
Abstract
Glioblastoma (GBM) poses significant therapeutic challenges due to its hypoxic and immunosuppressive tumour microenvironment (TME), low immunogenicity and physical barriers. While combining photodynamic therapy (PDT) with immunotherapy holds promise, its efficacy is hampered by insufficient immune activation. In this study, we develop a multifunctional photodynamic-enhanced biomimetic intelligent nanoplatform (FBFO@HM@aOPN) responsive to the TME. The nanoplatform consists of a dual-enzyme nanozyme encapsulated in a prokaryotic-eukaryotic hybrid membrane, further modified with a pH-sensitive tumor-targeting antibody. After systemic administration, FBFO@HM@aOPN selectively accumulates in the GBM through vascular regulation and extracellular matrix (ECM) remodelling while generating oxygen to alleviate hypoxia. Crucially, the platform concurrently induces immunogenic death in tumour cells and reprograms protumoral macrophages to antitumor phenotypes. This dual action robustly activates both innate and adaptive immunity, significantly inhibiting GBM growth. Furthermore, when combined with anti-PD1 immunotherapy, the nanoplatform dramatically boosts the treatment effect and effectively prevents postsurgical tumour recurrence. Therefore, our work offers a multimodal platform for stimulating anti-tumour immunity, with potential applicability for GBM patients.
Similar content being viewed by others

Introduction
Immunotherapy has reshaped the treatment landscape for many types of cancer, and in recent years, it has become an attractive therapeutic modality for the treatment of cancer1,2. The tumour microenvironment (TME) of glioblastoma (GBM) is a highly complex environment composed of multicellular components, extracellular matrix (ECM) proteins, various secretory molecules, and an intricate network of blood and lymphatic vessels that play crucial roles in tumorigenesis and progression3,4,5. Photodynamic therapy (PDT), with its unique direct tumour cytotoxicity and ability to stimulate a robust immune response, has advantages in terms of control and safety, exhibiting great potential to overcome the current shortfalls in cancer immunotherapy6,7. Thus, combining PDT with strategies targeting the tumour immune landscape is expected to become a new “golden key” for anticancer therapy, opening a new avenue for cancer treatment. However, the intricate hypoxic TME, which further exacerbates immunosuppression, largely limits PDT-dependent and immune-dependent therapeutic effects8,9. Thus, alleviating hypoxia is a promising strategy to improve the outcomes of combined PDT and immunotherapy.
Among the various components of the GBM TME, tumour-associated myeloid macrophages (TAMs) are among the most abundant tumour-infiltrating populations. TAMs are commonly classified into two main subsets: antitumour M1-like TAMs and tumour-promoting M2-like TAMs10,11. M2-like TAMs can effectively protect tumours from the suppression of adaptive immune responses by expressing or secreting proteases, immunosuppressive factors, etc12,13., whereas M1-like TAMs play a central role in antitumour immunity. They secrete proinflammatory cytokines and simultaneously upregulate antigen-presenting and costimulatory molecules to enhance antigen cross-presentation ability, thereby activating CD8+ T cells11,14. In addition, M1-like TAMs can recruit effector T cells to infiltrate the tumour site by secreting chemokines such as CXCL9/10, thereby breaking the local immune suppression barrier15. Considering the high abundance of TAMs in the GBM TME compared with other APCs, such as dendritic cells (DCs), a strategy that promotes the direct polarisation of infiltrating monocytes or the repolarization of M2-like TAMs to an M1-like phenotype, increasing their antigenic cross-presentation capacity to activate CD8+ T cells in the tumour site would be a more direct and effective immunotherapeutic approach.
Biomimetic hybrid membranes from different sources can mimic the structure and function of parent cells, providing multiple functions, such as targeted delivery, specific recognition, and immunostimulation, that revolutionise the design of drug delivery systems16,17. M1-like macrophage-derived exosomes (M1EVs), which inherit the inflammation-directed tumour-homing ability of M1-like TAMs and can penetrate the blood‒brain barrier (BBB) via membrane proteins (integrins) on their surface18, have great potential for enhancing targeted drug delivery and antitumour effects. Bacterial outer membrane vesicles (OMVs) are highly immunogenic nanovesicles that effectively stimulate macrophages to adopt an M1-like phenotype and have become a commonly used delivery vehicle for tumour immunotherapy19,20. The hybridisation of M1EVs and OMVs to decorate NPs could achieve both precise targeted delivery of therapeutics and TAM repolarization. On this basis, we proposed a hybrid membrane (HM)-driven strategy for precise cotargeting to activate local innate and adaptive immunity within the GBM microenvironment.
The TME is a highly structured ecosystem containing cancer cells surrounded by multiple nonmalignant cell types that are coembedded in a dysfunctional tumour vascular system and a dynamically shifting ECM, ultimately imposing major structural and biochemical barriers to effector T-cell infiltration and effective tumour control21,22. Accordingly, antiangiogenic agents (e.g., VEGF inhibitors such as bevacizumab) are now being used to treat GBM, with variable clinical success23. Thus, there is an urgent need for new approaches to reprogram the vascular–immune interface. Recently, many studies have shown that TAMs can inhibit the immunotherapeutic effect of immune checkpoint inhibitors (ICIs) by secreting large amounts of OPN (encoded by the SPP1 gene), a multifunctional and complex secretory protein that can interact with CD44 or integrin molecules in tumour stromal cells, leading to abnormal proliferation and disordered distribution of the vascular system and the formation of tumour immune barrier (TIB) structures, which in turn prevent the tumour-targeted delivery of lymphocytes and nanomedicines24,25,26,27. Moreover, OPN can interact with integrin β1 molecules on the surface of T cells to mediate T-cell dysfunction28. Therefore, blocking the OPN pathway may destroy the TIB and promote CD8 + T-cell infiltration and activation, thereby increasing the efficacy of combined PDT and immunotherapy.
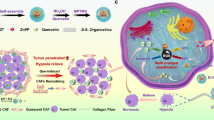
Here, we show a multimodal biomimetic nanoplatform (FBFO@HM@aOPN) designed to enhance PDT-immunotherapy synergy in GBM. The nanoplatform encapsulates a nanozyme (Fe3O4@BiFeO3, FBFO) with dual enzyme-like activity within a hybrid membrane (HM) formed by the fusion of OMVs and M1EVs, which are then conjugated with a pH-sensitive anti-OPN (aOPN) antibody to obtain FBFO@HM@aOPN. Upon intravenous administration, FBFO@HM@aOPN accumulates in GBM tissues, where acidic TME-triggered aOPN release increases vascular permeability and promotes ECM remodelling. Concurrently, the FBFO nanozyme generates oxygen to alleviate hypoxia, increasing the efficacy of PDT. The HM further directs the nanoplatform to tumour cells and TAMs to induce ferroptosis (neoantigen release) and repolarizes TAMs towards an M1-like phenotype. This dual action activates both innate and adaptive immunity, synergises with checkpoint inhibitors, and suppresses tumour progression and recurrence. Overall, our platform provides a minimally invasive strategy to overcome TME-driven resistance in GBM therapy.
Results
SPP1+ M2-like macrophages promoted GBM progression through hypoxia-mediated angiogenesis and immune evasion
To elucidate the functional roles and contributions of TAMs in GBM progression and immune modulation, we analysed GBM single-cell (scRNA) data and found that M2-like TAMs, which can effectively contribute to epithelial mesenchymal transition (EMT) and tumour angiogenesis (Supplementary Fig. 1) and inhibit antitumour immunity through the expression or secretion of proteases and immunosuppressive factors, are the most abundant TAM subpopulation29. We also found that M1-like TAMs have antigen-presenting cell (APC) potential (Supplementary Fig. 1c), suggesting that M1-like TAMs may act as a bridge between innate and adaptive immunity.
We then combined scRNA-seq and spatial transcriptomics data to identify the specific spatial structures in GBM. We found that SPP1 was expressed mainly by M2-like TAMs (Supplementary Fig. 1b), and the same results were obtained with scRNA-seq data from mice (Supplementary Fig. 1d, e). Further scRNA-seq and spatial distribution mapping revealed that the M2-like TAM subpopulation was highly enriched in hypoxic and angiogenic tumour regions, and SPP1 and its ligands (Cd44 and Itgb1), as well as VEGFA, were similarly highly expressed in both M2-like BMDMs and hypoxic regions (Supplementary Fig. 2a–e). We further performed immunofluorescence analysis of CT2A tumour-bearing mice and found that the regions with high expression of the myeloid marker f4/80 also showed high expression of OPN, with a clear boundary. Moreover, the expression of its ligands (Cd44 and Itgb1) was greater than that in paracancerous tissues (Supplementary Fig. 2f). On the basis of these findings, we hypothesised that targeting OPN and its associated pathways could increase the efficacy of combined PDT and immunotherapy.
Preparation and characterization of FBFO@HM@aOPN nanoparticles
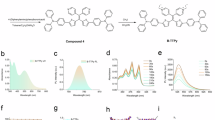
The development of dot photocatalysts has been examined as a viable method for broadening the light absorption range and facilitating the separation of photoinduced hole–electron pairs30. First, the solvothermal method was adopted to fabricate Fe3O4 nanoparticles (NPs). Subsequently, Bi3+ and PVP were added to the Fe3O4 NPs to obtain FBFO NPs via in situ growth (Fig. 1a). Transmission electron microscopy (TEM) revealed that the average diameter of the FBFO NPs was approximately 30 nm (Fig. 2a). Further analysis by scanning TEM (STEM) coupled with energy dispersive X-ray spectroscopy (EDX) and energy dispersive spectroscopy (EDS) demonstrated the presence of Bi, Fe and O in the FBFO NPs, as illustrated in Fig. 2b. The FBFO NPs were also confirmed through X-ray diffraction (XRD), as depicted in Fig. 2c. These findings collectively validated the successful synthesis of FBFO NPs. The optical properties are the major factors affecting the photocatalytic activity of FBFO NPs. UV‒vis/NIR spectra revealed that the doping of Bi and Fe3O4 NPs broadens the light absorption range of FBFO (Fig. 2d), increases the separation efficiency of photogenerated carriers, and greatly enhances the photocatalytic activity of the nanomaterials. As proposed in the scheme in Fig. 2e, the efficient separation of hole (h+)–electron (e−) pairs could be achieved via irradiation with a 660 nm laser (Equation 1), followed by combination with water and oxygen, which can generate abundant hydroxyl radicals (·OH) and superoxide radicals (·O2−) for type-I PDT (Equations 2 and 3). With the further transition of Fe2+ to Fe3+, H2O2 is then converted to ·OH (Equations 4, mimicking peroxidase, POD), which further increases the generation of reactive oxygen species (ROS). The generated h+ can also oxidise H2O2 to produce O2 (Equations 5, mimicking catalase (CAT)) to sustain photodynamic therapy in the hypoxic TME. Next, we evaluated the ability of the NPs to produce ROS and O2 via methylene blue (MB) and dissolved oxygen analyser assays, respectively, under irradiation at 660 nm (0.5 W/cm2). Compared with that in the Fe3O4 group and other groups, the absorption of MB clearly significantly decreased, whereas the O2 production ability significantly increased in the FBFO and Laser groups (Fig. 2f, g). Furthermore, the generation of ROS (·OH and ·O2-) through FBFO NP-mediated photodynamics was verified by electron spin resonance (ESR) spectroscopy (Fig. 2h). Therefore, the FBFO NPs exhibited excellent catalytic activity for the generation of ROS (·OH and ·O2−) and O2, indicating the great potential of the FBFO NPs for application in antitumour PDT.

a Schematic diagram of the synthetic route of FBFO NPs. b Preparation of a prokaryotic-eukaryotic hybrid vesicle membrane (HM) formed by the fusion of bacterial outer membrane vesicles (OMVs) and M1 macrophage-derived exosomes (M1EVs). c Preparation of anti-OPN (aOPN) attached to a pH-sensitive benzoic acid‒imide bond. d Schematic illustration of FBFO@HM@aOPN NPs for immunity-remodelled photodynamic therapy against GBM in vivo.
To further increase the safety, stability and multifunctionality of FBFO and expand its application value, we encapsulated FBFO with HMs from M1 macrophage-derived exosomes (M1EVs) and bacterial membrane vesicles (OMVs) to obtain FBFO@HM NPs (Fig. 1b). Azide-choline serves as a substrate for cells to synthesise phosphatidylcholine, a crucial component of cell membranes31. In this study, Raw264.7 cells (MΦs) were incubated with azide-choline to generate azide (N3)-modified M1 MΦs. Confocal laser scanning microscopy (CLSM) results demonstrated the successful incorporation of N3 groups into the M1 macrophage membrane (Supplementary Fig. 3a). M1EVs were isolated from the cell culture supernatant of N3-modified M1 macrophages by ultracentrifugation and thus carried N3 groups on their surface, which was verified by flow cytometry (Fig. 2i and Supplementary Fig. 3b). To prepare OMVs, the K12-derived E. coli strain W3110 was genetically engineered to harvest low-endotoxin vesicles (Supplementary Fig. 3c), which have minimal side effects on the circulation32. Secreted OMVs were added to M1EVs at a 1:1 ratio and incubated for 2 h, followed by sonication for 10 min and freeze‒thawing in liquid nitrogen 5 times to obtain M1EV–OMV HMs. Afterwards, the HMs and FBFO NPs underwent 10 cycles of extrusions via a liposome extruder, resulting in the formation of relatively homogeneous FBFO@HM NPs. CLSM confirmed that the surfaces of the FBFO@HM NPs were coated with N3-modified M1EVs (Fig. 2j). Furthermore, M1EV–OMV fusion was visualised via CLSM. The M1EVs and OMVs were labelled with Cy5.5-DBCO and DiI, respectively. The two vesicles morphed into new lipid vesicles that colocalized with Cy5.5 and DiI signals, wrapped on FITC-labelled FBFO NPs (Fig. 2k). Moreover, as shown in Fig. 2l, Western blotting revealed that the HMs inherited components from M1EVs (TSG101, CD63, CD81 and integrins α4β1, as well as the chemokine receptor CXCR4) and OMVs (FtsZ).

a Transmission electron microscopy (TEM) images of FBFO NPs of uniform size and morphology. Scale bar, 100 nm. b Element mapping of the FBFO NPs. Scale bar, 100 nm. c X-ray diffraction (XRD) patterns of the Fe3O4 and FBFO NPs. d UV–vis diffuse reflectance spectra of the Fe3O4 and FBFO NPs. e Schematic illustration of the working mechanisms of FBFO. f Degradation of methylene blue (MB) by the Fe3O4 and FBFO NPs. g Oxygen (O2) generation curves of Fe3O4 and FBFO NPs. h Electron spin resonance (ESR) spectra of different groups. i Representative flow cytometry and quantification analysis of azide (N3)-modified M1EVs after incubation with DBCO-Cy5.5 (n = 3 independent experiments). The data are presented as the means ± SDs. p values were calculated via Student’s t test for two-group comparisons; ****p < 0.0001. j Confocal laser scanning microscopy (CLSM) analysis of azide (N3)-modified M1EVs after incubation with DBCO-Cy5.5. Scale bar, 50 nm. k CLSM analysis showing the colocalization of M1EVs and OMVs. Scale bar, 50 nm. l Western blotting revealed that the HMs inherited components from M1EVs (TSG101, CD63, CD81 and integrins α4β1, as well as the chemokine receptor CXCR4) and OMVs (FtsZ). m CLSM image analysis showing the colocalization of HM and aOPNs. Scale bar, 100 nm. n TEM images of FBFO@HM@aOPN NPs of uniform size and morphology. Scale bar, 100 nm. o ELISA quantification of anti-OPN release at different pH values (n = 3 independent experiments). The data are presented as the means ± SDs. p values were calculated via Student’s t test for two-group comparisons; ****p < 0.0001. p Degradation of MB and q O2 generation curves of FBFO@HM@aOPN under the indicated conditions. Source data are provided as a Source Data file.
Thereafter, FBFO@HM NPs were conjugated with aOPN antibodies to obtain FBFO@HM@aOPN. Specifically, first, the benzaldehyde-PEG2000 − NHS linker (BD-PEG2000-NHS) reacted with the amino terminus of aOPN to form aOPN-PEG2000-BD. Then, aOPN-PEG2000-BD was linked to DBCO-PEG5-NH2, forming an acid-sensitive benzoic-imine bond to synthesize aOPN-PEG2000-DBCO (Fig. 1c), which allowed the controlled release of aOPN in the acidic TME at pH 6.531. The successful conjugation of aOPN to FBFO@HM was confirmed by the colocalization of the Dil-labelled HM and Alexa Fluor 647-labelled aOPN (Fig. 2m). TEM images revealed that FBFO@HM@aOPN exhibited a uniform spherical nanostructure with an inner core and an outer shell (Fig. 2n). Afterwards, the responsiveness of the benzoic-imine bond between aOPN and FBFO@HM to acidic environments was validated by ELISA measurement of aOPN antibody levels in the dialysate, which revealed greater release of aOPN antibodies at pH 6.5 than at pH 7.4 (Fig. 2o). Furthermore, the generation of ROS (·OH and ·O2-) through FBFO@HM@aOPN NP-mediated photodegradation was verified by ESR (Fig. 2h) and MB degradation experiments (Fig. 2p). The ability of FBFO@HM@aOPN NPs to catalyse the decomposition of H2O2 into O2 across various pH values was subsequently investigated. Like FBFO, FBFO@HM@aOPN NPs produced ROS (Fig. 2p) and O2 (Fig. 2q) under laser irradiation. These observations indicated that we successfully prepared FBFO@HM@aOPN biohybrid systems with photoenhanced dual enzyme-like activities, which could enhance the efficacy of PDT.
In vitro catalytic activity and therapeutic effect
The optimal safe concentration of FBFO NPs for MΦ culture was determined by the CCK8 assay to be 50 µg/mL (Supplementary Fig. 4a). To evaluate the PDT efficacy of FBFO@HM@aOPN NPs, we conducted a series of in vitro experiments using the CT2A GBM cell line. In the subsequent experiments, the subjects were divided into 7 groups (I: PBS, II: aOPN, III: FBFO, IV: FBFO@OMV, V: FBFO@M1EV, VI: FBFO@HM, and VII: FBFO@HM@aOPN). Multiple interacting molecules on the M1EV membrane surface, including integrin α4β1, can interact with Icam1 or Vcam1 ligand molecules, which are highly expressed on endothelial cells33 and GBM cells34,35,36, to facilitate the passage of membrane-encapsulated nanoparticles across the blood‒brain barrier (BBB) as well as the active targeting of tumour cells. We then used Cy5.5-labelled FBFO to evaluate its internalisation by CT2A GBM cells. The fluorescence intensity (FI) of Cy5.5 was greater in the GBM cells treated with FBFO@HM@aOPN NPs, and a similar trend of fluorescence signals was observed in the groups treated with FBFO@M1EV-NPs or FBFO@HM-NPs. In addition, the internalisation efficiency sharply decreased when blocked by anti-Icam1/Vcam1 antibodies (Fig. 3a and Supplementary Fig. 4b), suggesting that M1EVs endowed the NPs with active tumour cell-targeting capabilities. We then assessed the production of intracellular ROS using 2’,7’-dichlorofluorescein diacetate (DCFH-DA) as a probe. As depicted in Fig. 3b, the green fluorescence intensity was slightly greater in the FBFO@HM@aOPN treatment group than in the control group. Notably, the FBFO@HM@aOPN combined with laser irradiation group presented the strongest green fluorescence intensity among all the groups. These results suggested that FBFO@HM@aOPN can efficiently deliver FBFO into tumour cells with the help of tumour-targeting M1EVs and generate a substantial ROS surge under laser irradiation, increasing the susceptibility of cancer cells to oxidative damage.

a Flow cytometry analysis showing the internalisation of Cy5.5-labelled FBFO by CT2A GBM cells. The right image shows the quantification of Cy5.5-positive cells (n = 3 independent experiments). b CLSM image of CT2A GBM cells stained with DCFH-DA to indicate NP-induced ROS generation. Scale bar, 200 μm. Cell viability of CT2A GBM cells treated with different formulations (c) with/without 660 nm irradiation. (d) Under hypoxia/normoxia conditions (n = 3 independent experiments). e Annexin V-FITC/PI apoptosis analysis of CT2A GBM cells treated with different formulations under 660 nm irradiation by flow cytometry (n = 3 independent experiments). f Lipid peroxidation level of CT2A GBM cells treated with different formulations under 660 nm irradiation, as determined with a malondialdehyde (MDA) assay kit (n = 3 independent experiments). g CLSM image of CALR and Hoechst costained CT2A cells treated with different formulations under 660 nm irradiation. Scale bar, 50 μm. Released h HMGB1 and i ATP detected in the cell culture supernatant of CT2A GBM cells treated with different formulations under 660 nm irradiation (n = 3 independent experiments). j Schematic illustration of ICD induced by FBFO@HM@aOPN and the potential mechanism by which it enhances immunotherapy. All the data are presented as the means ± SDs. The p values were determined via two-tailed one-way ANOVA with a Tukey post hoc test (a, c–f, h, i), p > 0.05, no significance (ns), *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001. Source data are provided as a Source Data file.
To assess the cytotoxic effect of FBFO@HM@aOPN, a standard CCK8 assay was performed on CT2A GBM cells. The viability of the tumour cells was significantly lower in the FBFO@HM@aOPN combined with laser irradiation group than in the other groups (Fig. 3c). Hypoxia has profound effects on the proliferation, migration, and epithelial‒mesenchymal transition (EMT) of cancer cells, thereby strongly influencing their biological behaviour and malignancy37. Thus, alleviating hypoxic conditions could inhibit the antioxidative ability of tumour cells and promote oxidative damage. As shown in Fig. 3d, GBM cells treated under normoxic conditions presented a much greater inhibition rate than those treated under hypoxic conditions did. Even under hypoxic conditions, the viability of the tumour cells decreased significantly in the FBFO@HM@aOPN combined with laser irradiation group, indicating that the PDT of FBFO@HM@aOPN could overcome the limitations of the hypoxic microenvironment. Furthermore, the photoinduced killing efficiency of FBFO@HM@aOPN NPs was dramatically greater than that of the other treatments (Fig. 3e). These results suggested that FBFO@HM@aOPN NPs possessed excellent in vitro catalytic PDT efficiency.
High accumulation of ROS (·OH) in the intracellular environment can increase the lipid peroxidation of membranes, inducing cellular ferroptosis38. To validate this hypothesis, we assessed intracellular lipid peroxidation by measuring malondialdehyde (MDA) levels, which are indicative of the early phase of ferroptosis. As demonstrated in Fig. 3f, FBFO@HM@aOPN treatment significantly increased lipid peroxidation, and subsequent laser irradiation further intensified lipid peroxidation. The cells exhibited swelling, bubbling, and eventually bursting (Fig. 3g), which were indicative of ferroptosis.
Ferroptosis is considered a form of immunogenic cell death (ICD) and is characterised by the release of damage-associated molecular patterns (DAMPs), which include membrane calreticulin (CALR), high mobility group protein 1 (HMGB1), and adenosine 5′-triphosphate (ATP). Consistent with our expectations, CRT expression on the cell membrane was obviously increased in the FBFO@HM@aOPN group (Fig. 3g). Additionally, FBFO@HM@aOPN resulted in increased ATP and HMGB1 secretion by GBM cells (Fig. 3h, i), thus inducing ICD and providing abundant antigens for APCs, thereby promoting T-cell infiltration and activation (Fig. 3j).
FBFO@HM@aSPP NPs significantly polarised M0- and M2-like macrophages towards the M1-like phenotype in vitro
Fe3O4 NPs are promising candidates for TAM repolarization, which can induce high intracellular ROS levels, thus activating the downstream NF-κB pathway, a key pathway regulating M1 macrophage polarisation39,40,41. OMVs are highly immunogenic nanovesicles that can strongly stimulate macrophages to polarise towards the M1 phenotype due to their high surface lipopolysaccharide expression19,42. We next systematically explored the effects of FBFO@HM@aOPN on macrophages. Notably, specific proteins on M1EV membranes, including integrin α4β1, are prerequisites for GBM cell targeting (Fig. 2l) via the ICAM1 or VCAM1 pathway43,44. Our mouse scRNA-seq analysis revealed that TAMs also highly expressed Icam1 or Vcam1 (Supplementary Fig. 5a). To investigate whether FBFO@HM@aOPN NPs can increase the cellular internalisation of FBFO by macrophages through integrin α4β1-Icam1/Vcam1-mediated targeting, we used Cy5.5-labelled FBFO to study its uptake by MΦ cells. The Cy5.5 fluorescence intensity was greater in the HM-encapsulated NPs, which could also be blocked by a neutralising Icam1/Vcam1 antibody, suggesting that the M1EVs contributed to the delivery of the FBFO NPs to MΦs (Fig. 4a). Moreover, the fluorescence intensity of DCFH-DA was slightly greater in the FBFO NP treatment group than in the control group. However, the FBFO HM-encapsulated NPs presented the most robust green fluorescence intensity (Fig. 4b and Supplementary Fig. 5b), indicating that FBFO NPs can be efficiently delivered to macrophages to generate ROS under laser irradiation.

a Flow cytometry analysis showing the internalisation of Cy5.5-labelled FBFO by Raw264.7 cells (n = 3 independent experiments). b CLSM image of Raw264.7 cells stained with DCFH-DA to indicate NP-induced ROS generation. Scale bar, 200 μm. c Volcano plot showing the expression profile related to DEGs related to macrophage polarisation in Raw264.7 cells treated with PBS or FBFO@HM@aOPN. d GO BP and (e) GSEA enrichment analyses of the DEGs between the PBS- and FBFO@HM@aOPN-treated Raw264.7 cells. Flow cytometry analysis of the proportions of f CD80+CD86+ (n = 3 independent experiments), g MHC I+ (n = 4 independent experiments), h H-2Kb/SIINFEKL+ (n = 4 independent experiments), and i CD206+ (n = 3 independent experiments) among Raw264.7 cells treated with different formulations under 660 nm irradiation. j Establishment of BMDMs. Flow cytometry analysis of the proportions of k CD80+CD86+, l MHC I+, m H-2Kb/SIINFEKL+, and n CD206+ cells in BMDMs treated with different formulations under 660 nm irradiation (n = 3 independent experiments). o GSEA enrichment analyses of the DEGs between the PBS- and FBFO@HM@aOPN-treated Raw264.7 cells. p Western blot analysis of STING-IRF3 and NF-κB p65 phosphorylation in Raw264.7 cells and BMDMs treated with different formulations under 660 nm irradiation. q Schematic diagram of the mechanism by which FBFO@HM@aOPN reprograms macrophages to the M1-like phenotype. All the data are presented as the means ± SDs. The p values were determined via two-tailed one-way ANOVA with a Tukey post hoc test (f–i, k–n), p > 0.05, no significance (ns), *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001. Source data are provided as a Source Data file.
To explore the polarisation status of macrophages treated with the nanoplatform in detail, the transcriptomes of MΦ cells treated with PBS (control) or FBFO@HM@aOPN were first mapped via RNA sequencing (RNA-seq) analysis, and the results revealed significant differences between the two groups (Fig. 4c). A total of 4161 differentially expressed genes (DEGs) were identified, among which 1566 DEGs were upregulated and 2595 DEGs were downregulated (Supplementary Data 1). A volcano plot revealed that M1 macrophage markers (Cd86, Cd80, Tnf and Il6) were obviously upregulated, whereas M2 macrophage markers (Cd206, which is encoded by the Mrc1 gene, and Arg1) were markedly downregulated (Fig. 4c). In addition, Gene Ontology (GO) analysis revealed that the enriched biological process (GO BP) terms associated with the upregulated genes were associated mainly with immune processes, including the innate immune response, cellular responses to type I interferon (Ifn-β) and type II interferon (IFN-γ), positive regulation of the immune effector process, and the canonical NF-kappaB (NF-κB) signal transduction pathway. The downregulated genes were associated primarily with BPs such as matrix remodelling (Fig. 4d). Additionally, gene set enrichment analysis (GSEA) revealed that both the antigen-presenting pathway and the T-cell chemotaxis pathway were enriched in the upregulated genes (Fig. 4e). Overall, transcriptomic analysis revealed that the majority of DEGs from the FBFO@HM@aOPN-treated group were highly related to M1 macrophage polarisation and to IFN-mediated innate immune responses.
Next, we assessed the effects of different formulations on macrophage differentiation in vitro. As shown in Supplementary Fig. 5c, the results of the qPCR assays revealed that the FBFO@HM@aOPN group presented significantly increased mRNA levels of M1-like markers (Il6 and Thf), type I interferons (Ifn-β), and T-cell chemokines (Ccl5 and Cxcl10). Furthermore, the expression of M1 macrophage markers (Cd80 and Cd86) in MΦs after various treatments was detected via flow cytometry. Compared with the control treatment, the FBFO@OMV and FBFO@M1EV treatments both significantly upregulated these markers, and the FBFO@HM@aOPN-treated group presented the strongest effect (Fig. 4f and Supplementary Fig. 5d). These results confirmed the ability of FBFO@HM@aOPN NPs to promote the polarisation of macrophages from the M0 type to the M1 type. During antigen cross-presentation, tumour antigens are internalised by APCs, such as macrophages, and are subsequently presented to major histocompatibility complex class I (MHC-I) molecules, leading to CD8+ T-cell activation45. Our flow cytometry analysis revealed elevated major histocompatibility complex I (MHC-I) molecule expression in FBFO@HM@aOPN-treated macrophages (Fig. 4g and Supplementary Fig. 5e). Furthermore, when CT2A-overexpressing ovalbumin antigen (CT2A-OVA) was used as a model system, flow cytometry quantification revealed that the MHC-I-associated SIINFEKL peptide was significantly upregulated in FBFO@HM@aOPN-stimulated macrophages (Fig. 4h and Supplementary Fig. 5f), thereby establishing a foundation for modulating antigen cross-presentation and subsequently triggering the activation of specific CD8+ T cells by these FBFO@HM@aOPN-stimulated TAMs.
Since antitumour immunity involves the repolarization of M2 macrophages into M1 macrophages, we further investigated the ability of FBFO@HM@aOPN NPs to polarise M2-like macrophages into M1 macrophages. We cultured MΦ cells with conditioned medium (collected from CT2A cell culture 72 h after original seeding) to induce M2-like macrophage polarisation and then further cultured them with different formulations for 48 h. We showed that FBFO@HM@aOPN NPs significantly reduced the proportion of M2-like macrophages, as detected by Cd206 marker expression (Fig. 4i and Supplementary Fig. 5g), demonstrating that FBFO@HM@aOPN NPs exhibit a robust ability to repolarize M2-like macrophages.
To confirm the universality of the effect of FBFO@HM@aOPN NPs on macrophage polarisation, myeloid cells were isolated from C57BL/6 mice and further cultured and differentiated into bone marrow-derived macrophages (BMDMs) (Fig. 4j). The expression levels of M1 and M2 macrophage markers were detected via flow cytometric and qPCR analyses. As shown in Fig. 4k–m and Supplementary Fig. 6a–d, FBFO@HM@aOPN NPs caused significant upregulation of M1 macrophage markers and antigen-presenting processing-related factors. Moreover, the M2 macrophage marker CD206 was strongly downregulated in the FBFO@HM@aOPN group (Fig. 4n and Supplementary Fig. 6e). These results indicated that FBFO@HM@aOPN NPs could significantly polarise M0- and M2-like macrophages into the M1 phenotype in vitro.
To further understand how FBFO@HM@aOPN NPs promoted macrophage polarisation, we explored the underlying mechanisms involved. Given the robust association between IFN production and cGAS-STING activation46, we subsequently sought to confirm whether FBFO@HM@aOPN NPs modulate the immune response in macrophages through the cGAS-STING pathway. Our GSEA results also revealed that upregulated genes in the FBFO@HM@aOPN-treated group were significantly enriched in the cytosolic DNA sensing pathway via the Sting pathway (Fig. 4o). The Western blot results further confirmed that FBFO@HM@aOPN effectively increased the level of phosphorylated STING (p-STING) and its downstream induction of IRF3 transcriptional activity (Fig. 4p). Hypoxia and its induced products are strong inhibitors of STING activation in the TME47. Thus, FBFO@HM@aOPN-induced STING activation may be attributed to the CAT activity of FBFO. Moreover, the polarisation of M1 macrophages is also regulated by several signalling pathways, with the NF-κB factor being a key transcriptional regulator48, and NF-κB can be activated by multiple ROS and the LPS/TLR4 signal transduction pathway19. Our enrichment analysis also revealed that the NF-κB and Toll-like receptor signalling pathways were significantly enriched in genes whose expression was upregulated in the FBFO@HM@aOPN-treated group (Fig. 4d, o). As expected, the level of p65 phosphorylation was elevated in FBFO@HM@aOPN-treated macrophages (Fig. 4p). Our results showed that FBFO@HM@aOPN effectively promoted M1 macrophage polarisation via cGAS-STING and NF-κB activation (Fig. 4q). Collectively, these in vitro findings suggest that FBFO@HM@aOPN can overcome the current challenges in immunotherapy by promoting macrophage M1 polarisation, increasing the antigen-presenting capacity of TAMs, and synergistically alleviating TME immunosuppression, thus offering promising opportunities for enhancing antitumour immune responses in vivo.
Tumour suppression in a 3D tumour spheroid model
M1 macrophages are able to phagocytose tumour cells to inhibit malignant tumour progression47. To determine whether FBFO@HM@aOPN promotes macrophage phagocytosis, we cocultured tdtomato-labelled CT2A cells treated with different formulations with EGFP-labelled RAW264.7 cells pretreated with different formulations in vitro. CLSM and flow cytometry analysis revealed that RAW264.7 macrophages preincubated with NPs phagocytosed more cancer cells than control cells did (Fig. 5a, b). A 3D tumour model, namely, a multicellular tumour spheroid (MCTS) established by the coculture of M2-like macrophages and CT2A GBM cells, was subsequently used to investigate the ability of FBFO@HM@aOPN to penetrate the tumour (Fig. 5c). To determine whether FBFO@HM@aOPN enhances targeted FBFO delivery, MCTS was treated with Cy5.5-labelled FBFO for 6 h. CLSM in Z-stack scanning mode was utilised to assess the penetration effect (Fig. 5d). As shown in Fig. 5e, red fluorescence clearly diffused into the interior of the MCTS in the FBFO@HM@aOPN-treated group. However, in the FBFO-treated group, red fluorescence was mostly distributed on the periphery of the MCTS. These results indicated that in response to the TME, FBFO@HM@aOPN significantly promoted the deep penetration of PDT agents into tumour tissues, overcoming the natural physical barriers in solid tumours.

a CLSM image of the killing of CT2A GBM cells (red) by repolarized M1 macrophages (green). Scale bar, 100 μm. b Flow cytometry analysis of the killing of CT2A GBM cells by repolarized M1 BMDMs (n = 3 independent experiments). Schematic illustration of in vitro three-dimensional (3D) tumour model (c) preparation and (d) potential for assessing the penetration effect. e Z-stack CLSM images of the penetration of Cy5.5-labelled FBFO NPs in CT2A-BMDM multicellular spheroids after 6 h of incubation. Scale bar, 250 μm. f Z-stack CLSM images of CT2A-BMDM multicellular spheroids (MCTSs) dyed with hypoxia green reagent to indicate the degree of hypoxia. Scale bar, 250 μm. g Representative photographs of MCTSs at a certain time (left) and the volume histogram of MCTSs treated with different formulations on day 7 (right, n = 3 independent experiments). Scale bar, 200 μm. h Flow cytometry analysis of the proportions of CD206+ BMDMs in MCTSs treated with different formulations under 660 nm irradiation (n = 3 independent experiments). i Secreted IFN-β, IL-6, TNF-α, CCL5 and CXCL10 concentrations in the cell culture supernatants of MCTSs subjected to various treatments, as measured by ELISA (n = 3 independent experiments). All the data are presented as the means ± SDs. The p values were determined via two-tailed one-way ANOVA with a Tukey post hoc test (b, g–i), p > 0.05, no significance (ns), *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001. Source data are provided as a Source Data file.
Next, the potential of FBFO@HM@aOPN to remodel the abnormal TME was investigated by analysing the hypoxia level and macrophage phenotype in MCTS. After 12 h of FBFO@HM@aSPP treatment, the hypoxia level in the interior of MCTS was assessed via immunofluorescence staining with hypoxia green reagent. FBFO@HM@aSPP-treated tumour spheroids presented the weakest green fluorescence intensity, even in the core. In contrast, the PBS- or free FBFO-treated tumour spheroids displayed strong green fluorescence signals (Fig. 5f), confirming the effective alleviation of tumour hypoxia by FBFO@HM@aSPP. Moreover, the final volume of spheroids in the FBFO@HM@aOPN group was significantly lower than that in the PBS group (Fig. 5g). According to flow cytometry and ELISA analyses of the macrophage phenotype, FBFO@HM@aOPN treatment effectively promoted the polarisation of M2-like macrophages to the M1-like phenotype (Fig. 5h, i), indicating the ability of FBFO@HM@aOPN to modulate the immunosuppressive microenvironment in solid tumours. These results suggest that FBFO@HM@aOPN can remodel the hypoxic and immunosuppressive microenvironments while simultaneously successfully penetrating natural physical tumour barriers, thus providing promising opportunities to enhance antitumour immune responses in vivo.
Biodistribution, biocompatibility, and antitumour effects in vivo
Specific proteins on M1EV membranes, including integrins α4 and β1 (Fig. 2l), are known to be crucial for facilitating BBB penetration18,43. To assess the ability of the FBFO@HM@aOPN biohybrid delivery system to permeate the BBB, a TranswellTM coculture system was used in which bEnd.3 cells were cultured in the upper chamber and primary BMDMs and CTA2 cells were cultured in the lower chamber (Fig. 6a). The bEnd.3 cells cultured in the upper chamber formed an intact monolayer with a transendothelial electrical resistance (TEER) of 200 − 300 Ω·cm (Fig. 6b and Supplementary Fig. 7a). We observed that the ability of FBFO@HM@aOPN-NPs to cross the BBB was approximately the same as that of FBFO@M1EV NPs, but the latter could be blocked by anti-ICAM1/VCAM1 (Fig. 6c), suggesting that FBFO@HM@aOPN NPs can better penetrate the BBB.

a Schematic diagram of the in vitro BBB model. b The transendothelial electrical resistance (TEER, Ω·cm²) values in the in vitro BBB model at different time points after incubation with formulations (n = 3 independent experiments). c Transport ratios of different formulations determined by detecting Cy5.5 fluorescence intensity in the apical chamber and basolateral chamber after 4 h of incubation in the in vitro BBB model (n = 3 independent experiments). d In vivo Cy5.5 fluorescence images of CT2A-bearing mice at different time points after intravenous injection of different formulations. Ex vivo (e) Cy5.5 fluorescence images and (f) quantitative histogram of the fluorescence intensity of both the CT2A-bearing brain and major organs (including the heart, liver, spleen, lung, and kidney; n ≥ 3 mice for each group). g Experimental design of therapeutic FBFO@HM@aSPP intervention in a CT2A tumour model (n = 5 mice per group). h Representative bioluminescence images (left) and fluorescence intensity quantification (right), as well as (i) Kaplan–Meier survival curves of luciferase-expressing CT2A-bearing mice subjected to different treatments (n = 5 mice per group). j H&E staining of brain tumour tissues from CT2A tumour-bearing mice subjected to various treatments. Scale bar, 2 mm. CLSM images of (k) Ki67 (scale bar, 50 μm), (l) CD44 (scale bar, 20 μm) and (m) CALR (scale bar, 20 μm) staining of brain tumour tissues from CT2A tumour-bearing mice subjected to various treatments. All the data are presented as the means ± SDs. The p values were determined via two-tailed one-way ANOVA with a Tukey post hoc test (c, h). For i, p values are calculated via the log-rank (Mantel‒Cox) test. p > 0.05, not significant (ns), *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001. Source data are provided as a Source Data file.
Next, the in vivo biological effects of various nanoagents were investigated. To confirm whether M1EV membrane encapsulation promotes the accumulation of FBFOs in GBM tissue, a Cy5.5-labelled FBFO system was injected into mice expressing luciferase CT2A (CT2A-Luc), and Cy5.5 fluorescence was monitored in real time. Stronger red fluorescence signals were clearly observed in the brains of the mice treated with FBFO@HM@aOPN-NPs at 6 h postinjection, which remained detectable for up to 48 h (Fig. 6d), and a similar trend of fluorescence signals was observed at the tumour sites of the mice treated with FBFO@M1EV-NPs or FBFO@HM-NPs (Fig. 6d). Ex vivo fluorescence images (Fig. 6e) and quantitative fluorescence intensity analysis (Fig. 6f) of both the tumour-bearing brain and major organs (including the heart, liver, spleen, lung, and kidney) also revealed substantial accumulation of FBFO@HM@aOPN with respect to tumour size, indicating that FBFO@HM@aOPN can penetrate the BBB and accumulate in tumours. To further investigate biocompatibility, whole blood was collected from FBFO@HM@aOPN-treated and untreated mice for blood biochemistry and routine blood tests. Compared with the PBS treatment, the FBFO@HM@aOPN treatment had no discernible effect on blood indicators (Supplementary Fig. 8a), indicating suitable biosafety. The above results demonstrated that FBFO@HM@aOPN possessed suitable tumour targeting ability, tumour retention, and biocompatibility.
The antitumour efficacy of FBFO@HM@aOPN was further evaluated in vivo. The CT2A tumour-bearing mice were randomly divided into 7 groups on the sixth day after tumour inoculation and then subjected to various treatments (Fig. 6g). Tumour growth was continuously monitored via 7-day interval bioluminescence imaging to assess the antitumour effects. Compared with the rapid tumour growth in the PBS group, the monotherapy groups demonstrated moderate tumour inhibitory effects, whereas the FBFO@HM@aOPN group demonstrated a significant tumour inhibitory effect (Fig. 6h, i), which may be attributed to the combined effect of PDT-induced tumour cell killing and TME modulation. All the tumour-bearing mice were sacrificed at the end of the experiment, and haematoxylin and eosin (H&E), Ki67 and CD44 staining were performed on the tumour sections to investigate the antitumour effects in depth. The results revealed prominent tumour cell growth suppression and reduced ECM protein production in the FBFO@HM@aOPN treatment group (Fig. 6j–l). Notably, all groups of mice presented slightly increased body weights during antitumour therapy, and no discernible damage was observed in the H&E-stained images of major organs (heart, liver, spleen, lung, and kidney), further suggesting that these NPs had negligible side effects (Supplementary Fig. 8b). We also conducted a comprehensive analysis of systemic serum inflammatory cytokine levels (IL-6, TNF-α, and IL-1α/β) in treated mice (Supplementary Fig. 9a). Our results revealed that FBFO@HM@aOPN did not trigger cytokine syndrome, and the cytokine profiles remained within safe physiological ranges (Supplementary Fig. 9b). Moreover, the immunofluorescence images clearly revealed a notable increase in CALR exposure in tumours treated with FBFO@HM@aOPN (Fig. 6m), indicating the successful induction of immunogenic cell death in vivo. Overall, these results suggested that FBFO@HM@aOPN NPs have superior antitumour capacity in vivo.
ScRNA-seq analysis revealed increased immune remodelling
We further investigated the changes in the TME to elucidate the underlying antitumour mechanism of FBFO@HM@aOPN. ScRNA-seq was performed on CT2A tumours that underwent 3 rounds of FBFO@HM@aOPN treatment (Fig. 6g). Specifically, after quality control, filtration and cellular annotation, we obtained thirteen cell types (Fig. 7a and Supplementary Data 4). We found that the proportion of tumour cells was significantly decreased. Notably, a population of cells with high expression of IFN-regulated genes, such as CXCL10, ISG15 and interferon-induced proteins with tetratricopeptide repeats (IFITs), was identified and named the malignant-IFN population (Fig. 7a–c), suggesting that a strong interferon response was triggered in vivo. We then analysed the overall changes in the tumour cell populations in the FBFO@HM@aOPN-treated group. Differential gene analysis revealed that 618 and 890 genes were significantly upregulated in the malignant and malignant-IFN populations, respectively, in the FBFO@HM@aOPN-treated group (Fig. 7d). Interestingly, 234 of these genes, including IFN-regulated genes and T-cell-related chemokines (CXCL10 and CCL5), were significantly upregulated in both subpopulations (Fig. 7d). Subsequent KEGG enrichment analyses revealed that pathways such as ferroptosis, apoptosis and those related to the immune response, including the TNF signalling pathway, the cytosolic DNA-sensing pathway by sting, the Toll−like receptor signalling pathway, and the chemokine signalling pathway, were significantly enriched in the upregulated genes. However, pathways such as the HIF − 1 signalling pathway and focal adhesion were significantly enriched in the downregulated genes (Fig. 7e), indicating that FBFO@HM@aOPN could alleviate tumour hypoxia and matrix deposition.

a Uniform manifold approximation and projection (UMAP) plot showing the 13 major cell types. The dots represent individual cells, and the colours represent different cell populations. The samples were separated according to their origin (PBS- and FBFO@HM@aSPP-treated GBM). b Cell density distribution across two tissue groups. A high relative cell density indicates bright magma. c Bar graphs displaying the relative cellular fractions of the major cell types across the two tissue groups. d Differential gene expression analysis showing up- and downregulated genes across malignant and malignant IFN subpopulations in the treated group compared with the PBS group. e Bar plot showing the results of the KEGG enrichment analyses of the DEGs between FBFO@HM@aSPP-treated and PBS-treated malignant cells. f UMAP plot showing the 3 BMDM subpopulations. The dots represent individual cells, and the colours represent different cell populations. g Cell density distribution across two tissue groups. A high relative cell density indicates bright magma. h Dot plot showing the gene expression of M1-like and M2-like markers. i Bar plot showing HALLMARK enrichment analyses of the DEGs between FBFO@HM@aSPP-treated and PBS-BMDM-treated cells. j GSEA showing that the upregulated genes were significantly enriched in antigen processing and presentation and positive regulation of phagocytosis in the treated group. k UMAP plot showing the 6 lymphocyte subpopulations. The dots represent individual cells, and the colours represent different cell populations. l Cell density distribution across two tissue groups. A high relative cell density indicates bright magma. m Cell density showing the expression of effector-related genes. A high relative cell density indicates bright magma. n Enriched pathways of DEGs across different lymphocyte subpopulations. Colours from blue to red indicate the absolute value of the DE score from low to high. o Dot plot showing the gene expression of immune checkpoints and effectors. p Visualisation of the main senders and receivers of cell clusters in the 2D space of the (CD80/CD86) and (MHC-I and MHC-II) pathways.
We next analysed the overall changes in tumour myeloid cells and infiltrating T-cell populations in the FBFO@HM@aOPN-treated group. Through dimensionality reduction, we divided BMDMs into three subpopulations: monocyte, M1 and M2 subpopulations (Fig. 7f). Our scRNA analysis revealed that, compared with those in the PBS group, the proportion of M1 macrophages among the tumour-infiltrating BMDMs was greater in the FBFO@HM@aOPN-treated group, with a corresponding decrease in the proportion of M2 macrophages (Fig. 7g). Moreover, the expression of M1 macrophage markers (Cd86, Tnf, Ifnb1), T-cell-related chemokines (Ccl5, Cxcl10/11), and antigen-presenting molecules (MHC complex) was upregulated, whereas the expression of M2 macrophage markers (Mrc1, Arg1, Il10 and Vegfa) was downregulated in the FBFO@HM@aOPN-treated group (Fig. 7h). Subsequent HALLMARK enrichment analyses revealed that the upregulated genes were related mainly to immune processes, including the innate immune response, cellular responses to interferon-β (IFN-β) and IFN-γ, positive regulation of inflammatory cytokines, and Tnfa signalling via the NF-κB pathway (Fig. 7i). Furthermore, GSEA revealed significant enrichment of pathways related to antigen presentation and phagocytosis in BMDMs in the treated group (Fi. 7j). Furthermore, we analysed the expression of the top ten genes associated with hypoxia (Supplementary Fig. 10a) and found that they were predominantly expressed in the M2 subpopulation and that their expression was downregulated in the treatment group (Supplementary Fig. 10b). Collectively, these findings suggested that FBFO@HM@aOPN synergistically promoted M1 macrophage polarisation, enhanced TAM antigen presentation and phagocytosis, and alleviated hypoxic immunosuppression in vivo.
Our scRNA analysis revealed that the proportion of lymphocytes in the tumour-infiltrating immune population was greater in the treated group than in the PBS group (Fig. 7c). To analyse the lymphoid compartment in more detail, we further divided the lymphocytes into six lymphocyte subpopulations, namely, CD8+ T cells, conventional CD4+ T cells (convCD4+ T cells), regulatory CD4+ T cells (Tregs), natural killer (NK) cells, naive T cells, and one cluster of proliferating lymphoid cells (Fig. 7k). STING activation leads to the activation of the type I interferon proinflammatory pathway by recruiting and activating immune cells and augmenting NK cell- and T-cell-mediated tumour killing, driving multifaceted antitumour immune functions in the TME and tumour-draining lymph nodes49,50. We found that FBFO@HM@aOPN treatment significantly induced the expansion and proliferation of CD8+ T cells and NK cell populations (Fig. 7l), which might be attributed to the activation of type I and II interferon pathways in vivo. We further analysed the expression of the nine genes most relevant to the effector function of lymphocytes and found that most of them were highly expressed in the CD8+ T and NK cell populations (Fig. 7m and Supplementary Fig. 10c). In addition, functional enrichment analysis revealed significant enrichment of pathways associated with T-cell activation among the upregulated genes, whereas pathways associated with immunosuppression were significantly enriched among the downregulated genes in the treatment group (Fig. 7n). A bubble diagram revealed that the expression of effector genes was upregulated in the treatment group, whereas the expression of most of the immune checkpoint genes tended to be downregulated in the treatment group (Fig. 7o). Finally, we analysed intercellular communication networks between immune cells and GBM cells via CellChat ligand−receptor analysis. Interestingly, we revealed that Cd80/86 costimulation and the antigen-presenting (MHC-I/II) signalling pathway are mediated predominantly by a subpopulation of BMDMs and CD8+ T cells in the treated group (Fig. 7p), indicating that BMDMs can act as activated APCs to promote antigen cross-presentation to effector T cells through antigen-presenting and costimulatory signals, which ultimately enhances the adaptive immune response in the treatment group. Collectively, these findings suggest that photoimmunotherapy utilising FBFO@HM@aOPN can potentially augment innate and acquired immune activation by promoting the intratumoral infiltration of M1 macrophages, cytotoxic T lymphocytes (CTLs), and NK cells, thereby facilitating the conversion of immune-cold to immune-hot tumours.
FBFO@HM@aOPN enhances antitumour immunity in vivo
We further performed a comprehensive immunoassay to further validate the results of the scRNA-seq analysis. At the end of the antitumour treatments, all the tumours were collected to analyse the tumour-infiltrating immune cells via flow cytometry. Consistent with the in vitro results, FBFO@HM@aOPN significantly increased the proportion of antitumour M1 macrophages and the level of cross-presenting antigens but reduced the proportion of protumour M2 macrophages in TAMs within tumours (Fig. 8a and Supplementary Fig. 11a, b), further indicating that FBFO@HM@aOPN can modulate TAM cross-presentation capabilities and stimulate a robust, antigen-specific T-cell immune response. To further demonstrate the activation of antitumour immune responses, we investigated infiltrating lymphocytes in tumours harvested from various groups. As shown in Fig. 8b and Supplementary Fig. 12a, b, the proportion of T cells significantly increased in the FBFO@HM@aOPN group. Moreover, consistent with the scRNA results, FBFO@HM@aOPN significantly increased the proportions of antitumorigenic cytotoxic lymphocytes, including NK cells (Fig. 8c and Supplementary Fig. 12c) and CTLs (Fig. 8d and Supplementary Fig. 12d). Furthermore, the increased proportion of granzyme B (GZMB)-positive CTLs in the FBFO@HM@aOPN group indicated that our nanotherapeutic platform has excellent ability to activate CTLs (Fig. 8e and Supplementary Fig. 12e). Long-term immune memory is essential for preventing tumour recurrence. Thus, we subsequently measured the proportions of central memory T cells (TCM, CD3+ CD44+ CD62L+) and effector memory T cells (TEMs, CD3+ CD44+ CD62L-) in the spleen. We found that both proportions increased significantly after treatment with FBFO@HM@aOPN (Fig. 8f, g and Supplementary Fig. 12f, g), suggesting that durable immunological memory was elicited by photoimmunotherapy.

a Flow cytometry analysis of the proportions of (left) CD86+ (middle) MHCI+ and (right) CD206+ among f4/80+ CD45+ cells from CT2A-bearing brain tissues treated with different formulations under 660-nm irradiation (n = 5 mice for each group). Flow cytometry analysis of the proportions of (b) CD3+ T cells, (c) NK1.1+ CD3- NK cells, (d) CD3+ CD8+ TILs, and (e) GZMB+ TILs among the CD45+ cells from CT2A-bearing brain tissues treated with different formulations under 660 nm irradiation (n = 5 mice for each group). Flow cytometry analysis of the proportions of (f) central memory T cells (TCM, CD3+ CD44+ CD62L+) and (g) effector memory T cells (TEM, CD3+ CD44+ CD62L-) in CT2A-bearing spleens treated with different formulations under 660-nm irradiation (n = 5 mice for each group). h Bubble chart showing the interactions between PD-1/PD-L1 activity (released by TAMs and the parasite) and PD-L1 receptor expression (expressed by lymphocyte cell subtypes) in treated and PBS-treated samples. i Schematic illustration of the experimental design for postsurgical recurrence challenge in CT2A GBM tumours. (j) Representative bioluminescence image and (k) fluorescence intensity quantification (right), as well as (l) Kaplan–Meier survival curve of luciferase-expressing CT2A-bearing mice subjected to different treatments under 660-nm irradiation (n = 5 mice in each group). m Flow cytometry analysis of the proportions of (left) CD3+ T cells, (middle) IFN-γ + T cells, and (right) GZMB+ TILs among CD45+ cells from CT2A-bearing brain tissues treated with different formulations under 660-nm irradiation (n = 5 mice for each group). All the data are presented as the means ± SDs. The p values were determined via two-tailed one-way ANOVA with a Tukey post hoc test (a–g, k, m). For l, p values are calculated via the log-rank (Mantel‒Cox) test. p > 0.05, not significant (ns), *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001. Source data are provided as a Source Data file.
FBFO@HM@aOPN and ICB combination therapy prevents postsurgical GBM recurrence
In clinical practice, surgical intervention is widely recognised as the primary treatment option for early-stage solid tumours. However, owing to the presence of residual tumour cells after surgery, GBM recurs around the resected lesion in almost all patients. FBFO@HM@aOPN promoted interferon-stimulated gene transcription, and both interferon gamma and type I interferon were able to upregulate the expression of PD-L1 on the surface of tumour cells and BMDMs, which increased the degree of immune escape of tumour cells51,52. As expected, the analysis of single-cell interaction data revealed that the PDL1-PD1 pathway between tumour cells and BMDM with T cells was upregulated during FBFO treatment (Fig. 8h). We subsequently explored whether FBFO@HM@aOPN, in combination with an anti-PD1 inhibitor, could delay tumour recurrence. The surgical procedure and treatment regimen are shown in Fig. 8i. The tumour resection cavity and the residual tumour foci were imaged after H&E staining (Supplementary Fig. 12a). Tumour recurrence was monitored by bioluminescence (BLI) signals from Luci+ CT2A cells. The BLI signal of the FBFO@HM@aOPN-treated tumours markedly decreased (Fig. 8j, k). Combination with an anti-PD1 (aPD1) antibody further increased the tumour suppression efficacy (Fig. 8j, k). Furthermore, the survival rate of the mice that received FBFO@HM@aOPN NPs and aPD1 antibody combination treatment was significantly greater than that of the mice treated with the other formulations (Fig. 8l). All the mice survived on day 100 after the FBFO@HM@aOPN and aPD1 antibody combination treatments (Fig. 8l). We then investigated infiltrating lymphocytes in tumours harvested from various groups. As shown in Fig. 8m and Supplementary Fig. 12b–d, the proportions of T cells, IFNγ-positive T cells, and GZMB-positive CTLs were significantly increased in the FBFO@HM@aOPN group and the combination group. Interestingly, FBFO@HM@aOPN combined with αPD1 antibody treatment resulted in the strongest resistance to tumour rechallenge. Together, these data suggest that the combination of FBFO@HM@aOPN and ICB therapy is a highly personalised therapeutic strategy that is conducive to preventing tumour recurrence.
Discussion
The hypoxic and immunosuppressive properties of the TME and the lack of precise drug delivery techniques pose significant challenges to improving the effectiveness of PDT and immunotherapy. In this study, we developed an efficient and precise synergistic antitumour therapeutic nanoplatform for highly effective deep GBM treatment, which consists of NPs with photodynamically catalysed POD-like and CAT-like dual enzyme-like enzymes (FBFO NPs) encapsulated within OMV-M1EV HMs loaded with an aOPN antibody to form FBFO@HM@aOPN NPs. First, the ability of M1EV to penetrate the BBB and stimulate inflammation facilitated the precise delivery of the treatment to the tumour site. The release of an anti-OPN neutralising antibody in the acidic TME effectively increased vascular permeability, regulated the composition of the ECM, reduced the hardness of the tumour, and facilitated the infiltration of the NPs and immune cells into the tumour. Moreover, the local hypoxic microenvironment was alleviated via the CAT-like enzyme activity of FBFO. HM modification endows FBFO@HM@aOPN with a more efficient active targeting effect on tumour cells and TAMs, which, on the one hand, provides abundant tumour-specific neoantigens for APCs by inducing the ferroptosis of tumour cells and, on the other hand, repolarizes TAMS to increase their antigen presentation and phagocytosis, which together activate innate and adaptive antitumour immunity, inhibiting GBM malignant progression. In addition, the platform was used in combination with anti-PD1 ICIs to enhance the therapeutic effect and effectively prevent tumour recurrence after GBM surgery.
Tumours use the immune and vascular systems to establish immunosuppressive ecological niches and evade recognition and eradication by T cells53. Accordingly, antiangiogenic agents (e.g., VEGF inhibitors such as bevacizumab) are now being used to treat GBM, with variable clinical success23. Thus, there is an urgent need for new approaches to reprogram the vascular–immune interface. Recent studies have reported that perivascular M2 macrophages produce large amounts of secreted OPN, which interacts with receptors such as CD44 and integrins on the surface of a variety of stromal cells, leading to aberrations in the tumour vasculature system and to the stimulation of extracellular matrix remodelling to promote the formation of TIB structures, thus limiting immune cell infiltration into the tumour core15,24,54. Preclinical studies have shown that blocking OPN in mice disrupts the TIB structure and sensitises tumours to immunotherapy27,55, which has become a new target for antitumour immunotherapy. Here, to increase the efficiency of the immune response, we coupled a therapeutic OPN-blocking antibody to the surface of a nanoplatform to enable its controlled release in the acidic TME, thereby increasing tumour vascular permeability and altering the ECM composition, which helped increase the penetration and accumulation of nanoenzymes in the TME, increase the tumour-killing abilities of lymphocytes within the tumour, and alleviate the local immunosuppressive TME.
In recent years, biomimetic strategies based on natural cell membranes have been used to functionalise nanoparticles (NPs) for targeted delivery of therapeutic agents56,57. In this study, we created a new nanotechnology to greatly improve the targeting and functionality of therapeutic platforms and expand their application value by fusing E. coli OMVs and M1EVs to form a prokaryotic and eukaryotic HM (PEHM) delivery platform. M1EVs have the advantages of penetrating the BBB and homing to the inflammatory microenvironment, and OMVs, as natural adjuvants, can provide stable targeted delivery and immune stimulation. Combination therapy with HM-coated NPs and PDT exerted enhanced antitumour effects.
This study has several limitations. First, while ultracentrifugation is a widely adopted method for EV isolation in preclinical research, its scalability for industrial production was not directly validated here. Although ultracentrifugation underpins current biologics manufacturing methods58,59,60, optimising yield and cost efficiency for large-scale hybrid membrane production requires the further integration of scalable technologies such as tangential flow filtration or bioreactor systems61. Second, the immunogenicity and long-term stability of prokaryotic‒eukaryotic hybrid membranes need systematic evaluation in large animal models to advance their clinical translation. Addressing these challenges will accelerate the translation of multifunctional nanoplatforms.
In summary, we generated an innovative and promising nanotherapeutic platform for GBM to increase the efficacy of PDT and immunotherapy combination treatment by addressing the inherent complexity of the TME from a different perspective. In vivo therapeutic results revealed that the system had synergistic antitumour effects on the hypoxic TME, normalising the vasculature and mediating the extracellular matrix composition, which in turn enhanced antitumour innate and adaptive immunity. In addition, we created a biocompatible nanodelivery platform using eukaryotic–prokaryotic membrane fusion, establishing a bionanomimetic strategy for enhanced photodynamic therapy and immunotherapy combinations.
Methods
Human and animal ethics declarations
The human primary GBM samples used in this study were obtained from Qilu Hospital of Shandong University according to a protocol approved by the Ethics Committee of Qilu Hospital of Shandong University. All participants provided written informed consent, and the study was approved by the Ethics Committee on Scientific Research of Shandong University Qilu Hospital (approval number: KYLL-2023(ZM)−657).
All procedures involving mice were approved by the Committee and Laboratory Animal Department of Qilu Hospital of Shandong University (Institutional Animal Care and Use Committee issue no. DWLL-2021-126). According to the ethics committee, the orthotopic brain tumours required an absolute tumour volume limit of ≤ 200 mm³ (spherical-equivalent diameter ≤ 7.3 mm), a threshold reflecting cranial cavity constraints and preceding neurological compromise. Throughout this study, no tumours exceeded 150 mm³, while the animal must not lose more than 20% of its body weight during the research. Sex was not taken into consideration in the study design. The mice were kept in a barrier environment with a constant temperature of 24 °C and a relative humidity of 50%. The mice were maintained under a 12-hour light and 12-hour dark cycle.
Materials
Iron trichloride hexahydrate, bismuth nitrate pentahydrate, polyvinyl pyrrolidone, sodium oleate, diphenyl ether and oleyl alcohol were purchased from Shanghai Macklin Biochemical Co., Ltd. (Shanghai). Ethanol and n-hexane were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai). DBCO-PEG5-NH2 was purchased from Dianhuatech Co., Ltd. (Xian, China), and benzaldehyde (BD)-PEG2000-NHS was obtained from Ponsure Co., Ltd. (Shanghai, China). DMEM and RPMI-1640 cell culture media and foetal bovine serum (FBS) were purchased from Cellmax (China), and 1× PBS was purchased from Servicebio (G4202, China). Azide-choline (AEcho) and DBCO-PEG5-NH2 were purchased from Dianhuatech Co., Ltd. (Xian, China), and benzaldehyde-PEG2000-NHS was obtained from Ponsure Co., Ltd. (Shanghai, China). InVivoMAb anti-mouse OPN (osteopontin/OPN antibody) was obtained from R&D Systems (AF808, USA). Azide-choline (AEcho, HY-W588725), lipopolysaccharides (LPS, HY-D1056), IL4 (HY-P70653), cyanine 5.5 (HY-D0924), and cyanine5 DBCO (HY-D1583) were purchased from MCE (China). The Cell Counting Kit-8 (CCK8, C0038), Annexin V-FITC Apoptosis Detection Kit (C1062S), Hoechst 33342 (C1029), DAPI-containing anti-fluorescence quencher (P0131), ATP assay kit (S0026), and Calcein/PI Cell Viability/Cytotoxicity Assay Kit (C2015M) were purchased from Beyotime (China). Recombinant murine M-CSF (AF-315-02) was purchased from PeproTech (USA). An RNA Extractor kit was purchased from Yeasen (China). Mouse interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), interferon-β (IFN-β), C-C motif chemokine ligand 5 (CCL-5), and C-X-C motif chemokine ligand 10 (CXCL0) enzyme-linked immunosorbent assay (ELISA) kits were obtained from MultiSciences Biotech Co., Ltd. (Hangzhou, China). The antibody information used in this study is listed in Supplementary Data 2.
Cell culture
CT2A mouse GBM cells were a gift from Prof. Chunsheng Kang of Tianjin Medical University. The mouse macrophage line (RAW 264.7) and brain endothelial cells (bEnd.3) were supplied by the American Type Culture Collection (ATCC). All the cell lines were cultured in DMEM supplemented with 10% FBS and maintained in a 37 °C humidified chamber with 5% CO2.
Synthesis of Fe3O4 NPs
Iron oleate was added to 200 mL of diphenyl ether and 50 mL of oleyl alcohol. The mixture was stirred and heated to 200 °C and then reacted for 30 min under a N2 atmosphere. The product was centrifuged, washed three times with ethanol, and dried, resulting in a brown powder. Iron oleate was prepared according to methods described in earlier reports62.
Synthesis of Fe3O4@BiFeO3 (FBFO)
The solvothermal method was employed to synthesize Fe3O4@BiFeO3 (FBFO) NPs. Initially, 2 mmol of bismuth nitrate pentahydrate and 8 μmol of polyvinyl pyrrolidone were dispersed in 20 mL of ethylene glycol with magnetic stirring at 600 rpm for 120 min. Separately, 2 mmol of Fe3O4 was dispersed in 20 mL of ethylene glycol and then added to the previously prepared mixture, with magnetic stirring at 800 rpm for 120 min. The resulting mixed solution was transferred to a Teflon-lined stainless steel autoclave and maintained at 200 °C for 20 h. Finally, the product was collected and washed three times alternately with ethanol and distilled water.
Preparation of M1 macrophage-derived exosomes (M1EVs)
Azide-choline (AECho) was first prepared according to our previous report. Then, 1×106 RAW264.7 cells were treated with 200 nM LPS and 200 μM AECho for 24 h. To obtain azide-modified M1EVs, the treated macrophages were cultured in exosome-free medium (DMEM containing exosome-depleted FBS) for another 48 h. Thereafter, the conditioned cell culture medium was collected, and the cells and debris were removed by centrifugation at 300 × g (10 min), 2000 × g (20 min), 0.22 μm filter membrane filtration, and 10,000 × g (30 min) at 4 °C. M1EVs were collected by centrifugation at 100,000 × g for 70 min via an Optima XPN ultracentrifuge with a type SW32 Ti rotor (Beckman). Transmission electron microscopy (TEM) was used to determine the morphology and size of the exosomes. To confirm the successful modification of M1EVs with azide (N3) groups, flow cytometry analysis was performed. M1EVs were isolated from the culture supernatant of RAW264.7 cells treated with azide-choline (AECho) and LPS. The exosomes were then stained with DBCO-Cy5.5, a fluorescent dye that specifically binds to azide groups via click chemistry. The concentration and size of the purified exosomes were determined via nanoparticle tracking analysis (NTA, Zeta View PMX 110, Meerbusch, Germany).
Preparation of bacterial membrane vesicles (OMVs)
The ΔmsbB/ΔpagP E. coli W3110 strain was purchased from BioSciBio (China). OMVs were extracted from the msbB mutant W3110 strain via differential centrifugation19,63. E. coli was grown on solid Luria broth (LB) media at 37 °C overnight. Next, a single colony was inoculated into liquid LB medium, cultured in a rotary shaker at 37 °C for 6 h and then refreshed with LB medium at a 1:100 dilution. The shaking flask cultures were grown until the OD600 value of the medium reached approximately 1.0, indicating the logarithmic growth phase. OMVs were collected as follows. Briefly, 500 ml of bacterial culture medium was centrifuged at 5,000 × g for 30 min to remove the bacteria, followed by filtering through a 0.45 μm polyethersulfone filter (Millipore, SHLP033RB). The filtrate was then concentrated via centrifugal filtration with a molecular weight cut-off of 100 kDa (Millipore, UFC910096). The concentrated medium was pelleted by ultracentrifugation at 150,000 × g for 3 h at 4 °C via an Optima XPN ultracentrifuge with a type SW32 Ti rotor (Beckman). Then, the pellet was suspended in sterile PBS and ultracentrifuged at 150,000 × g for 3 h at 4 °C. Finally, the pellet was resuspended in PBS as the stock OMV preparation and subjected to agar plating to ensure a lack of bacterial contamination. TEM was used to determine the morphology and size of the OMVs. The concentration and size of the purified OMVs were determined via nanoparticle tracking analysis (NTA, Zeta View PMX 110, Meerbusch, Germany).
Preparation and characterization of FBFO@HM@OPN
OMVs were added to the Cy5.5-labelled M1EVs at a concentration ratio of 1:1, followed by stirring at 37 °C for 2 h and sonication for 10 min, then by 5 cycles of freeze-thawing with liquid nitrogen to facilitate membrane fusion. The mixed solution was then mixed with FBFO NPs and physically extruded through a 100 nm polycarbonate membrane via a liposome extruder (Avanti®, 610000, USA) for ten passes to form the fused membrane-coated nanoparticles FBFO@HM. The uni-membrane-coated FBFO NPs were fabricated via the same method with OMVs or M1EVs. M1EVs were stained with DBCO-Cy5.5, FBFO was labelled with FITC, and OMVs were doped with DiI. The colocalization of Cy5.5 and DiI fluorescence in the FITC-labelled FBFO NPs was observed via CLSM, which indicated the successful preparation of FBFO@HM.
Anti-OPN antibodies (aOPN) were reacted with a 100-fold molar excess of BD − PEG2000 − NHS in 1× PBS for 4 h at 4 °C under gentle stirring, and the unreacted BD − PEG2000 − NHS ester was then removed by a centrifugal filter device (Millipore Co., Amicon Ultra0.5) at 10,000 × g and 4 °C for 10 min. The resulting antibodies were resuspended in 0.01 M citric acid buffer (pH 6.0), followed by the addition of a 100-fold molar excess of Sulfo DBCO-PEG4-Amine. The mixture was allowed to react for 3 h at 4 °C under mild stirring, and the product was purified by a centrifugal filter device and then suspended in PBS for future use to form aOPN-PEG2000-DBCO.
To prepare aOPN-decorated FBFO@HM (FBFO@HM@aOPN), the newly synthesised aOPN-PEG2000-DBCO was mixed with FBFO@HM NPs (2.4 μg aOPN per 100 μg HM) in 500 μL of PBS for click chemistry reactions at 4 °C overnight. After the removal of any unreacted antibodies by centrifugation (11000 × g, 20 min), the resulting product of FBFO@HM@aOPN NPs was obtained and resuspended in PBS. The colocalization of DiI-doped HM and aOPN was observed via CLSM, which confirmed the successful synthesis of FBFO@HM@aOPN. TEM was used to determine the morphology and size of the FBFO@HM@aOPN NPs.
To study the release of total aOPN, FBFO@HM@aOPN NPs were dialysed in 1× PBS (pH=7.4 or 6.5), and ELISA capture was used to measure the concentration of aOPN in the dialysate collected at different time intervals. The antibody concentration was subsequently calculated according to the standard curves.
Assessment of the Reactive oxygen species (ROS) generation capacity
ROS generation was detected by methyl blue (MB), which could oxidise MB and made it fade from blue. Electron Spin Resonance (ESR) was used to further verify the production of ROS by using a signal capture agent of 5,5-dimethyl-1-pyrroline-N-oxide (DMPO).
Intracellular ROS detection using the fluorescent probe DCFH-DA, which in turn detects the fluorescence of DCF by CLSM and flow cytometry.
Analysis of dissolved oxygen in solution
100 μM H2O2 was prepared using PBS buffer at pH = 7.4 and pH = 6.5, respectively. After treatment with various FBFO NPs, the oxygen concentrations in the solution were measured. All dissolved oxygen detection experiments were measured by a dissolved oxygen metre (JPB-607A).
Cell Viability Evaluation
RAW 264.7 cells and CT2A GBM cells were seeded into 96-well plates and allowed to grow to ~75% confluence at 37 °C with 5% CO2. For RAW 264.7 cells, the culture medium was replaced with fresh medium containing various concentrations FBFO for 6 h, the culture medium of each sample was refreshed following a single rinse with PBS buffer, and the cells were allowed to culture for an additional 20 h. For CT2A cells, the culture medium was replaced with fresh medium containing PBS, aOPN (3 µg/mL), and various FBFO formulation, including FBFO, FBFO@OMV, FBFO@M1EV, FBFO@HM and FBFO@HM@aOPN(50 µg/mL), and the concentrations of the treatments were quantified by the FBFO concentration. For groups subjected to laser treatment, after the addition of fresh medium, the cells were exposed to 660 nm laser at a power density of 0.5 W/cm2 for 10 min before another 20 h of incubation. Finally, the cells were further incubated with the fresh culture medium containing 10% CCK8 solution for 30 min, and cell viability was measured by a microplate reader (PerkinElmer EnSpire, USA) at 450 nm.
Cellular uptake assay
RAW 264.7 cells and CT2A GBM cells were seeded into 96-well plates and allowed to grow to ~75% confluence at 37 °C with 5% CO2. Then, the culture medium was replaced with fresh medium containing PBS, aOPN (3 µg/mL), and various cy5.5-labelled FBFO formulation, including FBFO, FBFO@OMV, FBFO@M1EV, FBFO@HM and FBFO@HM@aOPN (50 µg/mL), and the concentrations of the treatments were quantified by the FBFO concentration. After culturing for another 4 h, the cells were stained with Hoechst 33342 (50 µg/mL) for 10 min. Further, the cells were washed with PBS (pH = 7.4) three times. Finally, confocal laser scanning microscopy (CLSM) images and flow cytometry were taken to analyze the cellular uptake of various FBFO formulations.
Apoptosis assay
The cell apoptosis was measured using AnnexinV-FITC/PI apoptosis detection kit. CT2A GBM cells were seeded into 12-well plates and allowed to grow to ~75% confluence at 37 °C with 5% CO2. Then, the culture medium was replaced with fresh medium containing PBS, aOPN (3 µg/mL), and various cy5.5-labelled FBFO formulation, including FBFO, FBFO@OMV, FBFO@M1EV, FBFO@HM and FBFO@HM@aOPN (50 µg/mL), and the concentrations of the treatments were quantified by the FBFO concentration. For groups subjected to laser treatment, after the addition of fresh medium, the cells were exposed to 660 nm laser at a power density of 0.5 W/cm2 for 10 min before another 20 h of incubation. Then cells were washed 3 times with PBS, and stained with 10 μL iodide and 5 μL Annexin V-FITC, cultured for 15 min in the dark. Finally, cells were washed 3 times with PBS, and then resuspended in 500 μL PBS followed by analyzing with flow cytometry.
RNA-seq Analysis
M0-type Raw264.7 were treated with different conditions (PBS, FBFO@HM@aOPN + Laster). Total intracellular RNA was extracted using a Trizol reagent kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The NEBNext® UltraTM II RNA Library Prep Kit for Illumina® (New England Biolabs) was used to generate the sequencing library. The products were purified and quantified by the Agilent high-sensitivity DNA assay on a Bioanalyzer 2100 system (Agilent). Afterward, the sequencing library underwent sequencing by an Illumina NovaSeq 6000 platform. FastQC was used for raw data quality control64. The sequenced reads were aligned to the mouse reference genome using HISAT265. while the aligned reads were used to quantify messenger RNA expression using HTSeq66. The count data were analyzed for differential expression with DESeq267. And the criteria for identifying DEGs were as follows: expression difference multiple log2FC (Fold Change) > 1 and significant Padj <0.05. The pathway activity was conducted by GO and KEGG enrichment analyses. GSEA was performed by using GSEA software version 4.0.3, which uses predefined gene sets from the Molecular Signatures Database (MSigDB v7.1).
Real-time quantitative PCR
RT-PCR analysis was performed with a SuperScript RT-PCR system (Invitrogen) and recombinant Taq polymerase (Invitrogen). The primers used to detect the corresponding gene expression were listed in Supplementary Data 3.
Isolation of mouse primary BMDMs
BMDMs isolation was conducted following a standard protocol. Briefly, after euthanizing C57BL/6 mice, the hind legs were removed and placed in petri dishes (10 mm) containing sterile PBS. The bone marrow was flushed out with a syringe, and the collected bone marrow cells were washed with PBS. Red blood cells (RBC) Lysis Buffer (Invitrogen) was added, were incubated for 3 min. After the incubation, the cell suspension was centrifuged and resuspended in BMDMs complete medium (100 mL containing 82 mL of DMEM + M-CSF (10 ng/mL, AF-315-02, PeproTech) + 15 mL of FBS + 3 mL penicillin-streptomycin. The cells were then incubated at 37 °C with 5% CO2. Fresh medium was replaced on days 3 and 5, respectively. On day 6, the cells were used for experiments.
In Vitro Raw264.7 and BMDMs Cell Reprogramming
For M1-like polarisation, Raw264.7 or BMDMs were co-cultured with DMEM, aOPN (3ug/ml) various FBFO formulation, including FBFO, FBFO@OMV, FBFO@M1EV, FBFO@HM and FBFO@HM@aOPN (50 µg/mL), and the concentrations of the treatments were quantified by the FBFO concentration for 6 h, and the culture medium was replaced with fresh medium. Then, the cells were exposed to 660 nm laser at a power density of 0.5 W/cm2 for 10 min before another 12 h of incubation. The cells were harvested for flow cytometry. Before analysis, Fc receptors were blocked by preincubating cells with Anti-Mouse CD16/CD32 (5 μg/ml) at 4 °C for 10 min, and then incubated with FITC-conjugated F4/80 Monoclonal Antibody (BM8, eBioscience™), PE-conjugated MHC Class I (H-2Kd/H-2Dd) Monoclonal Antibody (34-1-2S, eBioscience™), PerCP-eFluor™ 710-conjugated CD86 (B7-2) Monoclonal Antibody (GL1, eBioscience™), and APC-conjugated CD80 (B7-1) Monoclonal Antibody (16-10A1, eBioscience™).
For M2-like repolarization, media was replaced by 50% complete DMEM and 50% conditioned media (collected from CT2A cell culture 72 h post original seeding) and maintained for an additional 2 days. Then, Raw264.7 or BMDMs were co-cultured with DMEM, aOPN (3ug/ml) and various FBFO formulations, including FBFO, FBFO@OMV, FBFO@M1EV, FBFO@HM and FBFO@HM@aOPN (50 µg/mL), and the concentrations of the treatments were quantified by the FBFO concentration for 6 h. Then, the culture medium was replaced with fresh medium. Then, the cells were exposed to 660 nm laser at a power density of 0.5 W/cm2 for 10 min before another 24 h of incubation. The immunosuppressive phenotype of in vitro-generated BMDM was determined by flow cytometry. Before analysis, Fc receptors were blocked by preincubating cells with Anti-Mouse CD16/CD32 (5 μg/ml) at 4 °C for 10 min, and then incubated with FITC-conjugated F4/80 Monoclonal Antibody (BM8, eBioscience™) and CD206 (MMR) Monoclonal Antibody (MR6F3, eBioscience™).
In vitro phagocytosis, and antigen presentation assay
For phagocytosis assay, CT2A-Tdtomato cells (treated with various formulations and Laster for 24 h prior to assay) were co-cultured with raw264.7-EGFP (treated with various formulations and Laster for 12 h prior to assay) for 6 h at 37 °C. Then, the cells were analyzed for CLSM or flow cytometry analysis. The phagocytosis index was calculated by the ratio of macrophages containing phagocytosed OPN cells/total number of TAMCs.
For antigen presentation, CT2A-OVA cells (treated with various formulations and Laster for 24 h prior to assay) were co-cultured at 1:2 ratio with raw264.7 (treated with various formulation and Laster for 12 h prior to assay). Cells were treated with various formulation and Laster for 24 h. OVA-peptide SIINFEKL presentation by MHC class I (H-2Kb) on macrophages was measured by flow cytometry.
Multicellular OPN Spheroids (MCTS) Experiments
Three-dimensional (3D) tumour spheroids of CT2A and primary macrophage cells (3:1) were prepared using the liquid overlay methods. To assess the ability of various FBFO formulations to overcome natural physical barrier, bicellular MCTS (≈200 μm in diameter) were treated with Cy5-labelled FBFO formulation for 6 h. Cy5.5 fluorescence signals in MCTS were detected by confocal microscopy (CLSM) Z-stack scanning.
For intracellular hypoxia measurement, MCTS were exposed to 660 nm laser at a power density of 0.5 W/cm2 for 10 min before another 20 h of incubation. Then, the 3D tumour spheroids were dyed with hypoxia green reagent for 15 min. Fluorescence signals in MCTS were detected by confocal microscopy (CLSM) Z-stack scanning.
For evaluation of the TME remodelling ability, MCTS were exposed to 660 nm laser at a power density of 0.5 W/cm2 for 10 min before another 48 h of incubation. All MCTS were digested with trypsin into single-cell suspensions, and stained with FITC-anti-F4/80, and PE-cy7-anti-CD206 for immunosuppressive phenotypic remodelling analysis following the protocols mentioned above. At the end of the culture, the supernatant was collected for the detection of Ifn-β, Il6, Tnf-α Ccl5 and Cxcl10 levels by ELISA kits (Lianke bio.), following the experimental procedures.
Chemotactic migration across the BBB
The in vitro BBB model was constructed with bEnd.3 cells using a TranswellTM cell culture system. Briefly, bEnd.3 cells (1×104 cells/well) were seeded onto the upper chamber of the TranswellTM in 24-well plates and primary macrophages and GBM tumour cells (CT2A) were cultured in the lower chamber. After incubation for several days, the integrity of the cell monolayer was examined by measuring the tight junctional protein (ZO-1) using CLSM. The endothelial resistance was recorded with a Millicell®ERS 3.0 digital voltmeter. After the transendothelial electrical resistance of this model reached greater than 200 Ω·cm2, the cy5.5-labelled FBFO, FBFO@OMV, FBFO@M1EV, FBFO@HM and FBFO@HM@aOPN were added into the apical chamber and cultured for 4 h, respectively. Neutralizing antibodies against Icam1(clone, 1A29) or Vcam1 (clone, 429) were used in antibody-blocking experiments. Then, cy5.5-labelled FBFO@HM@aOPN were preincubated with 10 µg/ml anti-Icam1, or anti-Vcam antibodies for 30 min before added to the upper chamber. The penetration efficiency was determined by collecting samples from the lower chamber at 4 h. Based on cy5.5 fluorescence to analyze the concentration of different formulations in the lower chamber. Additionally, to evaluate the BBB transport and uptake of membrane-decorated nanoparticles towards GBM cells and macrophages, the fluorescence intensity was measured with flow cytometry after 12 h incubation.
In vivo and ex vivo biodistribution
To investigate the in vivo pharmacokinetics of the cy5.5-labelled NPs before and after membrane coating, cy5.5-labelled FBFO, FBFO@OMV, FBFO@M1EV, FBFO@HM and FBFO@HM@aOPN (50 mg/ml), and the concentrations of the treatments were quantified by the FBFO concentration, were intravenously injected into CT2A-tumour-bearing mice through the tail vein. The fluorescence images of mice at different time points (1 h, 4 h, 8 h and 24 h) were acquired with the IVIS imaging system (PerkinElmer, USA). Mice were sacrificed at 24 h after injection and the major organs, including heart, liver, spleen, lungs, and kidneys as well as brain, were collected for ex vivo fluorescence imaging.
Evaluate the in vivo biosafety
To evaluate the in vivo biosafety of ME@C, BALB/c mice were intravenously administered with PBS, αOPN (100 µg), FBFO, FBFO@OMV, FBFO@M1EV, FBFO@HM and FBFO@HM@aOPN (50 mg/kg), and the concentrations of the treatments were quantified by the FBFO concentration, via the tail vein. 48 h post-injection, the mice were sacrificed, and the major organs (heart, liver, spleen, lung, and kidney) were collected for hematoxylin & eosin (H&E) staining. Additionally, blood serum was collected from the treated mice to evaluate the serum biochemistry.
Single-cell RNA (scRNA) sequencing
Freshly dissociated single cell suspensions were loaded into the microfluidic chip of Chip A Single Cell Kit v2.0 (MobiDrop, cat. no. S050100201) to generate droplets with MobiNova-100(MobiDrop, cat.no. A1A40001). Each cell/nucleus was involved into a droplet which contained a gel bead linked with up to millionsof oligos (cell unique barcode). After encapsulation, droplets suffer light cut by MobiNovaSP-100(MobiDrop, cat. no. A2A40001) while oligos diffuse into the reaction mix. The mRNAs were captured by gel beads containing oligo(dT) in droplets. Following reverse transcription, cDNAs with barcodes were amplified, and a library was constructed using the High Throughput Single Cell 3’RNA-Seq Kit v2.0 (MobiDrop (Zhejiang) Co., Ltd., cat. no. S050200201) and the 3’ Single Index Kit (MobiDrop (Zhejiang) Co., Ltd., cat. no. S050300201). The resulting libraries were sequenced on an Illumina NovaSeq 6000 System.
scRNA-seq data alignment and data processing
Raw reads of scRNA sequencing were processed with the cellranger pipeline (version 4.0.0) and mapped to GRCh38 reference genome to generate matrices of gene counts by cell barcodes. Gene-barcodes matrices were analyzed with the Seurat R package. Cells were used in subsequent analysis. All samples were merged into one Seurat object and filtered according to certain condition (nFeature_RNA > 200, nFeature_RNA < 7000 & percent.mt <10 & nCount_RNA < 60000 & percent.rb <20) for downstream analysis.
Clustering and dimension reduction
The filtered Seurat object was normalised and scaled by regressing out percentage of mitochondrial genes. The top 3000 most variable genes were found by using the “FindVariableFeatures” function and then used for principal component analysis (PCA). We used “FindNeighbors” in Seurat to get nearest neighbours for graph clustering based on PCs, and “FindCluster” to obtain cell subtypes. And the uniform manifold approximation and projection (UMAP) algorithm was applied to visualise cell subtypes. To eliminate the batch effect, we performed the harmony algorithm in the Harmony R package68 to remove batch correction before clustering analysis. Firstly, cells were partitioned into main cell types, then the clustered cells were subclustered into major subtypes for each main type in the second stage.
Analysis of differentially expressed genes
The function “FindAllMarkers” in Seurat was used to identify genes differentially expressed among clusters. The function “FindMarkers” in Seurat was used to find differentially expressed genes between two groups. The non-parametric Wilcoxon ranksum test was used to obtain p-values for comparison, as well as the adjusted p-values for all genes in the dataset based on the Bonferroni correction.
Analysis of differential pathway activities
Pathway analysis was conducted using the msigdbr69 and clusterProfiler packages70. Gene signatures from Hallmark and KEGG datasets were used as inputs to evaluate the pathway activity of different cell clusters.
Cell-cell communication analysis
The CellChat package (version 1.6.1) was used to assess cell–cell communication via interaction network analysis[6]. A Seurat object was used as the input for CellChat following the standard protocol described at https://github.com/sqjin/CellChat. Subsequently, we performed a comparison analysis to infer differentially enriched ligand–receptor interactions among the harvested mouse tumours. The interaction comparisons were performed using the compareInteractions and RankNet functions. We visualised the significant pathway LR interactions across all tumours using the netAnalysis signalingRole heatmap, netVisual bubble, and netAnalysis signalingRole scatter functions.
Analysis of spatial transcriptomics
Signature score derived from scRNA-seq dataset was added to “metadata” of ST dataset with “AddModulScore” function with default parameters in Seurat. Spatial feature expression plots were generated with the “SpatialFeaturePlot” function in Seurat package.
Orthotopic mouse models of GBM
The intracranial GBM mouse model was established with C57BL/6 J mice (6-8 weeks of age). Sex was not included as a biological variable in these studies. CT2A-Luci cells (5 × 104 cells per mouse) were injected into the right striatum of C57BL/6 J mice using a stereotactic fixation device with a mouse adapter (RWD Life Science, Shenzhen, China). The coordinates were 2.5 mm lateral to the midline, 3.0 mm anterior to bregma, and a depth of 3.0 mm. Luci fluorescence analysis was conducted using the IVIS imaging system (PerkinElmer, MA, USA). On the 7th day post inoculation, all mice were randomly divided into 7 groups, and each group was administered with PBS, aOPN (100 µg), FBFO, FBFO@OMV, FBFO@M1EV, FBFO@HM and FBFO@HM@aOPN (50 mg/kg), and the concentrations of the treatments were quantified by the FBFO concentration through intravenous injection every 3 days for 5 times in total. And, after 24 h of injection, the mice were irradiated by 660 nm laser for 10 min. Finally, at day 21, mice were euthanized, the tumours were obtained and sectioned into slices for further H&E, TUNEL, and immunofluorescent staining and immune cell analysis.
To create the post-surgery GBM mouse model, the GBM mouse model was established as described above, and a small circular portion of the skull (~4 mm in diameter) was removed from each mouse using a high-speed circle knife drill. On day 12 after inoculation, the tumour-bearing mice underwent surgical resection of GBM under microscopy at ×20 magnification. On the 7th day post-surgery, all mice were randomly divided into 4 groups, and each group was administered with PBS, InVivoMAb anti-mouse PD-1 (aPD1, 10 mg/kg), FBFO@HM@aOPN (50 mg/kg), and combination therapy (aPD1 + FBFO@HM@aOPN) through intravenous injection every 3 days for 5 times in total. And, after 24 h of injection, the mice were irradiated by 660 nm laser for 10 min. Finally, at day 33, mice were euthanized, and the tumours were obtained for immune cell analysis.
Isolation and analysis of GBM-infiltrating immune cells
The above mouse brain tumour tissues were obtained and single cell suspensions were prepared using the Brain Tumour Tissue Mild Enzymatic Digestion Kit (RWD Life) according to the manufacturer’s protocol. Then the cells were filtered through a 70 μm nylon filter strainer (352350, Falcon®), lysed with red blood cell lysate. Cells were stained with Fixable Viability Stain 510 (564406, BD Pharmingen) for viability analysis. Then, cells were blocked with anti-CD16/32 antibody (553141, BD Pharmingen) for 15 min at 4 °C. For macrophage detection, the cells were stained with FITC-anti-F4/80, APC-anti-CD80, PE-anti-MHCI, and PE-cy7-anti-CD206 antibodies following the protocols mentioned above. For CD8 T cells detection, the cells were stained with FITC-anti-CD3, percp-cy5.5-anti-CD8, PE-anti-Granzyme B. After that, the cells were washed with PBS followed by flow cytometry.
For memory T cell detection, spleens from mice with different treatments were collected and filtered through a 70-μm filter to obtain single-cell suspensions. After red blood cell lysis, spleen cells were blocked with anti-CD16/32 antibody (553141, BD Pharmingen) for 15 min at 4 °C. Then, cells were incubated with FITC-anti-CD3, PE-anti-CD44, APC-anti-CD62L antibodies, and the effector memory T cells (TEM) and central memory T cells (TCM) were detected by flow cytometry.
Statistics and reproducibility
All the statistical analyses and plots were performed via R (4.3.1) and GraphPad Prism (9.0.0). The log-rank test was used for Kaplan‒Meier survival analysis and for linear relationship evaluation. The Spearman correlation coefficient was utilised. Student’s t-test was used for two-group comparisons. For intergroup comparisons involving ≥3 conditions, one-way ANOVA followed by Tukey’s multiple comparisons test (α = 0.05) was applied via GraphPad Prism. The statistical parameters and the sample size subjects used per experiment are described in the figure legends. The methods for data collection and analysis were predetermined before the start of the experiments. Unless otherwise specified, all experiments were repeated at least three times to ensure that similar results were reproducible. All the data are presented as the means ± SDs. The p values were determined via two-tailed one-way ANOVA with a Tukey post hoc test. p > 0.05, not significant (ns); *p < 0.05; **p < 0.01; ***p < 0.001; and ****p < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mouse scRNA-seq data of GBM samples were obtained from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo; GSE163120) database. Spatial transcriptomics approaches for investigating recurrent GBM patients are available at Figshare at https://doi.org/10.6084/m9.figshare.22434341. The human scRNA-seq data supporting this study are available from the China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (https://ngdc.cncb.ac.cn; HRA009155). The mouse scRNA-seq data used in this study are available from the China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (https://ngdc.cncb.ac.cn; CRA027283). The RNAseq data used in this study are available from the NCBI SRA database (https://www.ncbi.nlm.nih.gov/sra/PRJNA1281716). The remaining data are available within the article, supplementary information or source data file. Source data are provided with this paper, also available at Figshare at https://doi.org/10.6084/m9.figshare.28559669. Source data are provided with this paper.
References
Griffin, M. E. et al. Enterococcus peptidoglycan remodeling promotes checkpoint inhibitor cancer immunotherapy. Science 373, 1040–1046 (2021).
Meric-Bernstam, F., Larkin, J., Tabernero, J. & Bonini, C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet 397, 1010–1022 (2021).
Sampson, J. H., Gunn, M. D., Fecci, P. E. & Ashley, D. M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 20, 12–25 (2020).
Venkataramani, V. et al. Disconnecting multicellular networks in brain tumours. Nat. Rev. Cancer 22, 481–491 (2022).
Wang, A. P. A review of glioblastoma and other primary brain malignancies. JAMA 330, 188–189 (2023).
Pham, T. C., Nguyen, V. N., Choi, Y., Lee, S. & Yoon, J. Recent strategies to develop innovative photosensitizers for enhanced photodynamic therapy. Chem. Rev. 121, 13454–13619 (2021).
Zhang, Y., Doan, B. T. & Gasser, G. Metal-based photosensitizers as inducers of regulated cell death mechanisms. Chem. Rev. 123, 10135–10155 (2023).
Singleton, D. C., Macann, A. & Wilson, W. R. Therapeutic targeting of the hypoxic tumour microenvironment. Nat. Rev. Clin. Oncol. 18, 751–772 (2021).
Wan, Y., Fu, L. H., Li, C., Lin, J. & Huang, P. Conquering the hypoxia limitation for photodynamic therapy. Adv. Mater. 33, e2103978 (2021).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Cassetta, L. & Pollard, J. W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904 (2018).
Miao, Y. R., Rankin, E. B. & Giaccia, A. J. Therapeutic targeting of the functionally elusive TAM receptor family. Nat. Rev. Drug Discov. 23, 201–217 (2024).
Ovais, M., Guo, M. & Chen, C. Tailoring nanomaterials for targeting tumor-associated macrophages. Adv. Mater. 31, e1808303 (2019).
Bill, R. et al. CXCL9:SPP1 macrophage polarity identifies a network of cellular programs that control human cancers. Science 381, 515–524 (2023).
Chen, H. Y. et al. Hybrid cell membrane-coated nanoparticles: a multifunctional biomimetic platform for cancer diagnosis and therapy. Acta Biomater. 112, 1–13 (2020).
Zhu, L. et al. Immune cell membrane-based biomimetic nanomedicine for treating cancer metastasis. Acta Pharm. Sin. B 13, 2464–2482 (2023).
Wu, T., Liu, Y., Cao, Y. & Liu, Z. Engineering macrophage exosome disguised biodegradable nanoplatform for enhanced sonodynamic therapy of glioblastoma. Adv. Mater. 34, e2110364 (2022).
Liu, G. et al. Bacteria-derived nanovesicles enhance tumour vaccination by trained immunity. Nat. Nanotechnol. 19, 387–398 (2024).
Qing, S. et al. Biomineralized bacterial outer membrane vesicles potentiate safe and efficient tumor microenvironment reprogramming for anticancer therapy. Adv. Mater. 32, e2002085 (2020).
de Visser, K. E. & Joyce, J. A. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell 41, 374–403 (2023).
Wang-Bishop, L. et al. STING-activating nanoparticles normalize the vascular-immune interface to potentiate cancer immunotherapy. Sci. Immunol. 8, eadd1153 (2023).
Khan, K. A. & Kerbel, R. S. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nat. Rev. Clin. Oncol. 15, 310–324 (2018).
Cheng, S. et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792–809.e723 (2021).
Zhang, L. et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181, 442–459.e429 (2020).
Song, Z. et al. Osteopontin takes center stage in chronic liver disease. Hepatology 73, 1594–1608 (2021).
Liu, Y. et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J. Hepatol. 78, 770–782 (2023).
Kilian, M. et al. MHC class II-restricted antigen presentation is required to prevent dysfunction of cytotoxic T cells by blood-borne myeloids in brain tumors. Cancer Cell 41, 235–251.e239 (2023).
Mantovani, A., Allavena, P., Marchesi, F. & Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 21, 799–820 (2022).
Zhang, D., Gokce, B. & Barcikowski, S. Laser synthesis and processing of colloids: fundamentals and applications. Chem. Rev. 117, 3990–4103 (2017).
Nie, W. et al. Responsive exosome nano-bioconjugates for synergistic cancer therapy. Angew. Chem. Int Ed. Engl. 59, 2018–2022 (2020).
Pan, J. et al. Bacteria-derived outer-membrane vesicles hitchhike neutrophils to enhance ischemic stroke therapy. Adv. Mater. 35, e2301779 (2023).
Davis, M. J., Earley, S., Li, Y. S. & Chien, S. Vascular mechanotransduction. Physiol. Rev. 103, 1247–1421 (2023).
Ghosh, D. et al. A cell-surface membrane protein signature for glioblastoma. Cell Syst. 4, 516–529.e517 (2017).
Liu, Y. S. et al. MiR-181b modulates EGFR-dependent VCAM-1 expression and monocyte adhesion in glioblastoma. Oncogene 36, 5006–5022 (2017).
Yoo, K. C. et al. Soluble ICAM-1 a pivotal communicator between tumors and macrophages, promotes mesenchymal shift of glioblastoma. Adv. Sci. (Weinh.) 9, e2102768 (2022).
Chen, Z., Han, F., Du, Y., Shi, H. & Zhou, W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct. Target Ther. 8, 70 (2023).
Zhang, X., Chen, X. & Zhao, Y. Nanozymes: versatile platforms for cancer diagnosis and therapy. Nanomicro Lett. 14, 95 (2022).
Zanganeh, S. et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 11, 986–994 (2016).
Gu, Z. et al. Mechanism of iron oxide-induced macrophage activation: the impact of composition and the underlying signaling pathway. J. Am. Chem. Soc. 141, 6122–6126 (2019).
Xia, Y. et al. Engineering macrophages for cancer immunotherapy and drug delivery. Adv. Mater. 32, e2002054 (2020).
Feng, Q. et al. Engineered bacterial outer membrane vesicles as controllable two-way adaptors to activate macrophage phagocytosis for improved tumor immunotherapy. Adv. Mater. 34, e2206200 (2022).
Wang, X. et al. Exploration and functionalization of M1-macrophage extracellular vesicles for effective accumulation in glioblastoma and strong synergistic therapeutic effects. Signal Transduct. Target Ther. 7, 74 (2022).
Xiao, T. et al. Macrophage membrane-camouflaged responsive polymer nanogels enable magnetic resonance imaging-guided chemotherapy/chemodynamic therapy of orthotopic glioma. ACS Nano 15, 20377–20390 (2021).
Xu, J. et al. A general strategy towards personalized nanovaccines based on fluoropolymers for post-surgical cancer immunotherapy. Nat. Nanotechnol. 15, 1043–1052 (2020).
Lv, M. et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res 30, 966–979 (2020).
Zhang, W. & Wu, Y. S JG. Membrane adhesion junctions regulate airway smooth muscle phenotype and function. Physiol. Rev. 103, 2321–2347 (2023).
Locati, M., Curtale, G. & Mantovani, A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev. Pathol. 15, 123–147 (2020).
Wang, Y. et al. Universal STING mimic boosts antitumour immunity via preferential activation of tumour control signalling pathways. Nat. Nanotechnol. 19, 856–866 (2024).
Dosta, P. et al. Investigation of the enhanced antitumour potency of STING agonist after conjugation to polymer nanoparticles. Nat. Nanotechnol. 18, 1351–1363 (2023).
Kalbasi, A. & Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 20, 25–39 (2020).
Kim, T. K., Vandsemb, E. N., Herbst, R. S. & Chen, L. Adaptive immune resistance at the tumour site: mechanisms and therapeutic opportunities. Nat. Rev. Drug Discov. 21, 529–540 (2022).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Hulsmans, M. et al. Recruited macrophages elicit atrial fibrillation. Science 381, 231–239 (2023).
Pittet, M. J., Michielin, O. & Migliorini, D. Clinical relevance of tumour-associated macrophages. Nat. Rev. Clin. Oncol. 19, 402–421 (2022).
Hu, C. M. et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature 526, 118–121 (2015).
Hu, C. M., Fang, R. H., Copp, J., Luk, B. T. & Zhang, L. A biomimetic nanosponge that absorbs pore-forming toxins. Nat. Nanotechnol. 8, 336–340 (2013).
Kimiz-Gebologlu, I. & Oncel, S. S. Exosomes: large-scale production, isolation, drug loading efficiency, and biodistribution and uptake. J. Control Release 347, 533–543 (2022).
Zhang, Q., Jeppesen, D. K., Higginbotham, J. N., Franklin, J. L. & Coffey, R. J. Comprehensive isolation of extracellular vesicles and nanoparticles. Nat. Protoc. 18, 1462–1487 (2023).
Zhao, X., Zhao, R. & Nie, G. Nanocarriers based on bacterial membrane materials for cancer vaccine delivery. Nat. Protoc. 17, 2240–2274 (2022).
Mizenko, R. R. et al. A critical systematic review of extracellular vesicle clinical trials. J. Extracell. Vesicles 13, e12510 (2024).
Park, J. et al. Ultra-large-scale syntheses of monodisperse nanocrystals. Nat. Mater. 3, 891–895 (2004).
Gujrati, V. et al. Bioengineered bacterial vesicles as biological nano-heaters for optoacoustic imaging. Nat. Commun. 10, 1114 (2019).
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet Journal 17, (2011).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Putri, G. H., Anders, S., Pyl, P. T., Pimanda, J. E. & Zanini, F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics 38, 2943–2945 (2022).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287 (2012).
Acknowledgements
We thank the surgeons and patients who participated in this study. Single-cell RNA sequencing was performed by CapitalBio, Beijing, China. This work was supported by grants from the National Natural Science Foundation of China (Nos. 82473403 to Gang Li, and 82273195 to Hao Xue), the Jinan Science and Technology Bureau of Shandong Province (2021GXRC029 to Gang Li), Fundamental Research Funds for the Central Universities (2022JC019 to Hao Xue), the Natural Science Foundation of Shandong Province of China (ZR2023LZL004 to Hao Xue), the Taishan Pandeng Scholar Program of Shandong Province (No. tspd20210322 to Gang Li) and the Youth Taishan Scholar Program of Shandong Province (tsqn202211316 to Hao Xue).
Author information
Authors and Affiliations
Contributions
R.R.Z., Y.H., B.Y.L. and Z.W.P. contributed equally to this work. G.L., H.X., W.J.Z. and H.L. conceived and designed the project. R.R.Z., Y.H., B.Y.L. and Z.W.P. performed all the experiments and analysed the data. J.W.Q., Y.H.Q., Q.T.W. and Z.H. were responsible for clinical sample collection and subsequent sample delivery. H.Y.Z. contributed to the data analyses. G.L., H.X., W.J.Z. and H.L. supervised the study. All the authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare competing interests.
Peer review
Peer review information
Nature Communications thanks Kebin Liu, Ferdinando Pucci, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, R., Hou, Y., Li, B. et al. Bioengineered hybrid dual-targeting nanoparticles reprogram the tumour microenvironment for deep glioblastoma photodynamic therapy. Nat Commun 16, 7672 (2025). https://doi.org/10.1038/s41467-025-63081-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63081-2
