
Abstract
This review focuses on three selected topics of current interest that are related to chronic fibrosing lung disorders and are important for pathologists. First, the clinical and pathologic features of smoking-related interstitial fibrosis (SRIF) are highlighted. SRIF is a common finding in smokers that has striking histologic changes but only mild associated clinical manifestations. It is characterized by marked alveolar septal fibrosis composed of a distinct form of hyalinized collagen deposition. The process is present mainly in subpleural and centrilobular parenchyma and is associated with emphysema and respiratory bronchiolitis. Second, important aspects of the pathogenesis and treatment of usual interstitial pneumonia (UIP) are reviewed. The current theory proposes that UIP is caused by tiny foci of acute lung injury (manifest pathologically by fibroblast foci) that occur and recur in the interstitium over many years. Inflammation may be present as a secondary phenomenon, but is not the primary cause, and therefore anti-inflammatory agents have little effect. The recurrent injury leads to permanent fibrosis, through a process that is considered to represent a form of abnormal wound healing. Multiple therapies have been attempted that are aimed largely at interrupting the fibrosing process, but none have been successful. The cause of the injury is unknown, but a role for aspiration due to gastroesophageal reflux is a popular current theory, and there is some evidence that anti-reflux therapy may be beneficial. Genetic predisposition has been implicated in the etiology of familial cases, and there is evidence that telomere shortening may be important in sporadic cases. Third, the use of transbronchial biopsy (TBB) in diagnosing UIP is reviewed. TBB can provide a surprising amount of information and is especially useful in certain situations, such as elderly or very sick patients in whom surgical lung biopsy carries increased morbidity and mortality.
Similar content being viewed by others
Main
The classification of the idiopathic interstitial pneumonias has been widely publicized in recent years and includes seven entities according to the ATS/ERS consensus statement of 20021 and four in the more concise classification that I prefer (Table 1).2 The clinical and pathologic features of these entities have been extensively reviewed elsewhere.1, 2, 3, 4, 5 This presentation concentrates instead on selected topics related to relatively new and sometimes controversial aspects of chronic fibrosing lung disorders.
Usual interstitial pneumonia (UIP) and non-specific interstitial pneumonia (NSIP) are the most common types of chronic interstitial fibrosis of unknown cause. There are other conditions that may also result in chronic interstitial fibrosis, and they are important to recognize because of differences in treatment and prognosis. Examples include scarred sarcoidosis, chronic hypersensitivity pneumonia, scarred Langerhans cell histiocytosis, and the recently described smoking-related interstitial fibrosis (SRIF).6 This review will present the pathologic features of SRIF with emphasis on its distinction from other types of fibrosing interstitial lung diseases, especially UIP and NSIP. Additional topics include an update on pathogenesis and therapy of UIP, and the use of transbronchial biopsy (TBB) in UIP.
Smoking-related interstitial fibrosis
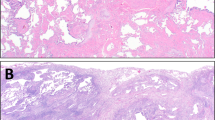
SRIF is a distinct form of chronic interstitial fibrosis that is common in cigarette smokers.6 It is characterized by uniform thickening of alveolar septa by collagen deposition with minimal associated inflammation (Figure 1). The process is accentuated in subpleural parenchyma where it is usually associated with emphysema, but affects deeper portions of parenchyma as well, especially in centrilobular areas. Emphysema is present in all cases, and is often a striking finding (Figure 2), but the fibrosis can also be found in non-emphysematous parenchyma (Figure 3). Typically, the fibrosis has a distinct hyalinized quality, with the collagen appearing in thick, ropey bundles, and there is scant, if any, associated inflammation (Figure 4). Clusters of smooth muscle fibers are often embedded within the collagen bundles. Fibroblast foci are occasionally seen, but usually only focally. Likewise, honey-comb change is generally not a feature, although rarely there are small poorly developed foci that likely are non-specific changes in the lung periphery. Respiratory bronchiolitis (RB), which is manifestation of cigarette smoking, not surprisingly, accompanies the fibrosis in all cases (Figure 5).7

SRIF. (a) Low magnification showing uniform appearing interstitial fibrosis involving subpleural parenchyma (pleural surface is at top). (b) High magnification of area from panel a showing dense collagen deposition within alveolar septa. (c) Low magnification showing centrilobular accentuation of the interstitial fibrosis which at higher magnification (d) is seen to be composed of relatively acellular collagen deposition. Mild associated emphysema is present in both fields.

SRIF associated with emphysema. (a) Low magnification shows the marked interstitial fibrosis associated with prominent centrilobular emphysema. Higher magnification (b) shows the typical hyalinized collagen of SRIF surrounding the enlarged emphysematous airspaces. (c) Low magnification shows SRIF associated with prominent emphysema in subpleural parenchyma. (d) High magnification shows distinct collagen deposition around the emphysematous spaces. Note, also, increased numbers of alveolar macrophages within some airspaces (RB, respiratory bronchiolitis).

SRIF involving non-emphysematous parenchyma. (a) Low magnification showing uniform appearing interstitial fibrosis surrounding normal sized airspaces without evidence of emphysema. (b) High magnification illustrating bland appearing collagen deposition within alveolar septa from same area. Lightly pigmented macrophages (RB) are prominent within airspaces.

Characteristic hyalinized collagen deposition in SRIF. (a) In this example, there is hyalinized collagen deposition with no associated inflammation. Lightly pigmented macrophages are present in adjacent airspaces. (b) Another example with typical collagen deposition along with a mild chronic inflammatory cell infiltrate. Intra-alveolar pigmented macrophages are prominent in adjacent airspaces.

SRIF and respiratory bronchiolitis (RB). (a) Low magnification showing RB with clusters of intra-alveolar pigmented macrophages in background of SRIF. (b) Higher magnification of the same area showing mucin and pigmented macrophages within an alveolar duct and surrounding airspaces that are characteristic of RB. Note the associated uniform and bland appearing interstitial fibrosis typical of SRIF. (c) Another case illustrating accumulation of pigmented macrophages within airspaces associated with the typical hyalinized alveolar septal fibrosis (d).
In a recent detailed histologic analysis of lobectomy specimens from smokers, we identified SRIF in 45% (9/20 cases).6 The mean age was 65 with a range from 52 to 81. Half were current and half were ex-smokers with mean pack years smoked of 39 and 38, respectively. Interestingly, neither pulmonary function abnormalities nor radiographic findings were identified that could be attributed to SRIF, although the lack of radiographic findings may be related to the fact that high-resolution CT examination was performed in only one case. Likewise, the fact that emphysema was present histologically in all patients may contribute to the absence of restrictive pulmonary function tests. Although follow-up was short (2–27 months, mean 16), there was no evidence of progressive interstitial lung disease in any patient.
Similar findings to SRIF were previously described by Yousem8 in biopsy specimens from nine smokers. He termed the changes ‘RB-associated interstitial lung disease with fibrosis’, and he also found similar changes in 4 of 30 (14%) lobectomy specimens from asymptomatic smokers with lung cancer. The mean age of the biopsied patients (44) was younger than those in our study with a range from 32 to 68, and the average pack years smoked (38) was identical to our patients. High-resolution CT exams were done in all cases, with bilateral micronodular infiltrates and ground glass opacities being the most common findings. Most patients presented with chronic shortness of breath and/or cough. Although follow-up was short (average 3.2 years, range 0.5–5.2 years), all patients were alive, and disease was stable in most (7/9). Progressive disease was noted in only two.
Kawabata et al9 examined 587 lobectomy specimens from smokers and correlated several pathologic findings with smoking index. They described airspace enlargement with fibrosis, which seems similar to SRIF in 6.5% of mild smokers and 21.1% of heavy smokers. This study, however, was based mainly on gross examination of specimens, with histologic corroboration in only a small proportion, and the findings, therefore, are difficult to compare.
Since SRIF is so common in smokers, yet interstitial lung disease so infrequently identified clinically, it seems likely that SRIF is masked by or has been included among other smoking-related disorders. Although the term, chronic obstructive pulmonary disease (COPD), classically encompasses cases of emphysema and chronic bronchitis, the term is often loosely applied to worsening respiratory function due to any cause in smokers and may include SRIF. Cases of combined emphysema and pulmonary fibrosis have been described in smokers, and most appear to represent the simultaneous occurrence of emphysema and UIP as evidenced by classic HRCT findings. The histologic findings in a few reported cases, however, have been labeled as ‘variant desquamative interstitial pneumonia (DIP)’,10 DIP, or unclassified interstitial pneumonia,11 and the photomicrographs from one such case appear identical to SRIF.10 It seems likely, therefore, that at least some cases of SRIF have been previously diagnosed as DIP, and it is also likely that some have been diagnosed as fibrosing NSIP. No doubt, a few cases have also been diagnosed as UIP.
Recently, several large radiographic studies have reviewed CT findings in smokers being followed for other reasons. Lederer et al12 reported functional and CT findings in 2563 smokers being screened for cardiac disease. Spirometric restriction was observed in 10% and increased with increasing pack years smoked. Areas of high attenuation on CT reflecting ground glass density and reticular opacities were noted in 2.2% and likewise increased with pack years smoked. Washko et al13 noted interstitial abnormalities on HRCT in 8% of 2416 smokers who were enrolled in a study of COPD. The incidence of radiographic abnormalities was influenced by the amount of smoking and the current smoking status. Sverzellati et al14 reviewed HRCT findings in 692 smokers being screened for lung cancer and found abnormalities in 158 (23%), including 3% with UIP-like changes, 3.8% with other interstitial pneumonia patterns, 15.7% with RB, and 3% with indeterminate changes. It seems likely that as subtle interstitial changes are becoming increasingly recognizable by improved radiographic techniques, lung biopsy may be utilized more often to clarify the process. Pathologists, therefore, need to be aware of SRIF in order to distinguish this seemingly relatively benign form of interstitial fibrosis from more ominous forms, mainly UIP and fibrosing NSIP.
Distinction of SRIF from UIP and Fibrosing NSIP
UIP is an inexorably progressive chronic fibrosing process with high mortality rates, a median survival of only 2–3 years, and no known effective therapy.5 Although, overall, NSIP has a significantly better prognosis than UIP with median survival of 9–13 years,15, 16 when fibrosis is prominent (fibrosing NSIP) the median survival decreases to <5 years, and when the fibrosis is severe, mortality rates approach that of UIP.17 Therefore, it is important to separate SRIF which has a relatively benign course from both UIP and fibrosing NSIP.
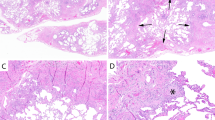
The main differentiating features among SRIF, UIP, and fibrosing NSIP are summarized in Table 2. The appearance of the collagen deposition in SRIF is the single most important feature in differentiating this entity from UIP, NSIP, and other fibrosing interstitial processes. In SRIF, the collagen has a deeply eosinophilic, hyalinized quality, and is composed of thick, ropey cords. In contrast, the collagen in other fibrosing conditions is lighter staining and lacks the dense hyalinized quality and the thick cords. The presence of this distinct hyalinized collagen is a prerequisite for diagnosing SRIF. A number of other features specific to UIP and NSIP further aid in their differentiation from SRIF. UIP is characterized by the combination of a patchwork pattern of lung involvement by the fibrosis, temporal variegation in which there is a mixture of active fibrosis (fibroblast foci) and collagen deposition, and architectural distortion with either parenchymal scarring or honey-comb change (Figure 6). In contrast, the fibrosis in SRIF involves the lung in a more even or uniform configuration, fibroblast foci are usually not present, and well-formed honey-comb change is usually absent. The fibrosis in SRIF preferentially affects subpleural parenchyma where it usually is combined with emphysema, whereas the distribution in UIP is random, and emphysema is usually not present.

Characteristic histologic features of UIP. (a) Low magnification showing the heterogeneous appearing patchwork pattern of parenchymal involvement with areas of interstitial scarring alternating with islands of normal lung. Temporal variability is indicated by the presence of a fibroblast focus (arrow). (b) Higher magnification of the fibroblast focus shown in panel a. There is the characteristic proliferation of spindle cells within myxoid stroma. The cells are arranged with their long axis parallel to the long axis of the interstitium, and their luminal surface is covered by epithelium. (c) Another area from the same case showing honey-comb change (top) alternating with normal lung (bottom). The honey-comb areas are characterized by enlarged airspaces lined by bronchiolar epithelium and containing a mucinous exudate (shown at higher magnification in (d)).
Fibrosing NSIP has a fairly uniform lung involvement without significant architectural distortion or temporal variability, and in this way is similar to SRIF (Figure 7). However, neither the subpleural distribution nor the association with emphysema that are typical of SRIF is seen. Furthermore, the interstitial fibrosis of NSIP appears less dense and lacks the characteristic hyalinized quality of SRIF, and some degree of chronic inflammation is invariably admixed with the fibrosis.

Contrasting features of fibrosing NSIP (a, b) and SRIF (c, d). At low magnification (a) NSIP is characterized by uniform, mild thickening of alveolar septa without background emphysema. At high magnification (b) the fibrosis is composed of lightly staining, delicate collagen deposition along with mild chronic inflammation. The appearance contrasts strikingly with the more advanced and severe fibrosis of SRIF (c), which is composed mainly of dense, hyalinized collagen deposition with minimal inflammation (d). Emphysema and RB are additionally present in SRIF.
Current concepts of pathogenesis and treatment of UIP
For many years, UIP was considered to be a primarily inflammatory disease that began with an ‘alveolitis’ (Table 3). According to this theory, a mixture of neutrophils and macrophages within alveoli released cytokines and other substances that attracted fibroblasts and stimulated them to produce collagen with subsequent development of fibrosis. This theory, however, failed to explain why only a minority of UIP patients responded to anti-inflammatory agents. Subsequently, in the mid 1990s when NSIP was separated from UIP, it was recognized that the inclusion of NSIP among the previously diagnosed UIP cases accounted for most of the responses to anti-inflammatory therapy, while veritable UIP cases failed to respond. This observation along with recognition of the importance of fibroblast foci in diagnosing UIP led to the current theory that acute lung injury is the primary event in the pathogenesis of UIP.2, 18, 19
Fibroblast foci are small aggregates of fibroblasts and myofibroblasts that are present within myxoid stroma along the interstitium (Figure 8).2 The spindle-shaped cells are arranged in layers parallel to the long axis of the alveolar septa beneath an epithelial lining composed of either hyperplastic type 2 pneumocytes or ciliated bronchiolar epithelium. Ultrastructural and immunohistochemical studies have shown that fibroblast foci represent a reaction to an area of acute lung injury. The process is analogous to diffuse alveolar damage (DAD), except that it involves only microscopic foci of parenchyma in contrast to larger, macroscopic areas in DAD. It is thought these minute foci of acute lung injury occur and recur over many years and eventually lead to irreversible fibrosis. A popular ancillary theory is that the subsequent development of fibrosis represents a form of abnormal wound healing related to abnormalities in fibroblast regulation. There is extensive ongoing investigation into the molecular mechanisms leading to the development of fibrosis that are beyond the scope of this review, and these are summarized elsewhere.18, 19 Although inflammation is no longer considered the primary event in the development of UIP, it is important to remember that some degree of inflammation does occur as a secondary phenomenon, and it may further drive the development of fibrosis.

Fibroblast focus. (a) Low magnification shows lightly staining fibroblast focus that is easily visible within background collagen in an area of honey-comb change. (b) Higher magnification showing spindle-shaped fibroblasts and myofibroblasts within lightly staining stroma. The cellular area contrasts sharply with adjacent collagen deposition (top), and the luminal surface is covered by alveolar epithelial lining cells.
While focal acute lung injury is widely accepted in the pathogenesis of UIP, the etiology of the acute lung injury remains elusive. Various factors have been postulated, such as cigarette smoking, autoimmunity, toxic inhalants, and gastroesophageal reflux disease, for example, but these entities are common and very few individuals develop UIP.18, 19, 20, 21 Mutations in TERT and TERC genes, and in genes coding for surfactant proteins A and C, have been detected in up to 20 and 3% of familial UIP patients, respectively, and telomere shortening has been noted in about one fourth of sporadic UIP in the absence of detectable genetic mutations.22, 23, 24, 25, 26, 27 It has been proposed that these abnormalities may predispose the lung epithelium to acute injury from environmental exposures that ordinarily would not be damaging in persons with normal genetic composition.23
Given the current focus on acute lung injury and abnormal wound healing in the pathogenesis of UIP, there are several potential treatment approaches, as summarized in Table 4.5 Several drugs have been utilized to interrupt the mechanism of fibrosis, but to date none have had proven efficacy. Another approach is to remove or neutralize the source of injury, and anti-oxidants have been tried. Some studies suggest that protein pump inhibitors and fundoplication may be useful, but the data are not all in yet.20, 21 Immunosuppressives are still being utilized in some cases, but supporting evidence is scant. There has been some success with treating secondary complications of UIP, such as cough and pulmonary hypertension, but such treatment has not prolonged life. Unfortunately, so far no treatment has been proven to be effective, short of lung transplant that currently offers the only possibility of cure.
Role of TBB in diagnosing UIP
Traditionally, pathologists and clinicians alike have discounted a role for TBB in diagnosing UIP. Both the ATS/ERS consensus and ‘evidence-based’ statements from 20021 and 20115, respectively, echo a similar sentiment against the use of TBB in diagnosing UIP. Although there is little data in the literature on the topic, some pathologists have suggested that ‘collective wisdom be followed and TBB not be used to diagnose IPF/UIP’,28 while some clinicians have suggested that the use of TBB in diagnosing UIP is ‘a step backward in disease management’.29 These negative attitudes are fueled partially by unwillingness of pathologists to consider that a small biopsy specimen can yield useful information and partially on misconceptions of clinicians about the types of information that can be expected on TBB. For example, it is generally not understood that the patchwork pattern typical of UIP can be appreciated on TBB, or that honey-comb change can be recognized. Figure 9 illustrates that, in fact, all the histologic pathologic changes of UIP can be present in even a single piece of lung sampled by TBB. While, admittedly, this situation is not common, when multiple pieces of lung tissue are provided, the characteristic histologic findings may be found surprisingly often. The fact that fibrosis and chronic inflammation are common non-specific findings in peribronchial parenchyma is often used as an argument against TBB in the setting of UIP. While it is true that fibrosis and chronic inflammation are common non-specific findings, when the fibrosis shows temporal variegation with fibroblast foci in addition to collagen deposition the findings become more useful.

Transbronchial biopsy specimen from UIP. This single tissue fragment obtained by TBB contains all the characteristic features necessary for diagnosing UIP. At low magnification (a) the typical variegated, patchwork pattern can be appreciated. There is a focus of honey-comb change (shown at high magnification in (b)), a fibroblast focus (arrow, and high magnification in (c)), and an area of relatively normal interstitium (arrowhead, and high magnification in (d)).
There are two papers in the literature that address the question about the usefulness of TBB in diagnosing UIP. Both are retrospective studies. Berbescu et al30 examined 22 cases and found changes diagnostic of or consistent with UIP in 9 (43%) and concluded that TBB may be more useful than generally appreciated. In contrast, Shim et al31 reviewed 32 patients and found changes consistent with UIP in only 3 (9.4%) and concluded that TBB is not more useful than generally appreciated. They suggested, however, that TBB may be helpful in elderly or weak individuals who cannot undergo surgical lung biopsy.
It has been our experience that, in the right clinical setting and with appropriate tissue sampling, TBB can support a diagnosis of UIP fairly often. It is imperative, however, that the pathologist be provided both a clinical history and a description of the HRCT findings. TBB should not be used to diagnose UIP in unusual clinical settings, such as young patients under 40, for example, or in individuals with unusual radiographic findings. Since HRCT findings are diagnostic by themselves without biopsy confirmation in about 50% of patients with UIP, one might question whether TBB can be used at all in the 50% of patients with ‘atypical’ HRCT findings who need biopsy confirmation. Unfortunately, there is no right and wrong answer to this question and each case needs to be individually assessed with regard to the overall clinical situation. It has been our experience, however, that many clinicians still perform surgical lung biopsies to confirm the diagnosis even though patients may have characteristic HRCT findings. Many such patients are elderly or have advanced fibrosis with respiratory failure. In my opinion, this is the setting in which TBB has the greatest utility, in that it can obviate surgical lung biopsy in these very sick individuals in whom there is significant mortality and morbidity from surgical lung biopsy.
References
American Thoracic Society/European Respiratory Society. International multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002;165:277.
Katzenstein A-LA . Surgical Pathology of Non-Neoplastic Lung Disease. Elsevier-Saunders: Philadelphia, 2006.
Katzenstein A-LA, Mukhopadhyay S, Myers JL . Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Hum Pathol 2008;39:1562.
Myers JL, Katzenstein A-LA . Beyond a consensus classification for idiopathic interstitial pneumonias: Progress and controversies. Histopathology 2009;54:90.
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824.
Katzenstein A-LA, Mukhopadhyay S, Zanardi C, Dexter E . Clinically occult interstitial fibrosis in smokers: classification and significance of a surprisingly common finding in lobectomy specimens. Hum Pathol 2010;41:316.
Fraig M, Shreesha U, Savici D, Katzenstein A-LA . Respiratory bronchiolitis. A clinicopathologic study in current smokers, ex-smokers, and never-smokers. Am J Surg Pathol 2002;26:647.
Yousem SA . Respiratory bronchiolitis-associated interstitial lung disease with fibrosis is a lesion distinct from fibrotic nonspecific interstitial pneumonia: a proposal. Mod Pathol 2006;19:1474.
Kawabata Y, Hoshi E, Murai K, et al. Smoking-related changes in the background lung of specimens resected for lung cancer: A semiquantitative study with correlation to postoperative course. Histopathology 2008;53:707.
Cottin V, Nunes H, Brillet P-Y, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J 2005;26:586.
Jankowich MD, Polsky M, Klein M, Rounds S . Heterogeneity in combined pulmonary fibrosis and emphysema. Respiration 2008;75:411.
Lederer DJ, Enright PL, Kawut SM, et al. Cigarette smoking is associated with subclinical parenchymal lung disease. The multi-ethnic study of atherosclerosis (MESA)-lung study. Am J Respir Crit Care Med 2009;180:407–414.
Washko GR, Hunninghake GM, Fernandez IE, et al. Lung volumes and emphysema in smokers with interstitial lung abnormalities. New Engl J Med 2011;364:897–906.
Sverzellati N, Guerci L, Giorgia R, et al. Interstitial lung diseases in a lung cancer screening trial. ERJ Express. Eur Respir J 2011;38:392–400.
Katzenstein A-LA, Fiorelli RF . Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol 1994;18:136.
Travis WD, Hunninghake G, King Jr TE, et al. Idiopathic nonspecific interstitial pneumonia. Report of an American Thoracic Society project. Am J Respir Crit Care Med 2008;177:1338.
Latsi PI, du Bois RM, Nicholson AG, et al. Fibrotic idiopathic interstitial pneumonia. The prognostic value of longitudinal functional trends. Am J Respir Crit Care Med 2003;168:531.
Coward WR, Saini G, Jenking G . The pathogenesis of idiopathic pulmonary fibrosis. Ther Adv Respir Dis 2010;4:367.
Strieter RM, Mehrad B . New mechanisms of pulmonary fibrosis. Chest 2009;136:1364.
Lee JS, Ryu JH, Elicker BM, et al. Gastroesophageal reflux therapy is associated with longer survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011, doi:10.1164/rccm.201101-0138OC; advance online publication 23 June 2011.
Raghu G, Yang ST-Y, Spada C, et al. Sole treatment of acid gastroesophageal reflux in idiopathic pulmonary fibrosis. A case series. Chest 2006;129:794.
Alder JK, Chen JJ-L, Lancaster L, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. PNAS 2008;105:13051.
Crossno PF, Polosukhin VV, Blackwell TS . Identification of early interstitial lung disease in an individual with genetic variations in ABCA3 and SFTPC. Chest 2010;137:969.
Diaz de Leon A, Cronkhite JT, Katzenstein A-LA, et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS One 2010;5:e10680.
Garcia CK . Idiopathic Pulmonary Fibrosis. Update on genetic discoveries. Proc Am Thorac Soc 2011;8:158–162.
van Moorsel CHM, van Oosterhout MFM, Barlo NP, et al. Surfactant protein mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a Dutch cohort. Am J Respir Crit Care Med 2010;182:1419.
Wang Y, Kuan PJ, Xing CX, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet 2009;84:52.
Churg A . Transbronchial biopsy and usual interstitial pneumonia. A new paradigm? Chest 2006;129:1117–1118.
Mukherjee S, Spiteri M . Transbronchial biopsy and usual interstitial pneumonia. A step backward in disease management? Chest 2006;130:1628.
Berbescu EA, Katzenstein A-LA, Snow JL, Zisman DA . Transbronchial biopsy in usual interstitial pneumonia. Chest 2006;129:1126.
Shim HS, Park MS, Park IK . Histopathologic findings of transbronchial biopsy in usual interstitial pneumonia. Pathol Int 2010;60:373.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Rights and permissions
About this article
Cite this article
Katzenstein, AL. Smoking-related interstitial fibrosis (SRIF), pathogenesis and treatment of usual interstitial pneumonia (UIP), and transbronchial biopsy in UIP. Mod Pathol 25 (Suppl 1), S68–S78 (2012). https://doi.org/10.1038/modpathol.2011.154
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.2011.154
Keywords
This article is cited by
-
Effect of the 2020 hypersensitivity pneumonitis guideline on the pathologic diagnosis of interstitial pneumonia
Scientific Reports (2023)
-
Pulmonary dust foci as rat pneumoconiosis lesion induced by titanium dioxide nanoparticles in 13-week inhalation study
Particle and Fibre Toxicology (2022)
-
Diffuse smoking-related lung diseases: insights from a radiologic-pathologic correlation
Insights into Imaging (2019)
-
Combined pulmonary fibrosis and emphysema predicts recurrence following surgery in patients with stage I non-small cell lung cancer
Medical Oncology (2018)
-
Smoking-related lung abnormalities on computed tomography images: comparison with pathological findings
Japanese Journal of Radiology (2018)
