
Abstract
Granulomatous nephritis can be triggered by diverse factors and results in kidney failure. However, despite accumulating data about granulomatous inflammation, pathogenetic mechanisms in nephritis remain unclear. The DNA-binding high-mobility group box-1 protein (HMGB1) initiates and propagates inflammation when released by activated macrophages, and functions as an ‘alarm cytokine’ signaling tissue damage. In this study, we showed elevated HMGB1 expression in renal granulomas in rats with crystal-induced granulomatous nephritis caused by feeding an adenine-rich diet. HMGB1 levels were also raised in urine and serum, as well as in monocyte chemoattractant protein-1 (MCP-1), a mediator of granulomatous inflammation. Injection of HMGB1 worsened renal function and upregulated MCP-1 in rats with crystal-induced granulomatous nephritis. HMGB1 also induced MCP-1 secretion through mitogen-activated protein kinase (MAPK) and phosphoinositide-3-kinase (PI3K) pathways in rat renal tubular epithelial cells in vitro. Hmgb1+/− mice with crystal-induced nephritis displayed reduced MCP-1 expression in the kidneys and in urine and the number of macrophages in the kidneys was significantly decreased. We conclude that HMGB1 is a new mediator involved in crystal-induced nephritis that amplifies granulomatous inflammation in a cycle where MCP-1 attracts activated macrophages, resulting in excessive and sustained HMGB1 release. HMGB1 could be a novel target for inhibiting chronic granulomatous diseases.
Similar content being viewed by others

Main
Granulomas are distinctive chronic inflammatory lesions characterized by aggregations of activated macrophages and marked fibrosis. In extreme cases, granulomas can lead to serious organ failure.1 Several lines of evidence indicate that cytokines such as monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor-α (TNF-α) function in the initiation and persistence of granulomatous inflammation.2, 3, 4 Various initiators give rise to granulomatous disease, including infections (such as tuberculosis and leprosy), particulate stimulants (as in silicosis and gout), and unidentified factors (as in sarcoidosis and Wegener's granulomatosis). Pathogenetic mechanisms have not been fully investigated, restricting the potential therapies available.5
High-mobility group box-1 protein (HMGB1) was originally described as a nuclear DNA-binding protein6, 7 that facilitates gene transcription by stabilizing nucleosome formation.8 HMGB1 can also be released extracellularly and acts as a pro-inflammatory cytokine or alarm signal for tissue damage.9 HMGB1 is released from cells through ‘passive release’ and ‘active secretion’. Passive release occurs as a result of cellular necrosis in most eukaryotic cells.10, 11 Active secretion from activated macrophages and monocytes occurs in response to inflammatory stimuli, such as lipopolysaccharide and TNF-α,12, 13 and can trigger a potent inflammatory response leading to severe tissue injury.14, 15 Extracellular HMGB1 is also involved in the progression of several inflammatory diseases, including septic shock,16 disseminated intravascular coagulation,17 acute lung injury,18, 19 as well as chronic inflammatory diseases such as rheumatoid arthritis20 and atherosclerosis.21, 22
We hypothesized that granulomas, which mainly comprise activated macrophages, could be a source of HMGB1. We therefore investigated the pathogenetic functions of HMGB1, focusing on its role in granulomatous inflammation using a crystal-induced nephritis model. We also proposed an association between HMGB1 expression and MCP-1 in granulomas, which could create a ‘vicious cycle’ that promotes chronic inflammation.
MATERIALS AND METHODS
Granulomatous Nephritis Rat Model
All animal studies were conducted according to the National Institutes of Health Guide, and reviewed and approved by the Committee on Animal Experimentation of Kagoshima University, Japan. All rats were kept under pathogen-free conditions and maintained under a 12-h light–dark cycle (lights on at 0700 hours) at 22±1°C.
2,8-dihydroxyadenine (DHA)-induced interstitial nephritis was generated using an established method.23 In brief, male Wistar rats, initially weighing approximately 60 g, were fed on a commercial diet (Type CE-2; CLEA Japan, Tokyo, Japan) for 1 week, and subsequently given ad libitum access to a powdered diet consisting of 18.0 g casein, 57.9 g α-cornstarch, 15.0 g sucrose, 2.0 g soybean oil, and 0.1 g choline chloride per 100 g. This was supplemented with 0.75, 0.5, 0.25, or 0% adenine (Wako Pure Chemical Industries, Osaka, Japan) for 5 weeks in the dose-dependent disease model, or with 0.25 or 0% adenine for 20 weeks in the time-dependent disease model. During the adenine-feeding period, serum samples were collected weekly in the dose-dependent model, and monthly in the time-dependent model. The 24-h urine samples were collected at 5 weeks in the dose-dependent model, and at 4, 12, 16, and 20 weeks in the time-dependent model. At the end of the feeding periods for both models, the rats were killed, and their urine, blood, and kidneys were collected.
HMGB1 Injection
Four-week-old rats were randomized into four groups: (1) rats that were injected with saline with normal food (control/saline); (2) rats that were injected with HMGB1 with normal food (control/HMGB1); (3) rats that were injected with saline with 0.75% adenine food (adenine/saline); (4) rats that were injected with HMGB1 with 0.75% adenine food (adenine/HMGB1). HMGB1 (500 μg/kg, a gift from Shino-Test, Sagamihara, Japan) was injected intraperitoneally in 500 μl injection volumes three times/week (M-W-F) for 5 weeks. Control animals received sterile saline injections. Five weeks after injection, the rats were killed, and their urine, blood, and kidneys were collected. The concentration of endotoxin in HMGB1 protein was less than 67.3 pg/μg HMGB1, using Limulus endotoxin assays (Wako Pure Chemical Industries).
Granulomatous Nephritis Model in Mice
The Hmgb1+/– mice used in this study were previously described.24 All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee at The Scripps Research Institute. The genotype of the mice was determined by polymerase chain reaction (PCR) analysis of tail DNA.25 Eight- to fifteen-week-old mice were separated into four groups (5–7 mice per group): Wild-type (WT) mice fed with normal food (WT/control); Hmgb1+/− mice fed with normal food (Hmgb1+/–/control); WT mice fed with 0.25% adenine food (WT/adenine); Hmgb1+/− mice fed with 0.25% adenine food (Hmgb1+/–/adenine). Four weeks after feeding, mice were killed, and their urine, blood, and kidneys were collected. Hmgb1+/− mice are on BALB/c background and BALB/c mice were thus used as controls.
Immunohistochemical Analysis
Kidney tissues were fixed in 10% neutral-buffered formalin and embedded in paraffin. Sections (4 μm) were deparaffinized, and endogenous peroxidase activity was blocked by treatment with 0.3% H2O2 solution for 15 min. Sections were then blocked with 2% goat serum for 30 min, except for those stained with the mAb ED1. The sections were incubated overnight in a humidified chamber with 150 μl of the rat cytokine-specific Abs, to give final concentrations of 1–5 μg/ml. The following Abs were used: anti-rat HMGB1 Ab (a gift from Shino-Test), mouse anti-rat CD68 mAb, ED1 (Serotec, Oxford, UK), and rat anti-mouse macrophage mAb (ab56297, Abcam, Cambridge, UK). For staining HMGB1 and ED1, the slides were washed and then incubated for 30 min with a biotin-labeled goat anti-rabbit (Lab Vision, Fremont, CA, USA) or horse anti-mouse (Vector Laboratories, Burlingame, CA, USA) secondary Ab, followed by 150 μl Vectastain avidin-biotin-horseradish peroxidase solution (Vectastain Elite, ABC-kit; Vector Laboratories). For staining mouse macrophages, the slides were washed and then incubated with Histofine Simple Stain Mouse MAX PO (Rat) (Nichirei Biosciences Tokyo, Japan). Then the slides were developed with 3′,3′-diaminobenzidine (Dako Cytomation, Carpinteria, CA, USA), washed with deionized water, counterstained with Mayer's hematoxylin, and mounted in buffered glycerol.
Quantitation of Tissue Staining
A semi-quantitative method was used to quantify macrophages stained by rat anti-mouse macrophage mAb as previously described.26 The number of labeled cells was counted in randomly selected areas of the same section in kidneys under high-power fields ( × 400) by means of a 0.02-mm2 graticule fitted in the eyepiece of a microscope, and expressed as cells per mm2. At least 10–20 lesions were examined for each mouse in the Ab-labeled tissue sections. All counting was performed on numerically coded slides.
Quantitative RT-PCR
The expression of selected genes was measured by quantitative real-time reverse transcription (RT)-PCR. Total RNA was extracted from rat kidneys using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Samples (2 μg) of total RNA were reverse transcribed using a First Strand complementary DNA synthesis kit for RT-PCR (Roche, Indianapolis, IN, USA). Aliquots of the cDNAs were amplified by real-time RT-PCR (Ct value 20–30 s cycles) using a 7300 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with gene-specific primers (Assay IDs: HMGB1, Rn00820665_g1; MCP-1, Rn00580555_m1; receptor for advanced glycation end products (RAGE), Rn00584249_m1; Toll-like receptor (TLR)4, Rn00569848_m1; glyceraldehyde-3-phosphate dehydrogenase [GAPDH], Rn99999916_s1). As ‘minus RT’ controls, samples containing total RNA instead of the cDNA were also examined.
The TaqMan technique was used for signal detection.27 All analyses were carried out in triplicate, and non-template controls and dissociation curves were used to ensure the specificity of template amplification. For each primer pair, serial dilutions of a control cDNA were used to construct standard curves, and those with R2>0.97 were then used to determine mRNA levels in individual samples. The mRNA levels for each gene of interest were normalized to the GAPDH mRNA levels in the same cDNA.
Cell Culture
The normal rat renal tubular epithelial cell line NRK-52E (American Type Culture Collection [ATCC] CRL-1571, Manassas, VA, USA) was maintained in plastic tissue culture flasks in high-glucose Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Grand Island, NY, USA) containing 5% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin. The cells were maintained at 37°C in a humidified 5% CO2 incubator, and the medium was changed twice weekly.
In vitro Stimulation with HMGB1
NRK-52E rat renal tubular epithelial cells (RTECs) were cultured with medium containing 2% heat-inactivated FBS in 12-well plates at 1.5 × 105 cells per well and were stimulated with 250 ng/ml HMGB1 for 0, 6, 12, 24, 36, or 48 h to examine the effects over time, or with 0, 50, 250, 500, or 1000 ng/ml HMGB1 for 24 h to examine dose-dependence of the effects. Supernatants were frozen at −80°C until enzyme-linked immunosorbent assay (ELISA) for MCP-1 was performed. To exclude any influence of endotoxin, we determined that the content was <0.5 pg/μg HMGB1 protein, using Limulus endotoxin assays (Wako Pure Chemical Industries); we also confirmed that MCP-1 release was abrogated by heating HMGB1 samples to 100°C for 2 h (data not shown).
Western Blotting
HMGB1 in rat urine collected over 24 h was analyzed by western blotting as previously described.20 Samples were prepared as follows: each urine sample (5–10 ml) was incubated with 50 μl HiTrap heparin HP beads (GE Healthcare, Uppsala, Sweden) for 24 h at 4°C. The beads were washed twice with 10 mM phosphate buffer, mixed with 50 μl sample buffer (50 mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate, 6% 2-mercaptoethanol, 10% glycerol, and 0.002% bromophenol blue), and boiled for 5 min. HMGB1-induced cell signaling was analyzed in NRK-52E RTECs stimulated with 500 ng/ml HMGB1 for 7.5, 15, 30, 60, 120, or 240 min. The cells were washed with sterile PBS, lysed by adding 100 μl sodium dodecyl sulfate sample buffer containing protease inhibitor (25 × cocktail, Roche; 100 mM phenylmethanesulfonyl fluoride) and 100 mM Na3VO4, and immediately placed on ice. Rabbit anti-rat HMGB-1 Ab (a gift from Shino-Test) and rabbit antibodies against phosphorylated extracellular signal-regulated kinase (ERK) (Thr202/Tyr204), c-Jun N-terminal kinase (JNK) (Thr183/Tyr185), p38 (Thr180/Tyr182), and phosphoinositide-3-kinase (PI3K)/Akt (Ser473; all from Cell Signaling Technology, Beverly, MA, USA) were used for primary incubation overnight at 4°C. Then the membranes were washed and incubated with horseradish peroxidase-conjugated anti-rabbit polyclonal immunoglobulin G (IgG; MP Biomedicals, Solon, OH, USA) at RT for 1 h. Labeled bands were visualized using an enhanced chemiluminescence system (GE Healthcare) and exposed to high-performance chemiluminescence film (GE Healthcare). The intensity of the protein bands on the western blots was quantified using the National Institutes of Health image 1.63 software.28
ELISA for HMGB1, MCP-1, BUN, and Creatinine
We measured cytokine and creatinine concentrations using commercially available ELISA kits. The kits for MCP-1 were purchased from BioSource (Camarillo, CA, USA), the HMGB1 kits were a gift from Shino-Test,29 and the kits for urine creatinine were purchased from Oxford Biomedical Research (Oxford, MI, USA). SPOTCHEM assays (Arkray, Kyoto, Japan) were used to measure the serum blood urea nitrogen (BUN) levels by the lipoprotein lipase—glycerol kinase—glycerol-3-phosphate-oxidase method, and to measure serum creatinine using colorimetry. In all ELISA, serum, urine and whole kidney samples from rats and mice, and stimulated tissue culture supernatants were measured in duplicate.
Inhibition of MAPK and PI3K/Akt-Signaling Pathways
To assess the contribution of mitogen-activated protein kinase (MAPK) and PI3K/Akt pathways to HMGB1-stimulated cytokine responses, we pre-incubated cells for 1 h with specific inhibitors of ERK (U0126; Promega, Madison, WI, USA), JNK (SP600125; Calbiochem, Darmstadt, Germany), p38 (SB203580; Calbiochem), and PI3K/Akt (LY294002; Calbiochem). The cells were then stimulated with 1 μg/ml HMGB1 for 24 h.
Statistical Analysis
The statistical significance of differences in values between the experimental groups and the controls was determined using a two-tailed Student's t-test. P-values <0.05 were considered significant.
RESULTS
Granuloma Formation Associated with High HMGB1 Expression in Adenine-Fed Rat Kidneys
This study used the adenine-fed rat model of human chronic renal failure, in which the deposition of crystals of the abnormal adenine metabolite DHA in the kidney induces nephritis with granulomas.23 In a time-dependent and dose-dependent pattern, adenine-fed rats gradually became cachectic with body weight reduction, showing inactivity, loss of skin tone, and ruffled hair (Supplementary Figure 1A). The kidneys in rats fed adenine for 5 weeks retained a normal shape, but became enlarged, edematous, and pale in a dose-dependent manner, peaking in rats fed 0.75% adenine (Supplementary Figure 1B). In cross-sections, a discrete, narrow white band appeared at the cortex–medulla boundary in the early stages, as abundant crystal deposits formed. The kidneys gradually developed a yellowish stippling and pitting between the cortex and medulla that progressed with time, finally becoming soft (Supplementary Figure 1C). The BUN levels increased further over time and in dose-dependent pattern. Creatinine levels in adenine-fed rats were also elevated at later time points in a dose-dependent manner (Table 1). Microscopic examination of periodic acid-Schiff (PAS)-stained kidney sections from the rats showed many granulomas at different stages (Figure 1a). As the size and number of granulomas increased with adenine dose and time, the nephrons became tapered showing mildly-to-severe dilatation of renal tubules as they were occupied by crystal deposits and/or granulomas, and also became inflamed (Supplementary Figure 2). The presence of macrophages was confirmed by immunostaining with the anti-rat macrophage monoclonal antibody (mAb) ED1. ED1-positive cells were observed around the needle-shaped crystals (Figure 1b and e), whereas such cell aggregates were not observed in control rats (Figure 1f).

High expression of HMGB1 in granulomas in adenine-fed rat kidneys. (a–c) Expression of HMGB1 in renal granulomas from rats fed 0.75% adenine for 5 weeks. Granulomas accumulated around the crystals (a). (b) Macrophages labeled with mAb ED1, accumulated around the site of crystal formation. (c) Increased expression of cytoplasmic and nuclear HMGB1 in granulomas. (d) HMGB1 expression in the cytoplasm of RTECs (arrows) and reduced nuclear staining in an expanded renal tubule from a rat fed 0.25% adenine for 16 weeks. Immunohistochemical analysis of the tissues of nephritis rats that were fed 0.25% adenine for 16 weeks (e and g) and of control rats that were fed 0% adenine for the same period (f and h). Accumulation of macrophages (e, arrows) around the crystals (asterisks) led to granuloma formation, whereas neither crystals nor granulomas were detected in control rats (f). HMGB1 was expressed in both the cytoplasmic and nuclear patterns in granulomas (g, arrows), although in control rats, HMGB1 was expressed only in the cell nuclei (h). Crystals are absent in C and G as a result of heat treatment during the staining procedure. Nuclei were counterstained with Mayer's hematoxylin. Original magnifications: × 400 (a–d), × 200 (e–h).
To test our hypothesis that for granulomas, mainly organized by activated macrophages, to be a source of HMGB1, we initially determined whether the protein was expressed in the severe nephritis model by immunostaining granulomatous nephritic tissue. HMGB1 was detected in the nuclei and diffusely in the cytoplasm in granulomas (Figure 1c and g), but in control rats HMGB1 was expressed only in nuclei (Figure 1h). In some RTECs in the distended tubuli, HMGB1 was expressed in the cytoplasm, especially where granulomatous inflammation was advanced (Figure 1d). It is reported that renal tubules are dilated and contain apoptotic cells, leading to tubular atrophy in severe obstructive nephropathy,30 and that HMGB1 is released during apoptotic cell death.31 We examined whether these cells, which expressed HMGB1 in the cytoplasm, were undergoing apoptotic cell death or not using double staining against the HMGB1 antibody with TUNEL assay. The result showed that a large number of renal tubular cells were positive for TUNEL, although those cells did not share HMGB1 in the cytoplasm (Supplementary Figure 3).
Upregulation of HMGB1 and its Receptors in Granulomatous Nephritis Tissue
As we observed HMGB1 expression in macrophage-rich granulomas in the kidneys, we next monitored the kinetics of HMGB1 mRNA expression by quantitative PCR in renal tissue from adenine-fed and control rats at 8, 12, and 16 weeks. HMGB1 mRNA levels in adenine-fed rats were significantly increased with time compared with control rats (Figure 2a). Similarly, we examined mRNA expression of MCP-1, which is a mediator of granuloma formation and the cause of organ impairment in granulomatous inflammation,32, 33 and found a time-dependent increase in granulomatous-inflamed rat kidneys, similar to the increase in HMGB1 (Figure 2b).

Upregulation of HMGB1 and its receptors as well as MCP-1 in kidneys from rats with granulomatous nephritis. The ratio of mRNA levels for HMGB1 (a), MCP-1 (b), RAGE (c), and TLR4 (d) increased compared with control rats as the period of adenine supplementation increased. Quantitative PCR was performed using mRNA isolated from whole kidneys of rats fed 0.25 and 0% adenine for 8, 12, or 16 weeks (n=3). *P<0.05, **P<0.01. –RT indicates RT (reverse transcriptase) minus control samples that contain total RNA instead of cDNA.
We found that the expression of RAGE and TLR4, which are receptors for HMGB1,34, 35 was increased in the kidneys of rats fed adenine in a time-dependent manner at the mRNA level (Figure 2c and d). The expression of these receptors was also increased at the protein level in kidneys from rats fed adenine for 16 weeks, compared with controls (Supplementary Figure 4).
HMGB1 and MCP-1 in the Urine and Serum of Rats with Granulomatous Renal Inflammation
We next examined the levels of the cytokines HMGB1 and MCP-1 in the urine of rats with adenine-induced nephritis. HMGB1 was detected after 4 weeks, increased sharply after 12 weeks, and remained at moderately increased levels until 20 weeks in the urine of 0.25% adenine-fed rats, whereas HMGB1 was undetectable in the urine of control rats. Accordingly, the results of densitometric analysis of protein signals were increased in the urine of adenine-fed rats compared with the control rats (Figure 3a, left panel). The urinary HMGB1 level tended to increase with the dose of adenine (Figure 3a, right panel). MCP-1 expression was also increased with the dose of adenine, peaking in rats fed 0.5% after 5 weeks (Figure 3b).

Increased expression of HMGB1 and MCP-1 in the urine and serum of granulomatous nephritis rats. (a) Western blotting shows increased HMGB1 levels in urine (5 or 10 ml samples concentrated using heparin beads) over time (left panel) and at different doses (right panel) in rats fed adenine. Urine HMGB1 levels peaked at 12 weeks. The protein signals are expressed as the ratio of the densitometric intensity of the urine HMGB1 concentration, which was normalized to urine creatinine. (b) ELISA shows increased urine MCP-1 levels in rats that were fed different doses of adenine for 5 weeks. n=3–15; **P<0.01. (c–e) ELISA shows increased serum HMGB1 in rats fed 0.25% adenine for 20 weeks (c), and a dose-dependent increase in serum MCP-1 (d) with a peak at 20 weeks in the time-dependent model (e). n=3–15; *P<0.05, **P<0.01.
The cytokine levels were also elevated in the serum of the adenine-fed rats. Serum HMGB1 increased significantly in the 0.25% adenine-fed rats after 20 weeks, but was undetectable in the controls (Figure 3c). The serum MCP-1 increased dose-dependently (Figure 3d), with significant levels in the 0.25% adenine-fed rats after 8 weeks, and then increased sharply after 20 weeks (Figure 3e).
Induction of MCP-1 Release by HMGB1 and its Signaling Pathways in rat RTECs in vitro
It has been reported that RTECs express RAGE36 and TLR4,37 which are the receptors of HMGB1. We then studied the effect of HMGB1 on the release of MCP-1 by the rat RTEC cell line in vitro. We first observed MCP-1 levels in the supernatant of RTEC cells with HMGB1 treatment for 24 h and found that MCP-1 secretion occurred significantly at doses above 250 ng/ml (data not shown). Then we examined the time course by using 250 ng/ml HMGB1; MCP-1 increased significantly between 6 and 48 h, peaking at 36 h (Figure 4a). Moreover, we investigated signaling pathways, focusing on three different MAPK pathways (ERK, JNK, and p38) and the PI3K/Akt pathway in the HMGB1-mediated secretion of MCP-1 in rat RTEC. Western blotting shows that phosphorylation of PI3K/Akt and ERK peaked 7.5 min after HMGB1 stimulation, decreasing gradually thereafter. Phosphorylation of JNK was detected after 7.5 min and peaked at 15 min (Figure 4b). Phosphorylation of p38 peaked 60 min after adding HMGB1 and this level persisted for at least 240 min. Inhibitors of ERK (U0126), JNK (SP600125), and p38 (SB203580), or PI3K/Akt (LY294002) reduced the MCP-1 secretion caused by HMGB1 stimulation (Figure 4c–f).

HMGB1-induced MCP-1 secretion and its signaling pathways in rat RTECs in vitro. (a) MCP-1 was measured by ELISA in supernatants from NRK-52E RTECs on stimulation with 250 ng/ml HMGB1 for 0, 6, 12, 24, 36, or 48 h. The MCP-1 levels increased significantly with time, peaking at four-fold above the control levels after 36 h. Each figure is representative of three separate experiments. n=3; **P<0.01. (b) Phosphorylation of PI3K/Akt and three different MAPKs in rat RTECs incubated with HMGB1 and determined by western blotting. Phosphorylation peaked at different times for the four kinases after HMGB1 addition. Each figure is representative of three separate experiments. (c–f) MCP-1 was measured by ELISA in supernatants from rat RTECs incubated with or without the protein kinase inhibitors LY294002 (which inhibits PI3K/Akt), U0126 (which inhibits ERK), SP600125 (which inhibits JNK), and SB203580 (which inhibits p38). The inhibitors partially but significantly reduced the HMGB1-induced MCP-1 secretion in rat RTEC. Each figure is representative of three separate experiments. **P<0.01 compared with HMGB1 stimulation alone.
HMGB1 Worsened Kidney Function with Increasing MCP-1 Expression in Rats with DHA-Induced Granulomatous Nephritis
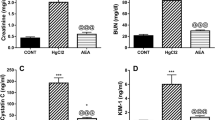
To further investigate whether HMGB1 could accelerate inflammation in rats with granulomatous nephritis, we examined renal function and MCP-1 levels in nephritic rats that were treated with HMGB1 by intraperitoneal injection. BUN and serum creatinine levels increased significantly in rats with DHA-induced nephritis that were injected with HMGB1 compared with nephritis rats injected with saline for 3 to 5 weeks (Figure 5a and b). MCP-1 mRNA expression in renal tissue from nephritis rats with HMGB1 injection for 5 weeks was increased significantly compared with nephritis rats with saline treatment (data not shown), and the serum MCP-1 levels were also significantly elevated after 4 and 5 weeks in the granulomatous nephritis rats with HMGB1 injection compared with nephritis rats injected with saline (Figure 5c). The rats that were injected with HMGB1 without adenine feeding did not show elevation of BUN, serum creatinine, and MCP-1 compared with control (no adenine, no HMGB1) rats (Figure 5a–c).

Intraperitoneal injection of HMGB1 increased MCP-1 expression and worsened kidney function in rats with DHA crystal-induced nephritis. HMGB1 injection aggravated renal failure with increased BUN (a) and serum creatinine (b) in DHA crystal-induced nephritis rats. (c) Significant MCP-1 increase in serum was detected at 4 and 5 weeks. Adenine-fed rats were compared with the HMGB1 injection and saline injection groups. Each data point represents the mean±s.d. for three rats; *P<0.05, **P<0.01: saline-injected rats vs HMGB1-injected rats in DHA crystal-induced nephritis rats.
Reduced MCP-1 Expression and Decreased Accumulation of Macrophages in Hmgb1+/− Mice with Adenine-Induced Nephritis
To confirm the role of HMGB1 in granulomatous nephritis, we analyzed the number of macrophages and MCP-1 levels in kidneys in Hmgb1-deficient mice with adenine-induced nephritis. As Hmgb1-homozygous knockout mice die within 24 h after birth,24 we used Hmgb1-heterozygous (Hmgb1+/−) mice for this study. The adenine-induced nephritis developed in a time- and dose-dependent manner in mice as observed in rats, but the mice that were treated with 0.75 and 0.5% adenine died after 14 days (data not shown). Thus, in four weeks we used —0.25% adenine-treated mice, whose kidney inflammation resembles that of 0.75% adenine-fed rats, and studied the following four groups: wild-type (WT) mice fed with normal food (WT/control); Hmgb1+/− mice fed with normal food (Hmgb1+/−/control); WT mice fed with 0.25% adenine food (WT/adenine); and Hmgb1+/− mice fed with 0.25% adenine food (Hmgb1+/−/adenine).
We found that there was no obvious change in the kidneys of the Hmgb1+/− control mice at both macroscopic and microscopic examinations compared with WT mice. The color of the kidneys in adenine-fed mice turned yellow because of the inflammatory change, and this was less severe in Hmgb1+/− adenine mice compared with WT mice (Figure 6). In microscopic examination, the basophilic areas that indicate inflammatory cell infiltration were more prominent in the kidney tissues of WT adenine mice compared with those of Hmgb1+/− adenine mice (Figure 6). There was no statistically significant difference in the number of crystals between both adenine-fed mouse groups (Supplementary Figure 5). MCP-1 expression in the kidneys and urine of Hmgb1+/− adenine-fed mice was significantly lower compared with that of WT adenine mice (Figure 7a and b). The number of macrophages in the kidneys was also decreased in Hmgb1+/− adenine mice (Figure 7c and d). In both WT control and Hmgb1+/− control groups, no macrophage infiltration was observed (Figure 7c).

Histological changes of kidneys in WT and Hmgb1+/− mice with DHA crystal-induced nephritis. The color change to yellow of the kidneys was more profound in WT adenine mice compared with Hmgb1+/− adenine mice. In histological observation, the basophilic areas that indicate inflammatory cell infiltration (arrows) were more remarkable in the kidney tissues of WT adenine mice compared with those of Hmgb1+/− adenine mice. The renal tissues of both control groups were intact. H.E. staining; Original magnifications: × 25.

Inflammatory changes and reduction in the number of macrophages in Hmgb1+/− mice with DHA crystal-induced nephritis. (a and b) In DHA crystal-induced nephritis mice, MCP-1 expression of Hmgb1+/− mice was significantly decreased in both whole kidney extracts and urine compared with that of WT mice. (c) Immunohistochemical analysis of macrophages in kidney tissues in adenine-fed mice and control mice. Macrophage aggregates were observed around the crystals (asterisks) in the kidney tissue of adenine-fed mice, whereas no crystals and few macrophages were seen in control mice. Black arrows indicate macrophages. Nuclei were counterstained with Mayer's hematoxylin. Original magnifications: × 400. (d) The number of macrophages (stained by rat anti-mouse mAb) was also decreased in adenine-fed Hmgb1+/− mice compared with that in adenine-fed WT mice. n=3–7; *P<0.05, **P<0.01.
DISCUSSION
We have shown that HMGB1 accelerates granulomatous inflammation in DHA crystal-induced nephritis in rats and mice. Our study generated two major findings in vivo and in vitro. First, HMGB1 was abundant and accumulated in granulomas in the kidney, and in the urine and blood of rats with crystal-induced granulomatous nephritis, whereas control rats showed little HMGB1 expression. Furthermore, the administration of HMGB1 worsened renal function and increased MCP-1 expression in nephritic rats. In Hmgb1+/− mice with the same adenine-induced nephritis, MCP-1 expression decreased significantly in kidneys and urine, and the number of macrophages in the kidneys also showed a significant decrease compared with congenic WT mice. Second, HMGB1 stimulated the release of MCP-1 mediated by MAPK and PI3K/Akt-signaling pathways in cultured RTECs in vitro.
This is the first evidence that HMGB1 is extensively expressed in renal granulomatous lesions and released into the urine. We observed an ongoing increase in HMGB1 mRNA expression in granulomatous nephritis tissue over several months, accompanying the inflammatory condition, and then HMGB1 was released from activated macrophages in the developing granulomas. In activated macrophages, upregulation of mRNA expression as well as an active release of HMGB1 in response to cytokine stimulation has been reported.12 We also found an unusual distribution of HMGB1 in distended RTECs, where it was clearly translocated from the nucleus to the cytoplasm. Although active secretion of HMGB1 was reported in innate immune cells,12 this observation showed that perturbed RTECs might secrete HMGB1 and represent another source of HMGB1 in granulomatous nephritis. In our hands, however, the number of cells that expressed HMGB1 in the cytoplasm was much higher in macrophages than in RTECs, suggesting that macrophages might be a major source of HMGB1 in this nephritis model (Supplementary Figure 6).
We also confirmed the expression of HMGB1 receptors RAGE and TLR4 at both the mRNA and protein levels in granulomatous nephritis tissue. Although we did not perform immunohistochemistry to identify the cell populations expressing RAGE, it is probably expressed by macrophages as well as interstitial and tubular cells.36, 38, 39 These results suggest that HMGB1 is a functional mediator, which participates in the induction of granulomatous nephritis by signaling through these receptors. Our results agree with previous reports that RAGE and TLR4 are abundantly expressed in pulmonary granulomas of patients with sarcoidosis or tuberculosis.40, 41 As monosodium urate crystal-induced inflammation was attenuated in TLR4-knockout mice,42 this receptor might also be involved in DHA crystal-induced inflammation.
Increases of MCP-1 expression have been reported in the serum of sarcoidosis patients, and in bronchoalveolar lavage fluid from pneumoconiosis patients.43, 44 MCP-1 was also increased in experimental pulmonary granulomas, and in lung tissue with granulomatous inflammation.32, 33 Our observations that mRNA and/or protein levels of MCP-1 were increased in the urine, serum, and kidney tissue from experimental rats with renal granulomatous nephritis are consistent with the previous reports concerning granulomas in other organs.32, 33
Our findings that the expression of HMGB1 and MCP-1 mRNAs increased in granulomatous nephritic tissue with time suggests that HMGB1 might interact with MCP-1 to effect the progression of this chronic granulomatous inflammation. Furthermore, we have shown for the first time that HMGB1 stimulates the release of MCP-1 in RTECs, and that both the MAPK and PI3K/Akt signal pathways were involved in that induction. Considering that p38 and JNK, which were described in the setting of cell response to stress45 were involved in this induction, it could suggest that HMGB1 can induce renal tubular cell injury or stress. In addition, the PI3K/Akt-signaling pathway, which is widely acknowledged for its anti-apoptotic role, was also involved in this HMGB1 stimulation. Some studies have shown that inflammation could be caused by HMGB1 through this signaling pathway to induce reactive oxygen species (ROS) and pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-8,46, 47 suggesting that the PI3K/Akt signal pathway may act in response to stress and cell damage.
Taking into account our findings of the relationship between HMGB1 and MCP-1, we propose that HMGB1 attracts macrophages by stimulating the release of MCP-1 from renal tubules, thereby worsening interstitial nephritis by expanding the inflamed areas in the kidney and promoting granuloma formation. In addition, we also confirmed that TNF-α was increased in the granulomatous nephritis rats in the kidney, serum, and urine (Supplementary Figure 7). In vitro studies showed that DHA crystal stimulation induced MCP-1 release by RTECs as well as TNF-α release by macrophages (Supplementary Figure 8), suggesting that DHA crystals trigger granuloma formation, and then induce MCP-1 release that activates resident macrophages in the kidney tissues.
Our results collectively suggest that granulomas forming in the kidney could act as reservoirs of HMGB1. Moreover, HMGB1 may promote granulomatous inflammation by inducing MCP-1, which in turn attracts additional macrophages as further sources of HMGB1 that is stimulated by TNF-α. We showed that the granulomatous nephritis rats injected with HMGB1 had elevated MCP-1 expression and worsened renal function compared with the nephritis rats without HMGB1 injection. Serum HMGB1 was elevated in patients with chronic kidney disease and correlates with the glomerular filtration rate as well as markers of inflammation.48 Considering the previous reports and our present results, HMGB1 could enhance inflammation, which results in renal dysfunction. More importantly, our study of the granulomatous nephritis model in Hmgb1+/− mice also provides direct evidence that HMGB1 caused further accumulation of macrophages with upregulating MCP-1, which is a hallmark of renal dysfunction.49 These findings suggest new therapeutic approaches for chronic kidney diseases and other granulomatous diseases whose pathological processes are not fully defined and whose therapies are lacking (such as sarcoidosis and Wegener's granulomatosis). Further studies are needed to show that inhibiting HMGB1 is beneficial for treating renal and other granulomatous diseases.
References
Williams GT, Williams WJ . Granulomatous inflammation—a review. J Clin Pathol 1983;36:723–733.
Kunkel SL, Chensue SW, Strieter RM, et al. Cellular and molecular aspects of granulomatous inflammation. Am J Respir Cell Mol Biol 1989;1:439–447.
Lu B, Rutledge BJ, Gu L, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med 1998;187:601–608.
Jones ML, Warren JS . Monocyte chemoattractant protein 1 in a rat model of pulmonary granulomatosis. Lab Invest 1992;66:498–503.
Kunkel SL, Strieter RM, Lukacs N, et al. Initiation and maintenance of the granulomatous response. Chest 1993;103:135S–137S.
Goodwin GH, Sanders C, Johns EW . A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur J Biochem 1973;38:14–19.
Muller S, Ronfani L, Bianchi ME . Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med 2004;255:332–343.
Bianchi ME . Significant (re)location: how to use chromatin and/or abundant proteins as messages of life and death. Trends Cell Biol 2004;14:287–293.
Ulloa L, Messmer D . High-mobility group box 1 (HMGB1) protein: friend and foe. Cytokine Growth Factor Rev 2006;17:189–201.
Scaffidi P, Misteli T, Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002;418:191–195.
Bianchi ME, Manfredi A . Chromatin and cell death. Biochim Biophys Acta 2004;1677:181–186.
Lotze MT, Tracey KJ . High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat rev 2005;5:331–342.
Jiang W, Li J, Gallowitsch-Puerta M, et al. The effects of CpG DNA on HMGB1 release by murine macrophage cell lines. J Leukoc Biol 2005;78:930–936.
Riedemann NC, Guo RF, Ward PA . The enigma of sepsis. J Clin Invest 2003;112:460–467.
Fiuza C, Bustin M, Talwar S, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 2003;101:2652–2660.
Wang H, Bloom O, Zhang M, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999;285:248–251.
Ito T, Kawahara K, Nakamura T, et al. High-mobility group box 1 protein promotes development of microvascular thrombosis in rats. J Thromb Haemost 2007;5:109–116.
Abraham E, Arcaroli J, Carmody A, et al. HMG-1 as a mediator of acute lung inflammation. J Immunol 2000;165:2950–2954.
Ueno H, Matsuda T, Hashimoto S, et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med 2004;170:1310–1316.
Taniguchi N, Kawahara K, Yone K, et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum 2003;48:971–981.
Inoue K, Kawahara K, Biswas KK, et al. HMGB1 expression by activated vascular smooth muscle cells in advanced human atherosclerosis plaques. Cardiovasc Pathol 2007;16:136–143.
Kalinina N, Agrotis A, Antropova Y, et al. Increased expression of the DNA-binding cytokine HMGB1 in human atherosclerotic lesions: role of activated macrophages and cytokines. Arterioscler, Thromb, Vasc Biol 2004;24:2320–2325.
Yokozawa T, Zheng PD, Oura H, et al. Animal model of adenine-induced chronic renal failure in rats. Nephron 1986;44:230–234.
Calogero S, Grassi F, Aguzzi A, et al. The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet 1999;22:276–280.
Taniguchi N, Yoshida K, Ito T, et al. Stage-specific secretion of HMGB1 in cartilage regulates endochondral ossification. Mol Cell Biol 2007;27:5650–5663.
Lan HY, Bacher M, Yang N, et al. The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J Exp Med 1997;185:1455–1465.
Heid CA, Stevens J, Livak KJ, et al. Real time quantitative PCR. Genome Res 1996;6:986–994.
Kawahara K, Setoyama K, Kikuchi K, et al. HMGB1 release in co-cultures of porcine endothelial and human T cells. Xenotransplantation 2007;14:636–641.
Yamada S, Inoue K, Yakabe K, et al. High mobility group protein 1 (HMGB1) quantified by ELISA with a monoclonal antibody that does not cross-react with HMGB2. Clin Chem 2003;49:1535–1537.
Chevalier RL . Obstructive nephropathy: towards biomarker discovery and gene therapy. Nat Clin Pract 2006;2:157–168.
Bell CW, Jiang W, Reich CF, et al. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol 2006;291:C1318–C1325.
Flory CM, Jones ML, Warren JS . Pulmonary granuloma formation in the rat is partially dependent on monocyte chemoattractant protein 1. Lab Invest 1993;69:396–404.
Chensue SW, Warmington KS, Lukacs NW, et al. Monocyte chemotactic protein expression during schistosome egg granuloma formation. Sequence of production, localization, contribution, and regulation. Am J Pathol 1995;146:130–138.
Hori O, Brett J, Slattery T, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem 1995;270:25752–25761.
Park JS, Svetkauskaite D, He Q, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 2004;279:7370–7377.
Oldfield MD, Bach LA, Forbes JM, et al. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J Clin Invest 2001;108:1853–1863.
Anders HJ, Banas B, Schlondorff D . Signaling danger: toll-like receptors and their potential roles in kidney disease. J Am Soc Nephrol 2004;15:854–867.
Chung AC, Zhang H, Kong YZ, et al. Advanced Glycation End-Products Induce Tubular CTGF via TGF-{beta}-Independent Smad3 Signaling. J Am Soc Nephrol, AQ1print copy in press 2009 Dec. Available from: URL: http://jasnasnjournals.org/cgi/rapidpdf/ASN.2009010018v1.
Abel M, Ritthaler U, Zhang Y, et al. Expression of receptors for advanced glycosylated end-products in renal disease. Nephrol Dial Transplant 1995;10:1662–1667.
Fenhalls G, Squires GR, Stevens-Muller L, et al. Associations between toll-like receptors and interleukin-4 in the lungs of patients with tuberculosis. Am J Respir Cell Mol Biol 2003;29:28–38.
Campo I, Morbini P, Zorzetto M, et al. Expression of receptor for advanced glycation end products in sarcoid granulomas. Am J Resp Crit Care Med 2007;175:498–506.
Liu-Bryan R, Scott P, Sydlaske A, et al. Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum 2005;52:2936–2946.
Iyonaga K, Suga M, Ichiyasu H, et al. Measurement of serum monocyte chemoattractant protein-1 and its clinical application for estimating the activity of granuloma formation in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 1998;15:165–172.
Boitelle A, Gosset P, Copin MC, et al. MCP-1 secretion in lung from nonsmoking patients with coal worker's pneumoconiosis. Eur Respir J 1997;10:557–562.
Tian W, Zhang Z, Cohen DM . MAPK signaling and the kidney. Am J Physiol Renal Physiol 2000;279:F593–F604.
Fan J, Li Y, Levy RM, et al. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J Immunol 2007;178:6573–6580.
Park JS, Arcaroli J, Yum HK, et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol 2003;284:C870–C879.
Bruchfeld A, Qureshi AR, Lindholm B, et al. High Mobility Group Box Protein-1 correlates with renal function in chronic kidney disease (CKD). Mol Med 2008;14:109–115.
Grandaliano G, Gesualdo L, Ranieri E, et al. Monocyte chemotactic peptide-1 expression in acute and chronic human nephritides: a pathogenetic role in interstitial monocytes recruitment. J Am Soc Nephrol 1996;7:906–913.
Acknowledgements
We thank Y Motomiya and T Yokozawa for their valuable advice. We thank S Yamada for providing us both recombinant HMGB1 protein and antibody. We thank N Miura, N Uto, T Nagasato, H Sameshima, T Kodama, M Souda, Y Jitouho, H Kawaguchi, S Kato, T Orichi, K Mougi, S Yunoue, and N Tanabe for their excellent technical assistance. This work was supported by the Health and Labor Sciences Research Grant from the Ministry of Health, Labor, and Welfare (to IM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
All of the authors have no financial conflict of interest.
Supplementary information
Rights and permissions
About this article
Cite this article
Oyama, Y., Hashiguchi, T., Taniguchi, N. et al. High-mobility group box-1 protein promotes granulomatous nephritis in adenine-induced nephropathy. Lab Invest 90, 853–866 (2010). https://doi.org/10.1038/labinvest.2010.64
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/labinvest.2010.64
Keywords
This article is cited by
-
Chronic kidney disease induces a systemic microangiopathy, tissue hypoxia and dysfunctional angiogenesis
Scientific Reports (2018)
-
Urinary MCP-1、HMGB1 increased in calcium nephrolithiasis patients and the influence of hypercalciuria on the production of the two cytokines
Urolithiasis (2017)
-
Urine levels of HMGB1 in Systemic Lupus Erythematosus patients with and without renal manifestations
Arthritis Research & Therapy (2012)
-
HMGB1 in Development and Diseases of the Central Nervous System
Molecular Neurobiology (2012)
-
High mobility group box 1 (HMGB1) and anti-HMGB1 antibodies and their relation to disease characteristics in systemic lupus erythematosus
Arthritis Research & Therapy (2011)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}