
Abstract
Acute kidney injury (AKI) can lead to chronic kidney disease (CKD), a transition driven by cellular senescence, a state of irreversible cell-cycle arrest. However, the molecular mechanisms promoting this pathological process remain unclear. Here we show that the channel protein Pannexin1 (Panx1) promotes this detrimental senescence and subsequent kidney fibrosis. We found that Panx1 functions in a noncanonical role as a calcium (Ca2+) leak channel within the endoplasmic reticulum (ER), a key intracellular calcium store. This Panx1-mediated leak occurs at contact sites between the ER and mitochondria, leading to mitochondrial calcium overload, dysfunction, and the generation of pro-senescence signals. Genetically deleting Panx1 in male mouse models of AKI attenuates renal senescence and fibrosis. These findings, validated in human kidney tissue, identify ER-resident Panx1 as a critical driver of kidney disease progression and a potential therapeutic target.
Similar content being viewed by others


Introduction
Acute kidney injury (AKI) is a common clinical issue, with a prevalence of 20–31.7% in hospitalized patients and 8.3% in ambulatory patients1. Unfortunately, AKI survivors are at increased risk of developing chronic kidney disease (CKD), a condition that affects up to 13.4% of the population and places a considerable burden on healthcare systems2. Therefore, it is essential to investigate the molecular mechanisms that are responsible for the transition from AKI to CKD, which has been conceptualized as acute kidney disease (AKD)3. However, a comprehensive understanding of this process is still lacking.
The transition from AKI to CKD is considered to result from maladaptive repair processes, characterized by an increase in inflammation, an increase in myofibroblasts, and an accumulation of the cellular matrix4. Recent studies have identified tubular epithelial cells (TECs) as a driving force in this process5. Although TECs can repair themselves, severe or repeated injury can result in failed repair6. This failure is due to cell cycle reprogramming, which is an acute stress response. Cell cycle reprogramming can prevent TECs death by arresting the cell cycle or cause the polyploidization of TECs to compensate for the loss of TECs7. However, fibrosis progression represents a trade-off of cell cycle reprogramming, as both cycle arrest and polyploidization can trigger the senescence of TECs, which is a key event in fibrosis progression8,9. Recent studies have shown that eliminating senescent TECs can promote renal recovery after AKI and slow the AKI-CKD transition10,11. Therefore, it is crucial to clarify the mechanism of senescence in TECs post-AKI to develop effective therapies.
Pannexins, a family of transmembrane channels comprising three isoforms, Panx1, Panx2 and Panx3, are widely expressed in various organs, including the kidney12. Panx1 in the plasma membrane is known to act as an ATP release channel, whereas Panx1 in the endoplasmic reticulum (ER) is believed to facilitate calcium (Ca2+) leakage13,14. While the role of Panx1 in the plasma membrane has been extensively studied in relation to various physiological and pathological processes, the impact of Panx1 as an ER Ca2+ leak channel remains largely unknown15. Recent research has revealed the role of Panx1 as a regulator of intracellular Ca2+ in hypertension and hypoxic pulmonary vasoconstriction16,17. However, these studies only examined the function of Panx1 in the plasma membrane and did not investigate the potential involvement of Panx1 in the ER. Given the importance of intracellular Ca2+ homeostasis in cellular senescence18,19, Panx1 in the ER may modulate intracellular Ca2+ homeostasis, thereby regulating senescence.
In this work, we examined the role of Panx1 in senescence during the AKI-CKD transition. We studied the influence of ER-resident Panx1 on stress-induced senescence and discovered that Panx1 functions as a Ca2+ leak channel in senescence. In addition, we examined the mechanism by which ER-resident Panx1 promotes senescence. Moreover, by utilizing two AKI-CKD murine models and Panx1 knockout mice, we examined the impact of Panx1 on senescence and fibrosis progression post-AKI.
Results
Expression of Panx1 in the kidneys of aging humans and mice
To investigate the role of Panx1 in renal senescence, Panx1 protein levels were assessed in kidney tissues obtained from elderly humans and mice (refer to Table S1 for individual characteristics). Our analysis revealed elevated Panx1 expression in the renal tissue of elderly humans and mice, as indicated in Fig. 1a, g as well as in Fig. S1a, S1d, along with an increase in the expression of senescence markers, including SA-β-Gal, as shown in Fig. 1b, h and Fig. S1b and S1e. A positive correlation between Panx1 expression and SA-β-Gal staining activity was observed (Fig. 1c, i). Co-staining of Panx1 and C12FDG confirmed their colocalization in renal tissue, as quantified by Pearson’s and Manders’ coefficients (Fig. 1d, j, Fig. S1c and S1f). The colocalization of Panx1 with aquaporin-1 (AQP-1, a marker for TECs) and p21 in renal tissue was subsequently examined (Fig. 1e, k). Significant increases in Panx1-positive cells in the renal epithelium were noted in elderly humans and mice, whereas no Panx1-positive cells were detected in the renal interstitium of either group. P21-positive cells were significantly increased in both the renal epithelium and interstitium in elderly humans and mice (Fig. 1f, l). Additionally, an RNAscope multiplexed in situ hybridization assay was used to determine the localization of Panx1 and Cdkn1a gene expression. This analysis revealed the colocalization of Panx1 and Cdkn1a mRNAs within the renal tubule epithelium and interstitium of elderly mice (Fig. S1g). The colocalization was more pronounced in the renal tubules than in the interstitium. These findings indicate high expression of Panx1 in renal TECs within aged renal tissue.

a–f Analysis of renal tissue from young (n = 6) and aged (n = 6) human individuals. a Representative immunohistochemical staining for Panx1. Scale bars, 20 µm. b Representative SA-β-Gal staining. Scale bars, 100 µm. c Correlation analysis between Panx1 expression and SA-β-Gal activity. d Representative images of Panx1 (red) and C12FDG (green) colocalization. Scale bars, 60 µm. e Representative immunofluorescence for Panx1 (green), p21 (red) and AQP-1 (purple). Scale bars, 60 µm. f Quantification of Panx1- and p21-positive cells. g–l Analysis of renal tissue from young (n = 6) and aged (n = 6) mice. g Representative immunohistochemical staining for Panx1. Scale bars, 20 µm. h Representative SA-β-Gal staining. Scale bars, 100 µm. i Correlation analysis between Panx1 expression and SA-β-Gal activity. j Representative images of Panx1 (red) and C12FDG (green) colocalization. Scale bars, 60 µm. k Representative immunofluorescence for Panx1 (green), p21 (red) and AQP-1 (purple). Scale bars, 60 µm. l Quantification of Panx1- and p21-positive cells. Asterisk: p21-positive epithelial cells. Arrow: p21-positive interstitial cells. Data in bar graphs are presented as means ± SD. P values were determined by a two-sided Student’s t-test (f, l) or Pearson’s correlation analysis (c, i). Exact P values are provided in the Source Data file. ***P < 0.001.
Enhanced Panx1 expression detected in the kidneys of the uIRI and RLDC murine models
We conducted a post hoc analysis of RNA sequencing data from the renal tissue of mice, comparing uninjured kidneys to stress-induced senescent kidneys. The data were obtained from GSE15786620. The analysis revealed a significant increase in Panx1 mRNA levels in the renal tissue of stress-induced senescent kidneys (Fig. S2a). Panx1 expression was analyzed in both unilateral ischemia‒reperfusion injury (uIRI) and repeated low-dose cisplatin (RLDC) models (Fig. S2b and S2c). Elevated levels of Panx1 were observed (Fig. S2d, S2e, S2j and S2k), accompanied by increased SA-β-Gal staining, which is indicative of senescence (Fig. S2f, S2g, S2l and S2m). The colocalization of Panx1 and C12FDG in renal tissue was confirmed, as quantified by Pearson’s and Manders’ coefficients (Fig. S2h, S2i, S2n and S2o). Significantly increased Panx1-positive and p21-positive cells were observed in the renal tubule epithelium in the uIRI and RLDC murine models (Fig. S2p and S2q). Additionally, RNAscope multiplexed in situ hybridization staining revealed more pronounced colocalization of Panx1 and Cdkn1a mRNAs within the renal tubule epithelium than within the interstitium in the uIRI and RLDC murine models (Fig. S2r and S2s, S2t and S2u). Furthermore, the colocalization of Panx1 with CD31 (an endothelial marker), F4/80 (a macrophage marker), and FSP1 (a fibroblast marker) in renal tissue was evaluated. (Fig. S2v). An RNAscope multiplexed in situ hybridization assay was employed to determine the localization of the Panx1 gene in endothelial cells, macrophages, fibroblasts within the renal tissue of the uIRI murine model (Fig. S2w). While Panx1 was detectable in these cell types, its presence was less pronounced than its distribution in the renal tubule epithelium. These results suggest a potential role of Panx1 in TECs in stress-induced renal senescence.
Panx1 mediates stress-induced senescence in tubular epithelial cells
Next, we examined the involvement of Panx1 in stress-induced senescence in human renal TECs. Exposure to irradiation and cisplatin, which are established triggers of cellular senescence, led to elevated Panx1 transcript levels in stressed TECs (Fig. S3a, S3b, S3f, and S3g). Panx1 knockdown significantly attenuated senescence induced by irradiation, as evidenced by decreased C12FDG and SA-β-Gal staining (Fig. 2a, Fig. S3d, and S3e); reduced expression of Cdkn2a, Cdkn1a, and Tp53 at both mRNA and protein levels (Fig. 2b, c); and downregulation of senescence-associated secretory phenotype (SASP) factors (IL-1β, IL-6, IFN-β, and TNF-β) at the mRNA level (Fig. 2d). Similar outcomes were observed in cisplatin-induced senescent TECs upon treatment with Panx1 siRNA (Fig. S3i, S3j, S3k, and S3l). Panx1 knockdown also facilitated the proliferation of the cisplatin-treated TECs (Fig. S3m). Moreover, Panx1 overexpression in TECs via Panx1-adenovirus suppressed proliferation (EdU positive) and promoted senescence (SA-β-Gal and C12FDG positive), underscoring the role of Panx1 in driving cellular senescence (Fig. 2e, Fig. S3p, S3q, and S3r). Panx1 overexpression resulted in an increased proportion of cells in the G0/G1 phase (Fig. 2f); upregulated the expression of Cdkn2a, Cdkn1a, and Tp53 at both mRNA and protein levels (Fig. 2g, h); and elevated the levels of SASP factors (IL-1β, IL-6, IFN-β, and TGF-β) at the mRNA level (Fig. 2i). Collectively, these findings underscore the significant role of Panx1 in promoting senescence in TECs and its capacity to induce senescence.

a–d Effects of Panx1 knockdown on irradiated HK-2 cells. a Representative images of C12FDG and SA-β-Gal staining. Scale bars, 30 µm (C12FDG); 100 µm (SA-β-Gal). b Relative mRNA levels of senescence markers (Cdkn2a, Cdkn1a, and Tp53). c Representative immunoblot and quantification of p16, p21, and p53. d Relative mRNA levels of SASP factors (IL-1β, IL-6, IFN-β, and TGF-β). For a, c images and blots are representative of n = 3 independent biological replicates. For b, d data are from n = 6 independent biological replicates. e–i Effects of Panx1 overexpression in HK-2 cells. e Representative images of EdU, C12FDG, and SA-β-Gal staining. Scale bars, 50 µm (EdU); 60 µm (C12FDG); 100 µm (SA-β-Gal). f Cell cycle distribution analysis. g Representative immunoblot and quantification of p16, p21, and p53. h Relative mRNA levels of senescence markers (Cdkn2a, Cdkn1a, and Tp53). i Relative mRNA levels of SASP factors (IL-1β, IL-6, IFN-β, and TGF-β). For e, f images are representative of n = 3 independent biological replicates. For g–i data are from n = 6 independent biological replicates. The qPCR and immunoblot results are shown as the fold change compared with the control group. All quantitative data are presented as means ± SD. P values were determined by one-way ANOVA with Dunnet’s correction for multiple comparisons (b–d) or a two-sided Student’s t-test (f–i). Exact P values are provided in the Source Data file. *P < 0.05, **P < 0.01, ***P < 0.001, NS not significant.
Panx1 deficiency attenuates renal senescence in AKI-CKD murine models
To assess the role of Panx1 in renal senescence in vivo, we generated Panx1 knockout (Panx1 K/O) mice, which were viable and fertile. Both wild-type (WT) and Panx1 K/O mice underwent uIRI or RLDC treatment, establishing models for studying AKI-CKD progression11. In the uIRI mouse model, we observed increased renal senescence in WT mice, as indicated by SA-β-Gal and C12FDG staining. Global Panx1 deletion significantly reduced renal senescence (Fig. 3a, b, Fig. S4a). This finding was consistent with reduced mRNA levels of Cdkn2a, Cdkn1a, and Tp53 (Fig. 3c) and decreased mRNA levels of SASP markers (IL-1β, IL-6, and TGF-β) (Fig. 3d) in Panx1 K/O mouse kidneys postinjury. Similar effects of Panx1 deletion on renal senescence were observed in the RLDC mouse model (Fig. S4b–g). These findings affirm the protective role of Panx1 deletion against renal senescence during the AKI-CKD transition

a Representative SA-β-gal staining in renal tissue. Scale bars, 50 µm. b Representative immunofluorescent images of C12FDG (green), Panx1 (red), and DAPI (blue) in kidneys. Scale bars, 40 µm. c qPCR analysis of senescence markers (Cdkn2a, Cdkn1a, Tp53). d Relative mRNA levels of SASP factors (IL-1β, IL-6, TGF-β). For all panels, images are representative, and data are from WT Sham (n = 6), K/O Sham (n = 6), WT d14-IRI (n = 8), K/O d14-IRI (n = 7), WT d28-IRI (n = 8), K/O d28-IRI (n = 8) mice. The qPCR results are shown as the fold change compared with the WT-sham group. All quantitative data are presented as means ± SD. P values were determined by one-way ANOVA with Dunnet’s correction for multiple comparisons. Exact P values are provided in the Source Data file. *P < 0.05, **P < 0.01, ***P < 0.001.
The pivotal role of Panx1 in the ER during cellular senescence
The impact of Panx1 in the ER on cellular homeostasis remains unclear15. To explore the role of ER-resident Panx1 in stress-induced senescence, we stimulated primary murine renal tubular cells with irradiation. Gradient separation analysis revealed the presence of Panx1 in both the ER and plasma membrane, with more obviously increased expression of ER-resident Panx1 (Fig. S5a). Similarly, irradiation or cisplatin treatment of HK-2 cells demonstrated Panx1 localization in the ER and plasma membrane, with a marked elevation of ER-resident Panx1 expression in TECs (Fig. S5c and S5d). Additionally, immunofluorescence staining of TECs with the ER marker Erp72 and Panx1 was performed, and confocal triple-immunofluorescence microscopy showed close proximity of the Panx1 and ERp72 signals (Fig. S5e and S5h). Colocalization was quantified via Pearson’s and Manders’ coefficients (Fig. S5f and S5g, S5i and S5j). Furthermore, Panx1 overexpression in TECs via Panx1 adenovirus resulted in increased ER-resident Panx1 expression, as confirmed by immunofluorescence microscopy and colocalization quantification (Fig. 4a–c). Immunoblot analysis also revealed elevated Panx1 levels in the ER protein fraction (Fig. 4d).

a–d Effects of Panx1 overexpression in HK-2 cells. a Representative images of Panx1 (red) and the ER marker ERp72 (green). Scale bars, 10 µm. b, c Quantification of Panx1 and ERp72 colocalization by Pearson’s coefficient and Manders’ coefficient. Data are from n = 9 cells per group. d Representative immunoblot of Panx1 in whole-cell lysates (WCL) and ER fractions. e–i Effects of overexpressing truncated Panx1 (Panx11–89) in Panx1 knockout HK-2 cells. e Representative immunoblot of Panx11–89. f Representative images of Panx1 (green) and ER-Tracker (blue). Scale bars, 30 µm. g Representative images of EdU, SA-β-Gal, and C12FDG staining. Scale bars, 60 μm (EdU); 100 μm (SA-β-gal); 30 μm (C12FDG). For d–g results are representative of n = 3 independent biological replicates. Relative mRNA levels of h, senescence markers (Cdkn2a, Cdkn1a, and Tp53) and i SASP factors (IL-1β, IL-6, and TGF-β). For h, i data are from n = 6 independent biological replicates. The qPCR results are shown as the fold change compared with the control group. All quantitative data are presented as means ± SD. P values were determined by a two-sided Student’s t-test. Exact P values are provided in the Source Data file. *P < 0.05, **P < 0.01, ***P < 0.001.
Given our findings on the involvement of ER-resident Panx1 in stress-induced senescence, we investigated whether specific upregulation of ER-resident Panx1 could induce senescence. Using CRISPR-Cas9 technology, we generated Panx1-null TECs and then transfected these cells with truncated Panx1 (Panx11–89), which is known to localize in perinuclear regions rather than the plasma membrane14,21. Successful Panx11–89 transfection was confirmed by immunoblotting (Fig. 4e) and confocal microscopy (Fig. 4f). Our results revealed that truncated Panx1 colocalized with the ER and induced decreased proliferation (EdU positive) and increased senescence (SA-β-Gal and C12FDG positive), suggesting a role in cellular senescence (Fig. 4g, Fig. S5k–m). Furthermore, we observed upregulated mRNA levels of Cdkn2a, Cdkn1a, and Tp53 (Fig. 4h) and increased mRNA levels of SASP factors (IL-1β, IL-6, and TGF-β) (Fig. 4i). These findings collectively demonstrate the significant role of ER-resident Panx1 in stress-induced cellular senescence.
A study has shown that stress can activate Panx1 in the plasma membrane, which leads to an increase in ATP release22. To explore this further, apyrase was utilized to effectively decrease extracellular ATP levels in Panx1-overexpressing TECs (Fig. S5n), resulting in reduced expression of C12FDG and SA-β-Gal, both of which were previously upregulated by Panx1 overexpression (Fig. S5o). The role of plasma membrane-resident Panx1 during senescence was subsequently examined via the 10Panx1 peptide, a specific inhibitor of the plasma membrane-resident Panx1 channel23. However, blocking with the 10Panx1 peptide did not significantly reduce C12FDG expression induced by Panx1 overexpression (Fig. S5p). In contrast, treatment with carbenoxelone (CBX), which is capable of blocking both membrane and ER Panx1 channels23, effectively reduced C12FDG expression upregulated by Panx1(Fig. S5q). This prompted a focused investigation into the specific contribution of ER-resident Panx1 to senescence. A comparison of the effects of CBX and the 10Panx1 peptide in Panx1 null TECs transfected with Panx11–89 to overexpress ER-resident Panx1 revealed that CBX, but not the 10Panx1 peptide, reduced the degree of senescence induced by ER-resident Panx1 overexpression (Fig. S5r and S5s). These results suggest that while plasma membrane-resident Panx1 is involved in senescence, ER-resident Panx1 plays a more prominent role.
Panx1 increases cytoplasmic calcium in senescent tubular epithelial cells
The ER plays a crucial role in intracellular Ca2+ homeostasis24, which is disrupted during senescence18. Panx1, an ER-resident protein, has been identified as a Ca2+ leak channel13. Here, we aimed to investigate the role of ER-resident Panx1 in Ca2+ homeostasis. We assessed cytoplasmic Ca2+ levels via Fura red AM (Fura red) and Fluo-4 AM (Fluo-4). Irradiation increased resting Fluo-4 signals, which were reduced by Panx1 siRNA (Fig. S6a). Irradiated TECs exhibited elevated resting levels of Fura red signals. Upon thapsigargin (TG) stimulation, the augmentation of Fura red signals was less obvious in irradiated TECs compared to controls, which was mitigated by Panx1 siRNA (Fig. 5a). Panx1 overexpression via adenoviral transfection increased resting Fluo-4 signals (Fig. S6b). TECs with Panx1 overexpression also demonstrated higher baseline Fura red levels, and the TG-induced increase in Fura red signals was attenuated in Panx1-overexpressing TECs (Fig. 5b). To rule out the involvement of plasma membrane-resident Panx1 in the increased cytoplasmic Ca2+ levels induced by Panx1 overexpression, we utilized the 10Panx1 peptide to block plasma membrane Panx1, which did not significantly affect the Fura red signals and Fluo-4 signals in TECs (Fig. S6c and S6d). Additionally, we used Panx1-null TECs transfected with a truncated Panx1 form (Panx11–89) to assess the role of ER-resident Panx1 in altering Fura red signals in TECs (Fig. S6e). These findings suggest that ER-resident Panx1 mediates Ca2+ transfer from the ER to the cytoplasm under stress conditions.

a, b Analysis of cytosolic Ca2+ levels (Fura Red) in HK-2 cells. a Shows results after Panx1 knockdown in irradiated cells; b shows results after Panx1 overexpression. The panels show the Fura red fluorescent trace over time, baseline signal levels, and the difference between maximal fluorescent signal and baseline in HK-2 cells. Line graphs show the mean trace ± SEM from n = 3 independent experiments. c–f Effects of the intracellular Ca2+ chelator BAPTA-AM on Panx1-induced senescence. Representative images of EdU (c), SA-β-Gal and C12FDG staining (d). Images are representative of three independent experiments. Scale bars, 60 μm (C12FDG); 50 μm (SA-β-gal and EDU); e, f Representative immunoblot and quantification of p16, p21, and p53. The blot in e is representative, and quantification in f is from n = 3 independent biological replicates. Data in bar graphs are presented as means ± SD. P values were determined by one-way ANOVA with Dunnet’s correction for multiple comparisons (a, f) or a two-sided Student’s t-test (b). Exact P values are provided in the Source Data file. *P < 0.05, **P < 0.01, ***P < 0.001.
To investigate the impact of cytoplasmic Ca2+ on stress-induced senescence, TECs were subjected to cisplatin or irradiation treatment with or without 1,2-bis(2-aminophenoxy) ethane-N, N, N’, N’-tetraacetic acid (BAPTA)-AM, an intracellular Ca2+ chelator. BAPTA-AM reduced resting cytoplasmic Ca2+ levels (Fig. S6f and S6h) and attenuated stress-induced senescence, as evidenced by reduced SA-β-Gal and C12FDG staining (Fig. S6g and S6i). Treatment with BAPTA-AM also lowered the resting cytoplasmic Ca2+ levels induced by Panx1 overexpression (Fig. S6j) and increased EdU incorporation (Figs. 5c and S6k) and decreased senescence biomarkers, including C12FDG expression and SA-β-Gal activity (Fig. 5d, Fig. S6l and S6m), along with reduced protein levels of p16, p21, and p53 (Fig. 5e, f). Taken together, these results indicate that Ca2+ release from the ER, facilitated by Panx1, plays a crucial role in driving stress-induced senescence.
Panx1 facilitates ER to mitochondria calcium transfer to induce senescence
Both the ER and mitochondria regulate cytoplasmic Ca2+ levels, with elevated cytoplasmic Ca2+ stimulating mitochondrial Ca2+ uptake25. We hypothesized that ER-localized Panx1 facilitates the release of Ca2+ absorbed by mitochondria. To assess the ER and mitochondrial Ca2+ levels, we utilized the calcium-sensitive dyes Mag-Fluo4 AM (Mag-Fluo4) for the ER and Rhodamine-2N AM (Rhod2) for the mitochondria. Irradiation decreased the resting Mag-Fluo4 signals in the ER and increased resting Rhod2 signals in the mitochondria (Fig. S7a and S7b). Specifically, baseline Mag-Fluo4 signals were lower in irradiated TECs and the decline in Mag-Fluo4 signals upon TG stimulation was less pronounced than in controls; both effects were mitigated by Panx1 siRNA (Fig. 6a). Conversely, baseline Rhod2 signals were higher in irradiated TECs, whereas the subsequent TG-induced increase in Rhod2 signals was less marked relative to controls; this effect was also alleviated by Panx1 siRNA (Fig. 6b). Similar alterations in ER-to-mitochondria Ca2+ transfer and the effect of Panx1 siRNA were also observed in cisplatin-treated cells (Fig. S7c–f). Panx1 overexpression decreased resting Mag-Fluo4 signals while increasing resting Rhod2 signals (Fig. S7g and S7h). The TG-induced decline in Mag-Fluo4 signals and rise in Rhod2 signals were both attenuated in Panx1-overexpressing TECs (Fig. 6c, d).

a, b Analysis of Ca2+ levels in irradiated HK-2 cells after Panx1 knockdown. a ER Ca2+ levels (Mag-Fluo4) showing the fluorescent trace over time, baseline signal, and the difference between signal and endpoint. b Mitochondrial Ca2+ levels (Rhod2) showing the fluorescent trace over time, baseline signal, and the peak-to-baseline difference. Analysis of Ca2+ levels in Panx1-overexpressing HK-2 cells, showing c ER Ca2+ levels, and d mitochondrial Ca2+ levels. e Mitochondrial Ca2+ levels after MCU knockdown in Panx1-overexpressing cells. For a–e, line graphs show the mean trace ± SEM from n = 3 independent experiments. f Representative images of EdU and SA-β-Gal staining after MCU knockdown. Images are representative of n = 3 independent biological replicates. Scale bars, 50 µm (EdU); 100 µm (SA-β-Gal). g Relative mRNA levels of senescence markers (Cdkn2a, Cdkn1a, and Tp53). h Relative mRNA levels of SASP factors (IL-1β, IL-6, and TGF-β). For g, h data are from n = 6 independent biological replicates. Data in bar graphs are presented as means ± SD. P values were determined by one-way ANOVA with Dunnet’s correction for multiple comparisons (a, b, e, g, h) or a two-sided Student’s t-test (c, d). Exact P values are provided in the Source Data file. *P < 0.05, **P < 0.01, ***P < 0.001. NS, not significant.
We preliminarily ruled out the involvement of inositol 1,4,5-trisphosphate receptors (IP3Rs) in the ER, as the levels of their encoded mRNA (ITPR) remained unchanged following Panx1 overexpression (Fig. S7i). Additionally, blocking plasma membrane Panx1 with the 10Panx1 peptide had no effect on resting mitochondrial Ca2+ levels following Panx1 overexpression (Fig. S7j). Panx1-null TECs transfected with a truncated form of Panx1 (Panx11–89) presented increased mobilized mitochondrial Ca2+ levels induced by the overexpression of ER-resident Panx1 (Fig. S7k). These results indicate that stress can trigger Ca2+ transfer from the ER to the mitochondria, potentially through ER-resident Panx1.
To investigate the impact of mitochondrial Ca2+ influx on Panx1-mediated senescence, we conducted MCU (mitochondrial calcium uniporter) knockdown experiments (Fig. S7l and S7m). MCU, situated in the inner mitochondrial membrane, facilitates the uptake of Ca2+ into the mitochondrial matrix26. MCU knockdown reduced the Panx1-driven increase in resting mitochondrial Rhod2 signals (Fig. S7n) and attenuated the TG-induced rise in Rhod2 signals in Panx1-overexpressing TECs (Fig. 6e). Additionally, MCU suppression reversed the decrease in the mitochondrial membrane potential caused by Panx1 overexpression (Fig. S7o) and reduced the level of mitochondrial reactive oxygen species (ROS) (Fig. S7p). MCU knockdown prevented Panx1-induced senescence, as evidenced by increased EdU incorporation (Figs. 6f, S7q), a reduction in SA-β-Gal-positive cells (Figs. 6f, S7r), decreased mRNA expression of Cdkn2a, Cdkn1a, and Tp53 (Fig. 6g), and lower levels of SASP factors, including IL-1β, IL-6, and TGF-β (Fig. 6h). These findings confirm the critical role of mitochondrial Ca2+ accumulation in Panx1 overexpression-induced mitochondrial dysfunction and senescence.
Localization and regulation of Panx1 on MERCs in tubular epithelial cells
We aimed to investigate whether Panx1 is localized within mitochondria‒endoplasmic reticulum contacts (MERCs), known microdomains with elevated Ca2+ levels that facilitate rapid Ca2+ transfer between the ER and mitochondria27,28. By transfecting TECs with Panx1, we utilized confocal microscopy to observe the colocalization of Panx1 with ER and mitochondrial markers, as quantified by Pearson’s and Manders’ coefficients (Fig. 7a, b, Fig. S8a and S8b). Our findings revealed that Panx1 is associated with both ER and mitochondrial markers. By employing density gradient centrifugation, we isolated cellular fractions containing mitochondria, ER, and MERCs from TECs transfected with Panx1 adenovirus, revealing the substantial presence of Panx1 in the MERCs fractions, which were enriched with calnexin (ER marker) and voltage-dependent anion channel (VDAC, mitochondrial marker), indicative of MERCs (Fig. 7c). These results suggest that Panx1, which acts as an ER Ca2+ leak channel, specifically targets MERCs to facilitate Ca2+ transfer between the ER and mitochondria.

a–g Experiments in Panx1-overexpressing HK-2 cells. a Representative images co-stained for Panx1 (green), ER-Tracker (blue), and MitoTracker (red). Scale bars, 10 µm. b Quantification of Panx1 and MitoTracker colocalization by Pearson’s coefficient and Manders’ coefficient. Data are from n = 8 cells per group. c Immunoblot analysis of Panx1 in purified cellular fractions (WCL, whole-cell lysate, ER endoplasmic reticulum, P pure mitochondria, Cr crude mitochondria, M MERCs). The blot is representative of three independent experiments. d Representative TEM images of MERCs. Green triangle and dotted line: ER; red triangle and dotted line: mitochondria. Scale bars, 0.5 µm. e Quantification of outer mitochondrial membrane (OMM)-ER apposition. Data are from n = 6 fields per group, with each group containing 10–30 mitochondria. f Proximity ligation assay (PLA) for IP3Rs and VDAC. Scale bars, 20 µm. g Quantification of PLA-positive dots. Data are from n = 10 fields per group. h–l Experiments in kidneys from wild-type (WT) and Panx1 K/O mice post-unilateral IRI. h Representative TEM images of MERCs. Scale bars, 0.5 μm. i, j Quantification of OMM-ER apposition. Data are from WT Sham (n = 4), K/O Sham (n = 5), WT d14-IRI (n = 6), K/O d14-IRI (n = 5), WT d28-IRI (n = 6), K/O d28-IRI (n = 5) fields per group, with each group containing 10–30 mitochondria. k PLA for IP3Rs and VDAC. Scale bars, 50 µm. l Quantification of PLA-positive dots. For k, l images are representative, and data are from WT Sham (n = 6), K/O Sham (n = 6), WT d14-IRI (n = 8), K/O d14-IRI (n = 7), WT d28-IRI (n = 8), K/O d28-IRI (n = 8) mice. Data are presented as means ± SD. P values were determined by one-way ANOVA with Dunnet’s correction for multiple comparisons (i, j, l) or two-sided Student’s t-test (b, e, g). Exact P values are provided in the Source Data file. *P < 0.05, **P < 0.01, ***P < 0.001.
Furthermore, we explored the impact of Panx1 on MERCs and Ca2+ transfer. Electron microscopy analysis revealed an increase in typical MERCs and closer ER‒mitochondria contacts in TECs overexpressing Panx1 (Fig. 7d, e). A proximity ligation assay (PLA) targeting VDAC and IP3Rs revealed a significant increase in red PLA-positive puncta in Panx1-overexpressing cells (Fig. 7f, g). In the renal tissue of the AKI-CKD murine models, the number and closely apposed areas of MERCs increased significantly in the uIRI model, whereas Panx1 knockout attenuated these changes (Fig. 7h–j). Furthermore, PLA revealed an increase in the number of MERCs in the uIRI model, which was reversed in Panx1 knockout mice (Fig. 7k, l). Moreover, in the RLDC murine model, PLA analysis revealed an increase in MERCs, which was mitigated in Panx1-deficient mice (Fig. S8c and S8d). These findings collectively suggest that Panx1 upregulation may exert a regulatory effect on MERCs during stress-induced senescence.
Panx1 inhibition protects mitochondria in senescent tubular epithelial cells
Considering the influence of Panx1 on mitochondrial Ca2+ levels and the link between mitochondrial Ca2+ overload and impaired mitochondrial function, we investigated the impact of Panx1 on mitochondria. Panx1 overexpression in TECs led to mitochondrial vacuolization (Fig. S9a). In irradiation-stimulated TECs, Panx1 suppression mitigated the decrease in the mitochondrial membrane potential (Fig. 8a). An increase in mitochondrial Ca2+ levels and depolarization contribute to the generation of ROS, which is implicated in senescence. Immunofluorescence staining of MitoSOX revealed an increase in mitochondrial ROS production in irradiation-stimulated TECs, a response alleviated by Panx1 knockdown (Fig. 8b). Similar alterations in the mitochondrial membrane potential and mitochondrial ROS levels were observed in the cisplatin-treated TECs (Fig. S9b and S9c). In a uIRI murine model, renal IRI increased mitochondrial ROS levels and led to swollen mitochondria with disorganized cristae in the kidney. Panx1 knockout preserved normal mitochondrial structure (Fig. 8c) and reduced mitochondrial ROS production (Fig. 8d) in mice with renal IRI. Furthermore, in the RLDC murine model, Panx1 knockout also decreased mitochondrial ROS production in the kidneys (Fig. S9d). These findings indicate that Panx1 mediates mitochondrial impairment during stress-induced senescence.

a, b Mitochondrial function in irradiated HK-2 cells after Panx1 knockdown. a Representative images and quantification of mitochondrial membrane potential using JC-1 staining. b Representative images and quantification of mitochondrial ROS using MitoSOX staining. For a, b images are representative of n = 3 independent biological replicates. Scale bars, 30 µm. c, d Mitochondrial analysis in renal tissue from wild-type (WT) and Panx1 knockout (K/O) mice post-uIRI. c Representative TEM images and quantification of mitochondrial vacuolization. Swollen or vacuolated mitochondria are indicated with red asterisks. Data are from n = 3 fields per group, with each group containing 20–30 mitochondria. Scale bars, 1 µm. d Representative images and quantification of mitochondrial ROS (MitoSOX). Images are representative of WT Sham (n = 6), K/O Sham (n = 6), WT d14-IRI (n = 8), K/O d14-IRI (n = 7), WT d28-IRI (n = 8), K/O d28-IRI (n = 8) mice. Scale bars, 50 µm. Data are presented as means ± SD. P values were determined by one-way ANOVA with Dunnet’s correction for multiple comparisons. Exact P values are provided in the Source Data file. *P < 0.05, **P < 0.01, ***P < 0.001.
Mitochondrial protection attenuates Panx1 overexpression-induced senescence in tubular epithelial cells
Our study validates the role of Panx1 in instigating mitochondrial dysfunction, a pivotal element in cellular senescence. To elucidate the role of mitochondria in Panx1-triggered senescence, cells were treated with MitoQ, a mitochondria-targeted antioxidant, to counteract oxidative damage29. MitoQ effectively reversed the decrease in the mitochondrial potential caused by Panx1 overexpression (Fig. S9e) and reduced the mitochondrial ROS levels in Panx1-transfected TECs (Fig. S9f). Cells treated with MitoQ presented reduced levels of senescence markers, which included cell proliferation, decreased SA-β-Gal-positive cells, and decreased expression of Cdkn2a, Cdkn1a, and Tp53 (Fig. S9g and S9h). Similar anti-senescence effects were observed with N-acetyl cysteine (NAC), a widely recognized mitochondrial antioxidant (Fig. S9i–k). Overall, these findings confirm that Panx1-induced mitochondrial dysfunction significantly contributes to Panx1-mediated onset of senescence.
Panx1 deletion slows the progression of renal fibrosis in murine models
To determine the impact of Panx1 on senescence in AKI-CKD murine models, we explored its role in renal fibrosis post-AKI. In a uIRI murine model, we observed reduced mRNA levels of Collagen I, Fibronectin, and α-smooth muscle actin (α-SMA) in renal tissue in the absence of Panx1 (Fig. 9a). Additionally, the protein levels of Collagen I and α-SMA were reduced in renal tissue (Fig. 9b, c), and the areas of Sirius Red staining and positive Collagen I and Fibronectin staining were diminished in Panx1 knockout mice (Fig. 9d–g). In the RLDC murine model, Panx1 knockout mitigated the increase in blood urea nitrogen and serum creatinine levels induced by cisplatin (Fig. S10a and S10b). Immunohistochemical staining for Sirius Red and Collagen I, coupled with analysis of α-SMA and Fibronectin mRNA levels, further validated the anti-fibrotic effects of Panx1 knockout (Fig. S10c–f). These results collectively underscore the significant contribution of Panx1 to the progression of renal fibrosis following AKI.

a Relative mRNA levels of Collagen I, Fibronectin, and αSMA in the renal tissue. b, c Representative immunoblots and quantification of Collagen I and α-SMA in renal tissue. Representative images and quantification of d, e Sirius Red, f Collagen I, and g Fibronectin staining in renal tissue. Scale bars, 50 µm (Sirius Red); 20 µm (Collagen I, Fibronectin). For all panels, data are from WT Sham (n = 6), K/O Sham (n = 6), WT d14-IRI (n = 8), K/O d14-IRI (n = 7), WT d28-IRI (n = 8), K/O d28-IRI (n = 8) mice. Data are presented as means ± SD. P values were determined by one-way ANOVA with Dunnet’s correction for multiple comparisons. Exact P values are provided in the Source Data file. *P < 0.05, **P < 0.01, ***P < 0.001.
Upregulation of Panx1 in the renal tissue of AKD patients
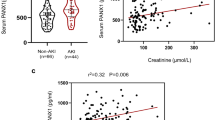
To investigate the link between Panx1 and human AKD, we analyzed Panx1 expression in renal tissue from nine AKD patients and eight control samples (IgA nephropathy or thin basement membrane nephropathy). The relevant patient characteristics are provided in the accompanying table (Table S2). AKD patients had significantly higher serum creatinine (Cr) levels and renal tubular injury scores (Fig. 10a, b). Immunohistochemical analysis revealed elevated Panx1 expression in AKD patient renal biopsies (Fig. 10c), accompanied by increased SA-β-Gal activity (Fig. 10d) and interstitial fibrosis (Fig. 10e). Correlation analysis revealed a negative correlation with the eGFR (Fig. 10f) and positive correlations between Panx1 expression, SA-β-Gal levels, and renal fibrosis (Fig. 10g, h). These findings suggest the potential role of Panx1 in renal senescence and fibrosis progression in human AKD.

Analysis of renal biopsies from control (Ctrl, n = 8 patients) and AKD (n = 9 patients) cohorts. a Serum creatinine (Cr) levels. b Representative PAS staining and quantification of tubular injury. Scale bars, 20 µm. c Representative immunohistochemistry and quantification of Panx1-positive area. Scale bars, 20 µm. d Representative SA-β-Gal staining and quantification. Scale bars, 100 µm. e Representative Sirius Red staining and quantification of fibrosis. Scale bars, 50 µm. For b–e images are representative of all patients analyzed in each group. Correlation analyses (n = 17 total patients) between renal Panx1 expression and f eGFR, g SA-β-Gal activity, and h Sirius Red staining. i Schematic illustrating the proposed mechanism of Panx1-mediated senescence. This schematic was created with Figdraw. Data are presented as means ± SD. P values for a–e were determined by a two-sided Student’s t-test. P values for f–h were determined by Pearson’s correlation analysis. Exact P values are provided in the Source Data file. ***P < 0.001.
Discussion
Panx1 is a widely distributed protein known for forming large conductance channels in plasma membranes and organelles30. While it plays a role in various physiological and pathological processes, such as inflammation, infection, and ischemia31,32,33, its connection to senescence remains unexplored. Our research revealed a correlation between Panx1 and senescence. We observed increased Panx1 expression in the kidney tissue of older individuals and aged mice as a natural consequence of aging. Moreover, we found that stress can upregulate Panx1, hastening senescence, whereas downregulation of Panx1 can mitigate stress-induced senescence. This relationship was confirmed through experiments on Panx1-deficient mice. Our findings align with a recent study on the role of Panx1 in the senescence of human endothelial progenitor cells34. We propose that Panx1 involvement in senescence may be linked to its ability to facilitate the transmembrane transport of essential elements such as ATP and Ca2+, which are crucial for maintaining cellular homeostasis. Stress-induced Panx1 upregulation may disrupt cellular homeostasis, serving as a driver of senescence.
Prior studies have primarily examined the role of Panx1 in the plasma membrane during various biological processes22,34,35. However, its involvement in intracellular organelles such as the ER remains underexplored in pathophysiological contexts. Given the pivotal role of the ER in maintaining cellular homeostasis, understanding Panx1 expression and function in the ER is crucial. In our study, we present evidence that Panx1 is upregulated in the ER during cellular stress in TECs. This ER retention of Panx1 can be attributed to its misfolding induced by stress, as Panx1 distribution is influenced by its anterograde transport from the ER to the plasma membrane36. To further investigate the impact of ER-resident Panx1 on senescence, we transfected a truncated ER-specific Panx1 construct into Panx1 knockout TECs. Our results demonstrate that increased levels of ER-resident Panx1 can induce senescence. These findings support Panx1 involvement in ER-related senescence, prompting us to explore the precise mechanisms underlying Panx1 regulation of senescence in the ER.
Our study provides evidence supporting the role of ER-localized Panx1 in stress-induced senescence through facilitating Ca2+ leakage from the ER. The ER serves as a critical reservoir for intracellular Ca2+ homeostasis, mainly through high luminal Ca2+ levels regulated by Ca2+ ATPases, inositol trisphosphate receptors (IP3Rs), ryanodine receptors (RyRs), and passive leak channels such as Panx137,38. Our findings align with previous studies identifying Panx1 in the ER as a Ca2+ leak channel13,14. This Ca2+ leakage affects intracellular Ca2+ levels, a key mechanism in stress-induced senescence. Increased intracellular Ca2+ is widely recognized as a driver of stress-induced senescence39, as shown by studies using Ca2+ chelators such as BAPTA-AM or IP3R knockdown to reverse senescence40,41. In our study, Panx1-induced senescence was reversed by reducing cytoplasmic Ca2+ with BAPTA-AM. These findings underscore the importance of understanding how ER-resident Panx1 modulates senescence by disrupting intracellular Ca2+ homeostasis.
Disruption of intracellular Ca2+ homeostasis is characterized by excessive cytoplasmic Ca2+ buildup and subsequent elevation of mitochondrial Ca2+ 42. Mitochondria-associated endoplasmic reticulum contact sites (MERCs) efficiently facilitate Ca2+ transfer from the ER to mitochondria43. MERCs, overlapping regions between the ER and mitochondria, rely on IP3Rs and RyRs on the ER side and VDACs on the mitochondrial side for Ca2+ transfer43. Other Ca2+ leak channels within MERCs may also contribute, as observed when IP3Rs are silenced in RyR-lacking cells44. Our investigation revealed Panx1 as a Ca2+ leak channel in the ER that is present in MERCs and potentially influences Ca2+ transfer from the ER to mitochondria. We observed a correlation between Panx1 expression, the abundance of MERCs, and the proximity of the ER–mitochondria. MERCs depend on direct interactions between the ER and outer mitochondrial membrane proteins, shaping their quantity and extent45. These interactions serve as the structural basis for tethering of MERCs46. Given the ability of Panx1 to interact with various intracellular proteins47,48, Panx1 may engage with mitochondrial proteins, potentially facilitating ER-mitochondrial membrane tethering. Further exploration is needed to fully understand the Panx1-mitochondria protein interaction. In summary, our results suggest that Panx1 in MERCs may contribute to both Ca2+ release and the promotion of MERCs.
Recent studies highlight the crucial role of MERCs in integrating senescence-inducing stress into subcellular compartments, including mitochondria49. MERCs respond to pro-senescence stress by modulating protein levels and quantity49. A positive correlation between the number of MERCs and the expression of senescence markers has been observed in liver and mouse embryonic fibroblasts, indicating their role in the regulation of senescence50. Furthermore, components of MERCs, including tethers and Ca2+ channels, influence senescence. For example, knocking out ITPR2, a Ca2+ leak channel in MERCs, reduces MERCs and decreases senescence50. However, the precise mechanism underlying the dysregulation of MERCs during cellular senescence remains unclear. It has been suggested that increased Ca2+ influx through MERCs may lead to mitochondrial dysfunction, contributing to MERCs-related cellular senescence41,50.
Excessive Ca2+ influx triggers mitochondrial dysfunction, leading to membrane depolarization, ROS generation, and cellular senescence51,52. Our research revealed that Panx1 knockdown under stressful conditions reduced mitochondrial ROS levels and senescence in TECs by restoring the membrane potential. This inhibition also protected mitochondria in knockout mice. MCU plays a crucial role in Ca2+ uptake, with released ER Ca2+ accumulating in mitochondria during senescence via the MCU channel41,53. Knocking down MCU in TECs reduced mitochondrial ROS and senescence caused by Panx1 overexpression. To further validate the role of mitochondria in Panx1-induced senescence, we used MitoQ, a mitochondria-targeted antioxidant29. MitoQ treatment effectively reduced Panx1-induced senescence. In summary, our findings indicate that Panx1-induced senescence results from mitochondrial dysfunction due to accumulated mitochondrial Ca2+.
Senescence plays a significant role in the progression of CKD following AKI54. Prior studies have established the role of senescence in the AKI-to-CKD transition10,11,55. Our study revealed that Panx1 knockout attenuated renal fibrosis and reduced renal senescence in both RLDC and uIRI murine models. Panx1 expression was detected in renal biopsies from AKD patients and was positively correlated with tubular injury, senescence markers, and fibrosis. Previous studies have explored the harmful effects of Panx1 in the early stages of injury. Jankowski et al. reported a link between Panx1-mediated ATP release and immune cell infiltration, which contributed to tubular injury in a murine renal ischemia‒reperfusion model56. Similarly, Lucas et al. investigated Panx1 in acute lung injury, highlighting its role in epithelial regeneration following acute injury57. Both studies focused on Panx1 involvement in the early injury phase, with immune cells playing a key role. However, the impact of Panx1 in the later stages of injury and fibrosis progression remains unexplored. Our study provides evidence for the detrimental role of Panx1 in the later stages of injury, suggesting that Panx1-mediated renal senescence contributes to renal fibrosis progression. Consequently, targeting Panx1 could be a potential therapeutic approach for CKD following AKI.
Our study has several limitations and unresolved questions. First, the use of global deletion strategy, rather than targeting TECs specifically, raises concerns that the observed renal benefits may be influenced by the deletion of Panx1 in other cell types. Although Panx1 expression in interstitial cells, such as endothelial cells, is minimal and suggests a limited role in senescence and renal fibrosis, the contribution of infiltrating macrophages influenced by Panx1 activation, should not be underestimated58. Furthermore, while our investigation focused primarily on the role of Panx1 in the ER during senescence, we recognize the necessity for a more thorough exploration of Panx1 at the PM, particularly its involvement in intracellular Ca2+ regulation and senescence. The potential activation of PM-Panx1 through stromal interaction molecules (STIM)1/2 mechanisms after ER Ca2+ depletion highlights a critical regulatory pathway that warrants further investigation59. Previous research has shown that TNF-α stimulation can elevate intracellular Ca2+ levels in endothelial cells via PM-Panx1 channels60. Elevated cytoplasmic Ca2+ levels can activate PM-Panx1, leading to ATP release and subsequent promotion of senescence61,62. We recognize that the use of Panx1 knockout mice leads to the loss of Panx1 from both the ER and PM, which may limit our ability to specifically interpret the functional role of ER-Panx1 in the reduced senescence and fibrosis observed in vivo. Future studies employing TEC-specific or ER-specific Panx1 knockout models will be essential for elucidating the distinct roles of Panx1 in various cellular compartments concerning senescence and renal fibrosis.
In summary, our research reveals a novel role for ER-localized Panx1 channels in stress-induced TEC senescence. Panx1 channels mediate ER Ca2+ release and transport to mitochondria via MERCs, promoting senescence. Deletion of Panx1 protects against renal senescence and fibrosis following AKI. Targeting Panx1 may offer therapeutic potential for AKI-associated renal senescence and CKD.
Methods
Mice and AKI-CKD murine model
This study used male C57BL/6 mice that were 8–12 weeks old and weighed 20–25 g. Only male mice were used to minimize potential experimental variability arising from the hormonal fluctuations of the female estrous cycle. The mice were obtained from the Guangdong Medical Laboratory Animal Center in Guangdong, China. Ethical review was conducted, and all animal experiments were approved by the Institutional Animal Care and Use Committee of Southern Medical University. The study was conducted in accordance with the National Institute of Health (NIH) Guide for the Care and Use of Laboratory Animals and the ARRIVE guidelines. The mice were maintained in a pathogen-free environment with laminar flow and were treated according to the guidelines of the institutional animal care and use committee. All mice were housed under a controlled 12-h light-dark diurnal cycle with regulated temperature (20–25 °C) and relative humidity (40–60%). The mice were anesthetized with 1% pentobarbital sodium (10 μl/g) and euthanized with pentobarbital sodium (150 mg/kg). The kidneys of the mice were processed in a variety of ways; they were embedded in paraffin for immunohistology, frozen in OCT solution for cryosectioning and senescence-associated β-galactosidase (SA-β-Gal) staining or homogenized for RNA and protein extraction. The Laboratory Animal Center was responsible for the disposal of all deceased animals, and the mice were randomly assigned to either the control or experimental groups at the beginning of each experiment.
To determine the expression of Panx1 in renal tissue, six young 12-week-old mice and six aged 96-week-old mice were humanely euthanized. Unilateral renal ischemia/reperfusion injury (uIRI) was performed on male mice as previously described63. The mice were anesthetized via the intraperitoneal administration of pentobarbital sodium. The mice were placed on a thermostatic station, and the body temperature was maintained at 37 °C. The left kidney was externalized, and the renal artery was clamped for 35 min, while the right kidney was left untouched. Sham-operated mice underwent the same operation without clamping of the left renal artery. The mice were humanely euthanized at either 14 or 28 days after surgery (Fig. S1a). The repeated low-dose cisplatin (RLDC) model was created following Katagiri’s protocol64. The mice were given three doses of cisplatin at 10 mg/kg via intraperitoneal injection at 0, 1, and 3 weeks. The mice were then humanely euthanized 8 weeks after the first cisplatin injection (Fig. S1b).
Panx1 knockout mice
Panx1 knockout mice were generated via a CRISPR/Cas9-based approach. Specifically, two single guide RNAs (sgRNAs) were designed via the CRISPR design tool from the Sanger Institute (http://www.sanger.ac.uk/) to target the exon 2–5 region. These sgRNAs were then assessed for on-target activity via a Universal CRISPR Activity Assay (UCA) provided by Biocytogen Pharmaceuticals (China). To facilitate this process, the T7 promoter sequence was incorporated into the Cas9 or sgRNA template through in vitro PCR amplification. Subsequently, Cas9 mRNA and sgRNAs were coinjected into the cytoplasm of fertilized C57BL/6 N eggs at the one-cell stage. The zygotes were injected and subsequently transferred into the oviducts of Kunming pseudopregnant females to produce F0 mice. The F0 mice, whose genotype was confirmed through tail genomic DNA PCR and sequencing, were then bred with C57BL/6 N mice to establish F1 heterozygous mice whose genetic information could be transmitted to future generations. The genotypes of the F1 heterozygous mice were determined through tail genomic PCR and DNA sequencing. For specific verification reports of F1 heterozygous mice, refer to Fig. S11.
Cell culture and treatment
Human proximal tubular cells (HK-2) were obtained from the American Type Culture Collection (ATCC, CRL-2190). HK-2 Cells were grown in 5% CO2 in DMEM/Ham’s F12 with 10% fetal bovine serum at 37 °C. For the cisplatin-induced senescence study, HK-2 cells were seeded at 10 × 104 cells/cm2, incubated with DMEM supplemented with 2% fetal bovine serum (FBS) in the presence of 50 μM cisplatin (Sigma–Aldrich, P4394, China) for 24 h and then harvested. Irradiation was conducted via an X-ray irradiator (Faxitron Multi Rad225, 225 kV, 17.8 mA), and the cells were exposed to a dose of 8 Gy at a rate of 1 Gy/min and harvested after 72 h.
To determine the role of cytoplasmic Ca2+, the cells were treated with BAPTA-AM (Selleck, S7534, USA). To block the Panx1 channel in the plasma membrane, 10Panx peptides (R&D Systems, 3348/1, USA) were used. To dehydrolize the extracellular ATP, Apyrase (Sigma‒Aldrich, A6410, USA) was applied. To protect the mitochondria, the cells were treated with Mitoquinol (MitoQ; Cayman, 89950, USA) or N-acetyl cysteine (NAC; Sigma‒Aldrich, A9165).
Isolation and culture of primary tubular epithelial cells
Primary tubular epithelial cells were isolated and cultured using modified procedures as previously described65. Briefly, kidneys from 8- to 12-week-old male mice were finely minced and digested in pre-warmed collagenase IV solution (Gibco, 17104-019, USA) for 10–15 min. To remove undigested tissue fragments, the suspension was passed through a 100 μm mesh sieve. The cell suspension was collected and centrifuged at 500 × g for 3 min at room temperature. Then the resulting pellet was resuspended in 32% Percoll solution (Marlborough, 17089101, USA) and subjected to centrifugation at 3000 × g for 10 min at 4 °C. After centrifugation, the pellet was resuspended with DMEM/ F-12 medium. Finally, the freshly isolated tubules were cultured in DMEM/F12 containing 10% FBS at 37 °C. After 72 h of culturing, primary tubular epithelial cells were identified using E-cadherin immunofluorescence staining (1:200, Proteintech, 20871-1-AP, China).
Cell transfection of Panx1 adenovirus and truncated Panx1 adenovirus
An adenovirus vector carrying Panx1 cDNA (Vigene Bioscience, China) was constructed to overexpress Panx1. Another adenovirus vector carrying the truncated Panx1 cDNA (Vigene Bioscience, China), which was designed on the basis of Furlow’s study, was constructed to overexpress Panx11–89 21. Vehicle recombinant adenovirus was used as a control. To transfect adenoviruses, cells in six-well plates were incubated with a mixture of adenovirus and Opti-MEM for 48 h. The effect of Panx1 transfection was determined by qPCR and immunoblotting after 48 h of transfection.
Cell transfection of siRNAs
To knockdown Panx1, cells in six-well plates were incubated with a mixture of Panx1 siRNA, Lipofectamine 3000, and Opti-MEM for 48 h, after which cell stimulation was performed. The control cells were treated with scrambled siRNA and transfection reagent. MCU silencing was performed via the same method described above. The siRNAs for Panx1 and MCU were designed and provided by RiboBio, China. The following Panx1 siRNA sequences were used: F: 5ʹ-GTGGCTGCATAAGTTTTTCCCC-3ʹ, R: 5ʹ-TCTCTCATGTCAAGGTCACGC-3ʹ. The following MCU siRNA sequences were used: F: 5’-TACACCAGAGGATCGCTTC-3’, R: 5’-TCATCAGAGGGCACAACA-3’.
Construction of the Panx1 knockout HK-2 cell line
The Panx1 knockout HK-2 cell line was produced by Vigene Biosciences (China). The CRISPR/Cas9 technique was used to create a knockout cell line. Briefly, small guide RNAs (sgRNAs) targeting the genome sequence of Panx1 were cloned and inserted into the sgRNA vector, which carried the spCas9 protein-encoding gene and the purinomycin resistance gene. The sgRNA sequences used were as follows: sgRNA1, GGTGTTACCGAGAACTTAGG; sgRNA2, CCTGTACCTGCCCCCGCTGT.
Reverse transcription and qPCR analyses
Total RNA was extracted from kidney tissues and HK-2 cells via RNAIso Plus (Takara, Japan), and 2 μg was subjected to reverse transcription via the PrimeScriptRT reagent kit (Takara). Subsequently, quantitative real-time polymerase chain reaction (qRT‒PCR) was performed via a SYBR Green Supermix kit (Takara), and the relative expression was normalized to the expression level of β-actin.
Histological and immunohistochemical staining
For renal histology, renal tissues were immersed and fixed in 4% paraformaldehyde (PFA) for 24 h before being embedded in paraffin to obtain 2 μm thick histological sections. Periodic acid–Schiff staining was used for visualization. For immunohistochemical microscopy, renal tissue was perfusion fixed in 4% PFA and embedded in paraffin, with 2-μm paraffin sections mounted on slides. Following deparaffinization and dehydration, antigen retrieval and blocking with 1% bovine serum albumin (BSA) were performed before incubation with primary antibodies overnight at 4 °C, followed by incubation with secondary antibodies conjugated with peroxidase. Finally, the sections were stained with a DAB kit (ZSGB-BIO, China) and counterstained with hematoxylin. Hydrogen peroxide (H2O2, 3% in water) was used to quench endogenous peroxidase activity. The primary antibodies used for immunohistochemical staining were Panx1 (1:200, Cell Signaling, 91137S, USA), Collagen I (1:200, Boster, BA0325, China), and Fibronectin (1:200, Abcam, ab268020, China). The secondary antibodies used were HRP-goat anti-mouse IgG (H + L) (1:200, Proteintech, SA00001-1, China) and HRP-goat anti-rabbit IgG (H + L) (1:200, Proteintech, SA00001-2). Isotype control primary antibodies or no primary antibodies (secondary antibodies alone) were used as negative controls for immunohistochemical staining. Semiquantitative analysis of Panx1 staining was performed via previously described methods65. Briefly, digital images were captured at × 400 magnification with an Olympus BX‒53 F microscope (Olympus, Japan). Images of randomly selected fields were taken, and the Panx1 staining scores were calculated via ImageJ software and expressed as the ratio of integrated optical density (IOD) to the observed area (IOD/area), which represents the relative expression level of Panx1.
Immunofluorescence staining
For immunofluorescent analysis, kidneys were excised post-euthanasia, sectioned to 2 mm, and placed in cassettes. The tissues were fixed in 4% paraformaldehyde for 4 h, followed by 2 h in sucrose for cryoprotection. The sections were then embedded in optimal cutting temperature (OCT) compound and stored at −80 °C.
Frozen kidney sections or HK-2 cells were postfixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 for 30 min and blocked with 5% BSA (bovine serum albumin) for 1 h. The sections or cells were stained with primary antibodies followed by secondary antibody staining via Alexa Fluor 488 or 594. Sections or cells were counterstained with DAPI (ZSGB-BIO). Fluorescence imaging was performed via confocal microscopy (Olympus, FV1000, Japan). The primary antibodies used for immunofluorescence staining were as follows: Panx1 (1:500, Cell Signaling, 91137S), ERp72 (1:50, Abcam, ab155800), and TOM20 (1:200, Proteintech, 11802-1-AP). The secondary antibodies used were as follows: CoraLite594-goat anti-rabbit IgG (H + L) (1:200, Proteintech, SA00013-4), CoraLite594-goat anti-mouse IgG (H + L) (1:200, Proteintech, SA00013-3), CoraLite488-goat anti-rabbit IgG (H + L) (1:200, Proteintech, SA00013-2), and CoraLite488-goat anti-mouse IgG (H + L) (1:200, Proteintech, SA00013-1). Isotype control primary antibodies or no primary antibodies (secondary antibodies alone) were used as negative controls for immunofluorescence staining.
Multiplex immunofluorescent analysis
We applied fluorescence multiplex immunofluorescence analysis via a multiplex fluorescence kit (RS0035, Immunoway, China) to colocalize p21 and Panx1 in renal TECs and Panx1 or p21 in other renal cells. First, we adhered to the manufacturer’s guidelines and cut paraffin-embedded renal tissues into 4 μm sections. The kidney sections were then subjected to dewaxing and rehydration, followed by antigen retrieval solution application as provided by the manufacturer. Upon successful preparation, we conducted protein blocking via a blocking solution for 30 min before each of the three staining cycles. Primary antibodies were applied to the renal sections at room temperature for 2 h. These antibodies, which target Panx1 (1:600, Cell Signaling, 91137 s), p21 (1:1000, Cell Signaling, 37543 s), and AQP1 (1:500, Proteintech, 20333-1-AP) or laminin (1:250, Sigma‒Aldrich, L9393), were used to identify renal TECs. For the identification of other renal cells, we used CD31 (1:1000, Cell Signaling, 77699 s) for endothelial cells, F4/80 (1:1000, Cell Signaling, 70076 s) for macrophages, and FSP1 (1:500, Proteintech, 20886-1-AP) for fibroblasts. For detection, we applied horseradish peroxidase (HRP)-labeled rabbit secondary antibodies to the renal sections for 20 min, followed by incubation with a tyramide signal amplification (TSA) opal fluorophore (Opal 488, 594 or 647) for 10 min at room temperature. We then performed heat-mediated antigen retrieval via Tris/EDTA buffer (pH 9.0) to remove the antibody-TSA complex during each staining cycle. Subsequently, counterstaining with DAPI was performed, and immunofluorescence images were captured via a confocal microscope (Leica, SP8, USA). As a negative control for immunofluorescence staining, we administered isotype control primary antibodies or secondary antibodies alone.
SA-β-Gal staining assay and C12FDG staining
For SA-β-Gal staining of renal tissue, 4 μm thick cryosections embedded in OCT compound were initially fixed with 1% formaldehyde at room temperature. The sections were subsequently incubated overnight at 37 °C with a staining solution from the SA-β-Gal staining kit (Cell Signaling). HK-2 cells were washed with PBS (pH 7.2), fixed with 0.4% PFA in PBS, and then stained with X-Gal solution for 16 h at 37 °C. Senescent cells were also detected by staining with C12FDG (a fluorogenic substrate of β-galactosidase) (Thermo Fisher Scientific). The cells were incubated for 2 h with 33 μm C12FDG. Images were acquired with a confocal microscope (Olympus), and semiquantitative analysis was performed with ImageJ software (version 1.53, USA).
EdU staining assay
EdU staining was conducted by culturing the cells in medium supplemented with 50 μM EdU for 2 h at 37 °C. After labeling, the cells were washed with PBS and fixed with formaldehyde. The staining mixture from RiboBio (China) was used to stain the cells for 30 min. The images were then obtained with a confocal microscope and analyzed via ImageJ software.
Flow cytometry analysis
For analysis of the cell cycle distribution, the cells were prepared for propidium iodide (Sigma‒Aldrich) staining according to a standard protocol. The DNA content was determined with a FACS Calibur system (BD Biosciences, USA).
Immunoblot
Proteins were extracted from frozen kidney tissue via RIPA lysis buffer (Cell Signaling) supplemented with phenylmethanesulfonyl fluoride (Cell Signaling). The resulting sample was subjected to centrifugation at 12,000 × g for 15 min to isolate the tissue debris, and the resulting supernatant was collected and prepared for immunoblot analysis. HK-2 cells were lysed in RIPA lysis buffer, and the protein concentration was determined via a BCA assay kit (Pierce, USA). A total of thirty micrograms of protein were subsequently loaded onto an SDS‒PAGE gel and separated via electrophoresis, followed by transfer onto a PVDF membrane (Millipore, China). The membranes were blocked with a 5% nonfat powdered milk solution for 1 h at ambient temperature. The blocked membranes were subsequently subjected to an overnight incubation at 4 °C with a primary antibody. The membranes were subsequently incubated with either anti-rabbit or anti-mouse Ig conjugated with horseradish peroxidase for a period of 1–2 h at ambient temperature. The visualization of antibody labeling on the membranes was accomplished via the Immobile Western Chemiluminescence HRP substrate kit (Millipore). Densitometric analysis was conducted via ImageJ software. The primary antibodies used for immunoblotting were as follows: Panx1 (1:1000, Cell Signaling, 91137S), p16 (1:1000, Cell Signaling, 18769S), p21 (1:1000, Cell Signaling, 37543S), p53 (1:1000, Cell Signaling, 2524S), AQP-1 (1:1000, 20331-AP, Proteintech), β-Actin (1:3000, Proteintech, 66009-1), ERp72 (1:100, Santa Cruz, sc-376230), VDAC1 (1:1000, Abcam, ab14734), Calnexin (1:1000, Cell Signaling, 2679S), Cyt-c (1:1000, Cell Signaling, 4272S), and MCU (1:1000, Cell Signaling, 14997S). The primary antibodies (anti-mouse) used for immunoblotting were as follows: Panx1 (1:1000, Cell Signaling, 91137S),p53 (1:1000, Cell Signaling, 2524S), GAPDH (1:3000, Proteintech, 60004-1), Collagen I (1:1000, Boster, BA0325, China), α-SMA (1:1000, Sigma‒Aldrich, A5228), and β-Actin (1:3000, Proteintech, 66009-1). The secondary antibodies used were HRP-conjugated AffiniPure goat anti-rabbit IgG (H + L) (1:3000, Proteintech) or HRP-conjugated AffiniPure goat anti-mouse IgG (1:3000, Proteintech).
Calcium imaging
HK-2 cells were cultured on coverslips and incubated for 30 min at 37 °C in Hank’s balanced salt solution (HBSS, Thermo Fisher Scientific, 14025092) containing Ca2+, Mg2+, devoid of phenol red, and supplemented with 0.2% Pluronic F127 (Thermo Fisher Scientific, P3000MP). The cells were loaded with 5 μM Fluo-4 AM Ca2+ tracer (Thermo Fisher Scientific, F14201) or 5 μM Fura Red AM Ca2+ tracer (MedChemExpress, HY-D1903, USA). Following incubation, the cells were rinsed twice with dye free HBSS buffer to clear any residual dye nonspecifically adhering to their surface. Resting cytosolic Ca2+ fluorescence was recorded via a confocal microscope (Fluo-4 AM: excitation, 488 nm; emission, 520 nm). For the evaluation of resting Ca2+ fluorescence in the ER or mitochondria, the cells were stained with 1 μM ER Ca2+ tracer (Mag-Fluo4-AM, Thermo Fisher Scientific, M14206) and 1 μM mitochondrial Ca2+ tracer (Rhod2-AM, Thermo Fisher Scientific, R1244), respectively. Fluorescence imaging was performed with a confocal microscope (Mag-Fluo4: excitation 488 nm, emission 520 nm; Rhod2: excitation 540 nm, emission 580 nm).
Moblized Ca2+ imaging was performed according to previously described methods66. Cells were loaded with the Ca2+ tracer and rinsed with an HBSS-Ca2+ deficient solution to eliminate any indicator dyes. The Ca2+ agonist thapsigargin (TG; Sigma‒Aldrich, T9033) in HBSS-Ca2+ deficient buffer was subsequently introduced to ensure that Ca2+ was released from the ER. Fura Red emission ratios (F420/488) were acquired at 660 nm, followed by excitation at 420 and 488 nm. The total Ca2+ measurement time frame ranged from 0 to 600 s, whereas the baseline Ca2+ concentration was measured from 0 to 100 s. At 101 s, TG stimulation was started. The F420/488 ratio was used to measure changes in the level of cytosolic Ca2+. To quantify the fluctuating fluorescence intensity of Mag-Fluo4 and Rhod2, the formula (F1-F0)/F0 was applied, where F0 represents the mean fluorescence from 0 to 100 s and F1 represents the real-time fluorescence at each specific time point. This ratio was used to measure changes in the levels of Ca2+ within the ER and mitochondria.
Isolation of plasma membrane, ER, mitochondria, and MERCs fractions
The plasma membrane fractions of TECs were obtained following the instructions of the Plasma Membrane Protein Extraction Kit (Bestbio, BB-31161, China). The isolation of ER, mitochondria and MERCs fractionations was performed according to previously described methods67. To detach the cells, 1 ml of a 0.25% trypsin solution per 10 cm2 sample was added, and the mixture was incubated for 5 min at 37 °C. Subsequently, 6 ml of DMEM supplemented with 10% FBS was added before the cells were pelleted at 600 × g for 5 min. The resulting supernatant was then subjected to centrifugation at 7000 × g for 10 min at 4 °C. The resulting supernatant was collected as the cytosolic fraction, which contained lysosomes and microsomes. To obtain purified ER proteins, the supernatant was diluted with ice-cold buffer 1 (225 mM mannitol, 75 mM sucrose, 0.1 mM EGTA, and 30 mM Tris–HCl pH 7.4) and subsequently subjected to centrifugation at 100,000 × g for 60 min at 4 °C. The pellet obtained from the preceding 7000 g spin, which contained mitochondria and MERCs, was resuspended in 20 mL of ice-cold buffer 2 (225 mM mannitol, 75 mM sucrose, and 30 mM Tris–HCl pH 7.4) and subjected to two additional rounds of centrifugation (once at 7000 × g and once at 10,000 × g for 10 min at 4 °C) to eliminate microsomal contamination. The supernatant was discarded, and the crude mitochondrial pellet was gently resuspended in 2 ml of ice-cold mitochondrial resuspension buffer containing 250 mM mannitol, 5 mM HEPES (pH 7.4), and 0.5 mM EGTA. Subsequently, 8 ml of Percoll medium, composed of 225 mM mannitol, 25 mM HEPES (pH 7.4), 1 mM EGTA, and 30% Percoll, was added to a 14 ml thin wall, polyallomer ultracentrifuge tube. The tube was then subjected to centrifugation at 95,000 × g for 30 min at 4 °C. Following centrifugation, a dense band consisting of purified mitochondria was observed at the bottom of the ultracentrifuge tube, just above a Percoll pellet. Additionally, the presence of MERCs was evident as a diffuse white band located above the mitochondria. Both were collected from the Percoll gradient with a Pasteur pipette, diluted with mitochondria resuspending buffer, and recentrifuged at 100,000 × g for 60 min at 4 °C to remove Percoll contamination.
Transmission electron microscopy
Perfused renal tissue or HK-2 cells were fixed with 4% PFA, postfixed in 1% osmium tetroxide, dehydrated in a graded series of alcohol, and embedded in Epon. Ultrathin sections were mounted on 200 mesh copper grids, contrasted with uranyl acetate and lead citrate, and examined via transmission electron microscopy (TEM, HITACHI HT7700, Japan). MERCs were determined as the distance below 50 nm between the ER and the outer mitochondrial membrane (OMM)68. ImageJ software was used to measure the mean distances between the ER and OMM and the surface area of the mitochondria. The distance of MERCs in the TEM images was quantified as previously described69. The ER‒OMM distance was measured. ER-OMM apposition was measured considering two subsets of ER-OMM distances: <50 nm or <30 nm. The quantification of the length of the ER adjacent to the mitochondria was normalized to the mitochondrial perimeter.
ER-Tracker and MitoTracker staining
HK-2 cells were plated on glass coverslips at 60% confluence and incubated for 24 h. The cells were then rinsed with Hank’s buffered salt solution (HBSS) and incubated in prewarmed staining solution for 20 min at 37 °C. The staining solution contained HBSS with 1 μM ER-Tracker Blue (Thermo Fisher, E12353) and 100 nM MitoTracker Deep Red FM (Thermo Fisher, M7512). After staining, the cells were rinsed three times with HBSS and fixed. Fluorescence imaging was performed via confocal microscopy (Olympus).
Mitochondrial membrane potential and MitoSOX staining assay
The mitochondrial membrane potential was evaluated by exposing HK-2 cells to 1 μg/ml JC-1 (Cayman) for 30 min at 37 °C and 5% CO2, after which the cells were washed extensively, and fluorescence emission was recorded via confocal microscopy. The accumulation of JC-1 in mitochondria was used to discriminate energized and de-energized mitochondria. When the dye was concentrated, the fluorescence shifted from green (488 nm) to red (590 nm). Mitochondrial depolarization is indicated by a decreased red/green fluorescence ratio. Increased fluorescence of MitoSOX Red (Thermo Fisher, M36008) was used to assess mitochondrial superoxide production. HK-2 cells were incubated with 1 μM MitoSOX Red for 10 min and counterstained with DAPI. The fluorescence was detected by using a confocal microscope (Olympus). The fluorescence intensity was quantified via ImageJ software.
In situ proximity ligation assay
For the analysis of ER-mitochondria contacts, an in situ proximity ligation assay (Sigma‒Aldrich) was applied by targeting voltage-dependent anion channels (VDAC, Abcam, ab14734) at the mitochondrial membrane and IP3Rs (1:50, Santa Cruz, sc-377518) at the ER membrane, as previously reported. Coverslips seeded with HK-2 cells or renal sections were permeabilized with 0.1% Triton X-100 in PBS for 10 min, blocked with 5% BSA for 1 h, and incubated overnight at 4 °C with primary antibodies. Then, the kidney sections or cells were rinsed with Wash Buffer A and incubated with the PLUS and MINUS PLA probes, both rabbit and mouse, for 1 h. Next, the sections or cells were washed with Wash Buffer A and incubated with Duolink ligation solution (Sigma‒Aldrich, DU082049) for 30 min, followed by Duolink amplification solution (Sigma‒Aldrich, DU092002, DU092004) for 100 min. Nuclei were stained with DAPI. Images were captured via a confocal microscope. The signals of the PLA dots were quantified via ImageJ software.
RNA in situ hybridization assay
Paraffin-embedded kidney sections (4 μm) were prepared and subjected to RNAscope hybridization following the manufacturer’s instructions via the RNAscope Multiplex Fluorescent Reagent Kit v2 from Advanced Cell Diagnostics (ACD, 323100, USA). Briefly, the sections were dehydrated and incubated with hydrogen peroxide for 10 min at room temperature. After the samples were washed with distilled water, antigen retrieval was performed. A hydrophobic barrier was drawn around the sections via an ImmEdge pen (Vector Laboratories). RTCs were identified by incubating the sections overnight at 4 °C with a primary antibody against PAX8 (1:50, Proteintech, 10336-1-AP). For the identification of other renal cell types, the sections were incubated overnight at 4 °C with FITC-LTL (Vector Laboratories, FL-1321-2) and primary antibodies against F4/80 (1:50, Cell Signaling Technology, 70076S), αSMA (1:50, Proteintech, 14395-1-AP), or CD31 (1:50, Cell Signaling Technology, 77699 s). The following day, the sections were washed with 1× PBST. They were subsequently fixed with 10% neutral-buffered formalin for 30 min and treated with Protease Plus for 20 min at 40 °C in a hybridization oven. The sections were then hybridized with target-specific probes for Panx1 (Mm-Panx1, ACD, 316321) and Cdkn1a (Mm-Cdkn1a, ACD, 408551) for 2 h at 40 °C, followed by a series of signal amplification steps. A probe targeting the housekeeping gene PPIB (ACD, 320881) was used as the positive control, while a probe targeting DapB (ACD, 320871) served as the negative control. After RNAscope, the sections were stained with a secondary antibody for 30 min at room temperature. Finally, the sections were sealed with mounting medium containing DAPI (ZSGB-BIO, ZLI-9600). Imaging was performed via a laser scanning confocal microscope (Olympus, FV1000, Japan).
Renal function
Blood samples from C57BL/6 mice were spun at 800 × g for 10 min at 4 °C, and the serum creatinine (Cr) and blood urea nitrogen (BUN) concentrations were determined via an autoanalyzer (Beckman DXC600, USA).
Sirius red staining assay
Paraffin-embedded kidney tissue sections (4 μM) were deparaffinized, hydrated, and exposed to a picrosirius red solution (Sigma‒Aldrich). The sections were subsequently dehydrated and mounted, and the percentage of the Sirius Red-positive area was subsequently calculated via ImageJ software.
Human kidney biopsy studies
All human kidney biopsy samples were obtained from the Department of Nephrology, Nanfang Hospital, from 2012 to 2018. The collection of human kidney biopsy samples was approved by the Institutional Review Board of Nanfang Hospital. All study participants provided written informed consent. Due to the limited sample size of the patient cohorts, a sex-based analysis of the data was not performed.
For the study of renal Panx1 expression in aged individuals, six young patients and six aged patients were included. Young patients, defined as those under 30 years of age, underwent renal biopsies due to microscopic hematuria and were pathologically diagnosed with either thin basement membrane nephropathy (TBMN) or no specific pathological alterations. In contrast, renal tissue from older patients aged over 60 years was collected from non-affected areas of the kidney during nephrectomy procedures performed for renal carcinoma.
To study renal Panx1 expression in AKD patients, eight patients who were diagnosed with TBMN or no specific pathological alterations were designated the control group. Nine patients who were diagnosed with acute tubular necrosis composed the injury group. Acute tubular necrosis was induced by drugs or ischemia, and patients underwent renal biopsy between 7 and 90 days after an AKI episode. Patients with a CKD history or who were over 60 years old were excluded. The degree of acute tubular injury was assessed with a scoring system based on the percentage of renal tubules affected by the loss of the tubule epithelial cell brush border and tubular necrosis (0 = no lesion, 1 + = less than 25%, 2 + = 25-50%, 3 + = 50-75%, 4+ over 75%).
Statistics
All the data are presented as means ± SD. The statistical significance of differences among 3 or more groups was determined via one-way ANOVA, followed by Dunnet’s correction for multiple comparisons. To determine the statistical significance of differences between 2 groups, a two-sided t test was conducted. Correlations were performed via Pearson’s analysis. Statistical calculations were performed via GraphPad Prism version 8.0 (GraphPad, USA). Statistical significance was accepted at p < 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data sets generated for this study are in the Gene Expression Omnibus (GEO) under accession number GSE157866. Source data are provided with this paper. All data supporting the findings described in this manuscript are available in the article and in the Supplementary Information and from the corresponding author upon request. Source data are provided with this paper.
References
Kellum, J. A. et al. Acute kidney injury. Nat. Rev. Dis. Prim. 7, 52 (2021).
Chawla, L. S., Eggers, P. W., Star, R. A. & Kimmel, P. L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 371, 58–66 (2014).
Kellum, J. A., Ronco, C. & Bellomo, R. Conceptual advances and evolving terminology in acute kidney disease. Nat. Rev. Nephrol. 17, 493–502 (2021).
Venkatachalam, M. A., Weinberg, J. M., Kriz, W. & Bidani, A. K. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J. Am. Soc. Nephrol. 26, 1765–1776 (2015).
Liu, B. C., Tang, T. T., Lv, L. L. & Lan, H. Y. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 93, 568–579 (2018).
Naved, B. A. et al. Kidney repair and regeneration: perspectives of the NIDDK (Re) building a kidney consortium. Kidney Int. 101, 845–853 (2022).
De Chiara, L., Conte, C., Antonelli, G. & Lazzeri, E. Tubular cell cycle response upon AKI: revising old and new paradigms to identify novel targets for CKD prevention. Int. J. Mol. Sci. 22, 11093 (2021).
Romagnani, P. Mechanisms and trade-offs of kidney repair: consequences for the nephrology clinician. Nephrol. Dial. Transplant. 37, 1046–1048 (2022).
De Chiara, L. et al. Tubular cell polyploidy protects from lethal acute kidney injury but promotes consequent chronic kidney disease. Nat. Commun. 13, 5805 (2022).
Li, S. et al. Tubular cell senescence promotes maladaptive kidney repair and chronic kidney disease after cisplatin nephrotoxicity. JCI Insight 8, e166643 (2023).
Li, C., Shen, Y., Huang, L., Liu, C. & Wang, J. Senolytic therapy ameliorates renal fibrosis postacute kidney injury by alleviating renal senescence. FASEB J. 35, e21229 (2021).
Penuela, S., Gehi, R. & Laird, D. W. The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 1828, 15–22 (2013).
Vanden, A. F. et al. Functional implications of calcium permeability of the channel formed by pannexin1. J. Cell Biol. 174, 535–546 (2006).
Lee, N. S. et al. Focused ultrasound stimulates ER localized mechanosensitive PANNEXIN-1 to mediate intracellular calcium release in invasive cancer cells. Front. Cell Dev. Biol. 8, 504 (2020).
D’Hondt, C. et al. Pannexin channels in ATP release and beyond: an unexpected rendezvous at the endoplasmic reticulum. Cell. Signal. 23, 305–316 (2011).
DeLalio, L. J. et al. Pannexin 1 channels in renin-expressing cells influence renin secretion and blood pressure homeostasis. Kidney Int. 98, 630–644 (2020).
Grimmer, B. et al. Pannexin 1: a novel regulator of acute hypoxic pulmonary vasoconstriction. Cardiovasc. Res. 118, 2535–2547 (2022).
Martin, N. & Bernard, D. Calcium signaling and cellular senescence. Cell Calcium 70, 16–23 (2018).
Farfariello, V., Iamshanova, O., Germain, E., Fliniaux, I. & Prevarskaya, N. Calcium homeostasis in cancer: a focus on senescence. Biochim. Biophys. Acta 1853, 1974–1979 (2015).
O’Sullivan, E. D. et al. Single-cell analysis of senescent epithelia reveals targetable mechanisms promoting fibrosis. JCI Insight 7, e154124 (2022).
Furlow, P. W. et al. Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat. Cell Biol. 17, 943–952 (2015).
Chekeni, F. B. et al. Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 467, 863–867 (2010).
Koval, M., Schug, W. J. & Isakson, B. E. Pharmacology of pannexin channels. Curr. Opin. Pharmacol. 69, 102359 (2023).
Park, S. J., Li, C. & Chen, Y. M. Endoplasmic reticulum calcium homeostasis in kidney disease: pathogenesis and therapeutic targets. Am. J. Pathol. 191, 256–265 (2021).
Bravo-Sagua, R. et al. Calcium transport and signaling in mitochondria. Compr. Physiol. 7, 623–634 (2017).
Samanta, K., Douglas, S. & Parekh, A. B. Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PLoS ONE 9, e101188 (2014).
Giorgi, C. et al. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 22, 995–1019 (2015).
Janikiewicz, J. et al. Mitochondria-associated membranes in aging and senescence: structure, function, and dynamics. Cell Death Dis. 9, 332 (2018).
Smith, R. A., Hartley, R. C. & Murphy, M. P. Mitochondria-targeted small molecule therapeutics and probes. Antioxid. Redox Signal. 15, 3021–3038 (2011).
Esseltine, J. L. & Laird, D. W. Next-generation Connexin and Pannexin cell biology. Trends Cell Biol. 26, 944–955 (2016).
Rusiecka, O. M., Tournier, M., Molica, F. & Kwak, B. R. Pannexin1 channels-a potential therapeutic target in inflammation. Front. Cell Dev. Biol. 10, 1020826 (2022).
D’Amico, D., Valdebenito, S. & Eugenin, E. A. The role of Pannexin-1 channels and extracellular ATP in the pathogenesis of the human immunodeficiency virus. Purinergic Signal 17, 563–576 (2021).
Good, M. E. et al. Endothelial pannexin 1 regulates cardiac response to myocardial infarction. Circ. Res. 128, 1211–1213 (2021).
Tien, T. Y. et al. Pannexin 1 modulates angiogenic activities of human endothelial colony-forming cells through IGF-1 mechanism and is a marker of senescence. Arterioscler. Thromb. Vasc. Biol. 43, 1935–1951 (2023).
Adamson, S. E. & Leitinger, N. The role of pannexin1 in the induction and resolution of inflammation. FEBS Lett. 588, 1416–1422 (2014).
Gehi, R., Shao, Q. & Laird, D. W. Pathways regulating the trafficking and turnover of pannexin1 protein and the role of the C-terminal domain. J. Biol. Chem. 286, 27639–27653 (2011).
Sammels, E. et al. Intracellular Ca2+ storage in health and disease: a dynamic equilibrium. Cell Calcium 47, 297–314 (2010).
Lemos, F. O., Bultynck, G. & Parys, J. B. A comprehensive overview of the complex world of the endo- and sarcoplasmic reticulum Ca2+-leak channels. Biochim. Biophys. Acta Mol. Cell Res. 1868, 119020 (2021).
Ahumada-Castro, U., Puebla-Huerta, A., Cuevas-Espinoza, V., Lovy, A. & Cardenas, J. C. Keeping zombies alive: The ER-mitochondria Ca2+ transfer in cellular senescence. Biochim. Biophys. Acta Mol. Cell Res. 1868, 119099 (2021).
Borodkina, A. V. et al. Calcium alterations signal either to senescence or to autophagy induction in stem cells upon oxidative stress. Aging8, 3400–3418 (2016).
Wiel, C. et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat. Commun. 5, 3792 (2014).
Nicholls, D. G. Intracellular calcium homeostasis. Br. Med. Bull. 42, 353–358 (1986).
Marchi, S. et al. Endoplasmic reticulum-mitochondria communication through Ca2+ signaling: the importance of Mitochondria-Associated Membranes (MAMs). Adv. Exp. Med. Biol. 997, 49–67 (2017).
Mendes, C. C. et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J. Biol. Chem. 280, 40892–40900 (2005).
Rowland, A. A. & Voeltz, G. K. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625 (2012).
Csordas, G. et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 174, 915–921 (2006).
Sayedyahossein, S. et al. Pannexin 1 binds beta-catenin to modulate melanoma cell growth and metabolism. J. Biol. Chem. 296, 100478 (2021).
Bhalla-Gehi, R., Penuela, S., Churko, J. M., Shao, Q. & Laird, D. W. Pannexin1 and pannexin3 delivery, cell surface dynamics, and cytoskeletal interactions. J. Biol. Chem. 285, 9147–9160 (2010).
Ziegler, D. V., Martin, N. & Bernard, D. Cellular senescence links mitochondria-ER contacts and aging. Commun. Biol. 4, 1323 (2021).
Ziegler, D. V. et al. Calcium channel ITPR2 and mitochondria-ER contacts promote cellular senescence and aging. Nat. Commun. 12, 720 (2021).
Peng, T. I. & Jou, M. J. Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 1201, 183–188 (2010).
Miwa, S., Kashyap, S., Chini, E. & von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 132, e158447 (2022).
Koval, O. M. et al. Loss of MCU prevents mitochondrial fusion in G(1)-S phase and blocks cell cycle progression and proliferation. Sci. Signal. 12, eaav1439 (2019).
Lin, X., Heng, J., Chai, Y. & Shou, S. Cellular senescence and acute kidney injury. Pediatr. Nephrol. 37, 3009–3018 (2022).
Mylonas, K. A.-O. et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci. Transl. Med. 13, eabb0203 (2021).
Jankowski, J. et al. Epithelial and endothelial pannexin1 channels mediate AKI. J. Am. Soc. Nephrol. 29, 1887–1899 (2018).
Lucas, D. C. et al. Pannexin1 drives efficient epithelial repair after tissue injury. Sci. Immunol. 7, eabm4032 (2022).
Liu, C., Shen, Y., Huang, L. & Wang, J. TLR2/caspase-5/Panx1 pathway mediates necrosis-induced NLRP3 inflammasome activation in macrophages during acute kidney injury. Cell Death Discov. 8, 232 (2022).
Patil, C. S. et al. ER-resident STIM1/2 couples Ca2+ entry by NMDA receptors to pannexin-1 activation. Proc. Natl. Acad. Sci. USA 119, e2112870119 (2022).
Yang, Y. et al. Endothelial pannexin1 channels control inflammation by regulating intracellular calcium. J. Immunol. 204, 2995–3007 (2020).
López, X. et al. A physiologic rise in cytoplasmic calcium ion signal increases pannexin1 channel activity via a C-terminus phosphorylation by CaMKII. Proc. Natl. Acad. Sci. USA 118, e2108967118 (2021).
Zwerschke, W. et al. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 376, 403–411 (2003).
Le Clef, N., Verhulst, A., D’Haese, P. C. & Vervaet, B. A. Unilateral renal ischemia‒reperfusion as a robust model for acute to chronic kidney injury in mice. PLoS ONE 11, e0152153 (2016).
Katagiri, D. et al. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int. 89, 374–385 (2016).
Luo, C. et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J. Am. Soc. Nephrol. 29, 1238–1256 (2018).
Tu, H. et al. Presenilins Form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 126, 981–993 (2006).
Wieckowski, M. R., Giorgi, C., Lebiedzinska, M., Duszynski, J. & Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 4, 1582–1590 (2009).
Giacomello, M. & Pellegrini, L. The coming of age of the mitochondria-ER contact: a matter of thickness. Cell Death Differ. 23, 1417–1427 (2016).
D’Eletto, M. et al. Transglutaminase type 2 regulates ER-mitochondria contact sites by interacting with GRP75. Cell Rep. 25, 3573–3581.e3574 (2018).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81270826, 81470972, 82070708 to J.W.), the GuangDong Basic and Applied Basic Research Foundation (2025A1515012031 to J.W.), the Natural Science Foundation of the Xinjiang Uyghur Autonomous Region of China (2021D01C012 to J.G.), the Presidential Foundation of Nanfang Hospital (2023B028 to C.L.), the Program of Introducing Talents of Discipline to Universities, 111 Plan (D18005 to F. F. H.), Guangdong Provincial Clinical Research Center for Kidney Disease (2020B1111170013 to F. F. H.), Key Technologies R&D Program of Guangdong Province (2023B1111030004 to F. F. H.), and National Natural Science Foundation of China (Key Program) (82030022 to F. F. H.).
Author information
Authors and Affiliations
Contributions
J.W., Liuwei.H. and Y.S. conceptualized the research design. Liuwei.H, Y.S., X.P., Lanlan.H., C.L. and S.H. developed the methodology. J.L., X.Z., K.L., J.Guo, L.R., and J.Geng performed the investigation. J.W. and Liuwei.H. drafted the paper. F. F. H. and J.W. supervised the work, reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Valerio Farfariello, Natalija Filipović, Anette Melk, Khaoula Talbi and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, L., Shen, Y., Pan, X. et al. Noncanonical function of Pannexin1 promotes cellular senescence and renal fibrosis post-acute kidney injury. Nat Commun 16, 7699 (2025). https://doi.org/10.1038/s41467-025-63152-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63152-4
This article is cited by
-
Autophagy–senescence interplay in kidney disease: mechanistic insights and therapeutic potential
Molecular Biology Reports (2026)
