
Abstract
Myocardial infarction-associated transcript (MIAT), an intergenic long noncoding RNA (lncRNA), is conserved between rodents and humans and is directly linked to maladaptive cardiac remodeling in both patients and mouse models with various forms of heart failure (HF). We previously reported attenuation of cardiac stress, apoptosis, and fibrosis in a murine model of myocardial infarction (MI) with global MIAT ablation. Our transcriptomic profiling and mechanistic studies further revealed MIAT-induced activation of maladaptive genes, such as Hoxa4, Fmo2, Lrrn4, Marveld3, and Fat4. However, the source of MIAT and its contribution to MI and HF remain unknown. In this study, we generate a novel cardiomyocyte (CM)-specific MIAT conditional knockout mouse model, which exhibits improved cardiac function after MI. We further report that CM-specific MIAT ablation is sufficient to reduce cardiac damage, apoptosis, and fibrosis following chronic MI. Mechanistically, CM-specific MIAT deletion in mice leads to decreased expression of proapoptotic and pathological profibrotic genes, such as p53, Bak1, Col3a1, Col6a1, Postn, and Snail1 after chronic MI. These results enable us to begin to dissect cell-specific contributions to MIAT signaling and bolster the idea that MIAT plays a direct pathological role in CMs after MI.
Similar content being viewed by others

Introduction
Because cardiomyocyte (CM) renewal is largely absent in adult mammalian hearts, damaged and dead CMs resulting from ischemic stress are constantly replaced by scar tissue, leading to progressive left ventricular (LV) remodeling and chronic heart failure (HF) [1, 2]. Although the significance of CM dropout in myocardial infarction (MI) has been established, identification of the specific effectors underlying CM loss remains incomplete.
Long noncoding RNAs (lncRNAs) are a class of noncoding RNAs over 200 nucleotides in length that have emerged as important regulators of HF [3, 4]. Crosstalk among lncRNAs, microRNAs (small noncoding RNAs; miRNAs or miRs), and messenger RNAs (mRNAs) represents an important regulatory mechanism underlying the pathogenesis of HF. Specifically, lncRNAs function as competing endogenous RNAs (ceRNAs) that sponge miRs, thus activating target mRNAs of miRs [5]. Clinical applications of lncRNAs in HF have also been reported [6, 7].
In previous work, we observed that a lncRNA called myocardial infarction-associated transcript (MIAT) downregulated carvedilol/β1-adrenergic receptor/β-arrestin-responsive miR-150-5p (miR-150) in mouse hearts [8]. Using global knockout (KO) and transgenic (TG) mouse models, we demonstrated that MIAT gain-of-function exacerbated maladaptive post-MI remodeling whereas MIAT loss-of-function protected hearts against MI. Additionally, miR-150 overexpression attenuated maladaptive post-MI remodeling caused by MIAT [8], directly establishing their in vivo functional relationship during HF. These results suggest that MIAT is a functionally important upstream negative regulator of miR-150 in the heart. Furthermore, MIAT was upregulated in mouse models of MI, angiotensin II (AngII)- and isoproterenol (ISO)-induced cardiac hypertrophy, and diabetic cardiomyopathy [9,10,11,12,13]. Lentivirus-mediated systemic knockdown of MIAT in rodents improved cardiac function and structure in post-MI [11] and in diabetic hearts [9] as well as reduced ischemia/reperfusion (I/R)-induced myocardial infarct size and apoptosis [10]. Systemic MIAT loss in mice also attenuated AngII- and transverse aortic constriction (TAC)-induced HF, in part, by blunting a CM hypertrophic gene program and enhancing CM contractility [14]. Our unbiased transcriptomic profiling, filtering, validation, and mechanistic studies further revealed that the deleterious effects of MIAT in ischemic hearts were attributed to the activation of maladaptive genes, including Hoxa4, Fmo2, Lrrn4, Marveld3, and Fat4 [8]. In addition, we showed that MIAT was upregulated in CMs isolated from MI hearts [8]. MIAT was also upregulated in cultured rodent CMs subjected to AngII, ISO, high glucose (HG), or hypoxia/reoxygenation (H/R) [9,10,11,12,13]. MIAT knockdown was also shown to inhibit neonatal rat ventricular cardiomyocyte apoptosis induced by HG [9] and H/R-induced embryonic rat myoblast apoptosis [10]. Other work showed that MIAT suppressed miR-150 expression, thereby acting as a positive regulator of CM hypertrophy [12, 13]. Cumulatively, these findings establish the role of MIAT in cardiac remodeling during HF and suggest that MIAT derived from CMs could play a key cell-specific role in promoting maladaptive remodeling in HF.
Notably, a clinical study showed an association between gain-of-function single nucleotide polymorphisms in MIAT and an increased risk of MI [15, 16]. MIAT was also overexpressed in patients with Chagas cardiomyopathy [17] and was directly linked to maladaptive cardiac remodeling in patients with type 2 diabetes [18]. Of note, the mouse and human MIAT genes have almost identical genomic organization, suggesting the evolutionary conservation of MIAT’s regulation and roles. These findings underscore the clinical relevance and potential therapeutic implications of deciphering the functional requirement of MIAT in murine models of HF. However, there is a lack of definitive studies using appropriate mouse models to address cell type-specific actions of MIAT. Specifically, the role of MIAT expressed selectively in CMs in regulating the response to MI remains to be elucidated.
In the current study, we utilize a novel CM-specific MIAT conditional knockout (cKO) mouse model. As evident in our global MIAT KO mice [8], CM-specific MIAT deletion is sufficient to attenuate cardiac dysfunction as well as damage, apoptosis, and fibrosis in murine hearts following MI. These new findings provide the direct and first evidence that CM is a major cellular component responsible for the maladaptive effects mediated by MIAT post-MI. Thus, our results highlight the crucial role of CM-derived MIAT as a key mediator of ischemic HF.
Results
MIAT selectively in cardiomyocytes exacerbates cardiac dysfunction after myocardial infarction
We previously found that MIAT was upregulated in CMs isolated from MI mice [8]. Additionally, MIAT was upregulated in CMs subjected to AngII, ISO, HG, or H/R [9,10,11,12,13], and systemic MIAT knockdown inhibited rat CM apoptosis in vitro [9, 10] and cardiac apoptosis post-I/R [10]. However, the in vivo role of CM-derived MIAT in the heart remained unclear. To address this gap, we generated a novel CM-restricted MIAT cKO mouse line by breeding αMHC-Cre mice [19] with MIATfl/fl mice (Fig. 1A). We previously validated αMHC-Cre mice that induced CM-specific cKO [20]. We first successfully demonstrate a significant downregulation of MIAT expression (0.28-fold) in the left ventricles of MIAT cKO mice compared to MIATfl/fl mice (Fig. 1B). We also confirm that a known cardiac target of MIAT repression, miR-150-5p [8], is upregulated in MIAT cKO mouse hearts (Fig. 1C), whereas a known direct target of miR-150-5p repression and a proapoptotic and profibrotic marker, Sprr1a [20, 21], is downregulated in MIAT cKO mouse hearts (Fig. 1D).

A Targeting scheme, mouse crossing, and establishment of cardiomyocyte (CM)-restricted conditional knockout (cKO) of MIAT in vivo. Top, Targeting scheme and mouse crossing. Bottom, Representative genotyping results of MIATfl/fl and αMHC-Cre mice. Target (tg) and control (ctrl) bands are shown. Neg.= negative control and Pos.= positive control. B–D QRT-PCR analyses of MIAT (B), miR-150-5p (a known direct target of MIAT; C), and Sprr1a (a known direct target of miR-150-5p; D) in left ventricles from adult MIATfl/fl or MIAT cKO (i.e., MIATfl/fl;αMHC-Cre) mice. N = 3–4 per group. Data are presented as mean ± SEM. Unpaired 2-tailed t-test. *P < 0.05 or **P < 0.01 vs. MIATfl/fl mice.
Mice aged 8–16 weeks were then subjected to MI, and we subsequently evaluated post-MI cardiac function in four groups: sham and MI groups of MIATfl/fl and MIAT cKO mice. First, CM-restricted MIAT cKO mice exhibit normal cardiac function at baseline (Supplementary Table 1and Fig. 2). Despite exhibiting normal cardiac function at baseline, CM-restricted MIAT cKO mice respond differently to ischemic cardiac injury induced by permanent ligation of the left anterior descending (LAD) artery.

A–F Transthoracic echocardiography was performed on the four experimental groups (sham and MI of MIATfl/fl and CM-specific MIAT cKO) at 0–28 days (d) post-myocardial infarction (MI). Quantification of left ventricular (LV) ejection fraction (EF: A), fractional shortening (FS: B), end-diastolic volume (LVEDV: C), end-systolic volume (LVESV: D), internal diameter in diastole (LVIDd: E), and internal diameter in systole (LVIDs: F) is shown. N = 18–20 per group. Data are presented as mean ± SD. Two-way repeated-measures ANOVA with Bonferroni’s post hoc test. ***P < 0.001 vs. Sham of same genotype (denoted by different colors for sham within same group); #P < 0.05, ##P < 0.01, or ###P < 0.001 vs. MI MIATfl/fl.
At 1 week post-MI, CM-restricted MIAT cKO mice show a significant improvement in cardiac function compared to MIATfl/fl controls, as evidenced by increased ejection fraction (EF) and fractional shortening (FS) along with decreased end-systolic volume (ESV) and left ventricular internal diameter in systole (LVIDs) (Supplementary Table 2 as well as Fig. 2A, B, D, and F). This improvement persists at 2 weeks post-MI, characterized by increased EF and FS coupled with decreased ESV and LVIDs (Supplementary Table 3 as well as Fig. 2A–B, D, and F). By 4 weeks post-MI, cardiac function of CM-restricted MIAT cKO mice further improves, with significant increases in EF and FS as well as significant decreases in end-diastolic volume (EDV), ESV, left ventricular internal diameter in diastole (LVIDd), and LVIDs (Supplementary Table 4 and Fig. 2).
In contrast, MIATfl/fl control mice exhibit greater functional impairment at all time points following MI, similar to that normally seen in wild-types (Supplementary Tables 2–4 and Fig. 2). Our morphometric data also show that CM-specific MIAT cKO mice have a significant decrease in the ratio of left ventricular weight to body weight (LVW/BW) at 4 weeks after MI, compared to MIATfl/fl controls (Supplementary Table 4). CM-restricted MIAT cKO mice thus display attenuated cardiac dysfunction post-MI compared to MIATfl/fl control mice (Fig. 2 and Supplementary Tables 2–4), similar to findings observed in systemic MIAT KO mice [8]. To visualize the variability between groups and to differentiate between males and females, figures for EF, FS, ESV, and LVIDs at 4 weeks post-MI were made in a dot plot format. We observe that there is no sex difference in cardiac function within the same group (Supplementary Fig. 1), supporting our conclusion from cardiac function results as well as our unbiased and appropriate representative sample selection for downstream assays. These data collectively suggest that CM-restricted deletion of MIAT is sufficient to improve cardiac function following murine MI in both sexes.
Cardiomyocyte-restricted MIAT deletion mitigates cardiac damage and apoptosis after chronic myocardial infarction
In our previous study, we demonstrated that systemic MIAT KO mouse hearts exhibited a significant decrease in cardiac damage, apoptosis, and fibrosis during chronic MI [8]. To assess whether the ablation of MIAT specifically from CMs leads to beneficial remodeling after MI, we utilized CM-specific MIAT cKO mice and compared post-MI remodeling in these mice with that of MIATfl/fl controls.
We observe that hearts from CM-specific MIAT cKO mice display smaller and less disorganized structures compared to hearts from MIATfl/fl MI mice, as evidenced by histological analysis at 4 weeks post-MI (Fig. 3A). Consistently, mRNA levels of HF markers, such as Nppa, Nppb, and Myh7, are also significantly reduced in CM-specific MIAT cKO hearts after 4 weeks of MI (Fig. 3B–D). Furthermore, we examined the expression of proinflammatory Il-1b in hearts of CM-specific MIAT cKO mice at 4 weeks post-MI. We find decreased expression of Il-1b in these hearts compared to MIATfl/fl controls after chronic MI (Fig. 3E).

A Representative hematoxylin and eosin (H&E) staining of heart sections of the peri-ischemic border area at 4 weeks post-MI shows a decrease in disorganized structure in CM-specific MIAT cKO hearts compared to MIATfl/fl controls. Scale bars: 100μm. B–D QRT-PCR analysis of Nppa, Nppb, and Myh7 expression representing cardiac damage in ischemic areas from CM-specific MIAT cKO hearts compared to MIATfl/fl controls at 4 weeks post-MI. E QRT-PCR analysis of Il-1b expression for cardiac inflammation in ischemic areas from CM-specific MIAT cKO hearts compared to MIATfl/fl controls at 4 weeks post-MI. N = 3 per group. QRT-PCR data are shown as fold induction of gene expression normalized to Gapdh. Data are presented as mean ± SEM. Two-way ANOVA with Tukey’s multiple comparison test. *P < 0.05, **P < 0.01, or ***P < 0.001 vs. sham of same genotype; #P < 0.05 or ##P < 0.01 vs. MI MIATfl/fl.
Additionally, we investigated apoptosis in the hearts at 4 weeks post-MI using cleaved-caspase 3 staining. CM-specific MIAT cKO hearts exhibit significantly fewer cleaved caspase 3-positive cells compared to MIATfl/fl hearts after chronic MI (Fig. 4A–B). Moreover, the expression of proapoptotic p53 and Bak1 is decreased in CM-specific MIAT cKO hearts compared to controls at 4 weeks post-MI (Fig. 4C–D). Overall, these findings suggest that CM-derived MIAT plays a deleterious role in murine MI by regulating apoptosis and post-MI remodeling.

A–B Representative cleaved-caspase 3 staining images in heart sections of the peri-ischemic border area at 4 weeks post-MI (A) and quantification of apoptosis in six 20x fields (B). Scale bars: 50μm. C–D, QRT-PCR analysis of proapoptotic p53 and Bak1 expression in the ischemic areas from CM-specific MIAT cKO hearts compared to MIATfl/fl controls at post-MI 4 weeks. QRT-PCR data are shown as fold induction of gene expression normalized to Gapdh. N = 3–7 per group. Data are presented as mean ± SEM. Two-way ANOVA with Tukey’s multiple comparison test. *P < 0.05, **P < 0.01, or ***P < 0.001 vs. sham of same genotype; #P < 0.05, ##P < 0.01, or ###P < 0.001 vs. MI MIATfl/fl.
Cardiomyocyte-specific deletion of MIAT in mice reduces cardiac fibrosis following chronic myocardial infarction
Crosstalk between fibroblasts and CMs plays a key role in cardiac remodeling, and CMs have been implicated in promoting cardiac fibrosis [22,23,24]. In our investigation of fibrosis post-MI, we employed Masson’s Trichrome staining to assess fibrotic areas in the hearts at 4 weeks post-MI. We observe large regions of fibrosis in MIATfl/fl hearts, whereas hearts from CM-specific MIAT cKO mice exhibit significantly smaller fibrotic areas (Fig. 5). Consistently, mRNA levels of fibrotic markers Col3a1, Col6a1, Postn, and Snail1 are significantly downregulated in CM-specific MIAT cKO hearts compared to controls at 4 weeks post-MI (Fig. 6). These findings align with our previous mechanistic studies, which showed that systemic MIAT deletion in mice decreased cardiac fibrosis and the expression of profibrotic genes, such as Ctgf and Hoxa4 [8]. Collectively, our current findings demonstrate for the first time that selective knockdown of MIAT in CMs is sufficient to attenuate pathology during post-ischemic cardiac structural and functional remodeling and that CM-expressed MIAT is required for CM-fibroblast crosstalk during pathological remodeling.

Representative Masson’s Trichrome staining (A–B) in heart sections from the four experimental groups at 4 weeks post-MI and fibrosis quantification (C). Fibrosis histology images from whole heart sections (A, Scale bars: 1 mm) and zoomed in images of the peri-ischemic border area (B, Scale bars: 100 μm). N = 6–7 per group. Data are presented as the mean ± SEM. Two-way ANOVA with Tukey’s multiple comparison test. *P < 0.05 or ***P < 0.001 vs. sham of same genotype; ##P < 0.00 vs. MI MIATfl/fl.

QRT-PCR analysis of profibrotic Col3a1 (A), Col6a1 (B), Postn (C), or Snail1 (D) expression in ischemic areas from MIATfl/fl and CM-specific MIAT cKO mouse left ventricles at 4 weeks post-MI. Data are shown as the fold induction of gene expression normalized to Gapdh. N = 3 per group. Data are presented as the mean ± SEM. Two-way ANOVA with Tukey’s multiple comparison test. *P < 0.05, **P < 0.01, or ***P < 0.001 vs. sham of same genotype; #P < 0.05 or ##P < 0.01 vs. MI MIATfl/fl.
Discussion
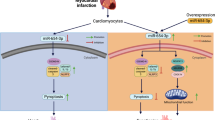
The current study identifies CM-expressed MIAT as a critical mediator of ischemic injury including cardiac dysfunction and remodeling. Mice with restrictive deficiency of MIAT in CMs show a similar level of protection to those with systemic MIAT deficiency [8]. Specifically, CM-specific MIAT cKO mice exhibit lower sensitivity to MI, characterized by reduced cardiac damage, apoptosis, and fibrosis, along with improved LV function. Mechanistically, ischemic hearts lacking MIAT in CMs exhibit significantly reduced expression of HF-associated genes (Nppa, Nppb, and Myh7), proapoptotic genes (p53 and Bak1), and profibrotic genes (Col3a1, Col6a1, Postn, and Snail1). These data support our prior unbiased transcriptomic and mechanistic studies in systemic MIAT KO mice, showing that MIAT activated other maladaptive genes, including Hoxa4, Fmo2, Lrrn4, Marveld3, Fat4, and Ctgf and that the decreased expression of these deleterious markers in mice lacking MIAT thus led to reduced cardiac damage, apoptosis, and fibrosis during ischemia [8].
It is worth noting that our previous study using MIAT TG mice showed that systemic MIAT overexpression led to compromised cardiac function and maladaptive remodeling by increasing cardiac damage, apoptosis, and fibrosis [8]. Other groups have also shown that systemic MIAT knockdown can inhibit cardiac apoptosis post-I/R [10] and cardiac fibrosis post-MI [11]. Moreover, systemic MIAT loss in mice was shown to attenuate AngII- and TAC-induced HF, partly by blunting a CM hypertrophic gene program and enhancing CM contractility [14]. These findings collectively suggest that maladaptive post-MI remodeling mediated by MIAT may be due to its synergistic actions on multiple cell types. Despite these insights, our understanding of MIAT’s overall actions remained incomplete, partly due to the lack of studies dissecting its cell type-specific actions. To address this gap in this study, we reveal a defining role of MIAT specifically in CMs under in vivo conditions.
A mitochondrial membrane protein, translocator protein (TSPO) has been suggested as a potential downstream mechanism explaining the deleterious actions of MIAT in the ischemic heart [25]. TSPO was previously shown to directly interact with MIAT, leading to mitochondria damage and triggering the mitochondrial death pathway [25]. Moreover, MIAT’s actions as a ceRNA that sponges miRs have been implicated as underlying mechanisms of MIAT’s maladaptive effects following ischemic injury. Specifically, MIAT was reported to sponge miR-10-5p, thereby activating the target of this miR, the proapoptotic EGR2 that promotes CM apoptosis and cardiac dysfunction [26]. MIAT has been, moreover, identified as a profibrotic lncRNA in post-infarct hearts, functioning as a ceRNA for miR-24 and subsequently increasing FURIN, an activator of TGF-β1 [11]. Our previous research also highlighted that MIAT promoted cardiac fibrosis following ischemic stress in part by blocking the inhibitory effect of antifibrotic miR-150-5p on profibrotic HOXA4 via its ceRNA action [8]. Our current data (Figs. 1C–D, 4, and 6) suggest that MIAT loss in CMs activates a key mediator of CM survival [20], miR-150-5p as well as suppresses proapoptotic and profibrotic genes, including Sprr1a, p53, Bak1, Col3a1, Col6a1, Postn, and Snail1. Taken together, our findings indicate that the activation of multiple apoptotic and fibrotic genes is a plausible downstream mechanism by which CM-derived MIAT regulates the response to MI.
Circulating MIAT, presumably released by CMs, has been shown to be a superior biomarker for HF compared to clinically used markers such as BNP and cTnT [18, 27]. Our previous transcriptomic and mechanistic studies identified HOXA4 as a key target of MIAT’s maladaptive actions in ischemic adult mouse hearts and primary human cardiac fibroblasts [8]. Additionally, we reported that Hoxa4 exhibited profibrotic and maladaptive effects in a mouse model of MI [8]. Given our findings that HOXA4 was a crucial target of MIAT [8] and several apoptotic and fibrotic genes are activated by CM-derived MIAT (Figs. 1D, 4, and 6), circulating levels of MIAT in post-MI patients could be used to guide current and future targeted treatment options by modulating the downstream targets of MIAT.
Limitations
Although we demonstrate that MIAT expression in CMs is a necessary and sufficient mediator of MI, MIAT derived from other cell types may also exhibit important roles during this pathology. Our previous findings support this possibility, as MIAT expression was upregulated in both CMs and cardiac myofibroblasts isolated from ischemic myocardium [8]. Understanding how MIAT from different sources acts together (via paracrine signaling) or independently (via autocrine signaling) will be important but is beyond the scope of the current study. Notably, despite our current data suggesting that CM-restricted MIAT loss reduces several proapoptotic and profibrotic markers (Figs. 1D, 4, and 6), the exact functional downstream targets of CM-derived MIAT that regulate cardiac pathology also remain elusive.
Although we reported that MIAT KO and TG mice had similar cardiac rupture and mortality rates post-acute MI as WT littermate controls [8], further immunohistochemical assessments and gene expression studies at earlier time points than 4 weeks post-MI are needed to fully understand the sequence of events. MIAT KO mice did not have obvious developmental defects [14], and our prior studies showed that MIAT KO mice had no baseline cardiac phenotypes [8]. These studies suggest that prenatal deletion of MIAT using αMHC-Cre mice may not cause developmental alterations. However, to address potential developmental issues and validate our findings, an inducible cKO mouse model where MIAT deletion in CMs is induced in adult MIATfl/fl;αMHC-MerCreMer mice by 7-day tamoxifen injections can be helpful for future investigations. Finally exploring additional in vivo injury models, such as I/R and TAC, along with detailed studies on the roles of MIAT in various cell types and their interactions, are warranted. These steps are essential before considering MIAT as a viable therapeutic strategy.
Conclusions
Our findings using a novel cKO mouse model demonstrate that MIAT loss specifically in CMs is sufficient to protect the heart from ischemic injury in mice. Interestingly, a previous study showed that systemic MIAT knockdown protected against murine MI by activating antifibrotic miR-24 and subsequently suppressing profibrotic FURIN [11]. Additionally, our earlier research using MIAT KO and TG mouse models indicated that MIAT promoted cardiac dysfunction and maladaptive cardiac remodeling by repressing antifibrotic miR-150-5p and subsequently activating profibrotic HOXA4 during chronic MI [8]. Although these systemic studies highlighted MIAT’s roles in fibroblast function after ischemic injury, our novel CM-restricted cKO studies clearly delineate that MIAT in CMs itself is sufficient to drive adjacent cardiac myofibroblast pathology during chronic MI. Given that MIAT upregulation is also implicated in other forms of heart disease [17, 18], the maladaptive role of MIAT in CMs may be relevant across various stress settings. Thus, strategies aimed at reducing MIAT levels, such as using antisense oligonucleotide-based lncRNA knockdown, could represent promising adjunctive approaches to enhance therapeutic benefits.
Materials and Methods
Cardiomyocyte-restricted knockout of MIAT in mice
To establish a novel CM-restricted MIAT cKO mouse model, we generated a new MIATflox/flox (MIATfl/fl) mouse line (Shanghai Model Organisms Center, Inc., Project #: 20170525-1) using the embryonic stem cell (ESC) strategy as shown in Fig. 7A. In brief, the targeting vector was constructed using infusion technology. The plasmid contains 5’ homologous arm (3.0 kb), flox region (5.2 kb), 3’ homologous arm (3.0 kb), and MC1-TK-polyA for negative selection. The final targeting vector was digested by appropriate restriction enzymes and was sequenced for confirmation purposes. The MIAT gene targeting vector was linearized and used in ESC targeting. One hundred forty-four resistant ESC clones were obtained after selecting by neomycin and ganciclovir, and six positive homologous recombinant ESC clones were identified by long-PCR identification as shown in Fig. 7B. The following PCR primers were used to identify 5’ homologous recombinant ES clones: Forward (5’-AGGGAAGCCAAGATATGTAACTGA-3’) and reverse (5’-CCAGAAAGCGAAGGAGCAAAGC-3’). For identifying 3’ homologous recombinant ES clones, the following PCR primers were used: Forward (5’-CAAACGTTCTGACACAATCCAATC-3’) and reverse (5’-CAGACATCACTGGATGACTCTGG-3’).

A Targeting strategy of MIAT conditional knockout mouse model. Flox region is hypothetical exon 1–3. B Genotyping strategies to screen embryonic stem (ES) cells. 5’ homologous arm: 9.1 kb fragment should be amplified in the homologous recombinant ES cell clones, and none of fragment should be amplified in the negative ES cell clones. 3’ homologous arm: 5.7 kb fragment should be amplified in the homologous recombinant ES cell clones, and 12.4 kb fragment should be amplified in the negative ES cell clones. As shown in agarose gel electrophoresis of PCR products, six positive homologous recombinant ES clones were identified. C–D Genotyping strategies to screen F1 MIATfl/+ mice. The chimeric male mice were crossed with Flp mice to generate F1 mice. 5’ homologous arm: 8.6 kb fragment should be amplified in the homologous recombinant F1 mice, and 11.7 kb fragment should be amplified in the negative F1 mice. 3’ homologous arm: 3.9 kb fragment should be amplified in the homologous recombinant F1 mice, and 12.4 kb fragment should be amplified in the negative F1 mice. By long-PCR identification, seven heterozygous F1 mice were identified. All positive PCR products were confirmed by sequencing. Regions 1 and 2 were for identifying the 5’ homologous recombination, and regions 3 and 4 were for identifying the 3’ homologous recombination (C). For genotyping the offspring, short-PCR was used to identify heterozygous (HE) and wild-type (WT) mice. M: DNA size marker shown in the right (D). Genotyping PCR images show germline transmission of the targeted MIAT floxed allele. Agarose gel electrophoresis of PCR products is shown in the bottom (C–D).
The positive ESC clones were injected into 120 blastocysts and transferred into eight recipients. Three mice were born, all of which were all chimeric males with more than 50% chimeric ratio. These chimeric male mice were crossed with Flp mice to generate F1 mice. By long-PCR identification and sequencing, seven heterozygous mice were identified. The genotyping strategy is shown in Fig. 7C, and the following PCR primers were used to screen positive F1 mice: Forward (5’-AGGGAAGCCAAGATATGTAACTGA-3’) and reverse (5’-CCCCGAGCCAAGGAAGGTGAGAC-3’) for identifying 5’ homologous recombinant F1 mice. Forward (5’-CAAACGTTCTGACACAATCCAATC-3’) and reverse (5’-CAGACATCACTGGATGACTCTGG-3’) for identifying 3’ homologous recombinant F1 mice. The four key regions of PCR product were sequenced for confirmation purposes. The sequencing regions were shown in Fig. 7C. Regions 1 and 2 were for identifying the 5’ homologous recombination, and regions 3 and 4 were for identifying the 3’ homologous recombination. The following primers for Flp were used to amplify a 725 bp gene product specific for the transgene gene: Forward (5’-cactgatattgtaagtagtttgc-3’) and reverse (5’-ctagtgcgaagtagtgatcagg-3’). The genotyping result of this long-PCR method was confirmed by the short-PCR method as shown in Fig. 7D and described below, to exclude any potential random inserts in homologous recombinant F0 mice.
The MIATflox/+ mice, harboring an allele of loxP-flanked MIAT, were backcrossed to C57BL/6 J for five generations and then bred to generate MIATflox/flox mice. αMHC-Cre mice [19], in which the expression of Cre recombinase is controlled by the promoter of the cardiac-specific marker gene, αMHC, were then intercrossed with MIATflox/flox mice to generate MIATflox/+;αMHC-Cre offspring. The MIATflox/+;αMHC-Cre mice were subsequently crossed back to MIATflox/flox mice to obtain the MIAT conditional (cardiac-specific) knockout (cKO) mice (MIATflox/flox;αMHC-Cre), as shown in Fig. 1A. Mice were maintained on a C57BL/6 J background, and genetically matched Cre-negative MIATflox/flox littermates were used as controls. Genotyping for αMHC heterozygous mice was done using primers (5’-ATGACAGACAGATCCCTCCTATCTCC-3’ and 5’-CTCATCACTCGTTGCATCATCGAC-3’) to amplify a 300 bp gene product specific for the transgene gene. Genotyping for MIAT floxed mice was done with the primers of 5’-GCCCTCCGCCATCTCCCT-3’ and 5’-GTGGTCCTTTCTCAGTCTCCCA-3’, resulting in band sizes of 236 bp for the wild-type allele and 401 bp for the floxed allele.
Ethics committee approval
The use of mice in this study was conformed to the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Mice were euthanized by asphyxiation with CO2 to minimize the pain and time needed for cessation of life. A secondary method of euthanasia, including bilateral thoracotomy, cervical dislocation, exsanguination, or decapitation, was then applied under 1–4% inhalant isoflurane. These methods are consistent with the recommendations of the Panel on Euthanasia of the American Veterinary Medical Association. All experiments with mice were conducted according to the protocols approved by the Institutional Animal Care and Use Committee at Indiana University (approval #21189). Eight to sixteen-week-old C57BL/6 J mice of both sexes were used for this study. Genotype- and sex-matched mice were randomly assigned to experimental groups to mitigate the cage effect. The genotypes of the mice were masked from researchers until the end of the analysis.
Statistics
Data are reported as mean ± SEM (except in Fig. 2, where SD is used because no clear variation bars are shown otherwise) from independent experiments with different biological samples per group. Given our retrospective data for experimental assays, we use a total of 18–20 mice of both sexes per group in echocardiography, a total of 3 mice of both sexes per group in expression analysis, and a total of 6–7 mice of both sexes per group in histology analysis, which would be adequately powered. The number of used mice in the current study is based on a valid statistical model with a one-tailed (α1) hypothesis each and 80% power. All animals were included for the analysis. The exact sample size for each experimental group/condition is given as a number in the figure/table legend. To ensure the robustness of the data and to facilitate the direct evaluation of the data distribution, graphical data (except in Fig. 2) are presented as scatter/dot plots. Normality was assessed using the Kolmogorov-Smirnov test. Statistical significance was determined by unpaired two-tailed t-test for comparisons between two groups, two-way ANOVA with Tukey’s multiple comparison test for comparisons between two groups with different treatments, and two-way repeated-measures ANOVA with Bonferroni’s post hoc test for comparisons between two groups over time. Sample sizes were 18–20 for echocardiographic analysis and 3–7 for other downstream analyses. A P value of <0.05 was considered statistically significant. P values are indicated as follows: * or #P < 0.05; ** or ##P < 0.01; and *** or ###P < 0.001.
Data availability
All data are included in the manuscript and Supplementary Information. The analytical methods and study materials will be made available to other researchers for the purposes of reproducing the results or replicating the procedures. Additional methods are provided in Supplementary Information.
References
Takemura G, Fujiwara H. Role of apoptosis in remodeling after myocardial infarction. Pharm Ther. 2004;104:1–16. https://doi.org/10.1016/j.pharmthera.2004.07.005.
Nijmeijer KrijnenPA, Meijer R, Visser CJ, Hack CA, Niessen CE. HW. Apoptosis in myocardial ischaemia and infarction. J Clin Pathol. 2002;55:801–11.
Archer K, Broskova Z, Bayoumi AS, Teoh JP, Davila A, Tang YL, et al. Long non-coding RNAs as master regulators in cardiovascular diseases. Int J Mol Sci. 2015;16:23651–67. https://doi.org/10.3390/ijms161023651.
Thum T, Condorelli G. Long Noncoding RNAs and MicroRNAs in cardiovascular pathophysiology. Circ Res. 2015;116:751–62. https://doi.org/10.1161/Circresaha.116.303549.
Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–66. https://doi.org/10.1146/annurev-biochem-051410-092902.
Meessen J, Bar C, di Dona FM, Staszewsky LI, Di Giulio P, Di Tano G, et al. LIPCAR is increased in chronic symptomatic HF patients. A sub-study of the GISSI-HF trial. Clin Chem. 2021;67:1721–31. https://doi.org/10.1093/clinchem/hvab197.
Koch PF, Garcia-Hidalgo MC, Labus J, Biener M, Thum T, de Gonzalo-Calvo D, et al. Circulating long noncoding RNA PDE4DIPP6: A novel biomarker for improving the clinical management of non-ST-segment elevation myocardial infarction. Clin Chim Acta. 2024;561:119840 https://doi.org/10.1016/j.cca.2024.119840.
Aonuma T, Moukette B, Kawaguchi S, Barupala NP, Sepulveda MN, Frick K, et al. MiR-150 attenuates maladaptive cardiac remodeling mediated by long noncoding RNA MIAT and directly represses profibrotic Hoxa4. Circ Heart Fail. 2022;15:e008686 https://doi.org/10.1161/CIRCHEARTFAILURE.121.008686.
Zhou X, Zhang W, Jin M, Chen J, Xu W, Kong X. LncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22-3p in diabetic cardiomyopathy. Cell Death Dis. 2017;8:e2929 https://doi.org/10.1038/cddis.2017.321.
Chen L, Zhang D, Yu L, Dong H. Targeting MIAT reduces apoptosis of cardiomyocytes after ischemia/reperfusion injury. Bioengineered. 2019;10:121–32. https://doi.org/10.1080/21655979.2019.1605812.
Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S, et al. MIAT is a pro-fibrotic long non-coding RNA governing cardiac fibrosis in post-infarct myocardium. Sci Rep. 2017;7:42657 https://doi.org/10.1038/srep42657.
Zhu XH, Yuan YX, Rao SL, Wang P. LncRNA MIAT enhances cardiac hypertrophy partly through sponging miR-150. Eur Rev Med Pharm Sci. 2016;20:3653–60.
Li Z, Liu Y, Guo X, Sun G, Ma Q, Dai Y, et al. Long noncoding RNA myocardial infarction-associated transcript is associated with the microRNA-150-5p/p300 pathway in cardiac hypertrophy. Int J Mol Med. 2018;42:1265–72. https://doi.org/10.3892/ijmm.2018.3700.
Yang L, Deng J, Ma W, Qiao A, Xu S, Yu Y, et al. Ablation of lncRNA Miat attenuates pathological hypertrophy and heart failure. Theranostics. 2021;11:7995–8007. https://doi.org/10.7150/thno.50990.
Ohnishi Y, Tanaka T, Yamada R, Suematsu K, Minami M, Fujii K, et al. Identification of 187 single nucleotide polymorphisms (SNPs) among 41 candidate genes for ischemic heart disease in the Japanese population. Hum Genet. 2000;106:288–92. https://doi.org/10.1007/s004390051039.
Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A, et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet. 2006;51:1087–99. https://doi.org/10.1007/s10038-006-0070-9.
Frade AF, Laugier L, Ferreira LR, Baron MA, Benvenuti LA, Teixeira PC, et al. Myocardial infarction-associated transcript, a long noncoding RNA, is overexpressed during dilated cardiomyopathy due to chronic Chagas disease. J Infect Dis. 2016;214:161–5. https://doi.org/10.1093/infdis/jiw095.
de Gonzalo-Calvo D, Kenneweg F, Bang C, Toro R, van der Meer RW, Rijzewijk LJ, et al. Circulating long-non coding RNAs as biomarkers of left ventricular diastolic function and remodelling in patients with well-controlled type 2 diabetes. Sci Rep. 2016;6:37354 https://doi.org/10.1038/srep37354.
Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, et al. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–45. https://doi.org/10.1161/01.RES.0000215985.18538.c4.
Aonuma T, Moukette B, Kawaguchi S, Barupala NP, Sepulveda MN, Corr C, et al. Cardiomyocyte microRNA-150 confers cardiac protection and directly represses proapoptotic small proline-rich protein 1A. JCI Insight. 2021;6:e150405 https://doi.org/10.1172/jci.insight.150405.
Kawaguchi S, Moukette B, Sepulveda MN, Hayasaka T, Aonuma T, Haskell AK, et al. SPRR1A is a key downstream effector of MiR-150 during both maladaptive cardiac remodeling in mice and human cardiac fibroblast activation. Cell Death Dis. 2023;14:446 https://doi.org/10.1038/s41419-023-05982-y.
Flores-Vergara R, Olmedo I, Aranguiz P, Riquelme JA, Vivar R, Pedrozo Z. Communication between cardiomyocytes and fibroblasts during cardiac ischemia/reperfusion and remodeling: Roles of TGF-beta, CTGF, the renin angiotensin axis, and non-coding RNA molecules. Front Physiol. 2021;12:716721 https://doi.org/10.3389/fphys.2021.716721.
Yang J, Yu X, Xue F, Li Y, Liu W, Zhang S. Exosomes derived from cardiomyocytes promote cardiac fibrosis via myocyte-fibroblast cross-talk. Am J Transl Res. 2018;10:4350–66.
Asensio-Lopez MDC, Lax A, Fernandez Del Palacio MJ, Sassi Y, Hajjar RJ, Pascual-Figal DA. Pharmacological inhibition of the mitochondrial NADPH oxidase 4/PKCalpha/Gal-3 pathway reduces left ventricular fibrosis following myocardial infarction. Transl Res. 2018;199:4–23. https://doi.org/10.1016/j.trsl.2018.04.004.
Bai X, Yang C, Jiao L, Diao H, Meng Z, Wang L, et al. LncRNA MIAT impairs cardiac contractile function by acting on mitochondrial translocator protein TSPO in a mouse model of myocardial infarction. Signal Transduct Target Ther. 2021;6:172 https://doi.org/10.1038/s41392-021-00538-y.
Cao X, Ma Q, Wang B, Qian Q, Liu N, Liu T, et al. Silencing long non-coding RNA MIAT ameliorates myocardial dysfunction induced by myocardial infarction via MIAT/miR-10a-5p/EGR2 axis. Aging (Albany NY). 2021;13:11188–206. https://doi.org/10.18632/aging.202785.
Azat M, Huojiahemaiti X, Gao R, Peng P. Long noncoding RNA MIAT: A potential role in the diagnosis and mediation of acute myocardial infarction. Mol Med Rep. 2019;20:5216–22. https://doi.org/10.3892/mmr.2019.10768.
Acknowledgements
We thank the Immunohistochemistry Core at Indiana University for histopathological and immunohistochemical analyses.
Funding
This work was supported by the American Physiological Society 2023–2024 Postdoctoral Fellowship to TH, the American Heart Association Career Development Award 931621 to MNS, Department of Defense HT9425-24-1-0209 and the National Institutes of Health (NIH) R01HL160941 to AAD, and NIH R01HL146481 to IK.
Author information
Authors and Affiliations
Contributions
TH, SK, MNS, JT, BM, TA, and IK designed research studies, directed the study, and wrote the manuscript. TH, SK, MNS, JT, BM, and TA conducted the experiments, acquired the data, analyzed the data, and prepared the figures. IK supervised the study and provided financial support. MSM, AAD, SL, and SJC helped to analyze the data and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hayasaka, T., Kawaguchi, S., Sepúlveda, M.N. et al. Cardiomyocyte-restricted MIAT deletion is sufficient to protect against murine myocardial infarction. Cell Death Discov. 11, 70 (2025). https://doi.org/10.1038/s41420-025-02352-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-025-02352-9
