. E., Cox, T. R. & Erler, J. T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 12, 540–552 (2012).
Lyu, C. et al. Advanced glycation end-products as mediators of the aberrant crosslinking of extracellular matrix in scarred liver tissue. Nat. Biomed. Eng. 7, 1437–1454 (2023).
Fan, W. et al. Matrix viscoelasticity promotes liver cancer progression in the pre-cirrhotic liver. Nature 626, 635–642 (2024).
Maller, O. et al. Tumour-associated macrophages drive stromal cell-dependent collagen crosslinking and stiffening to promote breast cancer aggression. Nat. Mater. 20, 548–559 (2021).
Schuppan, D., Ruehl, M., Somasundaram, R. & Hahn, E. G. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 21, 351–372 (2001).
Somasundaram, R. & Schuppan, D. Type I, II, III, IV, V, and VI collagens serve as extracellular ligands for the isoforms of platelet-derived growth factor (AA, BB, and AB). J. Biol. Chem. 271, 26884–26891 (1996).
Schuppan, D. et al. Collagens in the liver extracellular matrix bind hepatocyte growth factor. Gastroenterology 114, 139–152 (1998).
Ruehl, M. et al. The epithelial mitogen keratinocyte growth factor binds to collagens via the consensus sequence glycine-proline-hydroxyproline. J. Biol. Chem. 277, 26872–26878 (2002).
Somasundaram, R. et al. Collagens serve as an extracellular store of bioactive interleukin 2. J. Biol. Chem. 275, 38170–38175 (2000).
Somasundaram, R. et al. Interstitial collagens I, III, and VI sequester and modulate the multifunctional cytokine oncostatin M. J. Biol. Chem. 277, 3242–3246 (2002).
Rafaeva, M. et al. Modeling metastatic colonization in a decellularized organ scaffold-based perfusion bioreactor. Adv. Health. Mater. 11, e2100684 (2022).
Karsdal, M. A. et al. The good and the bad collagens of fibrosis—their role in signaling and organ function. Adv. Drug Deliv. Rev. 121, 43–56 (2017).
Maquart, F. X. & Monboisse, J. C. Extracellular matrix and wound healing. Pathol. Biol. 62, 91–95 (2014).
Karsdal, M. A. et al. Novel insights into the function and dynamics of extracellular matrix in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G807–G830 (2015).
Sun, K. et al. Endotrophin triggers adipose tissue fibrosis and metabolic dysfunction. Nat. Commun. 5, 3485 (2014).
O’Reilly, M. S. et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88, 277–285 (1997).
Yamaguchi, Y. et al. A peptide derived from endostatin ameliorates organ fibrosis. Sci. Transl. Med 4, 136ra71 (2012).
Hamano, Y. & Kalluri, R. Tumstatin, the NC1 domain of alpha3 chain of type IV collagen, is an endogenous inhibitor of pathological angiogenesis and suppresses tumor growth. Biochem Biophys. Res Commun. 333, 292–298 (2005).
Cox, T. R. The matrix in cancer. Nat. Rev. Cancer 21, 217–238 (2021).
Lu, P., Takai, K., Weaver, V. M. & Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 3, a005058–a005058 (2011).
Brown, G. T. & Kleiner, D. E. Histopathology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Metabolism 65, 1080–1086 (2016).
Bissell, M. J. & Hines, W. C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med 17, 320–329 (2011).
Hynes, R. O. The extracellular matrix: not just pretty fibrils. Science (1979) 326, 1216–1219 (2009).
Engler, A. J., Sen, S., Sweeney, H. L. & Discher, D. E. Matrix elasticity directs stem. Cell Lineage Specif. Cell 126, 677–689 (2006).
Discher, D. E., Janmey, P. & Wang, Y. Tissue cells feel and respond to the stiffness of their substrate. Science 310, 1139–1143 (2005).
Massagué, J. & Sheppard, D. TGF-β signaling in health and disease. Cell 186, 4007–4037 (2023).
Huang, J. et al. Fibulin-4 deficiency results in ascending aortic aneurysms. Circ. Res 106, 583–592 (2010).
McLaughlin, P. J. et al. Lack of fibulin-3 causes early aging and herniation, but not macular degeneration in mice. Hum. Mol. Genet 16, 3059–3070 (2007).
Massam-Wu, T. et al. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGFβ. J. Cell Sci. 123, 3006–3018 (2010).
Klingberg, F. et al. The ED-A domain enhances the capacity of fibronectin to store latent TGF-β binding protein-1 in the fibroblast matrix. J. Cell Sci. https://doi.org/10.1242/jcs.201293 (2018).
Horiguchi, M., Ota, M. & Rifkin, D. B. Matrix control of transforming growth factor- function. J. Biochem. 152, 321–329 (2012).
Schaefer, L. et al. Small proteoglycans in human diabetic nephropathy: discrepancy between glomerular expression and protein accumulation of decorin, biglycan, lumican, and fibromodulin. FASEB J. 15, 559–561 (2001).
Ferrara, N. Binding to the extracellular matrix and proteolytic processing: two key mechanisms regulating vascular endothelial growth factor action. Mol. Biol. Cell 21, 687–690 (2010).
Birch, H. L. Extracellular matrix and ageing. 169–190. https://doi.org/10.1007/978-981-13-2835-0_7 (2018).
Jiang, D. et al. Injury triggers fascia fibroblast collective cell migration to drive scar formation through N-cadherin. Nat. Commun. 11, 5653 (2020).
Correa-Gallegos, D. et al. Patch repair of deep wounds by mobilized fascia. Nature 576, 287–292 (2019).
Baer, J. M. et al. Fibrosis induced by resident macrophages has divergent roles in pancreas inflammatory injury and PDAC. Nat. Immunol. 24, 1443–1457 (2023).
Dey, S. et al. Loss of miR-29a/b1 promotes inflammation and fibrosis in acute pancreatitis. JCI Insight 6, (2021).
Panizo, S. et al. Fibrosis in chronic kidney disease: pathogenesis and consequences. Int. J. Mol. Sci. 22, 408 (2021).
Tanwar, S., Rhodes, F., Srivastava, A., Trembling, P. M. & Rosenberg, W. M. Inflammation and fibrosis in chronic liver diseases including non-alcoholic fatty liver disease and hepatitis C. World J. Gastroenterol. 26, 109–133 (2020).
Furman, D. et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med 25, 1822–1832 (2019).
Zhao, X., Kwan, J. Y. Y., Yip, K., Liu, P. P. & Liu, F.-F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 19, 57–75 (2020).
Wynn, T. A. & Ramalingam, T. R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18, 1028–1040 (2012).
Henderson, N. C., Rieder, F. & Wynn, T. A. Fibrosis: from mechanisms to medicines. Nature 587, 555–566 (2020).
Simon, A. & Tanaka, E. M. Limb regeneration. WIREs Dev. Biol. 2, 291–300 (2013).
Godwin, J., Kuraitis, D. & Rosenthal, N. Extracellular matrix considerations for scar-free repair and regeneration: insights from regenerative diversity among vertebrates. Int J. Biochem Cell Biol. 56, 47–55 (2014).
Singh, S. et al. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 13, 643–654.e9 (2015).
Raghu, G. et al. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 198, e44–e68 (2018).
Siddiqui, M. S. et al. Vibration-controlled transient elastography to assess fibrosis and steatosis in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 17, 156–163.e2 (2019).
Sanyal, A. J. et al. Diagnostic performance of circulating biomarkers for non-alcoholic steatohepatitis. Nat. Med 29, 2656–2664 (2023).
Bihlet, A. R. et al. Clinical drug development using dynamic biomarkers to enable personalized health care in COPD. Chest 148, 16–23 (2015).
Vali, Y. et al. Biomarkers for staging fibrosis and non-alcoholic steatohepatitis in non-alcoholic fatty liver disease (the LITMUS project): a comparative diagnostic accuracy study. Lancet Gastroenterol. Hepatol. 8, 714–725 (2023).
Rasmussen, D. G. K. et al. Endotrophin is a risk marker of complications in CANagliflozin cardioVascular Assessment Study (CANVAS): a randomized controlled trial. Cardiovasc. Diabetol. 21, 261 (2022).
Borisov, A. N., Kutz, A., Christ, E. R., Heim, M. H. & Ebrahimi, F. Canagliflozin and metabolic associated fatty liver disease in patients with diabetes mellitus: new insights from CANVAS. J. Clin. Endocrinol. Metab. 108, 2940–2949 (2023).
Harrison, S. A. et al. Effects of resmetirom on noninvasive endpoints in a 36‐week phase 2 active treatment extension study in patients with NASH. Hepatol. Commun. 5, 573–588 (2021).
Mehal, W. Z., Iredale, J. & Friedman, S. L. Scraping fibrosis: expressway to the core of fibrosis. Nat. Med. 17, 552–553 (2011).
Roh, H. C. et al. Adipocytes fail to maintain cellular identity during obesity due to reduced PPARγ activity and elevated TGFβ-SMAD signaling. Mol. Metab. 42, 101086 (2020).
Zhao, Z. et al. TGF-β promotes pericyte-myofibroblast transition in subretinal fibrosis through the Smad2/3 and Akt/mTOR pathways. Exp. Mol. Med. 54, 673–684 (2022).
Zou, F., Li, Y., Zhang, S. & Zhang, J. DP1 (Prostaglandin D 2 Receptor 1) activation protects against vascular remodeling and vascular smooth muscle cell transition to myofibroblasts in angiotensin II–induced hypertension in mice. Hypertension 79, 1203–1215 (2022).
Little, K. et al. Macrophage to myofibroblast transition contributes to subretinal fibrosis secondary to neovascular age-related macular degeneration. J. Neuroinflammation 17, 355 (2020).
Xu, R. et al. Mesenchymal stem cells reversibly de-differentiate myofibroblasts to fibroblast-like cells by inhibiting the TGF-β-SMAD2/3 pathway. Mol. Med. 29, 59 (2023).
Lovisa, S. et al. Endothelial-to-mesenchymal transition compromises vascular integrity to induce Myc-mediated metabolic reprogramming in kidney fibrosis. Sci. Signal 13, 998–1009 (2020).
Lovisa, S. et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 21, 998–1009 (2015).
Younesi, F. S., Miller, A. E., Barker, T. H., Rossi, F. M. V. & Hinz, B. Fibroblast and myofibroblast activation in normal tissue repair and fibrosis. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-024-00716-0 (2024).
Ezzo, M. & Hinz, B. Novel approaches to target fibroblast mechanotransduction in fibroproliferative diseases. Pharm. Ther. 250, 108528 (2023).
Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 47, 54–65 (2015).
Sobierajska, K., Wawro, M. E. & Niewiarowska, J. Oxidative stress enhances the TGF-β2-RhoA-MRTF-A/B axis in cells entering endothelial-mesenchymal transition. Int. J. Mol. Sci. 23, 2062 (2022).
Alharthi, A., Verma, A., Sabbineni, H., Adil, M. S. & Somanath, P. R. Distinct effects of pharmacological inhibition of stromelysin1 on endothelial‐to‐mesenchymal transition and myofibroblast differentiation. J. Cell Physiol. 236, 5147–5161 (2021).
Shiju, T. M., Sampaio, L. P., Martinez, V. V., Hilgert, G. S. L. & Wilson, S. E. Transforming growth factor beta-3 localization in the corneal response to epithelial-stromal injury and effects on corneal fibroblast transition to myofibroblasts. Exp. Eye Res. 235, 109631 (2023).
Kolosova, I., Nethery, D. & Kern, J. A. Role of Smad2/3 and p38 MAP kinase in TGF‐β1‐induced epithelial–mesenchymal transition of pulmonary epithelial cells. J. Cell Physiol. 226, 1248–1254 (2011).
Zhou, X. et al. Amelioration of fibrotic remodeling of human 3‐dimensional full‐thickness skin by transglutamase 2 inhibition. Arthritis Rheumatol. 75, 1619–1627 (2023).
Levental, K. R. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906 (2009).
Nguyen, X.-X. et al. Lysyl oxidase directly contributes to extracellular matrix production and fibrosis in systemic sclerosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 320, L29–L40 (2021).
Coelho, N. M. et al. Discoidin domain receptor 1 mediates myosin-dependent collagen contraction. Cell Rep. 18, 1774–1790 (2017).
Zhan, L. & Li, J. The role of TRPV4 in fibrosis. Gene 642, 1–8 (2018).
Parichatikanond, W., Duangrat, R. & Mangmool, S. Gαq protein-biased ligand of angiotensin II type 1 receptor mediates myofibroblast differentiation through TGF-β1/ERK axis in human cardiac fibroblasts. Eur. J. Pharm. 951, 175780 (2023).
Razinia, Z. et al. Stiffness-dependent motility and proliferation uncoupled by deletion of CD44. Sci. Rep. 7, 16499 (2017).
Cain, S. A., Woods, S., Singh, M., Kimber, S. J. & Baldock, C. ADAMTS6 cleaves the large latent TGFβ complex and increases the mechanotension of cells to activate TGFβ. Matrix Biol. 114, 18–34 (2022).
Rose, K. W. J., Taye, N., Karoulias, S. Z. & Hubmacher, D. Regulation of ADAMTS proteases. Front. Mol.Biosci. 8, (2021).
Noda, K. et al. Latent TGF-β binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proc. Natl. Acad. Sci. USA 110, 2852–2857 (2013).
Camarena, V. et al. ADAMTSL2 mutations determine the phenotypic severity in geleophysic dysplasia. JCI Insight 110, 2852-2857 (2024).
Corey, K. E. et al. ADAMTSL2 protein and a soluble biomarker signature identify at-risk non-alcoholic steatohepatitis and fibrosis in adults with NAFLD. J. Hepatol. 76, 25–33 (2022).
Dagoneau, N. et al. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am. J. Hum. Genet. 75, 801–806 (2004).
Aoki, F. & Kojima, I. Therapeutic potential of follistatin to promote tissue regeneration and prevent tissue fibrosis. Endocr. J. 54, 849–854 (2007).
Innis, C. A. & Hyvönen, M. Crystal structures of the heparan sulfate-binding domain of follistatin. Insights into ligand binding. J. Biol. Chem. 278, 39969–39977 (2003).
Kiagiadaki, F. et al. Activin-A causes Hepatic stellate cell activation via the induction of TNFα and TGFβ in Kupffer cells. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 891–899 (2018).
Patella, S., Phillips, D. J., Tchongue, J., de Kretser, D. M. & Sievert, W. Follistatin attenuates early liver fibrosis: effects on hepatic stellate cell activation and hepatocyte apoptosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 290, G137–G144 (2006).
Nordholm, A. et al. Activin A inhibition reduces kidney fibrosis and normalizes bone abnormalities in AKI. J. Am. Soc. Nephrol. 35 (2024).
Maeshima, A. et al. Follistatin, an activin antagonist, ameliorates renal interstitial fibrosis in a rat model of unilateral ureteral obstruction. Biomed. Res. Int. 2014, 376191 (2014).
Mendell, J. R. et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 23, 192–201 (2015).
Di Rocco, M. et al. Garetosmab in fibrodysplasia ossificans progressiva: a randomized, double-blind, placebo-controlled phase 2 trial. Nat. Med 29, 2615–2624 (2023).
Hoeper, M. M. et al. Phase 3 trial of sotatercept for treatment of pulmonary arterial hypertension. N. Engl. J. Med. 388, 1478–1490 (2023).
Nusse, R. & Varmus, H. E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 31, 99–109 (1982).
Somanader, D. V. N., Zhao, P., Widdop, R. E. & Samuel, C. S. The involvement of the Wnt/β-catenin signaling cascade in fibrosis progression and its therapeutic targeting by relaxin. Biochem Pharm. 223, 116130 (2024).
Froidure, A. et al. Chaotic activation of developmental signalling pathways drives idiopathic pulmonary fibrosis. Eur. Respir. Rev. 29, 190140 (2020).
Skronska-Wasek, W., Gosens, R., Königshoff, M. & Baarsma, H. A. WNT receptor signalling in lung physiology and pathology. Pharm. Ther. 187, 150–166 (2018).
Kneidinger, N. et al. Activation of the WNT/β-catenin pathway attenuates experimental emphysema. Am. J. Respir. Crit. Care Med. 183, 723–733 (2011).
Blyszczuk, P. et al. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur. Heart J. ehw116, https://doi.org/10.1093/eurheartj/ehw116 (2016).
Gurney, A. et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. USA 109, 11717–11722 (2012).
Li, S.-S. et al. Targeting the Wnt/β-catenin signaling pathway as a potential therapeutic strategy in renal tubulointerstitial fibrosis. Front. Pharmacol. 12 (2021).
Gehrke, A. R. et al. Acoel genome reveals the regulatory landscape of whole-body regeneration. Science 363, eaau6173 (2019).
Mitoh, S. & Yusa, Y. Extreme autotomy and whole-body regeneration in photosynthetic sea slugs. Curr. Biol. 31, R233–R234 (2021).
Gerber, T. et al. Single-cell analysis uncovers convergence of cell identities during axolotl limb regeneration. Science 362, eaaq0681 (2018).
Wei, X. et al. Single-cell Stereo-seq reveals induced progenitor cells involved in axolotl brain regeneration. Science 377, eabp9444 (2022).
Poss, K. D., Wilson, L. G. & Keating, M. T. Heart regeneration in zebrafish. Science (1979) 298, 2188–2190 (2002).
Stewart, S., Tsun, Z.-Y. & Belmonte, J. C. I. A histone demethylase is necessary for regeneration in zebrafish. Proc. Natl. Acad. Sci. USA 106, 19889–19894 (2009).
Brown, A. C. et al. Fibrin network changes in neonates after cardiopulmonary bypass. Anesthesiology 124, 1021–1031 (2016).
Leung, A., Crombleholme, T. M. & Keswani, S. G. Fetal wound healing. Curr. Opin. Pediatr. 24, 371–378 (2012).
Xu, L. et al. Transforming growth factor β3 attenuates the development of radiation-induced pulmonary fibrosis in mice by decreasing fibrocyte recruitment and regulating IFN-γ/IL-4 balance. Immunol. Lett. 162, 27–33 (2014).
Singh, K., Sachan, N., Ene, T., Dabovic, B. & Rifkin, D. Latent transforming growth factor β binding protein 3 controls adipogenesis. Matrix Biol. 112, 155–170 (2022).
Klingberg, F. et al. Prestress in the extracellular matrix sensitizes latent TGF-β1 for activation. J. Cell Biol. 207, 283–297 (2014).
Xia, H. et al. Tissue repair and regeneration with endogenous stem cells. Nat. Rev. Mater. 3, 174–193 (2018).
Mascharak, S. et al. Preventing Engrailed-1 activation in fibroblasts yields wound regeneration without scarring. Science 372, eaba2374 (2021).
Monaghan-Benson, E., Aureille, J. & Guilluy, C. ECM stiffness regulates lung fibroblast survival through RasGRF1-dependent signaling. J. Biol. Chem. 301, 108161 (2025).
Jiang, C. et al. Collagen signaling and matrix stiffness regulate multipotency in glandular epithelial stem cells in mice. Nat. Commun. 15, 10482 (2024).
Paranjpe, S. et al. Combined systemic elimination of MET and epidermal growth factor receptor signaling completely abolishes liver regeneration and leads to liver decompensation. Hepatology 64, 1711–1724 (2016).
Michalopoulos, G. K. Liver regeneration. J. Cell. Physiol. 213, 286–300 (2007).
French, B. A. & Holmes, J. W. Implications of scar structure and mechanics for post-infarction cardiac repair and regeneration. Exp. Cell Res. 376, 98–103 (2019).
Rudolph, K. L. et al. Differential regulation of extracellular matrix synthesis during liver regeneration after partial hepatectomy in rats. Hepatology 30, 1159–1166 (1999).
Drixler, T. A. et al. Plasminogen mediates liver regeneration and angiogenesis after experimental partial hepatectomy. Br. J. Surg. 90, 1384–1390 (2003).
Zhou, B. et al. Matrix metalloproteinases-9 deficiency impairs liver regeneration through epidermal growth factor receptor signaling in partial hepatectomy mice. J. Surg. Res. 197, 201–209 (2015).
Sparrelid, E. et al. Serial assessment of growth factors associated with liver regeneration in patients operated with associating liver partition and portal vein ligation for staged hepatectomy. Eur. Surg. Res. 59, 72–82 (2018).
Nejak-Bowen, K., Orr, A., Bowen, W. C. & Michalopoulos, G. K. Conditional genetic elimination of hepatocyte growth factor in mice compromises liver regeneration after partial hepatectomy. PLoS ONE 8, e59836 (2013).
Matchett, K. P. et al. Multimodal decoding of human liver regeneration. Nature https://doi.org/10.1038/s41586-024-07376-2 (2024).
Volk, A., Michalopoulos, G., Weidner, M. & Gebhardt, R. Different proliferative responses of periportal and pericentral rat hepatocytes to hepatocyte growth factor. Biochem. Biophys. Res. Commun. 207, 578–584 (1995).
Wei, Y. et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science 371, eabb1625 (2021).
Gallai, M. et al. Proteoglycan gene expression in rat liver after partial hepatectomy. Biochem. Biophys. Res. Commun. 228, 690–694 (1996).
Lassailly, G. et al. Bariatric surgery provides long-term resolution of nonalcoholic steatohepatitis and regression of fibrosis. Gastroenterology 159, 1290–1301.e5 (2020).
Klein, S. et al. Gastric bypass surgery improves metabolic and hepatic abnormalities associated with nonalcoholic fatty liver disease. Gastroenterology 130, 1564–1572 (2006).
Lefere, S. et al. Bariatric surgery and the liver—mechanisms, benefits, and risks. Obes. Rev. 22, e13294 (2021).
Marcellin, P. et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 381, 468–475 (2013).
Rockey, D. C. & Friedman, S. L. Fibrosis regression after eradication of hepatitis C virus: from bench to bedside. Gastroenterology 160, 1502–1520.e1 (2021).
Harrison, S. A. et al. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N. Engl. J. Med. 390, 497–509 (2024).
Sanyal, A. J. et al. A phase 2 randomized trial of survodutide in MASH and fibrosis. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2401755 (2024).
Loomba, R. et al. Randomized, controlled trial of the FGF21 analogue pegozafermin in NASH. N. Engl. J. Med. 389, 998–1008 (2023).
Roehlen, N. et al. A monoclonal antibody targeting nonjunctional claudin-1 inhibits fibrosis in patient-derived models by modulating cell plasticity. Sci. Transl. Med. 14, eabj4221 (2022).
Vali, Y. et al. Enhanced liver fibrosis test for the non-invasive diagnosis of fibrosis in patients with NAFLD: A systematic review and meta-analysis. J. Hepatol. 73, 252–262 (2020).
Nielsen, M. J. et al. Plasma Pro-C3 (N-terminal type III collagen propeptide) predicts fibrosis progression in patients with chronic hepatitis C. Liver Int. 35, 429–437 (2015).
Harrison, S. A. et al. Effects of resmetirom on noninvasive endpoints in a 36-week phase 2 active treatment extension study in patients with NASH. Hepatol. Commun. 5, 573–588 (2021).
Garbern, J. C. & Lee, R. T. Heart regeneration: 20 years of progress and renewed optimism. Dev. Cell 57, 424–439 (2022).
Vujic, A., Natarajan, N. & Lee, R. T. Molecular mechanisms of heart regeneration. Semin. Cell Dev. Biol. 100, 20–28 (2020).
Bergmann, O. et al. Evidence for cardiomyocyte renewal in humans. Science 324, 98–102 (2009).
Gourdie, R. G., Dimmeler, S. & Kohl, P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat. Rev. Drug Discov. 15, 620–638 (2016).
van den Borne, S. W. M. et al. Myocardial remodeling after infarction: the role of myofibroblasts. Nat. Rev. Cardiol. 7, 30–37 (2010).
Roche, P. L., Filomeno, K. L., Bagchi, R. A. & Czubryt, M. P. Intracellular signaling of cardiac fibroblasts. in Comprehensive Physiology 721–760. https://doi.org/10.1002/cphy.c140044 (Wiley, 2015).
Grotendorst, G. R. & Duncan, M. R. Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J. 19, 729–738 (2005).
Aghajanian, H. et al. Targeting cardiac fibrosis with engineered T cells. Nature 573, 430–433 (2019).
Maisel, A. S. & Choudhary, R. Biomarkers in acute heart failure—state of the art. Nat. Rev. Cardiol. 9, 478–490 (2012).
Ahmad, T., Fiuzat, M., Felker, G. M. & O’Connor, C. Novel biomarkers in chronic heart failure. Nat. Rev. Cardiol. 9, 347–359 (2012).
Chirinos, J. A. et al. Endotrophin, a collagen VI formation-derived peptide, in heart failure. NEJM Evid. 1, EVIDoa2200091 (2022).
Lederer, D. J. & Martinez, F. J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 378, 1811–1823 (2018).
Chambers, R. C. & Mercer, P. F. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann. Am. Thorac. Soc. 12, S16–S20 (2015).
Xu, J. et al. Use of senescence-accelerated mouse model in bleomycin-induced lung injury suggests that bone marrow-derived cells can alter the outcome of lung injury in aged mice. J. Gerontol. A Biol. Sci. Med. Sci. 64 A, 731–739 (2009).
Kathiriya, J. J. et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5+ basal cells. Nat. Cell Biol. 24, 10–23 (2022).
Melo-Narváez, M. C., Stegmayr, J., Wagner, D. E. & Lehmann, M. Lung regeneration: implications of the diseased niche and ageing. Eur. Respir. Rev. 29, 200222 (2020).
Wu, H. et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem. Cells Cell 180, 107–121.e17 (2020).
Knüppel, L. et al. A novel antifibrotic mechanism of nintedanib and pirfenidone. inhibition of collagen fibril assembly. Am. J. Respir. Cell Mol. Biol. 57, 77–90 (2017).
Finnerty, J. P., Ponnuswamy, A., Dutta, P., Abdelaziz, A. & Kamil, H. Efficacy of antifibrotic drugs, nintedanib and pirfenidone, in treatment of progressive pulmonary fibrosis in both idiopathic pulmonary fibrosis (IPF) and non-IPF: a systematic review and meta-analysis. BMC Pulm. Med. 21, 411 (2021).
Wind, S. et al. Clinical pharmacokinetics and pharmacodynamics of nintedanib. Clin. Pharmacokinet. 58, 1131–1147 (2019).
Flaherty, K. R. et al. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 381, 1718–1727 (2019).
Bhatt, S. P. et al. Dupilumab for COPD with type 2 inflammation indicated by eosinophil counts. N. Engl. J. Med. 389, 205–214 (2023).
Bhatt, S. P. et al. Dupilumab for COPD with blood eosinophil evidence of type 2 inflammation. N. Engl. J. Med. 390, 2274–2283 (2024).
Balzer, M. S. et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat. Commun. 13, 4018 (2022).
Doke, T. et al. Single-cell analysis identifies the interaction of altered renal tubules with basophils orchestrating kidney fibrosis. Nat. Immunol. 23, 947–959 (2022).
Fioretto, P., Steffes, M. W., Sutherland, D. E. R., Goetz, F. C. & Mauer, M. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N. Engl. J. Med. 339, 69–75 (1998).
Tajima, K. et al. An organ-derived extracellular matrix triggers in situ kidney regeneration in a preclinical model. NPJ Regen. Med. 7, 18 (2022).
Ruiz-Ortega, M., Rayego-Mateos, S., Lamas, S., Ortiz, A. & Rodrigues-Diez, R. R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 16, 269–288 (2020).
Perkovic, V. et al. Effects of semaglutide on chronic kidney disease in patients with type 2 diabetes. N. Engl. J. Med. 391, 109–121 (2024).
Greenberg, J. H. et al. Urine biomarkers of kidney tubule health, injury, and inflammation are associated with progression of CKD in children. J. Am. Soc. Nephrol. 32, 2664–2677 (2021).
Sparding, N. et al. Unique biomarkers of collagen type III remodeling reflect different information regarding pathological kidney tissue alterations in patients with IgA nephropathy. Biomolecules 13, 1093 (2023).
Zhang, X.-Q. et al. Serum lysyl oxidase is a potential diagnostic biomarker for kidney fibrosis. Am. J. Nephrol. 51, 907–918 (2020).
Zewinger, S. et al. Dickkopf-3 (DKK3) in urine identifies patients with short-term risk of eGFR loss. J. Am. Soc. Nephrol. 29, 2722–2733 (2018).
Genovese, F. et al. Collagen type III and VI remodeling biomarkers are associated with kidney fibrosis in lupus nephritis. Kidney360 2, 1473–1481 (2021).
Sparding, N. et al. Circulating levels of endotrophin are prognostic for long-term mortality after AKI. Kidney360 3, 809–817 (2022).
D’Alessio, S. et al. Revisiting fibrosis in inflammatory bowel disease: the gut thickens. Nat. Rev. Gastroenterol. Hepatol. 19, 169–184 (2022).
Domislović, V. et al. Differences in extra cellular matrix remodelling in highly active Crohn’s disease and ulcerative colitis. J. Crohns Colitis 14, S156–S157 (2020).
Mortensen, J. H. et al. The intestinal tissue homeostasis–the role of extracellular matrix remodeling in inflammatory bowel disease. Expert Rev. Gastroenterol. Hepatol. 13, 977–993 (2019).
van Haaften, W. T. et al. Misbalance in type III collagen formation/degradation as a novel serological biomarker for penetrating (Montreal B3) Crohn’s disease. Aliment Pharm. Ther. 46, 26–39 (2017).
Mortensen, J. H. et al. Fragments of citrullinated and MMP-degraded vimentin and MMP-degraded type III collagen are novel serological biomarkers to differentiate Crohn’s disease from ulcerative colitis. J. Crohns Colitis 9, 863–872 (2015).
Bourgonje, A. R. et al. Serological biomarkers of type I, III and IV collagen turnover are associated with the presence and future progression of stricturing and penetrating Crohnʼs disease. Aliment. Pharmacol. Ther. 1–19. https://doi.org/10.1111/apt.17063 (2022).
Alexdottir, M. S. et al. Serological biomarkers of extracellular matrix turnover and neutrophil activity are associated with long-term use of vedolizumab in patients with Crohn’s disease. Int J. Mol. Sci. 23, i468–i469 (2022).
van Haaften, W. T. et al. Serological biomarkers of tissue turnover identify responders to anti-TNF therapy in Crohn’s disease: a pilot study. Clin. Transl. Gastroenterol. 11, e00217 (2020).
Mortensen, J. H. et al. The citrullinated and MMP-degraded vimentin biomarker (VICM) predicts early response to anti-TNFα treatment in Crohn’s disease. J. Clin. Gastroenterol. 55, 59–66 (2021).
Alexdottir, M. S. et al. Serological biomarkers of intestinal collagen turnover identify early response to infliximab therapy in patients with Crohn’ s disease. Int. J. Mol. Sci. 23, 8137 (2022).
Manon-Jensen, T. et al. Elevated ectodomain of type 23 collagen is a novel biomarker of the intestinal epithelium to monitor disease activity in ulcerative colitis and Crohn’s disease. United Eur. Gastroenterol. J. https://doi.org/10.1177/2050640620977371 (2020).
Jensen, C. et al. Serum type XVI collagen is associated with colorectal cancer and ulcerative colitis indicating a pathological role in gastrointestinal disorders. Cancer Med. 1–8. https://doi.org/10.1002/cam4.1692 (2018).
Sun, S. et al. Serological assessment of the quality of wound healing processes in Crohn’s disease. J. Gastrointest. Liver Dis. 28, 175–182 (2019).
Pehrsson, M., Alexdóttir, M. S., Karsdal, M. A., Thakker, P. & Mortensen, J. H. Novel fibro-inflammatory biomarkers associated with disease activity in patients with Crohn’s disease. Expert Rev. Gastroenterol. Hepatol. 00, 1–13 (2023).
Yoo, J. H., Holubar, S. & Rieder, F. Fibrostenotic strictures in Crohn’s disease. Intest Res 18, 379–401 (2020).
De Bruyn, J. R. et al. Development of fibrosis in acute and longstanding ulcerative colitis. J. Crohns Colitis 9, 966–972 (2015).
Gordon, I. O. et al. Fibrosis in ulcerative colitis is directly linked to severity and chronicity of mucosal inflammation. Aliment. Pharm. Ther. 47, 922–939 (2018).
De Bruyn, J. R. et al. Intestinal fibrosis is associated with lack of response to infliximab therapy in Crohn’s disease. PLoS ONE 13, 1–13 (2018).
Lawrance, I. C. et al. Cellular and molecular mediators of intestinal fibrosis. J. Crohns Colitis j.crohns.2014.09.008, https://doi.org/10.1016/j.crohns.2014.09.008 (2015).
Neurath, M. F. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol. 20, 970–979 (2019).
Stolfi, C., Troncone, E., Marafini, I. & Monteleone, G. Role of TGF-beta and Smad7 in gut inflammation, fibrosis and cancer. Biomolecules 11, 17 (2020).
Gundersen, M. D. et al. Fibrosis mediators in the colonic mucosa of acute and healed ulcerative colitis. Clin. Transl. Gastroenterol. 10, e00082 (2019).
Rieder, F., Brenmoehl, J., Leeb, S., Scholmerich, J. & Rogler, G. Wound healing and fibrosis in intestinal disease. Gut 56, 130–139 (2007).
Hamilton, A. L. et al. Serologic antibodies in relation to outcome in postoperative Crohn’s disease. J. Gastroenterol. Hepatol. 32, 1195–1203 (2017).
Zhao, Z., Cheng, W., Qu, W., Shao, G. & Liu, S. Antibiotic alleviates radiation-induced intestinal injury by remodeling microbiota, reducing inflammation, and inhibiting fibrosis. ACS Omega 5, 2967–2977 (2020).
Ellermann, M. et al. Yersiniabactin-producing adherent/invasive escherichia coli promotes inflammation-associated fibrosis in gnotobiotic Il10−/− Mice. Infect. Immun. 87, e00587-19 (2019).
Mortensen, J. H. et al. A specific calprotectin neo-epitope [CPa9-HNE] in serum from inflammatory bowel disease patients is associated with neutrophil activity and endoscopic severity. J. Crohns Colitis 1–14. https://doi.org/10.1093/ecco-jcc/jjac047 (2022).
Chrysanthopoulou, A. et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 233, 294–307 (2014).
Chokr, D. et al. Adherent invasive Escherichia coli (AIEC) strain LF82, but not Candida albicans, plays a profibrogenic role in the intestine. Gut Pathog. 13, 5 (2021).
O’Donnell, S., O’Sullivan, M., O’Morain, C. A. & Ryan, B. M. The clinical significance of antimicrobial serologic responses within an Irish Crohn’s disease population. Eur. J. Gastroenterol. Hepatol. 25, 1464–1469 (2013).
Dubinsky, M. C. et al. Serum immune responses predict rapid disease progression among children with Crohn’s disease: immune responses predict disease progression. Am. J. Gastroenterol. 101, 360–367 (2006).
Park, J.-S. et al. Lactobacillus acidophilus improves intestinal inflammation in an acute colitis mouse model by regulation of Th17 and treg cell balance and fibrosis development. J. Med Food 21, 215–224 (2018).
Braat, H. et al. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin. Gastroenterol. Hepatol. 4, 754–759 (2006).
Zheng, J. et al. Noninvasive, microbiome-based diagnosis of inflammatory bowel disease. Nat. Med 30, 3555–3567 (2024).
Sandborn, W. J. et al. Efficacy and safety of upadacitinib in a randomized trial of patients with Crohn’s disease. Gastroenterology 158, 2123–2138.e8 (2020).
Roblin, X. et al. Effects of JAK1-preferential inhibitor filgotinib on circulating biomarkers and whole blood genes/pathways of patients with moderately to severely active Crohn’s disease. Inflamm. Bowel Dis. 28, 1207–1218 (2022).
Wang, J. et al. Preventing fibrosis in IBD: update on immune pathways and clinical strategies. Expert Rev. Clin. Immunol. 20, 727–734 (2024).
Sakers, A., De Siqueira, M. K., Seale, P. & Villanueva, C. J. Adipose-tissue plasticity in health and disease. Cell 185, 419–446 (2022).
Bonfante, I. L. P. et al. Combined training increases thermogenic fat activity in patients with overweight and type 2 diabetes. Int J. Obes. 46, 1145–1154 (2022).
Finlin, B. S. et al. Adipose tissue mast cells promote human adipose beiging in response to cold. Sci. Rep. 9, 8658 (2019).
Funcke, J.-B. & Scherer, P. E. Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J. Lipid Res 60, 1648–1697 (2019).
Meyer, L. K., Ciaraldi, T. P., Henry, R. R., Wittgrove, A. C. & Phillips, S. A. Adipose tissue depot and cell size dependency of adiponectin synthesis and secretion in human obesity. Adipocyte 2, 217–226 (2013).
Van Heek, M. et al. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. J. Clin. Investig. 99, 385–390 (1997).
Kelley, D. E., Thaete, F. L., Troost, F., Huwe, T. & Goodpaster, B. H. Subdivisions of subcutaneous abdominal adipose tissue and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 278, E941–E948 (2000).
Vishvanath, L. & Gupta, R. K. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J. Clin. Investig. 129, 4022–4031 (2019).
Sun, K., Tordjman, J., Clément, K. & Scherer, P. E. Fibrosis and adipose tissue dysfunction. Cell Metab. 18, 470–477 (2013).
Henriksen, K. et al. Endotrophin, a key marker and driver for fibroinflammatory disease. Endocr. Rev. 45, 361–378 (2024).
Zhu, Q. & Scherer, P. E. Immunologic and endocrine functions of adipose tissue: implications for kidney disease. Nat. Rev. Nephrol. 14, 105–120 (2018).
Ma, L.-J. et al. Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity. Am. J. Physiol. Ren. Physiol. 300, F1203–F1213 (2011).
Griendling, K. K., Minieri, C. A., Ollerenshaw, J. D. & Alexander, R. W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 74, 1141–1148 (1994).
Lim, C. C. et al. Elevated serum leptin, adiponectin and leptin to adiponectin ratio is associated with chronic kidney disease in Asian adults. PLoS ONE 10, e0122009 (2015).
Wolf, G. et al. Leptin stimulates proliferation and TGF-β expression in renal glomerular endothelial cells: potential role in glomerulosclerosis. Kidney Int 56, 860–872 (1999).
Cui, W., Maimaitiyiming, H., Qi, X., Norman, H. & Wang, S. Thrombospondin 1 mediates renal dysfunction in a mouse model of high-fat diet-induced obesity. Am. J. Physiol.-Ren. Physiol. 305, F871–F880 (2013).
Mao, R. et al. The mesenteric fat and intestinal muscle interface: creeping fat influencing stricture formation in Crohn’s disease. Inflamm. Bowel Dis. 25, 421–426 (2019).
HALPRIN, K. M. Epidermal ‘turnover time’–a re-examination. Br. J. Dermatol 86, 14–19 (1972).
Lorenz, H. P. et al. Scarless wound repair: a human fetal skin model. Development 114, 253–259 (1992).
Watt, F. M. The stem cell compartment in human interfollicular epidermis. J. Dermatol. Sci. 28, 173–180 (2002).
Gomes, M. L. N. P., Krijnen, P. A. J., Middelkoop, E., Niessen, H. W. M. & Boekema, B. K. H. L. Fetal skin wound healing: key extracellular matrix components and regulators in scarless healing. J. Investig. Dermatol. https://doi.org/10.1016/J.JID.2024.05.027 (2024).
Canady, J., Karrer, S., Fleck, M. & Bosserhoff, A. K. Fibrosing connective tissue disorders of the skin: molecular similarities and distinctions. J. Dermatol Sci. 70, 151–158 (2013).
Bailey, J., Schwehr, M. & Beattie, A. Management of keloids and hypertrophic scars. Am. Fam. Physician 110, 605–611 (2024).
Wang, Q., Ren, Z., Jin, W. & Jin, Z. Real-world effectiveness and safety of bleomycin in patients with keloids and hypertrophic scars: a systematic review and meta-analysis. Arch. Dermatol. Res. 317, 170 (2025).
Pérez, L. A., Leyton, L. & Valdivia, A. Thy-1 (CD90), Integrins and syndecan 4 are key regulators of skin wound healing. Front. Cell Dev. Biol. 10, https://doi.org/10.3389/fcell.2022.810474 (2022).
Hayn, E. Successful treatment of complex traumatic and surgical wounds with a foetal bovine dermal matrix. Int. Wound J. 11, 675–680 (2014).
Dobrota, R. et al. Circulating collagen neo-epitopes and their role in the prediction of fibrosis in patients with systemic sclerosis: a multicentre cohort study. Lancet Rheumatol. 3, e175–e184 (2021).
Tomcik, M. et al. S100A4 amplifies TGF-β-induced fibroblast activation in systemic sclerosis. Ann. Rheum. Dis. 74, 1748–1755 (2015).
Leask, A., Naik, A. & Stratton, R. J. Back to the future: targeting the extracellular matrix to treat systemic sclerosis. Nat. Rev. Rheumatol. 19, 713–723 (2023).
Steen, V. D. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 35, 35–42 (2005).
Graf, S. W. et al. South Australian Scleroderma Register: autoantibodies as predictive biomarkers of phenotype and outcome. Int. J. Rheum. Dis. 15, 102–109 (2012).
Walker, J. G. & Fritzler, M. J. Update on autoantibodies in systemic sclerosis. Curr. Opin. Rheumatol. 19, 580–591 (2007).
Walker, U. A. et al. Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials And Research group database. Ann. Rheum. Dis. 66, 754–763 (2007).
Reveille, J. D. et al. Evidence-based guidelines for the use of immunologic tests: anticentromere, Scl-70, and nucleolar antibodies. Arthritis Rheum. 49, 399–412 (2003).
Steen, V. D., Powell, D. L. & Medsger, T. A. Clinical correlations and prognosis based on serum autoantibodies in patients with systemic sclerosis. Arthritis Rheum. 31, 196–203 (1988).
Hesselstrand, R., Scheja, A., Shen, G. Q., Wiik, A. & Åkesson, A. The association of antinuclear antibodies with organ involvement and survival in systemic sclerosis. Rheumatol. (Oxf.) 42, 534–540 (2003).
Hanke, K. et al. Diagnostic value of anti-topoisomerase I antibodies in a large monocentric cohort.Arthritis Res. Ther. 11, R28 (2009).
Abignano, G. et al. The enhanced liver fibrosis test: a clinical grade, validated serum test, biomarker of overall fibrosis in systemic sclerosis. Ann. Rheum. Dis. 73, 420–427 (2014).
Jurisic, Z. et al. Relationship between interleukin-6 and cardiac involvement in systemic sclerosis. Rheumatology 52, 1296–1302 (2013).
Codullo, V. et al. An investigation of the inflammatory cytokine and chemokine network in systemic sclerosis. Ann. Rheum. Dis. 70, 1115–1121 (2011).
Puccetti, A. & Migliorini, P. Human and murine anti-DNA antibodies induce the production of anti-idiotypic antibodies with autoantigen-binding properties (epibodies) through immune-network interactions. J. Immunol. 15, 4229–4237 (1990).
Khanna, D. et al. A 24-week, phase iia, randomized, double-blind, placebo-controlled study of ziritaxestat in early diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. 75, 1434–1444 (2023).
Sheng, X. R. et al. Biomarkers of fibrosis, inflammation, and extracellular matrix in the phase 3 trial of tocilizumab in systemic sclerosis. Clin. Immunol. 254, 109695 (2023).
Baka, J. L. Ce. S. et al. Stiff skin syndrome: long-term follow-up. Bras. Dermatol 99, 597 (2024).
Fusco, C. et al. Pro-fibrotic phenotype in a patient with segmental stiff skin syndrome via TGF-β signaling overactivation. Int J. Mol. Sci. 21, 5141 (2020).
Anstee, Q. M., Reeves, H. L., Kotsiliti, E., Govaere, O. & Heikenwalder, M. From NASH to HCC: current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 16, 411–428 (2019).
Gofton, C., Upendran, Y., Zheng, M.-H. & George, J. MAFLD: how is it different from NAFLD? Clin. Mol. Hepatol. 29, S17–S31 (2023).
Sanyal, A. J. et al. Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N. Engl. J. Med. 385, 1559–1569 (2021).
Konyn, P., Ahmed, A. & Kim, D. Causes and risk profiles of mortality among individuals with nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 29, S43–S57 (2023).
Cypess, A. M. Reassessing human adipose tissue. N. Engl. J. Med. 386, 768–779 (2022).
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Nagai, A. et al. Overview of the BioBank Japan Project: study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Hardy, T. et al. The European NAFLD Registry: a real-world longitudinal cohort study of nonalcoholic fatty liver disease. Contemp. Clin. Trials 98, 106175 (2020).
Wilhelm, M. et al. Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587 (2014).
Kim, M.-S. et al. A draft map of the human proteome. Nature 509, 575–581 (2014).
Oh, H. S.-H. et al. Organ aging signatures in the plasma proteome track health and disease. Nature 624, 164–172 (2023).
Niu, L. et al. Noninvasive proteomic biomarkers for alcohol-related liver disease. Nat. Med. 28, 1277–1287 (2022).
Govaere, O. et al. A proteo-transcriptomic map of non-alcoholic fatty liver disease signatures. Nat. Metab. 5, 572–578 (2023).
Eldjarn, G. H. et al. Large-scale plasma proteomics comparisons through genetics and disease associations. Nature 622, 348–358 (2023).
Schuppan, D., Myneni, S. & Surabattula, R. Liquid biomarkers for fibrotic NASH—progress in a complex field. J. Hepatol. 76, 5–7 (2022).
Huan, T. et al. Integrative analysis of clinical and epigenetic biomarkers of mortality.Aging Cell 21, e13608 (2022).
Deelen, J. et al. A metabolic profile of all-cause mortality risk identified in an observational study of 44,168 individuals. Nat. Commun. 10, 3346 (2019).
Jørgensen, J. T. The current landscape of the FDA approved companion diagnostics. Transl. Oncol. 14, 101063 (2021).
Nielsen, M. J. et al. The neo-epitope specific PRO-C3 ELISA measures true formation of type III collagen associated with liver and muscle parameters. Am. J. Transl. Res. 5, 303–315 (2013).
Jansen, C. et al. PRO-C3-levels in patients with HIV/HCV-Co-infection reflect fibrosis stage and degree of portal hypertension. PLoS ONE 9, e108544 (2014).
Genovese, F. et al. Plasma levels of PRO-C3, a type III collagen synthesis marker, are associated with arterial stiffness and increased risk of cardiovascular death. Atherosclerosis 388, 117420 (2024).
Nielsen, M. J. et al. Serological markers of extracellular matrix remodeling predict transplant-free survival in primary sclerosing cholangitis. Aliment Pharm. Ther. 48, 179–189 (2018).
Lichtinghagen, R. et al. The enhanced liver fibrosis (ELF) score: normal values, influence factors and proposed cut-off values. J. Hepatol. 59, 236–242 (2013).
Parkes, J. et al. Enhanced liver fibrosis test can predict clinical outcomes in patients with chronic liver disease. Gut 59, 1245–1251 (2010).
Mayo, M. J. et al. Prediction of clinical outcomes in primary biliary cirrhosis by serum enhanced liver fibrosis assay. Hepatology 48, 1549–1557 (2008).
Sun, K., Park, J., Kim, M. & Scherer, P. E. Endotrophin, a multifaceted player in metabolic dysregulation and cancer progression, is a predictive biomarker for the response to PPARγ agonist treatment. Diabetologia 60, 24–29 (2017).
Luther, D. J. et al. Absence of type VI collagen paradoxically improves cardiac function, structure, and remodeling after myocardial infarction. Circ. Res 110, 851–856 (2012).
Lin, S.-N. et al. Human intestinal myofibroblasts deposited collagen VI enhances adhesiveness for T cells – A novel mechanism for maintenance of intestinal inflammation. Matrix Biol. 113, 1–21 (2022).
Stolz, D. et al. Systemic biomarkers of collagen and elastin turnover are associated with clinically relevant outcomes in COPD. Chest 151, 47–59 (2017).
Rønnow, S. R. et al. Endotrophin, an extracellular hormone, in combination with neoepitope markers of von Willebrand factor improves prediction of mortality in the ECLIPSE COPD cohort. Respir. Res. 21, 202 (2020).
Kerbert, A. J. C. et al. Biomarkers of extracellular matrix formation are associated with acute-on-chronic liver failure. JHEP Rep. 3, 100355 (2021).
Sparding, N. et al. Endotrophin levels are associated with allograft outcomes in kidney transplant recipients. Biomolecules 13, 792 (2023).
Nissen, N. I. et al. Prognostic value of blood-based fibrosis biomarkers in patients with metastatic colorectal cancer receiving chemotherapy and bevacizumab. Sci. Rep. 11, 865 (2021).
Nissen, N. I. et al. Collagen biomarkers quantify fibroblast activity in vitro and predict survival in patients with pancreatic ductal adenocarcinoma. Cancers 14, 819 (2022).
Leeming, D. J. et al. Endotrophin, a pro-peptide of Type VI collagen, is a biomarker of survival in cirrhotic patients with hepatocellular carcinoma. Hepat. Oncol. 8, HEP32 (2021).
Pilemann-Lyberg, S. et al. Markers of collagen formation and degradation reflect renal function and predict adverse outcomes in patients with type 1 diabetes. Diab. Care 42, 1760–1768 (2019).
Rasmussen, D. G. K. et al. Higher collagen VI formation is associated with all-cause mortality in patients with type 2 diabetes and microalbuminuria. Diab. Care 41, 1493–1500 (2018).
Tougaard, N. H. et al. Endotrophin as a marker of complications in a type 2 diabetes cohort. Diab. Care 45, 2746–2748 (2022).
Genovese, F. et al. The fibroblast hormone endotrophin is a biomarker of mortality in chronic diseases. Matrix Biol. https://doi.org/10.1016/j.matbio.2024.06.003 (2024).
Schuppan, D., Ashfaq-Khan, M., Yang, A. T. & Kim, Y. O. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. 68–69, 435–451 (2018).
Kingwell, K. NASH field celebrates ‘hurrah moment’ with a first FDA drug approval for the liver disease. Nat. Rev. Drug Discov. https://doi.org/10.1038/d41573-024-00051-1 (2024).
Friedman, S. L., Sheppard, D., Duffield, J. S. & Violette, S. Therapy for fibrotic diseases: nearing the starting line. Sci. Transl. Med. 5, 167sr1 (2013).
Schuppan, D. & Kim, Y. O. Evolving therapies for liver fibrosis. J. Clin. Investig. 123, 1887–1901 (2013).
Bay-Jensen, A. C. et al. Ankylosing spondylitis is characterized by an increased turnover of several different metalloproteinase-derived collagen species: a cross-sectional study. Rheumatol. Int. 32, 3565–3572 (2012).
Willumsen, N. et al. Extracellular matrix specific protein fingerprints measured in serum can separate pancreatic cancer patients from healthy controls. BMC Cancer 13, 554 (2013).
Dooling, L. J., Saini, K., Anlaş, A. A. & Discher, D. E. Tissue mechanics coevolves with fibrillar matrisomes in healthy and fibrotic tissues. Matrix Biol. 111, 153–188 (2022).
Pehrsson, M. et al. An MMP-degraded and cross-linked fragment of type III collagen as a non-invasive biomarker of hepatic fibrosis resolution. Liver Int. 42, 1605–1617 (2022).
Rasmussen, D. G. K. et al. NAFLD and NASH biomarker qualification in the LITMUS consortium—lessons learned. J. Hepatol. 78, 852–865 (2023).
Author information
Authors and Affiliations
Contributions
A.E.M.-G. wrote and edited the manuscript and prepared Figs. 1–4. D.J.L. wrote and edited the manuscript. K.H. wrote and edited the manuscript. J.H.M. wrote and edited the manuscript. Q.M.A. wrote and edited the manuscript, A.J.S. wrote and edited the manuscript. M.A.K. conceived, wrote and edited the manuscript. D.S. wrote and edited the manuscript. S.H.N. wrote and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
A.E.M.-G., D.J.L., K.H., J.H.M., and M.A.K. are employees of Nordic Bioscience A/S. D.J.L., K.H., J.H.M., S.H.N., and M.A.K. are Nordic Bioscience A/S stockholders. A.J.S.: has stock options in Durect, Inversago, Tiziana, Rivus, Exhalenz, Genfit. He has served as a consultant to Intercept, Gilead, Takeda, Meck, Eli Lilly, Novo Nordisk, Astra Zeneca, Boehringer Ingelheim, Alnylam, Regeneron, Histoindex, Path AI, Pfizer, 89Bio, Altimmune, Northsea, Akero, Madrigal, Salix, Myovant, Poxel, Surrozen, Hanmi, Aligos, Promed, Zydus. His institution has received grants from Intercept, Novo Nordisk, Eli Lilly, Boehringer Ingelheim, Echosens, Hanmi, Madrigal, Gilead, Salix, Meck, Takeda. He receives royalties from Elsevier and Wolter Kluwers. D.S.: is CMO, co-CEO/CSO for ImmuneNTech, and consults for Falk Pharma, Takeda, Boehringer-Ingelheim, Resalis, Immunic, Sanofi, Northsea Bioscirences.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Mayorca-Guiliani, A.E., Leeming, D.J., Henriksen, K. et al. ECM formation and degradation during fibrosis, repair, and regeneration. npj Metab Health Dis 3, 25 (2025). https://doi.org/10.1038/s44324-025-00063-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44324-025-00063-4
This article is cited by
-
FibroTrack: a standalone deep learning platform for automated fibrosis quantification in muscle and cardiac histology
Skeletal Muscle (2026)
-
Cardioprotective mechanisms of D-Ribose-L-Cysteine against isoprenaline-induced myocardial infarction in male Wistar rats: Experimental and computational insights
Nutrire (2026)
-
Overcoming tumor microenvironment barriers: transformable and bioinspired nanomedicine strategies for deep tumor penetration
Journal of Nanobiotechnology (2026)
-
Magneto-mechanics in mechanobiology: enabling remote force transmission to cells and extracellular matrix
Biophysical Reviews (2025)
