
Abstract
Conventional histopathological analysis focuses on single-gene mutations and struggles to capture pathway-level dysregulation driving cancer. To address this, we propose LCG-HGNN, a Logic-Constrained Gene-Pathway Heterogeneous Graph Neural Network that enables collaborative recognition of gene groups and infers signaling pathway alterations from whole-slide images. By integrating a gene-pathway graph structure, dynamic edge weighting within our proposed KePathGraph framework, which also incorporates logical clauses, our framework achieves superior prediction accuracy and clinical interpretability over single-gene and multi-label baselines. This work establishes a pathway-oriented paradigm for histopathological interpretation, providing deeper insights into the mechanisms underlying cancer initiation and progression.
Similar content being viewed by others

Introduction
Cancer exhibits complex functional mechanisms, and conventional histopathological examination is often hard to determine specific molecular subtypes, necessitating genomic testing to guide personalized therapy1,2. Current genomic assays typically focus on single-gene alterations, yet carcinogenesis usually involves coordinated dysregulation of multiple genes that collectively drive abnormal activation or inactivation of signaling pathways3. Single-gene analysis, therefore, cannot systematically capture the intricate mechanisms of multi-gene cooperative regulation at the pathway level. Notably, gene group aberrations frequently induce dysregulation of cancer-related signaling pathways; for example, PI3K-AKT pathway dysfunction commonly co-occurs with mutations in TP53 and KRAS, among others. Such pathway-level abnormalities subsequently manifest as histopathological phenotypes in whole-slide images (WSIs)4.
Recent advances in deep learning have enabled indirect inference of genomic biomarkers from histopathological images. Initial approaches employed convolutional networks such as ResNet to detect abnormalities of individual genes5. To address multiple genes, some scholars use graph learning methods to independently predict gene mutations6,7. Subsequent studies adopted multi-label learning methods to jointly predict gene group alterations, recognizing the frequent co-occurrence of mutations across genes8. Recently published PEaRL also achieves the prediction of continuous pathway expression in spatial transcriptomics9 (details in Supplementary Information). However, existing methods treat genes as independent labels and hard to incorporate functional relationships within gene groups during prediction. This limitation leads to an incomplete understanding of coordinated gene dysregulation in cancer and hinders progress in pathway-level histopathological interpretation1,10.
Recent studies propose modeling inter-label relationships in multi-label learning to address functional dependencies among genes within a gene group, thereby connecting otherwise isolated gene predictions11. However, gene-gene interactions are highly complex and difficult to model accurately in a purely data-driven manner. Fortunately, molecular biology research establishes signaling pathways as central functional units of gene activity, with pathway dysregulation showing strong association with cancer histopathological phenotypes12,13. For example, computational pathway extraction is validated by distinct morpho-molecular drivers, such as PI3K-Akt-mTOR-induced solid adenocarcinomas and PI3K/SOX2-mediated squamous phenotypes14,15 (details in Supplementary Information). Analyzing gene group aberrations within the context of signaling pathways thus provides deeper insight into cancer progression16. Moreover, because fully automatic label association methods may yield associations of questionable clinical relevance, incorporating established biological and clinical knowledge as constraints is necessary to enhance the clinical interpretability and utility of prediction results17.
To better capture the relationship between signaling pathways and histopathological images, we propose Logic-Constrained Gene-Pathway Heterogeneous Graph Neural Network (LCG-HGNN), a graph neural network (GNN) based framework for collaborative gene group recognition. The model first constructs a hierarchical gene-pathway graph structure using pathway-gene associations from the DAVID database, thereby linking isolated gene-level predictions into a biologically coherent system. Second, a dynamic edge weighting mechanism subsequently adjusts connection strengths during graph convolution according to gene-level prediction errors. The mechanism down-weights low-confidence genes to suppress noise and up-weights clinically validated lung cancer driver genes from the Cancer Gene Census to align with established molecular oncology knowledge. Third, the framework incorporates biological priors through KePathGraph, a logic clause-based constraint module that enforces established rules such as mutual exclusivity (e.g., EGFR and KRAS) and pathway hierarchy (e.g., PI3K activation downstream of EGFR), ensuring predictions conform to known molecular mechanisms. This integration enhances both the accuracy and clinical interpretability of pathway-level inference. The main contributions of this work are as follows:
(1) The framework overcomes the limitation of traditional single-gene prediction by enabling collaborative recognition of gene groups, thereby capturing gene-gene interactions and their integrated effects in complex biological processes, and offering a more comprehensive perspective on gene group-driven disease mechanisms.
(2) We further extend the analytical scope to signaling pathway-level alterations, allowing the model to uncover higher-order biological functions and regulatory networks, and thus providing deeper insights into the mechanisms underlying cancer initiation and progression.
(3) LCG-HGNN incorporates logical clauses into KePathGraph to encode prior knowledge about relationships among genes, between genes and pathways, and across patient cases, using this structured biological information to constrain and refine pathway recognition in histopathological samples, thereby enhancing both clinical relevance and predictive performance.
Results
Overall framework structure
Although existing methods can predict mutations of gene group from histopathological image, the resulting predictions often hard to reveal the underlying mechanisms within signaling pathways. To accurately capture the associations between phenotypic characteristics observed in pathology images and signaling pathways, we propose a gene-pathway co-identification framework, termed LCG-HGNN, based on GNN. This approach simultaneously models the co-variation of gene functional groups and pathway-level regulatory dynamics.
By integrating genomic and signaling pathway data, the GNN utilizes multi-level message passing mechanisms to dynamically aggregate node features, thereby capturing functional synergy among genes within signaling pathways. The model employs an adaptively weighted Gene-Pathway Heterogeneous Graph Structure, enabling deep fusion of multi-gene mutation predictions with pathway-level logic. This design allows pathway-level predictions to be dynamically optimized according to the mutational states of individual genes.
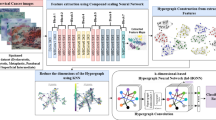
Furthermore, gene nodes are interconnected via edges that enforce mutual constraints, mitigating biases inherent in single-gene prediction paradigms. As a result, the model produces pathway abnormality predictions that are both biologically plausible and consistent with observed histopathological phenotypes. To ensure biologically consistent pathway predictions, we introduce a logic constraint module that encodes pathway rules into logical clauses, imposing structured constraints on gene-level predictions to align with established biological knowledge. The schematic framework structure is shown in Fig. 1, and overall pipeline of the procedure is in Fig. S1.

(Left) Pathology patches form a graph for gene and pathway prediction. (Middle) WSI features and similarity-basededges are constructed. (Bottom-left) Biological priors are converted into logical clauses. (Right) Logical constraintsenhance the pathway prediction network.
Data collection
To associate genes with their corresponding histopathological images, we utilize 1608 lung adenocarcinoma (LUAD) WSIs from TCGA with genomic mutation data from cBioPortal18. Patient-specific TCGA barcodes were use to link these datasets, enabling multi-omics correlation. This integration allows systematic exploration of the associations between genomic features and histopathological phenotypes in LUAD.
To enable gene group-based functional analysis of pathways, we further aggregated pathway annotations from four major databases: KEGG, Reactome, WikiPathways, and BioCarta, via the DAVID platform19,20. Using the top 100 most frequently mutated LUAD-associated genes as a filter, we identified 969 significantly enriched pathways (see Table 1 for details). This prioritization helps focus on the signaling pathways most relevant to LUAD.
Experimental design
To evaluate the impact of the LCG-HGNN framework on classification performance, we established a multi-dimensional assessment system based on the TCGA-LUAD21 cohort. The framework is evaluated in terms of its ability to identify individual gene mutations as well as to analyze gene groups. Specifically, we compared LCG-HGNN with single-gene binary classification methods and multi-gene multi-label classification approaches. For the single-gene comparison, we use ResNet-185 and spatially aware GNN6 to assess whether pathway-aware modeling improves upon conventional gene-level prediction. For the multi-label comparison, we employed DAMLN8 and ADD-GCN11 to evaluate LCG-HGNN’s ability to capture functional dependencies among genes within signaling pathways. In these experiments, the scalability and computational cost of our model are provided in Supplementary Information.
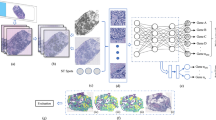
Here, we compare single-gene predictions with gene group analysis. Gene phenotypes are often associated with the combined effects of multiple genes. To achieve prediction results that better align with real-world biological manifestations, we propose a GNN-based gene group co-analysis pipeline within the LCG-HGNN framework. This framework models feature interactions in histopathological images using GNN methods and constructs a Gene-Pathway Heterogeneous Graph Structure based on inter-group relationships among signaling pathway-associated gene groups (see Fig. 2 for the detailed architecture). The predictions integrate pathway prior knowledge to assist in gene group inference and simultaneously yield preliminary insights into signaling pathway dysregulation.

A GNN-based signaling pathway identification framework: feature interactions in histopathological images are modeled using GNN methods, and a Gene–Pathway Heterogeneous Graph Structure is constructed based on inter-group relationships among signaling pathway-associated Gene groups, with prior knowledge leveraged to assist in the prediction of pathways and genes.
To evaluate the actual performance gain of the LCG-HGNN framework in single-gene identification, we compare LCG-HGNN against both traditional CNN-based single-gene classification models and GNN-based single-gene classification approaches5,6. More precisely, we select ResNet-18 as a representative of conventional CNN architectures. For the GNN-based single-gene classification comparison, we adopt a spatially aware GNN, which performs classification of genomic biomarkers via weakly supervised graph learning. The single-gene baseline models are summarized below.
We compared our framework against the following single-gene baseline models:
ResNet-18: As a representative traditional CNN, ResNet-18 effectively captures local spatial patterns in histopathological images and evaluates the feature representation and analysis capability of traditional CNNs under gene group settings5;
Spatially aware GNN: The model adopts a weakly supervised GNN as the backbone and constructs spatial adjacency relationships among tumor patches using geometric coordinates. This model enables effective validation of predictive discrepancies under gene group settings6.
We establish specific evaluation metrics for single-gene classification. To validate the LCG-HGNN framework, we define a set of evaluation metrics tailored to different task requirements. For single-gene classification tasks, we primarily adopt Accuracy (ACC) and Area Under the Receiver Operating Characteristic Curve (AUC) as core performance indicators, which assess the model’s ability to distinguish between positive and negative samples in binary classification settings. ACC evaluates overall single-gene classification performance by reflecting the proportion of correctly classified samples. AUC assesses model performance under class imbalance, quantifying discriminative ability across varying gene mutation rates22.
Here, we analyze gene groups and signaling pathways.
Pathways organize gene groups into functional units that reflect how genes cooperatively regulate cellular processes. Accurate modeling of gene group co-expression patterns is critical for uncovering disease mechanisms in pathway-level functional analysis. The LCG-HGNN framework first constructs a graph structure from predictions across multiple WSIs based on inter-case relationships. It then applies a knowledge enhancement module to inject logical prior knowledge after graph convolution, thereby imposing constraints on the predictions of both pathways and genes (see Fig. 3 for the detailed architecture)23,24.

The pipeline first constructs a graph structure from predictions across multiple WSIs based on inter-case relationships. A knowledge enhancement module then applies logical prior knowledge after graph convolution to impose constraints on the predictions of both signaling pathways and gene groups.
To better evaluate the performance of the proposed LCG-HGNN framework on the gene group (gene functional cluster) identification task, we designed a three-tier comparison and validation strategy: (1) performance comparison with non-graph-based and graph-based multi-label learning methods; (2) systematic evaluation of the adaptability of different graph learning architectures in pathway analysis tasks; (3) ablation experiments on key components of the proposed gene-pathway heterogeneous graph structure to verify the necessity and biological plausibility of the design.
We compare LCG-HGNN with two categories of multi-label classification frameworks.
Non-graph model: DAMLN is a weakly supervised multi-label model with dual attention for modeling feature importance and inter-label dependencies in gene group prediction8. Gene-DML predicts gene expression from histopathology images via dual-pathway multi-level discrimination, ensuring robust cross-modal alignment.
ADD-GCN model: ADD-GCN combines weakly supervised dynamic graph learning and attention to capture co-occurrence patterns among genes without a fixed topologies11.
To further assess the adaptability of different GNN architectures in pathway-level analysis, we conduct a systematic comparison of various graph learning methods in gene group identification. We evaluate the following four architectures under the same training protocol.
Graph Convolutional Networks (GCNs): GCNs use spectral convolution for smooth local aggregation, making it suitable for metabolic pathways with gradual signal propagation25.
Graph Attention Networks (GATs): GAT dynamically assigns attention weights to adjacent nodes within the graph structure, capturing heterogeneous regulator effects in signaling pathways26.
Graph Isomorphism Network (GINs): GIN employs MLP-based aggregation with Weisfeiler-Lehman expressivity to model complex gene regulatory interactions27.
Graph Sample and Aggregate (GraphSAGE): GraphSAGE combines neighbor sampling and LSTM aggregation to preserve topological fidelity in large-scale pathway graphs28.
We further validate the biological and architectural necessity of our gene-pathway heterogeneous graph through two targeted ablations.
Gene-pathway heterogeneous graph structure ablation: Our proposed gene-pathway heterogeneous graph enables joint reasoning between signaling pathways and associated genes, facilitating the identification of pathway-level abnormalities. To evaluate whether this structure truly aligns with biological pathway concepts, we perform an ablation study by replacing the graph module with a standard multi-label classification head.
Dynamic edge weighting mechanism ablation: Given that different gene mutations contribute unequally to signaling pathway activity, our framework introduces a dynamic edge weighting mechanism to quantify each gene’s influence on pathway predictions. To assess whether this mechanism reflects true biological significance, we conduct an ablation experiment by fixing all edge weights to 1, thereby removing the adaptive weighting capability.
We establish specific evaluation metrics for gene-group classification. When extended to the gene group prediction setting, the evaluation focuses on the overall performance of multi-label classification. We measure model performance using three metrics: Hamming loss, Mean Accuracy (Mean ACC), and Mean Area Under the ROC Curve (Mean AUC). Mean ACC and Mean AUC are computed by averaging per-gene performance to assess overall discriminative capability across parallel gene recognition tasks22. Hamming loss measures the average fraction of incorrectly predicted gene labels, reflecting the overall accuracy of gene group identification as cooperative functional units29.
Here, we evaluate the pathway logical constraint module. To analyze the impact of the pathway logical clause prior constraint module on LCG-HGNN, we perform an ablation study on the logical constraint component. This is achieved by comparing the pathway abnormality predictions with and without the logical constraints, thereby evaluating the effectiveness of the module in refining pathway-level predictions.
Due to the absence of a gold standard for pathway abnormality, we evaluate by comparing the similarity between predicted images and clinical histopathological images associated with gene group abnormalities in the corresponding pathways. Specifically, clinical histopathological images related to gene group abnormalities are collected from the Human Protein Atlas (HPA) database30. The predicted histopathological images and the HPA images are both input into a pretrained ResNet for feature extraction. The Euclidean distance between the extracted feature vectors is used to measure similarity and assess whether the predicted images conform to clinical standards.
In EGFR mutation prediction (Fig. 5), the pathway logical clause prior constraint module produces patches with the highest similarity to clinical references, exhibiting acinar morphology and bland nuclei strongly linked to EGFR mutations31,32. The lowest-similarity patch exhibits round nuclei and fine chromatin, features consistent with mucinous LUAD rarely associated with EGFR mutations33.
Comparison experiment
To evaluate the performance of our cancer signaling pathway-associated identification model in gene group identification, we conduct experiments using pathway-related graph-structured data constructed from the TCGA-LUAD dataset. Our proposed framework is compared with two categories of baseline methods: (1) single-gene classification approaches, including traditional deep learning models and spatially aware GNN, and (2) multi-label classification approaches, covering both non-graph-based and graph-based models.
In this study, we predict gene groups associated with specific signaling pathways in LUAD based on histopathological features extracted from WSIs. We focus on identifying abnormalities in the following three signaling pathways: (1) hsa04151: PI3K-Akt signaling pathway predicted gene group: TP53, KRAS, TNR, RELN, STK11, and EGFR, totaling 6 genes; (2) hsa04512: ECM-receptor interaction, predicted gene group: TNR, RELN, LAMA2, and COL6A3, totaling 4 genes; (3) R-HSA-8948216: Collagen chain trimerization, predicted gene group: COL11A1, COL22A1, and COL6A3, totaling 3 genes.
Here, we compare our framework with traditional deep learning models. To examine the differences in predictive performance between our cancer signaling pathway-associated identification model and conventional single-gene approaches, we conduct comparisons using gene groups as prediction labels. We first compare the gene group identification capability of our model with that of traditional CNN-based methods. In this comparison, we use ResNet-18 as a baseline model for gene group prediction.
To comprehensively assess the transition from single-gene modeling to gene group-level inference, we compare our model’s performance with ResNet-18 models individually fine-tuned on each gene-specific task. Our framework demonstrates significant performance improvements over ResNet-18 across multiple gene group identification tasks associated with different signaling pathways (See Fig. 4F for details).

Three components are ablated: the graph convolution operator, the gene-pathway heterogeneous graph structure, and the dynamic edge weighting mechanism within the heterogeneous graph. A–D Shows the ROC curves of the ablation variants on gene groups associated with the hsa04512 pathway; E presents the overall performance across all ablation settings. The results collectively demonstrate that the proposed components positively contribute to the framework’s performance; F shows the performance comparison of the LCG-HGNN framework (with GIN as the graph learning backbone) against current baseline methods on the gene functional cluster identification task. The height of each bar represents prediction performance, with higher values indicating better results. The plot shows that the proposed framework achieves overall superior identification performance.
In particular, for the hsa04512 (ECM-receptor interaction) related gene group identification task, our model achieves a 37.3% increase in Mean ACC and an 8.13% improvement in Mean AUC, compared to the ResNet-18 baseline. More detailed comparison results are presented in Table 2 and S1.
Here, we compare our approach with GNN-based single-gene identification methods. Predicting single-gene status using GNN enables better modeling of the spatial and contextual relationships among different regions in histopathological images, which aligns more closely with the design of our proposed identification framework. To compare the gene group prediction performance of our cancer signaling pathway-associated identification model with that of GNN-based single-gene identification methods, we retraining a spatially aware GNN individually for each gene.
The experimental results in Fig. 4F show that our model outperforms the spatially aware GNN baseline by achieving a 15.90% improvement in Mean ACC and a 10.19% improvement in Mean AUC, demonstrating its superior ability in capturing functional gene group patterns from histopathological images. Detailed comparison results are summarized in Table 2 and S1.
The above comparative experiments demonstrate that the proposed method outperforms existing single-gene prediction approaches in gene group identification tasks. The results confirm that the graph-based model effectively leverages functional associations within signaling pathways rather than relying solely on image features of individual genes. By incorporating gene-gene interdependencies as biological priors, the framework enhances both gene-level prediction accuracy and histopathological interpretability, thereby improving multi-label analysis at the pathway level.
Here, we compare our model with non-graph gene group identification methods. In real biological systems, phenotypes are typically governed by the cooperative effects of gene groups rather than by a single gene in isolation. Prediction based solely on individual genes fails to capture functional synergies among genes, prompting a comparison with multi-label learning approaches. We select DAMLN and Gene-DML as the baseline multi-label models, as they are specifically designed for histopathological image analysis.
In gene group evaluation, the cancer signaling pathway-associated identification model demonstrates clear advantages over DAMLN: performance is comparable in Mean ACC and Hamming loss, while Mean AUC improves by 22.57% on the hsa04512 pathway recognition task (see Table 2 and Fig. 4F). Compared to Gene-DML, LCG-HGNN demonstrates superior predictive accuracy, achieving a significantly higher Mean AUC of 0.9816 versus 0.7623. These observations indicate that the LCG-HGNN framework achieves more robust prediction under severe class imbalance in genomic data and is less affected by the skewed distribution of positive and negative samples.
Here, we compare our framework with the ADD-GCN graph-based multi-label baseline. Label dependencies among gene groups significantly influence the performance of multi-label recognition in pathway analysis. To account for this, we adopt ADD-GCN as a representative graph-based multi-label baseline that explicitly models inter-label relationships for gene group identification. Our LCG-HGNN framework uses a gene-pathway heterogeneous graph structure to explicitly encode functional relationships among genes.
Under the same training protocol, the LCG-HGNN framework achieves superior gene group recognition performance compared with ADD-GCN. On the hsa04512-related task, it improves by 40% relative to ADD-GCN in Mean ACC, Mean AUC, and Hamming loss (see Table 2 and Fig. 4F). This gain likely stems from the severe class imbalance in gene group data. The absence of pathway-level biological priors in ADD-GCN leads to less accurate feature learning and reduced discriminative capacity.
Ablation experiment
To evaluate the impact of different components of the LCG-HGNN framework on signaling pathway abnormality identification, we design three types of ablation experiments to assess the contribution of the following key elements: (1) the Effect of graph learning method selection on gene group identification performance; (2) the impact of the gene-pathway heterogeneous graph structure on prediction performance; (3) the enhancement of dynamic edge weight mechanism within the proposed heterogeneous graph structure for gene group identification. All experiments are conducted on the TCGA-LUAD dataset, with fixed hyperparameters across settings. The experiments are implemented using NVIDIA RTX A6000 GPU hardware, and the results are presented in Fig. 4A–E.
Here, we verify the gene-pathway heterogeneous graph structure. To verify whether the gene-pathway heterogeneous graph structure genuinely aligns with the biological pathway concept and effectively enhances identification performance, we directly ablate this structural component. After removing the gene-pathway heterogeneous graph from the framework, the gene group prediction capability of LCG-HGNN further declines.
In the relatively well-performing hsa04151 identification task, the three evaluation metrics of mean ACC, mean AUC, and Hamming loss are lower than those of the LCG-HGNN framework based on the GIN graph convolution operator. Detailed results are presented in the “AS_Graph” section of Table 2. This result shows that the gene-pathway heterogeneity graph structure is useful for associating pathway-related gene groups, and the association between gene groups and pathways can effectively improve the recognition accuracy.
Here, we validate the dynamic edge weight mechanism. In order to confirm that the dynamic learning process of weights has a positive impact on the LCG-HGNN framework, we replace the learnable weights with fixed uniform values (set to 1 in this case, indicating no weight modulation) and conduct ablation studies based on the GIN graph convolution operator.
In the hsa04512 pathway identification task, the use of fixed values leads to a decrease of 6.21% in Mean ACC and 3.85% in Mean AUC. This result indicates that static weights are difficult to adapt to the specific characteristics of gene nodes and signaling pathways. Detailed results are shown in the “AS_Weight” section of Table 2.
Here, we compare different graph learning architectures. To understand the impact of different graph learning methods on our constructed signaling pathway identification framework using histopathological images, we further analyze the differences in gene group recognition performance associated with the choice of graph operator. We change the structure of the core graph operator in the LCG-HGNN framework to compare the effectiveness of four graph learning methods, GAT, GIN, GCN, and GraphSAGE, for this task. In this ablatio, only the graph operator is modified. The results show that, except for GCN, GAT, GIN, and GraphSAGE variants, achieve good performance across different tasks. Specifically, in the hsa04512 pathway identification task, all three methods achieve a Mean AUC above 85%. Detailed results are presented in Table 2.
Because GraphSAGE randomly samples a fixed number of neighbors, it significantly reduces computational complexity in the histopathological image graph structure we constructed, thereby facilitates efficient feature interaction and fusion among nodes. This graph learning approach achieves a Mean AUC of 90.71%, with high computational efficiency, reasonable training time, and manageable memory usage.
The GAT method leverages an attention mechanism to assign distinct attention weights to neighboring nodes in the histopathological image graph structure. This approach is particularly suitable for high-precision multi-label graph classification tasks and achieves the highest Mean AUC of 98.16% in the hsa04512 pathway identification task. However, its high computational and memory requirements pose practical limitations in this study.
When using the GIN method, the Mean AUC reaches 85.18%. This is attributed to its strong ability to distinguish subtle differences in graph structures, enabling it to effectively capture complex phenotypic patterns under various genetic alterations. Given that each gene mutation phenotype is adequately represented by the graph structure, GIN can detect abnormalities by identifying minor structural variations within the graph.
Furthermore, we assess the impact of knowledge logical clauses. Following gene group prediction, the proposed framework leverages the gene-pathway heterogeneous graph structure together with pathway knowledge logical clause constraints in the KePathGraph to generate biologically plausible pathological image patches reflecting signaling pathway abnormalities. To investigate the impact of the pathway knowledge logical clause constraints on pathway-related pathological image recognition, an ablation is performed by removing this constraint module. In this ablation, feature vectors of clinical histopathological images (associated with gene group alterations in the target pathway) and those of the recognized image patches are extracted using a pretrained ResNet, and their similarity is quantified via Euclidean distance to evaluate recognition fidelity.
The recognition results collectively demonstrate that the KePathGraph constraint framework effectively guides the model to focus on biologically meaningful, pathway-relevant histopathological features. We illustrate how clauses influence prediction in Figs. S2 and S3 (details in Supplementary Information). As shown in Fig. 5, the first three columns present patches generated under logical constraints, whereas the last three show unconstrained predictions; a clear distinction emerges in the distribution of high-similarity (red) and low-similarity (blue) patches. In KRAS mutation prediction, high-similarity patches under logical constraints consistently exhibit KRAS mutant LUAD features such as solid or micropapillary structures, marked nuclear pleomorphism, and necrosis33. In contrast, low-similarity patches display well-differentiated, architecturally regular glands, suggesting low KRAS mutation likelihood. This contrast validates the efficacy of KePathGraph in pathway-level abnormality detection34.

The left panel shows clinical reference images with their corresponding mutated genes. The right panel displays the identified histopathological patches along with their similarity scores. In the right panel, red indicates high similarity to the clinical reference images, and blue indicates low similarity. Clinical reference images in the left panel are adapted from the Human Protein Atlas (available at proteinatlas.org) and used under the Creative Commons Attribution-ShareAlike 4.0 International License (CC BY-SA 4.0).
Further evidence comes from STK11 and TP53 mutation predictions, which confirm the framework’s generalizability. High-similarity STK11 patches show solid growth, clear cytoplasm, TTF-1 negativity, and a “cold tumor” microenvironment, features strongly associated with STK11 loss35,36. While low-similarity patches correspond to lepidic or acinar subtypes where STK11 mutations are rarely observed36. Similarly, in TP53 prediction, high-similarity patches display pronounced nuclear atypia, abundant mitotic figures, and necrosis, whereas low-similarity patches exhibit uniform nuclei and orderly glandular structures, consistent with TP53 wild-type status37. To quantitatively assess the differences in Fig. 5, we further conduct statistical test on their features, yielding significance distinctions across these visualized patches (details in Supplementary Information). These consistent observations across multiple gene groups indicate that KePathGraph not only performs well in single-mutation scenarios but also systematically enhances the model’s ability to recognize pathological patterns driven by coordinated gene group alterations within signaling pathways, thereby strongly supporting the central claim that biologically informed constraints improve histopathological interpretation.
To further demonstrate the correlation with transcriptomic-level pathway scores, we applied the Z-score mean method to calculate scores for gene groups. Both transcriptomic and predicted scores were normalized to a \(-1\) to 1 range, with values exceeding 0 defining abnormality. As illustrated in Fig. S4, our method’s predictions for the PI3K-Akt pathway show strong consistency with transcriptomic scoring. Additionally, the overlap analysis in Fig. S5 reveals a substantial 82.6% intersection between our predictions and transcriptomic abnormalities. These results confirm that the morphological features captured by LCG-HGNN are highly correlated with transcriptomic dysregulation, establishing our model as a robust proxy for molecular pathway activity (details in Supplementary Information).
Discussion
We propose LCG-HGNN, a GNN-based framework for joint gene-pathway recognition that overcomes the limitation of conventional histopathological analysis, which focuses on single-gene prediction. By constructing a gene-pathway heterogeneous graph, the model jointly models cooperative effects among multiple driver genes and directly infers signaling pathway dysregulation. Its core advantages are threefold: (1) it achieves the first collaborative recognition of gene groups from pathology images, aligning with the pathway-driven nature of cancer; (2) it elevates analysis to the pathway level, enabling deeper mechanistic insights into carcinogenesis; and (3) it integrates biological priors via logical clauses, enhancing clinical relevance and prediction consistency. Experiments confirm that LCG-HGNN significantly outperforms single-gene and multi-label baselines across multiple pathway-related tasks, validating the pathway-oriented modeling paradigm.
To assess clinical plausibility, we compare LCG-HGNN-predicted pathological regions with established clinical findings, focusing on the PI3K-Akt pathway (hsa04151). Abnormal activation of this pathway drives increased proliferation, invasion, and metastasis, manifesting histologically as large irregular nuclei, poor differentiation, and disorganized tissue architecture, features evident in the “Clinical Case” panel of Fig. 6. The model’s high-similarity patches under logical constraints closely match these characteristics (Fig. 6, “After logical knowledge constraints”), demonstrating that LCG-HGNN effectively captures pathway-relevant pathological phenotypes.

The “After logical knowledge constraints” panel shows histopathological images identified with logical clause constraints applied. The “No logical knowledge constraints” panel shows histopathological images identified without logical clause constraints. The “Clinical Case” panel shows a clinical histopathological image case with mutations in PI3K-Akt pathway–related genes. Images in the “Clinical Case” panel are adapted from the Human Protein Atlas (available at proteinatlas.org) and used under the Creative Commons Attribution-ShareAlike 4.0 International License (CC BY-SA 4.0).
Ablation experiments evaluate three key components: the gene-pathway heterogeneous graph, dynamic edge weighting, and logical knowledge constraints. Removal of any component causes significant performance degradation, confirming that the embedding of pathway-level biological priors is essential for modeling functional gene-pathway membership and capturing heterogeneous contributions of driver genes. Further analysis also confirms the model robustness across different resolutions, and other potential confounding factors such as tissue processing differences or staining variation (details in Supplementary Information). Together, they ensure the reliability of the predictions in both quantitative performance and biological plausibility.
The dynamic edge weighting mechanism adaptively modulates each gene’s influence on pathway output based on prediction confidence, suppressing noise from low-confidence genes. It further incorporates prior knowledge from the Cancer Gene Census (CGC)38, assigning higher initial weights to clinically validated lung cancer drivers. Given that pathway function is inherently coupled with the activity of key driver genes (details in Supplementary Information), this enables accurate representation of their functional dominance even at low mutation frequencies, where driver genes are explicitly modeled but dynamically regulated to prevent prediction dominance. Through a dual-channel strategy of “error-driven adjustment + knowledge-guided weighting,” the model balances data-driven flexibility with biological fidelity, significantly improving robustness and interpretability of pathway dysregulation inference.
Despite strong performance, LCG-HGNN has limitations in data scale and clinical validation. It is trained on 1608 TCGA-LUAD WSIs, covering only high-frequency driver events, and thus may miss rare or low-frequency alterations. Prospective validation is hindered by multi-center data access and ethical constraints, and the absence of a gold standard for pathway dysregulation prevents direct correlation with treatment response or survival outcomes. Current evaluation relies on indirect image similarity to public references. Future work requires external validation in larger, multi-cancer, multi-center cohorts and integration of longitudinal clinical data to enable real-world clinical decision support.
Methods
Data situation
To overcome the limitations of isolated single-gene analysis and improve label reliability, we integrated 1608 WSIs from TCGA with LUAD genomic mutation data from cBioPortal, supplemented by clinically validated cancer-associated variants from the CGC. Gene-pathway associations were derived from four databases (KEGG, Reactome, WikiPathways, BioCarta) via DAVID, where 969 significantly enriched pathways were selected based on the top 100 mutation-frequent gene groups to facilitate identification of core genomic alteration-driven pathways. Using CGC as a prior knowledge source to prevent potential model divergence and improve biological interpretability, we curated a set of 35 high-confidence driver genes (e.g., EGFR, KRAS, ALK, detailed in Table 3), covering key genomic alteration types in LUAD, including point mutations, gene fusions, and copy number amplifications.
By integrating functional annotations (oncogenic or tumor-suppressive roles) and clinical evidence, we construct a refined driver gene group. We provide detailed download instructions, dataset versions, and matching criteria for all primary sources, alongside standardized preprocessing steps for multi-omics data and images to ensure reproducibility (details in Supplementary Information). This gene set serves as a biologically informed, high-specificity knowledge base to guide the subsequent histopathology-signaling pathway association modeling.
Ethics statement
This study exclusively used publicly available, de-identified data. WSIs and associated genomic profiles for LUAD were obtained from TCGA, accessed through the Genomic Data Commons and cBioPortal. Clinical annotations and mutation calls were used in accordance with TCGA data use policies; no informed consent or institutional review board approval was required, as all data were anonymized and originally collected under IRB-exempt protocols approved by the NIH Office of Human Subjects Research Protections.
Clinically validated lung cancer driver genes were sourced from the Catalogue of Somatic Mutations in Cancer CGC; pathway annotations were retrieved from the DAVID Bioinformatics Database, which aggregates KEGG, Reactome, WikiPathways, and BioCarta. Reference histopathological images for pathway–mutation associations were obtained from the Human Protein Atlas, which uses tissue samples from consented donors and releases data under the Creative Commons CC-BY 4.0 license for non-commercial research. This work involved no human or animal experimentation, no new data collection, and no re-identification of individuals.
Data preprocessing method
To uncover the spatial phenotypic features of signaling pathways, we transform WSIs into graph-structured data connections based on morphological similarity and spatial proximity. Each WSI is segmented into 512×512 pixel patches (details in Supplementary Information), with morphological features extracted via an ImageNet-pretrained ResNet18 as node representations. To address the limitation of analyzing gene functional phenotypes across individual patches, we compute Euclidean distances between node features and connect each node to its top-10 nearest neighbors. This process generates WSI-level phenotypic clusters of morphological similarity, enabling a comprehensive representation of tissue architecture for downstream pathway-associated pattern recognition.
Graph data construction
In order to preserve the spatial heterogeneity of the tumor microenvironment, we incorporate spatial distance as a criterion for edge construction. Spatial distances are calculated based on patch coordinates, and an edge is established if the distance is less than a predefined threshold of 85 (details in Supplementary Information). By integrating edges based on morphological similarity and spatial proximity, the resulting hybrid graph representation effectively captures the spatial organization of organizational forms, serving as the foundational input for subsequent GNN-based pathology-pathway association modeling (details in Supplementary Information).
Pathway identification framework
To accurately establish the association between phenotypic characteristics in histopathological images and signaling pathways, the identification of gene groups serves as a critical first step. Instead of identifying individual genes in isolation, the pathway identification process should simultaneously recognize multiple genes, thereby enabling a comprehensive assessment for potential pathway prediction.
To achieve this objective, we design a network framework capable of jointly identifying gene group information and subsequently integrating this knowledge to infer pathway activity. The proposed framework comprises two main components: (1) a pathway and its corresponding gene group prediction network, and (2) logical clause constraints derived from multi-source data.
From single-gene analysis, we transition to gene group modeling. To address the limitation where early methods were limited to uncovering functional associations at the single-gene level, we propose a gene group co-analysis framework that enables simultaneous modeling of multi-gene interactions. We curate a refined set of LUAD-related genes\({{\mathcal{G}}}_{{list}}\) as identification labels and targets, integrating authoritative function annotations from the CGC database with TCGA mutation data using the following formulation:
where \({{\mathcal{G}}}_{\mathrm{CGC}}\) denotes the set of lung cancer-related genes selected from the CGC database, \({{\rm{N}}}_{\text{mut}}\left({{\rm{g}}}_{{\rm{i}}}\right)\) represents the number of samples in which gene \({g}_{i}\) is mutated within the cohort, and \({{\rm{N}}}_{\text{total}}\) indicates the total number of samples (e.g., TCGA-LUAD, N = 585). The operator \(\text{rank}(\cdot )\) refers to the mutation frequency ranking process, which selects the top 5 most frequently mutated genes. Additionally, \({{\mathcal{P}}}_{{\mathcal{j}}}\) signifies the full set of genes associated with the \({\rm{J}}\) pathway, and \({\rm{J}}\) denotes the total number of pathways considered in the analysis. This formulation ensures that the selected gene groups are both biologically meaningful and statistically representative of real-world genomic variation.
To enable cross-level modeling, we enhance the GIN-based network by introducing a hidden layer, Dropout, and skip-connection structures while extending the classification head into a multi-label framework. To address the long-tail distribution, we implement a mutation rate-guided stratified sampling strategy with a bidirectional elastic oversampling mechanism. This mechanism constrains the majority class sample size within 1.5 times that of the minority class and oversamples insufficient labels to a preset base number of 50. This strategy achieves cross-label balance while preserving the correlation structure among gene expressions, successfully extending the model into a multi-gene co-analysis network.
From gene group interactions, we extend the analysis to signaling pathways. Gene-Pathway Heterogeneous Graph Structure Discrimination: To infer pathway dysregulation by analyzing relationships among genes within the same pathway, we construct a gene-pathway heterogeneous graph that integrates gene group information. This process involves making comprehensive inference on pathway-level abnormalities by operating on the combined pre-activation values of individual genes output by the multi-gene co-analysis network. Specifically, we utilize pathway-gene membership information from the DAVID platform, aggregated across its constituent databases, to connect gene nodes \({{\mathcal{V}}}_{{\mathcal{G}}}=\{{{\rm{g}}}_{1},{{\rm{g}}}_{2},\ldots ,{{\rm{g}}}_{{\rm{N}}}\}\) and pathway nodes \({{\mathcal{V}}}_{{\mathcal{P}}}\) via membership edges, thereby constructing a gene-pathway heterogeneous graph structure. Gene node features are initialized as pre-activation values \({{\rm{a}}}_{{\rm{g}}}\in {{\rm{R}}}^{{\rm{d}}}\), while pathway node features are initialized to zero: \({{\rm{h}}}_{{\rm{p}}}^{\left(0\right)}=0\). During training, a GCN convolution layer propagates and aggregates information across the heterogeneous graph, iteratively updating node representations to produce the final pathway-level predictions \({{\rm{h}}}_{{\rm{p}}}\).
where \({{\rm{w}}}_{{\rm{g}}{\rm{p}}}\) is the initial edge weight based on DAVID annotations, and GCN denotes the information aggregation operation of the graph convolutional network.
Additionally, we design a dynamic edge weight refinement mechanism that adaptively updates edge weights in the graph during model training based on the discriminative error of each gene, \({\rm{e}}=|{\rm{predictor\; score}}-{\rm{label}}|\) (details in Supplementary Information). However, certain cancer driver genes show strong association with pathway dysregulation despite low mutation frequencies; mutation-based discrimination alone therefore underestimates their contribution.
Therefore, we propose a CGC database-based edge weight refinement strategy, which adjusts the gain coefficient \({{\rm{\gamma }}}_{{\rm{gp}}}\in {{\rm{R}}}^{\left|{\mathbb{E}}\right|}\) to double the edge weights of CGC-annotated lung cancer genes compared to non-priority genes. Building on this, we integrate error-driven updates with biological prior constraints into a unified edge weight update rule:
where λ = 0.03 controls the strength of preservation of biological priors. This mechanism operates via dual-channel constraints: the L2 regularization term \(\frac{{w}_{{gp}}}{|{w}_{{gp}}{|}_{2}}\) prevents edge weights from deviating excessively from known annotations, and \({{\rm{\gamma }}}_{{\rm{gp}}}\) amplifies the gradient signals of CGC lung cancer gene-related edges to enhance the influence of clinically critical driver genes.
Model training
To dissect cooperative regulatory effects of gene group alterations on signaling pathways, our cancer signaling pathway association recognition model utilizes a GIN-based multi-label graph classification framework augmented with a gene-pathway heterogeneous graph structure to associate predicted gene-level outputs with pathway-level outcomes for identifying signaling pathway dysregulation. The model is trained with a graph-level binary cross-entropy loss to supervise gene mutation type prediction, complemented by an L2 regularization term incorporating CGC-derived gene knowledge to constrain the learning process. To identify pathway dysregulation, the model employs global pooling to compute node-level importance based on Weisfeiler-Lehman color representations, where the top-n nodes with the highest structural importance are selected according to their Weisfeiler-Lehman color depth to reflect their contribution to the overall graph representation.
Hyperparameter settings: For fair comparison across models, identical hyperparameters are used throughout. All models employ the Adam optimizer with a batch size of 32, an initial learning rate of 1e−3, and a weight decay of 1e−5. Five-fold cross-validation is performed on the TCGA-LUAD dataset (details in Supplementary Information), comprising 585 WSIs in total. Each fold contains 117 WSIs, with 80% of the data use for training and the remaining 20% reserved for testing to evaluate model generalization.
A prior knowledge-based logical regularization method for pathway information
For the purpose of analyzing and exploring pathway-related genes, existing studies typically employ gene group enrichment analysis or differential gene expression analysis to reveal associations among genes across individuals, thereby identifying genes most relevant to specific signaling pathways. Based on the principle of analyzing association patterns among genes across individuals, we introduce KePathGraph, a logic-constrained strategy for gene groups within signaling pathways, as detailed below:
Constructing the inter-individual pathway graph structure. To obtain gene associations across individuals, we construct an inter-individual graph structure by first applying Global Sort Pooling to select the top 20 patches per WSI most relevant to the pathway (details in Supplementary Information), thereby reducing graph scale and mitigating interference from tissue microenvironment heterogeneity. Histopathological features extracted from these high-relevance patches via a pretrained ResNet-185 (details in Supplementary Information) form the basis for edge construction determined by Euclidean distances, while pre-activation values of pathway and gene predictions are assigned as node features to embed pathway information. Through this procedure, we obtain an inter-individual graph structure embedded with pathway information.
Representing prior knowledge in Disjunctive Normal Form (DNF). To formally integrate prior knowledge, we encoded relationships among genes, signaling pathways, and cases into DNF using logical clauses. First, to capture the functional redundancy and compensatory mechanisms within signaling pathways, we formulated the gene-pathway association clause
where the clause consists of unary atomic predicates, and the variable \(x\) denotes the WSI of case \(x\). \({{Pathway}}_{A}(x)\) indicates that signaling pathway A is functionally normal in case \(x\). \({{Gene}}_{An}(x)\) indicates that gene \(n\), which belongs to pathway \(A\), is not mutated in case \(x\). This clause posits that a signaling pathway A is functionally normal (¬Pathway A (x)) in case x only if all its constituent genes are non-mutated, thereby aligning model predictions with the molecular basis where multiple genes collectively sustain pathway activity.
Second, to leverage representational commonalities across individuals, we introduced inter-case association clauses (Eqs. 5 and 6) based on phenotypic convergence and functional consistency.
This type of clause consists of atomic binary predicates, where \(x\) and \(y\) denote the WSIs of two distinct cases, and \(\mathrm{similar}(x,y)\) indicates that cases \(x\) and \(y\) exhibit histopathological similarity. These constraints dictate that if distinct cases x and y share specific gene mutations (Clause (5)) or exhibit pathway dysfunction (Clause (6)), they must display histopathological similarity (similar(x, y)). A more detailed explanation of these clauses is provided in Supplementary Information additional explanation of clauses. By incorporating these logical constraints, we enforce that the model’s predictions for gene groups and signaling pathways remain consistent with established biological evidence regarding tumor evolution and phenotypic expression.
Implementing a logical clause-based graph structure constraint. To guide it toward learning desired features, we inject prior logical knowledge using a graph learning framework and a Knowledge Enhancer (KE) module. This module translates expert-defined logical rules into differentiable fuzzy logic operations, converting atomic predicates into continuous probabilities via fuzzy logic operators (e.g., sigmoid function):
where \(k\) is a slope coefficient controlling the softness/hardness of the logic, \(\sigma\) is the sigmoid function mapping values to [0,1], \(\tau\) and is a threshold (set to 0.5). When the feature value \({h}_{x}\left[{\text{Gene}}_{A}\right]\) exceeds \(\tau\), the sigmoid (or other fuzzy logic function) outputs a continuous probability compatible with neural network computation.
After converting atomic predicates, complex clauses are formed by combining atoms. For DNF, the max operator is used for aggregation. For example, the clause\(\,{{Gene}}_{A1}(x)\bigvee {{Gene}}_{A2}(x)\bigvee {\rm{\neg }}{{Pathway}}_{A}(x)\) is represented as:
Once prior knowledge is expressed as logical clauses, we directly embed these expressions into the model architecture by enhancing the graph message-passing process. Each clause operates on pre-activation values to guide information flow. Specifically, by modifying the adjacency matrix and adjusting feature aggregation weights to enable directional propagation.
To jointly analyze pathway alteration probabilities across multiple samples, we construct a knowledge-constrained graph structure, termed KePathGraph, and perform node message passing via a graph learning model. During propagation, edge weights are dynamically updated according to clause satisfaction. The update rule is:
where \({s}_{\text{clause}}\left(x,y\right)\) denotes clause satisfaction between nodes \(x\) and \(y\), \(A\) is the GNN adjacency matrix, and \({W}^{\left(l\right)}\) is a learnable weight matrix. Through this approach, clause-based prior knowledge constrains the model’s predictions in a differentiable and biologically informed manner.
Data availability
The CGC dataset provides clinically validated LUAD-related genes and is available from the COSMIC at https://cancer.sanger.ac.uk/census. TCGA-LUAD WSIs are accessible via the National Cancer Institute Genomic Data Commons (https://portal.gdc.cancer.gov/projects/TCGA-LUAD). The Human Protein Atlas supplies immunohistochemistry images for gene mutations at https://www.proteinatlas.org. cBioPortal provides gene-level molecular profiles corresponding to TCGA-LUAD samples at https://www.cbioportal.org. Pathway annotations are retrieved from the DAVID database (https://davidbioinformatics.nih.gov/), which integrates KEGG, Reactome, WikiPathways, and archived BioCarta annotations.
Code availability
All custom code is publicly available on GitHub at [https://github.com/xianyvxxx/LCG-HGNN]. Additional information required to reproduce this study is available from the lead contact upon reasonable request.
References
Unger, M. & Kather, J. N. Deep learning in cancer genomics and histopathology. Genome Med. 16, 44 (2024).
Swanton, C. et al. Embracing cancer complexity: hallmarks of systemic disease. Cell 187, 1589–1616 (2024).
Grimes, S. M. et al. Single-cell multi-gene identification of somatic mutations and gene rearrangements in cancer. NAR Cancer 5, zcad034 (2023).
Zhang, Y. et al. A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell 31, 820–832.e3 (2017).
He, K., Zhang, X., Ren, S. & Sun, J. Deep residual learning for image recognition. in Proc. 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR) 770–778 (IEEE, 2016).
Ding, K., Zhou, M., Wang, H., Zhang, S. & Metaxas, D. N. Spatially aware graph neural networks and cross-level molecular profile prediction in colon cancer histopathology: a retrospective multi-cohort study. Lancet Digit. Health 4, e787–e795 (2022).
Yu, Y., Shi, W., Xie, G. & Xi, J. Pathological tissue-level contour genomic profile interpretation of lung adenocarcinoma via spatial and morphological features co-action graph neural network. in Proc. 2023 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 958–964 (IEEE, 2023).
Wang, M., Wang, T., Cong, F., Lu, C. & Xu, H. Double-Tier Attention Based Multi-label Learning Network for Predicting Biomarkers from Whole Slide Images of Breast Cancer. in Medical Image Computing and Computer Assisted Intervention – MICCAI 2024 (eds M. G. Linguraru et al.) 91–101 Springer Nature Switzerland, (2024).
Majumder, S., Kapse, S., Bhattacharya, M., Xu, X., Yurovsky, A. & Prasanna, P. PEaRL: Pathway-Enhanced Representation Learning for Gene and Pathway Expression Prediction from Histology. in Proceedings of the IEEE/CVF Winter Conference on Applications of Computer Vision (WACV) 8052–8062 (2026).
Mostavi, M., Chiu, Y.-C., Huang, Y. & Chen, Y. Convolutional neural network models for cancer type prediction based on gene expression. BMC Med. Genom. 13, 44 (2020).
Ye, J., He, J., Peng, X., Wu, W. & Qiao, Y. Attention-driven dynamic graph convolutional network for multi-label image recognition. in Proc. Computer Vision – ECCV 2020 (eds Vedaldi, A., Bischof, H., Brox, T. & Frahm, J.-M.) 649–665 Springer International Publishing, (2020).
Talath, S. et al. An introduction to cell signalling pathways and their dysregulation in cancer. in Proc. Cell Signaling Pathways and Their Therapeutic Implication in Cancers (eds M. U. Rehman, M. S. Khan) 1–42 (Springer Nature, 2025).
You, M. et al. Signaling pathways in cancer metabolism: mechanisms and therapeutic targets. Signal Transduct. Target Ther. 8, 196 (2023).
Bilal, M. et al. Development and validation of a weakly supervised deep learning framework to predict the status of molecular pathways and key mutations in colorectal cancer from routine histology images: a retrospective study. Lancet Digit. Health 3, e763–e772 (2021).
Dong, Z.-Y. et al. Genetic and immune profiles of solid predominant lung adenocarcinoma reveal potential immunotherapeutic strategies. J. Thorac. Oncol. 13, 85–96 (2018).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173, 321–337.e10 (2018).
Gao, C., Goswami, M., Chen, J. & Dubrawski, A. Classifying unstructured clinical notes via automatic weak supervision. In Proc. 7th Machine Learning for Healthcare Conference 673–690 (PMLR, 2022).
Ceramil, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Sherman, B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
The Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Carrington, A. M. et al. Deep ROC analysis and AUC as balanced average accuracy to improve model selection, understanding and interpretation. IEEE Trans. Pattern Anal. Mach. Intell. 45, 329–341 (2023).
Daniele, A. & Serafini, L. Knowledge enhanced neural networks. in Proc. PRICAI 2019: Trends in Artificial Intelligence (eds A. C. Nayak, A. Sharma) 542–554 (Springer International Publishing, 2019).
Werner, L., Layaïda, N., Genevès, P. & Chlyah, S. Knowledge enhanced graph neural networks. In Proc. IEEE 10th International Conference on Data Science and Advanced Analytics (DSAA) 1–10 (IEEE, 2023).
Kipf, T.N. & Welling, M. Semi-Supervised Classification with Graph Convolutional Networks. In International Conference on Learning Representations (ICLR) (OpenReview.net, 2017).
Veličković, P. et al. Graph Attention Networks. in International Conference on Learning Representations (ICLR) (OpenReview.net, 2018).
Xu, K., Hu, W., Leskovec, J. & Jegelka, S. How Powerful are Graph Neural Networks? in International Conference on Learning Representations (ICLR) (OpenReview.net, 2019).
Hamilton, W. L., Ying, R. & Leskovec, J. Inductive representation learning on large graphs. In Proc. 31st International Conference on Neural Information Processing Systems 1025–1035 (Curran Associates Inc., 2017).
Wu, G. & Zhu, J. Multi-label classification: do Hamming loss and subset accuracy really conflict with each other? In Advances in Neural Information Processing Systems 3130–3140 (Curran Associates, Inc., 2020).
Uhlén, M. et al. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Pao, J. J. et al. Predicting EGFR mutational status from pathology images using a real-world dataset. Sci. Rep. 13, 4404 (2023).
Matsumura, M. et al. A histopathological feature of EGFR-mutated lung adenocarcinomas with highly malignant potential – an implication of micropapillary element. PLoS ONE 11, e0166795 (2016).
Federico, A. D. et al. Lung adenocarcinomas with mucinous histology: clinical, genomic, and immune microenvironment characterization and outcomes to immunotherapy-based treatments and KRASG12C inhibitors. Ann. Oncol. 36, 297–308 (2025).
Wang, J. et al. Effects of different KRAS mutants and Ki67 expression on diagnosis and prognosis in lung adenocarcinoma. Sci. Rep. 14, 4085 (2024).
Dziadziuszko, R. STK11 and KEAP1 mutations in lung adenocarcinoma: solving the puzzle continues. J. Thorac. Oncol. 17, 351–352 (2022).
Zheng, J., Deng, Y., Huang, B. & Chen, X. Prognostic implications of STK11 with different mutation status and its relationship with tumor-infiltrating immune cells in non-small cell lung cancer. Front. Immunol. 15, 1387896 (2024).
Mendoza, R. P. et al. Lung adenocarcinomas with isolated TP53 mutation: a comprehensive clinical, cytopathologic and molecular characterization. Cancer Med. 13, e6873 (2024).
Sondka, Z. et al. The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696 (2018).
Acknowledgements
The authors thank Xinchen Yuan and Yande Luo for their technical assistance. This study was funded by the Guangdong Basic and Applied Basic Research Foundation under Grant 2024A1515010851 and 2022A1515110001; in part by the National Natural Science Foundation of China under Grant 62202117; in part by the Tertiary Education Scientific Research Project of Guangzhou Municipal Education Bureau under Grant 2024312264; in part by Guangzhou Basic and Applied Basic Research Foundation under Grant SL2023A04J02440. The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
Y.Y. conceived and designed the study, developed the methodology, implemented the software, conducted validation and formal analysis, curated the data, and wrote the original draft; W.S. contributed to conceptualization and methodology, supervised the research, and acquired funding; J.X. contributed to conceptualization and methodology, provided overall supervision, and acquired funding; J.F. and S.H. assisted in methodology development and data curation; X.C. contributed to validation and data curation; X.B. contributed to data curation; S.Z. provided supervisory support. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, Y., Shi, W., Chen, X. et al. Inferring signaling pathway abnormalities from histopathological images via logic-constrained gene-pathway heterogeneous knowledge graph. npj Biomed. Innov. 3, 23 (2026). https://doi.org/10.1038/s44385-026-00078-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44385-026-00078-6
