
Abstract
Recent evidence has highlighted the importance of employing culture media designed to emulate the metabolic environment cells would be exposed to in vivo. Here, we utilize the physiologic medium Plasmax to examine the impact of nutrient availability on the human hepatocyte cell line, HepG2. Incubation of HepG2 cells in Plasmax suppressed a transcriptional program driven by Hepatocyte Nuclear Factor 4 (HNF4A), a master regulator of hepatocyte identity, leading to a dedifferentiated phenotype. Given that HepG2 cells were originally isolated from a patient with hepatoblastoma, this suggested reversion to the native state in physiologic medium. Importantly, exclusion of iron from Plasmax reinstated the HNF4A-driven transcriptional program. These studies suggest a relationship between iron availability and the fidelity of hepatocyte cell fate and highlight the importance of more faithfully recapitulating in vivo metabolite availability in vitro.
Similar content being viewed by others


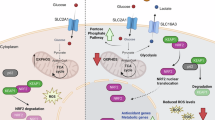
Introduction
In vitro cell culture models play a central role in biological and biomedical research. In recent years, efforts to improve the physiological relevance of these models have highlighted the importance of media composition on cellular phenotype and biological responses. For example, the dependence of some cancer cell lines on glutaminolysis is not observed in the in vivo context1. This has been attributed to the presence of high cystine concentrations in conventional in vitro culture conditions2. More generally, conventional media (e.g., Dulbecco’s Modified Eagle Medium, DMEM) poorly recapitulate metabolite levels that cells would be exposed to in vivo3,4. The increasing awareness and recognition that the composition of cell culture media is important in preserving biological processes in vitro has led to ongoing efforts to develop cell culture media that better represent physiological metabolite concentrations5,6,7. Studies employing physiologic media have demonstrated that media composition can alter cellular dependencies via metabolic and transcriptional changes5,6,8,9,10,11.
Hepatocytes are the major cell type in the liver and are widely modeled in vitro to study drug metabolism, drug-induced liver injury (DILI), and various liver diseases12,13. The most frequently used cell line in these contexts are HepG2 cells, which were originally derived from a hepatic tumor isolated from a 15-year-old male14,15. Although initially characterised as being derived from a hepatocellular carcinoma (HCC), subsequent histopathologic, genetic, and molecular analysis revealed that the origin of HepG2 cells was instead a hepatoblastoma (HB)16. Despite this, HepG2 cells are still widely utilized as a hepatocyte cell line as they exhibit the hallmarks of hepatocyte fate and function in vitro17,18,19,20,21.
In this study, we examined the impact of nutrient availability on HepG2 cells by comparing culture in conventional media with culture in a media previously developed to more faithfully reflect metabolite abundance in human plasma (Plasmax)5. We demonstrate that culturing HepG2 cells in Plasmax alters cell fate in a manner associated with loss of HNF4A, a transcription factor that regulates hepatocyte identity. Moreover, we demonstrate that iron availability contributes to the regulation of hepatic cell fate. This research suggests an important role for physiologic media in maintaining cell fate in vitro.
Results
HepG2 cells adopt a native hepatoblast-like state in physiologic medium
Typically, HepG2 cells are maintained in one of three conventional cell culture media: Eagle’s Minimal Essential Medium (EMEM), high glucose DMEM or low glucose DMEM22,23,24,25. To examine the impact of culture media on the transcriptional landscape, HepG2 cells were cultured for 4 days in each of the three conventional media types or in the physiologic medium Plasmax5. Plasmax mimics metabolite concentrations found in human plasma and has a dramatically different composition than conventional cell culture media (Fig. S1A). While minimal transcriptional changes were observed between cells cultured in any of the three conventional media, culture in Plasmax induced widespread transcriptional changes (Fig. 1A and Fig. S1B, C). Interestingly, Gene Set Enrichment Analysis (GSEA) revealed, amongst the top twenty differentially enriched signatures observed in the context of culture in Plasmax versus EMEM, suppression of pathways associated with sterol/steroid biosynthesis, a primary function of hepatocytes (Table S1). Consistent with this, downregulation of gene signatures associated with hepatocyte cell state and function was observed in the context of culture in Plasmax (Fig. 1B). Moreover, culture in Plasmax dramatically reduced the expression of HNF4A, a master regulator of hepatocyte identity (Fig. 1C). Loss of HNF4A is typically associated with dedifferentiation to a hepatoblast-like state26,27,28. Accordingly, in the context of culture in Plasmax, loss of genes controlled by HNF4A (Apolipoprotein H, APOH; Albumin, ALB; Transferrin, TF) was associated with a concomitant increase in the expression of hepatoblast-associated genes including the pioneer transcription factor SRY-Box Transcription Factor 4 (SOX4), a key regulator of hepatocyte dedifferentiation (Fig. 1D)29. Importantly, the transcriptional changes observed following culture in Plasmax were associated with a reduction in the capacity of HepG2 cells to store lipids in lipid droplets (Fig. 1E, F), a key housekeeping function of hepatocytes, and reduced sensitivity to the hepatotoxin ethanol (Fig. 1G). Interestingly, in the intrinsically differentiated hepatocyte cell line Hep3B, HNF4A protein expression was not altered by changes in culture conditions (Fig. S1D). Together, these data suggest that culture in physiologic media maintains the intrinsic cell fate fidelity of HepG2 cells in vitro.

A Volcano plot of differentially expressed genes (DEGs) identified from RNA-Seq analysis comparing HepG2 cells cultured in Plasmax with HepG2 cells cultured in EMEM, n = 3. Genes significantly downregulated and upregulated in Plasmax are highlighted in blue and red, respectively. B Gene set enrichment analysis (GSEA) plots derived from RNA-Seq analysis comparing HepG2 cells cultured in Plasmax with HepG2 cells cultured in EMEM demonstrating signatures associated with hepatocyte cell state and function. Hepatocyte signature, Gene set AIZARANI_LIVER_C14_HEPATOCYTES_2; HNF4A Target genes, Gene set OHGUCHI_LIVER_HNF4A_TARGETS_DN. C Representative immunoblot analysis of HNF4A expression in HepG2 cells cultured in EMEM or Plasmax. Actin is included as a loading control. D Heatmap of hepatocyte and hepatoblast gene expression in HepG2 cells culture in EMEM or Plasmax as determined by RNA-Seq analysis, n = 3. E Representative confocal images of HepG2 cells cultured in EMEM or Plasmax and stained with BODIPY (green) and DAPI (blue). White scale bars represent 50 µm. Yellow scale bars represent 10 µm. F Intensity of BODIPY staining per cell, determined by confocal microscopy, in HepG2 cells cultured in EMEM or Plasmax. Data are shown as median and interquartile ranges, n = 3. G Viability of HepG2 cells cultured in EMEM or Plasmax following treatment with ethanol for 24 h as determined by a CellTiter-Glo Assay. Data are shown as mean ± SEM, n = 4. For all experiments, *P < 0.05, ***P < 0.001, ****P < 0.0001.
Trace element availability regulates hepatocyte differentiation
To determine the components of Plasmax contributing to dedifferentiation of HepG2 cells, variants of Plasmax lacking either trace elements, which are severely underrepresented in EMEM/DMEM, or lacking metabolites absent from EMEM/DMEM (termed “supplemental metabolites”) were prepared (Fig. S1A)5. Withdrawal of trace elements, but not supplemental metabolites, restored HNF4A protein expression to levels observed in EMEM (Fig. 2A). Moreover, transcriptomic analysis revealed that trace element withdrawal restored hepatocyte gene expression and expression of signatures associated with hepatocyte cell state and hepatocyte function (Fig. 2B, C). Notably, SOX4 expression was suppressed and returned to levels observed in EMEM when cells were cultured in Plasmax devoid of trace elements (Fig. 2C, D).

A Representative immunoblot analysis of HNF4A expression in HepG2 cells cultured in EMEM, Plasmax, Plasmax devoid of trace elements (TE), or Plasmax devoid of supplemental metabolites (SM). Actin is included as a loading control. B GSEA plots derived from RNA-Seq analysis comparing HepG2 cells cultured in Plasmax with HepG2 cells cultured in EMEM or comparing HepG2 cells cultured in Plasmax-TE with HepG2 cells cultured in EMEM demonstrating signatures associated with hepatocyte cell state and function. Hepatocyte signature, Gene set AIZARANI_LIVER_C14_HEPATOCYTES_2; HNF4A Target genes, Gene set OHGUCHI_LIVER_HNF4A_TARGETS_DN. C Volcano plot of DEGs identified from RNA-Seq analysis comparing HepG2 cells cultured in Plasmax-TE with HepG2 cells cultured in Plasmax, n = 3. Hepatocyte differentiation markers are annotated and highlighted in maroon. Significantly downregulated and upregulated genes are highlighted in blue and red, respectively. D SOX4 transcript expression (counts per million, CPM) based on RNA-Seq analysis of HepG2 cells cultured in EMEM, Plasmax, or Plasmax-TE, n = 3. For all experiments, ns not significant, ***P < 0.001.
Multi-omic analysis reveals a relationship between trace element availability and metalloprotein expression
To gain insights regarding which trace element(s) was contributing to dedifferentiation in Plasmax, intracellular trace element abundance was quantified using inductively coupled plasma mass spectrometry (ICP-MS). Given that serum can be a source of trace elements, these species were also quantified in fresh culture media. As expected, the abundance of trace elements was higher in Plasmax compared to EMEM (Fig. S2). Interestingly, HepG2 cells cultured in Plasmax exhibited more than a twenty-fold increase in intracellular iron availability (Fig. 3A). A three-fold increase in intracellular copper was also observed (Fig. 3A). To explore the role of iron in hepatocyte differentiation, iron salts were added back to Plasmax devoid of trace elements. A multidimensional scaling (MDS) plot of the transcriptome revealed that while withdrawal of trace elements from Plasmax resulted in a transcriptome more similar to EMEM, reintroduction of iron resulted in a transcriptome that was more similar to Plasmax (Fig. 3B). This trend was recapitulated at the proteome level where reintroduction of iron restored a proteomic profile similar to HepG2 cells incubated in complete Plasmax (Fig. 3C). Interestingly, a correlation plot of the transcriptome and proteome of cells cultured in Plasmax lacking trace elements compared to complete Plasmax revealed proteins that were significantly downregulated at the level of protein, but not transcript, expression in response to removal of trace elements. Notably, amongst these proteins were selenoproteins (e.g. Selenoprotein P, SELENOP, and Glutathione Peroxidase 1, GPX1) and iron-regulated proteins (e.g. Ferritin Heavy Chain 1, FTH1) (Fig. 3D). These proteins belong to the metalloprotein superfamily and their translation is dependent on trace element availability30,31. Addition of iron salts to Plasmax devoid of trace elements was able to rescue the expression of iron-regulated proteins but not selenoproteins, demonstrating the importance of trace elements in the post-transcriptional regulation of protein abundance (Fig. 3E). Taken together, these results highlight a role for trace elements in the regulation of cellular phenotype and demonstrate the importance of utilizing a multi-omic approach to interrogate the impact of altered nutrient availability on cellular phenotype(s).

A Relative intracellular abundance of trace elements in HepG2 cells cultured in EMEM (blue) or Plasmax (red) n = 3. B MDS plot of RNA-Seq data from HepG2 cells cultured in EMEM, Plasmax, Plasmax-TE, or Plasmax-TE supplemented with iron salts (0.12 µM ferric nitrate and 1.04 µM ferric sulfate; Plasmax-TE+Fe), n = 3. C MDS plot of proteomic data from HepG2 cells cultured in EMEM, Plasmax, Plasmax-TE, or Plasmax-TE+Fe, n = 5. D Correlation between proteomic and transcriptomic data from HepG2 cells cultured in Plasmax-TE versus Plasmax. Significantly differentially downregulated and upregulated proteins whose transcripts were not significantly altered are highlighted in green and magenta, respectively. Metalloproteins are highlighted in blue. E Heatmap of metalloprotein expression in HepG2 cells cultured in EMEM, Plasmax, Plasmax-TE or Plasmax-TE+Fe as determined by proteomic analysis, n = 5. For all experiments, ns not significant, ****P < 0.0001.
Iron availability contributes to the regulation of hepatocyte cell fate
Given that intracellular levels of both iron and copper were significantly increased in HepG2 cells cultured in Plasmax versus EMEM (Fig. 3A), a role for these trace elements in modulating hepatocyte cell fate was examined. Interestingly, reintroduction of iron salts, but not copper salts, to Plasmax devoid of trace elements suppressed HNF4A expression (Fig. 4A). Moreover, re-introduction of iron salts suppressed signatures associated with the differentiated hepatocyte cell state (Fig. 4B, C). The majority of labile iron is incorporated in the cofactor heme32. The transcriptional repressor BTB and CNC Homolog 1 (BACH1) plays a central role in regulating intracellular heme homeostasis and is itself regulated by heme availability33,34,35. Interestingly, BACH1 has also been shown to contribute to the regulation of cell fate33,36. Notably, proteomics analysis revealed that BACH1 target genes were upregulated after reintroduction of iron salts to Plasmax devoid of trace elements (Fig. 4D). Finally, BACH1 protein expression was dramatically lower in HepG2 cells cultured in Plasmax versus EMEM (Fig. 4E). Taken together, these data suggest that the iron-dependent dedifferentiation of hepatocytes in Plasmax is mediated by the trace element iron through regulation of the transcription factor BACH1 (Fig. 4F).

A Representative immunoblot analysis of HNF4A expression in HepG2 cells cultured in Plasmax, Plasmax-TE, Plasmax-TE+Fe, Plasmax-TE supplemented with copper salts (0.005 µM cupric sulfate), or Plasmax-TE supplemented with iron and copper salts (0.12 µM ferric nitrate, 1.04 µM ferric sulfate, 0.005 µM cupric sulfate). Actin is included as a loading control. B GSEA plots derived from RNA-Seq analysis comparing HepG2 cells cultured in Plasmax-TE with HepG2 cells cultured in Plasmax and comparing HepG2 cells cultured in Plasmax-TE+Fe with HepG2 cells cultured in Plasmax demonstrating signatures associated with hepatocyte cell state and function. Hepatocyte signature, Gene set AIZARANI_LIVER_C14_HEPATOCYTES_2; HNF4A Target genes, Gene set OHGUCHI_LIVER_HNF4A_TARGETS_DN. C Heatmap of hepatocyte and hepatoblast gene expression in Plasmax, Plasmax-TE, or Plasmax-TE+Fe as determined by RNA-Seq analysis, n = 3. D Volcano plot of differentially expressed proteins identified from proteomic analysis comparing HepG2 cells cultured in Plasmax-TE+Fe with HepG2 cells cultured in Plasmax-TE, n = 5. BACH1 targets are highlighted in maroon. Significantly downregulated and upregulated proteins are highlighted in blue and red, respectively. E Representative immunoblot analysis of BACH1 and HNF4A expression in HepG2 cells cultured in EMEM or Plasmax. Actin is included as a loading control. F Schematic demonstrating the reciprocal relationship between iron availability, HNF4A activity, and hepatic fate.
Discussion
The liver biopsy from which HepG2 cells were isolated in 1979 was described as a well differentiated HCC14. It was not until 2009 when the histopathology and genetics of the original biopsy was revisited that it was reclassified as a HB16. Despite this correction, HepG2 cells are still widely used to model hepatocytes/HCC rather than hepatoblasts/HB17,18,19,20,21. This is reasonable as HepG2 cells cultured in conventional media exhibit characteristics consistent with differentiated hepatocytes. In this study, we have demonstrated that incubation in a physiologic medium, Plasmax, causes HepG2 cells to revert to their native hepatoblast state.
A previous study investigating the impact of cell culture conditions on HepG2 cells demonstrated that increased amino acid availability promoted the acquisition of a transcriptome similar to primary human hepatocytes37. In other cell lines, physiologically relevant metabolites that are underrepresented or absent in conventional media, such as urea and the trace element selenium, have been shown to drive significant changes in cell metabolism5,6. Here, we have shown that underrepresentation of the trace element iron in conventional media drives differentiation of HepG2 cells to a hepatocyte state. Maintenance of intracellular iron homeostasis has been shown to play a role in determining fate in other cell types including breast cancer cells, hematopoietic stem cells, and alveolar cells38,39,40. Although trace elements exist in minute quantities in plasma, these studies highlight the importance of considering trace element availability when employing cell culture models. Given the propensity of primary human hepatocytes to dedifferentiate in standard culture conditions, studies investigating the impact of physiologic media on cell fate fidelity in this cell type are warranted.
We demonstrate that removal of trace elements from Plasmax triggers the downregulation of proteins harboring iron response elements in the 5ʹ-UTR of the associated transcripts (IRE; e.g., ACO2, FTL, FTH1)41,42,43. IREs are recognized by IRE binding proteins, which in the absence of iron, repress protein expression by hindering the binding of translation initiation factors44. In addition to iron, the availability of other trace elements can dramatically impact the proteome. For example, the translation of selenoproteins, which contain the cysteine analog selenocysteine (Sec), is dependent on selenium availability30,45,46,47. Since the protein expression of IRE-containing proteins and selenoproteins are regulated at the translational level, changes in their expression resulting from altered trace element availability would not be detected at the transcriptional level. This underscores the importance of employing multi-omic approaches to investigate the consequences of altering metabolite availability.
BACH1 is a transcription factor that regulates iron homeostasis and is highly responsive to changes in iron availability48,49. Interestingly, in lung cancer, BACH1 has been shown to drive changes in cell plasticity via epithelial-to-mesenchymal transition50,51. Moreover, BACH1 has been shown to interact with transcription factors that regulate self-renewal (e.g. NANOG, SOX2, and OCT4) to repress transcription of mesodermal genes and promote pluripotency in human embryonic stem cells36. Our findings demonstrate that BACH1 is downregulated in HepG2 cells cultured in Plasmax. Future studies investigating the interplay between iron availability, BACH1, and cell fate are warranted.
In conclusion, our study adds to a growing body of literature highlighting the importance of more faithfully recapitulating the in vivo metabolic microenvironment when employing in vitro cell culture models. Considering these findings, we suggest that, in addition to providing an opportunity to identify novel modulators of cell fate, physiologic media should be more broadly examined for its capacity to impact cell fate fidelity and improve the in vivo relevance of in vitro cell culture models.
Methods
Cell culture
HepG2 cells were purchased from the European Collection of Authenticated Cell Cultures (ECACC) via CellBank Australia, and Hep3B cells were purchased from American Type Culture Collection (ATCC). Both cell lines were routinely assayed for mycoplasma contamination. Cells were cultured in high glucose DMEM (DMEM-HG; Thermo Fisher Scientific, 11965), low glucose DMEM (DMEM-LG; Thermo Fisher Scientific, 11885), Plasmax (prepared in-house as previously described5), or EMEM, which was prepared by supplementing MEM, NEAA, no glutamine (Thermo Fisher Scientific, 10370) with 1 mM sodium pyruvate and 2 mM glutamine. Cells were maintained in a humidified incubator at 37 °C with 5% CO2. For all experiments, 2 × 105 HepG2 cells and 8 × 104 Hep3B cells were seeded in 6-well plates in EMEM containing 10% heat-inactivated FBS (HyCloneTM, SH30084). 24 h after seeding, wells were washed with PBS and the appropriate media (DMEM-HG, DMEM-LG, Plasmax or EMEM) containing 10% heat-inactivated FBS was added. Media was refreshed after 48 h and cells were harvested 96 h after transition to the different media types.
RNA-Seq analysis
For RNA-Seq analysis, total RNA was extracted using the NucleoSpin RNA kit (Macherey-Nagel) as per manufacturer’s instructions. RNA quality was confirmed using an Agilent 4200 TapeStation System. Libraries were prepared using the QuantSeq 3ʹ mRNA-Seq kit (Lexogen) and sequenced with an Illumina NextSeq 500, with single-end 75 bp reads to a depth of 5 M reads per sample. FASTQ files were uploaded to the Galaxy web platform for quality control (FastQC), trimming (Cutadapt), alignment (RNA STAR), and counting (featureCounts)52. Reads were aligned to the human genome assembly (Ensembl hg19, GRCh37). Analysis of differentially expressed genes was performed with Limma-Voom (v3.40.6)53,54. Gene set enrichment analysis (GSEA) was performed and plotted in R using the ‘clusterProfiler’ package55. Heatmaps and volcano plots were generated in R using the ‘pheatmap’ and ‘ggplot’ packages, respectively56,57.
Immunoblotting
Cells were washed with PBS and lysed in SDS lysis buffer (1% SDS, 50 mM Tris-HCl, 10 mM EDTA) containing a protease inhibitor cocktail (Sigma-Aldrich) and PierceTM Universal Nuclease for Cell Lysis (Thermo Fisher Scientific). Lysates were resolved by SDS-PAGE and transferred to nitrocellulose membrane. Membranes were probed with primary antibodies recognising HNF4A (Cell Signaling Technology, 3113, 1:1000 dilution), BACH1 (Proteintech, 14018-1-AP, 1:500 dilution), and β-Actin (Cell Signaling Technology, 3700, 1:5000 dilution). Membranes were incubated with IRDye secondary antibodies (LI-COR, 926-68070 and 926-32211) and imaged using the Odyssey DLx imaging system (LI-COR).
Cell viability assay
2 × 104 HepG2 cells, cultured in EMEM or Plasmax for 96 h, were seeded in 96-well plates. The following day, cells were treated with 0, 250, 500, 750, 1000 or 1250 mM ethanol for 24 h. Total ATP was determined as a readout of cell viability using the luminesence-based CellTiter-Glo Assay (Promega) according to manufacturer’s instructions.
BODIPY staining and quantification
HepG2 cells, cultured in EMEM or Plasmax for 96 h, were seeded on coverslips coated with poly-L-lysine (Sigma-Aldrich). Two days after seeding, cells were fixed in 4% PFA for 10 min at room temperature and subsequently stained with BODIPYTM 493/503 (Thermo Fisher Scientific) and DAPI (Sigma-Aldrich) according to manufacturer’s instructions for 15 min in the dark. Cells were mounted on slides and imaged on an Olympus FV3000 confocal microscope. Images were collected from three biological replicates, acquiring five non-overlapping regions per replicate. Mean BODIPY staining intensity per cell was quantified using a CellProfiler58 pipeline.
Inductively coupled plasma mass spectrometry (ICP-MS)
ICP-MS was performed as previously described59. Cells were washed with PBS and whole cell lysates were collected in SDS lysis buffer. Media containing 10% FBS was used to determine trace element abundance in EMEM and Plasmax. Lysates or media were lyophilized prior to digestion in 65% nitric acid (Merck). Samples were heated at 90 °C for 20 min and subsequently diluted with 1% (v/v) nitric acid. Samples were run on an Agilent 8800 (ICPMS-QQQ-8800) system using a Helium reaction gas cell.
Proteomic analysis
Cells were washed with PBS and lysed in 200 µL of ice-cold RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 50 mM Tris-HCl, pH 7.5) containing a protease-inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitors (Roche). Protein was reduced by adding a final concentration of 10 mM TCEP (Sigma-Aldrich) and incubated at 37 °C for 45 min. Samples were then alkylated with 50 mM iodoacetamide (Sigma-Aldrich) and incubated in the dark for 45 min. After acidification with 2.5% phosphoric acid (Sigma-Aldrich), samples were passed through S-Trap micro columns (ProtiFi). Proteins were digested overnight at 37 °C with a 1:20 dilution of PierceTM Trypsin Protease (Thermo Fisher Scientific). Peptides were eluted sequentially with 50 mM TEAB (Sigma-Aldrich), 0.2% formic acid, and 50% acetonitrile/0.2% formic acid. Finally, the eluent was lyophilized and reconstituted in 2% acetonitrile/0.05% trifluoroacetic acid to make up the peptide solution for LC-MS/MS analysis by data-independent acquisition using a Thermo OrbiTrap Ascend mass spectrometer. Data analysis was performed using the software suite, DIA-NN60. Heatmaps and volcano plots were generated in R using the ‘pheatmap’ and ‘ggplot’ packages, respectively56,57.
Quantification and statistical analysis
Statistical analyses were performed with Prism 9 software (GraphPad Software). All statistical analyses for data comparing two groups were performed with an unpaired Student’s t test. One-way ANOVA with the Holm–Sidak method for multiple comparisons was used for comparison of more than two groups. All immunoblots are representative of results from at least three independent experiments. All other statistical details of experiments can be found in the figure legends.
Data availability
RNA-Seq datasets reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database (GSE287735 and GSE287739). Proteomic datasets reported in this paper have been deposited in the Proteomics IDEntifications Database (PXD060175). In addition, raw data from transcriptomics and proteomics analysis is included in the Extended Data file.
References
Davidson, S. M. et al. Environment impacts the metabolic dependencies of Ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016).
Muir, A. et al. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6, e27713 (2017).
Eagle, H. Amino acid metabolism in mammalian cell cultures. Science 130, 432–437 (1959).
Dulbecco, R. & Freeman, G. Plaque production by the polyoma virus. Virology 8, 396–397 (1959).
Vande Voorde, J. et al. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv. 5, eaau7314 (2019).
Cantor, J. R. et al. Physiologic medium rewires cellular metabolism and reveals uric acid as an endogenous inhibitor of UMP synthase. Cell 169, 258–272.e17 (2017).
Tardito, S. et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 17, 1556–1568 (2015).
Flickinger, K. M. et al. Conditional lethality profiling reveals anticancer mechanisms of action and drug-nutrient interactions. Sci. Adv. 10, eadq3591 (2024).
Apiz Saab, J. J. et al. Pancreatic tumors exhibit myeloid-driven amino acid stress and upregulate arginine biosynthesis. Elife 12, e81289 (2023).
Rawat, V. et al. Drug screening in human physiologic medium identifies uric acid as an inhibitor of rigosertib efficacy. JCI Insight 9, e174329 (2024).
Senkowski, W. et al. A platform for efficient establishment and drug-response profiling of high-grade serous ovarian cancer organoids. Dev. Cell 58, 1106–1121.e7 (2023).
Kaur, S. et al. In vitro models for the study of liver biology and diseases: Advances and limitations. Cell. Mol. Gastroenterol. Hepatol. 15, 559–571 (2023).
Yang, S., Ooka, M., Margolis, R. J. & Xia, M. Liver three-dimensional cellular models for high-throughput chemical testing. Cell Rep. Methods 3, 100432 (2023).
Aden, D. P., Fogel, A., Plotkin, S., Damjanov, I. & Knowles, B. B. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 282, 615–616 (1979).
Arzumanian, V. A., Kiseleva, O. I. & Poverennaya, E. V. The curious case of the HepG2 cell line: 40 years of expertise. Int. J. Mol. Sci. 22, 13135 (2021).
López-Terrada, D., Cheung, S. W., Finegold, M. J. & Knowles, B. B. Hep G2 is a hepatoblastoma-derived cell line. Hum. Pathol. 40, 1512–1515 (2009).
Liu, F. et al. Long non-coding RNA SNHG6 couples cholesterol sensing with mTORC1 activation in hepatocellular carcinoma. Nat. Metab. 4, 1022–1040 (2022).
Liu, Z. et al. Identification of targets of JS-K against HBV-positive human hepatocellular carcinoma HepG2.2.15 cells with iTRAQ proteomics. Sci. Rep. 11, 10381 (2021).
Lin, C.-Y. et al. Simultaneous induction of apoptosis and necroptosis by Tanshinone IIA in human hepatocellular carcinoma HepG2 cells. Cell Death Discov. 2, 16065 (2016).
Park, S. et al. A secretome profile indicative of oleate-induced proliferation of HepG2 hepatocellular carcinoma cells. Exp. Mol. Med. 50, 1–14 (2018).
Zhang, Z.-L. et al. Effect of PAK1 gene silencing on proliferation and apoptosis in hepatocellular carcinoma cell lines MHCC97-H and HepG2 and cells in xenograft tumor. Gene Ther. 25, 284–296 (2018).
Sanghvi, V. R. et al. The oncogenic action of NRF2 depends on De-glycation by fructosamine-3-kinase. Cell 178, 807–819.e21 (2019).
Chen, L. et al. Deep whole-genome analysis of 494 hepatocellular carcinomas. Nature 627, 586–593 (2024).
Jiang, X. et al. SLC7A14 imports GABA to lysosomes and impairs hepatic insulin sensitivity via inhibiting mTORC2. Cell Rep. 42, 111984 (2023).
Ong, A. J. S. et al. The KEAP1-NRF2 pathway regulates TFEB/TFE3-dependent lysosomal biogenesis. Proc. Natl. Acad. Sci. USA 120, e2217425120 (2023).
Parviz, F. et al. Hepatocyte nuclear factor 4alpha controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 34, 292–296 (2003).
Sekiya, S. & Suzuki, A. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 475, 390–393 (2011).
Merrell, A. J. & Stanger, B. Z. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 17, 413–425 (2016).
Katsuda, T. et al. Cellular reprogramming in vivo initiated by SOX4 pioneer factor activity. Nat. Commun. 15, 1761 (2024).
Copeland, P. R. Regulation of gene expression by stop codon recoding: selenocysteine. Gene 312, 17–25 (2003).
Addess, K. J., Basilion, J. P., Klausner, R. D., Rouault, T. A. & Pardi, A. Structure and dynamics of the iron responsive element RNA: Implications for binding of the RNA by iron regulatory binding proteins. J. Mol. Biol. 274, 72–83 (1997).
Ajioka, R. S., Phillips, J. D. & Kushner, J. P. Biosynthesis of heme in mammals. Biochim. Biophys. Acta 1763, 723–736 (2006).
Oyake, T. et al. Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 16, 6083–6095 (1996).
Ogawa, K. et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 20, 2835–2843 (2001).
Sun, J. et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 21, 5216–5224 (2002).
Wei, X. et al. Bach1 regulates self-renewal and impedes mesendodermal differentiation of human embryonic stem cells. Sci. Adv. 5, eaau7887 (2019).
Boon, R. et al. Amino acid levels determine metabolism and CYP450 function of hepatocytes and hepatoma cell lines. Nat. Commun. 11, 1393 (2020).
Kao, Y.-R. et al. An iron rheostat controls hematopoietic stem cell fate. Cell Stem Cell 31, 378–397.e12 (2024).
Zhuang, X. et al. Ageing limits stemness and tumorigenesis by reprogramming iron homeostasis. Nature 637, 184–194 (2025).
Müller, S. et al. CD44 regulates epigenetic plasticity by mediating iron endocytosis. Nat. Chem. 12, 929–938 (2020).
Aziz, N. & Munro, H. N. Iron regulates ferritin mRNA translation through a segment of its 5’ untranslated region. Proc. Natl. Acad. Sci. USA 84, 8478–8482 (1987).
Hentze, M. W. et al. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science 238, 1570–1573 (1987).
Shen, M., Goforth, J. B. & Eisenstein, R. S. Iron-dependent post transcriptional control of mitochondrial aconitase expression. Metallomics 15, mfac099 (2023).
Gray, N. K. & Hentze, M. W. Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J. 13, 3882–3891 (1994).
Zinoni, F., Birkmann, A., Stadtman, T. C. & Böck, A. Nucleotide sequence and expression of the selenocysteine-containing polypeptide of formate dehydrogenase (formate-hydrogen-lyase-linked) from Escherichia coli. Proc. Natl. Acad. Sci. USA 83, 4650–4654 (1986).
Chambers, I. et al. The structure of the mouse glutathione peroxidase gene: the selenocysteine in the active site is encoded by the “termination” codon, TGA. EMBO J. 5, 1221–1227 (1986).
Kryukov, G. V. et al. Characterization of mammalian selenoproteomes. Science 300, 1439–1443 (2003).
Kitamuro, T. et al. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J. Biol. Chem. 278, 9125–9133 (2003).
Chen, Y. et al. Oxidative stress induces mitochondrial iron overload and ferroptotic cell death. Sci. Rep. 13, 15515 (2023).
Wiel, C. et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 178, 330–345.e22 (2019).
Lignitto, L. et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 178, 316–329.e18 (2019).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44, W3–W10 (2016).
Smyth, G. K. limma: Linear Models for Microarray Data. in Bioinformatics and Computational Biology Solutions Using R and Bioconductor 397–420 (Springer-Verlag, New York, (2005).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Kolde, R. Pheatmap: pretty heatmaps. R package version (2012).
Wickham, H. Ggplot2: Elegant Graphics for Data Analysis. (Springer International Publishing, Cham, (2016).
Stirling, D. R. et al. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinforma. 22, 433 (2021).
Pyun, J. et al. Copper bis(thiosemicarbazone) complexes modulate P-glycoprotein expression and function in human brain microvascular endothelial cells. J. Neurochem. 162, 226–244 (2022).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020).
Acknowledgements
A.J.S.O., T.A.T. and A.J.G. are supported by Australian Government Research Training Program Scholarships. A.J.S.O. is also supported by a Peter MacCallum Cancer Centre Foundation Grant. K.K.B. is supported by National Health and Medical Research Council (NHMRC) Ideas Grants (GNT2004212 and GNT2012313) and a Victorian Cancer Agency Mid-Career Research Fellowship (MCRF17020). A.G.C. is supported by a NHMRC Investigator Grant (GNT1176650), and an Australian Research Council Discovery Project Grant (DP200102693). K.K.B and A.G.C. are also supported by the Peter MacCallum Cancer Foundation (Ted and Lila Seehusen Foundation). We acknowledge support from the Peter MacCallum Cancer Centre Foundation and the Australian Cancer Research Foundation. We acknowledge the Bio21 Melbourne Mass Spectrometry and Proteomics Facility (MMSPF) at the University of Melbourne and the Biometals Facility at the Florey Institute of Neuroscience and Mental Health, University of Melbourne for their training, support, and technical assistance. We extend our thanks to the Peter MacCallum Cancer Centre Core Facilities and their staff who provided support for this work; namely the Molecular Genomics Core (RRID:SCR_025695), the Flow Cytometry Core (RRID:SCR_025550), and the Research Laboratory Support Services Core (RRID:SCR_025699) Facilities. Finally, we thank members of the Cox Laboratory and Brown Laboratory (Peter MacCallum Cancer Center) for helpful discussions.
Author information
Authors and Affiliations
Contributions
A.J.S.O., A.G.C., and K.K.B. designed research; A.J.S.O., T.A.T., A.J.G. and J.M.R. performed research; A.J.S.O. analyzed data; A.G.C. and K.K.B. supervised the project; A.J.S.O., A.G.C., and K.K.B. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ong, A.J.S., Tigani, T.A., Gomes, A.J. et al. A multi-omic approach reveals iron availability influences cell fate fidelity. npj Metab Health Dis 4, 11 (2026). https://doi.org/10.1038/s44324-026-00102-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44324-026-00102-8
