
Abstract
The discovery of novel antimicrobial peptides (AMPs) against clinical superbugs is urgently needed to address the ongoing antibiotic resistance crisis. AMPs are promising candidates due to their broad-spectrum activity, rapid bactericidal mechanisms and reduced likelihood of inducing resistance compared with conventional antibiotics. Here, a pre-trained protein large language model (LLM), ProteoGPT, was established and further developed into multiple specialized subLLMs to assemble a sequential pipeline. This pipeline enables rapid screening across hundreds of millions of peptide sequences, ensuring potent antimicrobial activity and minimizing cytotoxic risks. Through transfer learning, we endowed the LLMs with different domain-specific knowledge to achieve high-throughput mining and generation of AMPs within a unified methodological framework. Notably, both mined and generated AMPs exhibited reduced susceptibility to resistance development in ICU-derived carbapenem-resistant Acinetobacter baumannii (CRAB) and methicillin-resistant Staphylococcus aureus (MRSA) in vitro. The AMPs also showed comparable or superior therapeutic efficacy in in vivo thigh infection mouse models compared with clinical antibiotics, without causing organ damage and disrupting gut microbiota. The mechanisms of action of these AMPs involve disruption of the cytoplasmic membrane and membrane depolarization. Overall, this study presents a generative artificial intelligence approach for the discovery of novel antimicrobials against multidrug-resistant bacteria, enabling efficient and extensive exploration of AMP space.
Similar content being viewed by others
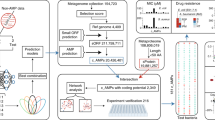

Main
The World Health Organization has released a list of multidrug-resistant bacteria, collectively known as ESKAPE1, necessitating urgent advancements in antimicrobial drug development. Topping the list is carbapenem-resistant Acinetobacter baumannii (CRAB). Carbapenem antibiotics, a ‘last resort’ treatment when all other options fail, are remarkably vulnerable to the emergence and dissemination of antimicrobial resistance (AMR)2,3,4. In light of this pressing issue, there is growing interest in exploring antimicrobial peptides (AMPs) as a promising alternative to conventional antibiotics, given their comparatively slower AMR development5,6,7.
Large language models (LLMs) have emerged as a transformative power in enhancing natural language comprehension, representing an important step toward artificial general intelligence. Typically, LLMs refer to those built on Transformer-based architectures with hundreds of millions (or even billions) of trainable parameters, trained on extensive textual corpora8. However, these existing general LLMs often struggle with scientific data, such as molecules, proteins and genes. To facilitate understanding of scientific languages, scientific LLMs have been devised, customized for various scientific domains and disciplines9,10,11,12,13,14,15,16,17,18,19.
In the present study, we achieved high-throughput mining and generation of AMPs by conducting multiple LLMs through transfer learning and validated the efficacy of these AMPs against clinical superbugs, specifically CRAB and methicillin-resistant Staphylococcus aureus (MRSA), from both in vitro and in vivo perspectives. Encouragingly, both the mined and generated AMPs demonstrated comparable or superior antimicrobial efficacy to clinical antibiotics, with additional anti-inflammatory effects and no detectable organ damage or disruption to intestinal homeostasis. Furthermore, they showed reduced susceptibility to AMR development of clinical superbugs, which is a critical aspect in the development of novel drugs. Taken together, our framework ensembled with LLMs can use data mining and text generation strategies to perform high-throughput discovery of efficient and safe AMPs as next-generation antibiotics against clinical superbugs.
Results
Development of ProteoGPT, a pre-trained LLM
A highly effective strategy in transfer learning involves pre-training models on extensive unlabelled data through meticulously designed self-supervised tasks. To tailor models specifically for mining and generating AMPs, we constructed a versatile pre-trained model named ProteoGPT, an LLM comprising more than 124 million parameters, achieved through knowledge transfer on protein sequence space (Fig. 1a).

a, Schematic of ProteoGPT. ProteoGPT comprises multiple Transformer blocks, stacked sequentially and categorized into three main modules: the input module, processing module and output module. The input module consists of embedding layers designed to map encoded sequence data into a continuous vector space, thereby facilitating the model’s comprehension. Positional encoding is also applied to introduce positional information. The processing module incorporates multihead self-attention layers and feed forward neural network layers. Its primary role involves the nonlinear transformation of vectors from the input module. This enables the model to focus on various segments of the sequences and establish contextual understanding. Each processing module includes residual connection and layer normalization to ensure the stable propagation of gradients and model training. The output module primarily comprises linear layers responsible for generating the probability distribution for the next token. b, Length distribution for protein sequences used in a. c, Sources distribution for protein sequences used in a. d, Training process of ProteoGPT. The blue solid line represents the raw loss values, while the black solid line depicts the smoothed loss values. The schematic in a was created with BioRender.com.
All 609,216 non-redundant canonical and isoform sequences were retrieved from the UniProtKB/Swiss-Prot database, with 96% of these sequences consisting of 1,000 amino acids (AAs) or fewer. In addition, there are 1,296 sequences exceeding 3,000 AAs in length (Fig. 1b). These proteins originate from various sources, including humans, animals, plants, microorganism and others (Fig. 1c). During the pre-training process, all protein sequences were initially partitioned into multiple data pieces and then fed into ProteoGPT to undergo processing by each functional component (Fig. 1a). Subsequently, we preserved the stabilized pre-trained model for future fine-tuning to accommodate more specific downstream tasks (Fig. 1d).
Compared to other Protein-LLMs20,21, a key distinguishing feature of ProteoGPT is its training foundation: the use of the Swiss-Prot database, a high-quality manually annotated collection representing a comprehensive array of protein sequences, rather than uncurated protein sequence data. This provides ProteoGPT with a more accurate and reliable data foundation, offering a pre-trained template that is biologically reasonable for downstream tasks involving functional peptides.
Development of transfer learning models
Following the pre-training of ProteoGPT, we proceeded with transfer learning to further refine its capabilities for text classification and generation tasks of AMPs. Three transfer subLLMs were developed by fine-tuning ProteoGPT on different datasets: AMPSorter, BioToxiPept and AMPGenix.
AMPSorter is a classifier designed for identifying AMPs by fine-tuning ProteoGPT with both AMP and non-AMP datasets (Fig. 2a). AMPSorter demonstrated robustness and offered an excellent fit without exhibiting signs of underfitting or overfitting throughout the training process, with loss and accuracy curves for both the training and validation sets consistently aligning across epochs (Extended Data Fig. 1a). On the test set, AMPSorter, while maintaining overall classification performance (Extended Data Fig. 1b, area under the curve (AUC) = 0.99), was able to accurately identify all true AMPs, reducing missed detections and avoiding overlooking a large number of true positives (Extended Data Fig. 1c, area under the precision recall curve (AUPRC) = 0.99). In both the training and predicting processes, AMPSorter can flexibly handle sequences with unnatural amino acids (UAAs), such as d-type (for example, r), unknown or arbitrary (X), non-canonical (for example, pyrrolysine, O; selenocysteine, U), as well as non-specific representations (for example, B, Z). The test set contains 194 sequences with UAAs, and AMPSorter achieved a precision of 96.43% on these sequences (Extended Data Fig. 1b,c and Supplementary Table 1), almost perfectly distinguishing between positive and negative samples.

a, Overview of transfer learning models derived from ProteoGPT. AMPSorter is employed to identify AMPs; BioToxiPept is applied for predicting the toxicity of peptides. Since ProteoGPT primarily functions as a generative model, an additional classification module was augmented to undertake the task of AMPs or toxicity recognition. The classification module consists of a classification head and an output layer. The classification head comprises a layer normalization, which takes the output representation from the final Transformer block of ProteoGPT, allowing for layer normalization before the model’s output. The output layer, in the form of a softmax layer, maps the output features from ProteoGPT to the final classification label space, thereby deriving probability scores for each category; AMPGenix is utilized for generating sequences of short peptides akin to AMPs. The main parameters involved in AMPGenix include temperature (which controls the randomness of the sequence generation), prefix (the initial token or AA that initiates the sequence), n_tokens (the number of tokens to be generated in the sequence) and n_samples (the number of sequences generated for a given prefix and n_tokens setting). b,c, AUCs (b) and AUPRCs (c) for different classification models used for AMP recognition on the benchmarking set. d,e, AUCs (d) and AUPRCs (e) for different classification models used for cytotoxicity recognition on the same test set. f, Comparison of the positive prediction rates for sequences generated by the pre-trained model (ProteoGPT) and the transfer submodel (AMPGenix) across different temperature parameter settings (0.5, 1, 2 and 3) using 6 high-performance AMP classification models. g, Comparison of the positive prediction rates for sequences generated by 5 external unconstrained AMP generation models and AMPGenix across different temperature parameter settings (0.5, 1, 2 and 3) using 6 high-performance AMP classification models.
To ensure the model’s generalizability and mitigate potential overfitting due to sequence similarity, we employed the CD-HIT clustering method to filter out sequences in the test set with >70% identity with those in the training and validation sets and sequences with UAAs, resulting in a more stringent benchmarking set (725 AMPs and 1,071 non-AMPs) for comparison with existing AMP classification algorithms. After applying CD-HIT, we observed a marked reduction in overlap between the training, validation and benchmarking sets (Extended Data Fig. 1d,e). The Fréchet ChemNet Distance (FCD) increased from 7.92 to 28.16 (Extended Data Fig. 1f), indicating a substantial difference in distribution between the benchmarking set and the training/validation sets. This approach aimed to test the model’s ability to handle novel, unseen sequences with lower similarity, ensuring that AMPSorter would be challenged to classify peptides that are more distantly related to the peptides it had been trained on, which is essential for real-world applications where novel AMPs may not perfectly match known sequences. In a comprehensive evaluation of AMP classification on the same benchmarking set, AMPSorter achieved an AUC of 0.97 and AUPRC of 0.96, outperforming all competing models22,23,24,25,26,27,28, including AMPlifyimbal and Macrel (Fig. 2b,c). AMPSorter reached 90.67% precision, 88.89% F1 score and 81.66% Matthews correlation coefficient (MCC) (Extended Data Table 1). These results indicate that AMPSorter has excellent capacity for accurately distinguishing AMPs from non-AMPs. Moreover, compared with other models, AMPSorter maintained a high level of balance between specificity (93.93%) and sensitivity (87.17%), demonstrating that AMPSorter not only effectively captures potential AMP sequences during the initial screening phase but also substantially reduces false positives. In addition, AMPSorter achieved 93.99% prediction precision on an entirely independent external validation dataset (Supplementary Table 1).
BioToxiPept, a classifier designed to identify the cytotoxicity of peptides, shares a similar model architecture with AMPSorter (Fig. 2a). During the fine-tuning phase, BioToxiPept was retrained and fine-tuned using datasets comprising both toxic and non-toxic short peptides. Similar to AMPSorter, BioToxiPept demonstrated excellent fit without signs of underfitting or overfitting (Extended Data Fig. 1g). BioToxiPept generally exhibited high classification performance comparable to those of ToxIBTL29 and ToxinPred2.0-RF30 within the same test set (Fig. 2d). Notably, the precision–recall (PR) curve indicated that BioToxiPept and ToxIBTL are more capable of recognizing genuinely toxic peptides, in contrast to their counterparts (Fig. 2e, AUPRC = 0.92). This capability substantially diminishes the rate of false negative, thereby considerably reducing the costs associated with experimental validation.
Without considering structure, short peptides can be regarded as permutations and combinations of 20 natural AAs (letters), forming functional domains (words) that convey specific functions (meanings). AMPGenix is an unconstrained sequence generator retrained on a dataset of AMPs based on ProteoGPT (Fig. 2a). During retraining, the weights of the underlying part of ProteoGPT were fine-tuned, resulting in the creation of the stable AMPGenix model with minimized loss, poised for generating potential AMPs (Extended Data Fig. 1h). AMPGenix can unconstrainedly generate a specific number of peptides with a defined number of tokens and specific prefix information (the initial token). We conducted an analysis of the first AA’s frequency of the collected AMPs and found that glycine (G), lysine (K), phenylalanine (F), arginine (R), alanine (A), leucine (L), isoleucine (I), valine (V), serine (S) and tryptophan (W) emerged as the most frequent, each exceeding a count of 400 (Supplementary Table 2). These high-frequency AAs were incorporated as prefix information for text generation based on different temperatures (0.5, 1, 2, 3), which control the randomness and diversity of the generated content. With token lengths ranging from 8 to 15, a set of 100 sequences for each prefix and token count was generated, totalling 8,000 sequences per temperature setting.
As the temperature increases, the randomness of the model output markedly rises (uniqueness increases from 0.81 to 1), leading to the generation of more unique sequences. For sequences containing only natural AAs, higher temperatures enhance the variety of generated sequences and increase the likelihood of producing distinct types (diversity increases from 0.97 to 0.99). Novelty remains relatively stable across the four temperature settings, suggesting that the novelty of generated sequences has reached saturation. In addition, as temperature rises, the generated sequences increasingly align with the characteristic distribution of real AMPs (FCD decreases from 13.61 to 9.26 and 9.56) (Extended Data Table 2). We then assessed these sequences using six high-performance AMP classification models, finding that AMPGenix-generated sequences consistently outperformed those from ProteoGPT across all temperature settings and classification models, with a substantially higher proportion classified as AMPs (Fig. 2f). This highlights the effectiveness of domain-specific fine-tuning through transfer learning, enabling AMPGenix to demonstrate an enhanced ability to capture the key characteristics of AMPs. With data alignment (including sequence prefix, quantity and length; Methods), we further compared the performance of AMPGenix with five external, unconstrained AMP generation models (HydrAMP31, Basic31, PepCVAE32, AMP-GAN33 and AMP-LM34). The results showed that AMPGenix-generated sequences achieved a higher AMP recognition rate across six classification models (Fig. 2g), highlighting its precision in capturing AMP-specific characteristics. Furthermore, AMPGenix-generated sequences exhibited a lower FCD compared with all other models (Extended Data Table 2). This indicates that AMPGenix successfully maintains diversity while producing sequences that more closely align with the distributional features of real AMP profiles. However, we observed that although the increased diversity at higher temperatures brings the sequences closer to the global characteristics of the real distribution, there is a slight decrease in classification positivity rate (Fig. 2g). This is due to the increased uniqueness and diversity of the sequences, which leads to deviations in some specific features. These changes make it more difficult for the classifier to accurately match the features of these sequences. This phenomenon highlights an inherent trade-off between diversity and effective classification in generative models.
Furthermore, AMPGenix generated a small number of short peptides containing UAAs, with over 70% of these sequences identified as AMPs by AMPSorter (Extended Data Fig. 1i). This ability provides AMPGenix with greater flexibility and creative potential in AMP design.
High-throughput mining and generation of AMPs
Integrated with transfer submodels, we proposed a sequential pipeline ensembled by LLMs (SPEL), which combines AMPSorter, BioToxiPept and wet-lab validation (Fig. 3b). In this study, we devised two high-throughput strategies for the discovery of novel AMPs: (1) data mining (m_AMPs): identifying encrypted AMPs from the complete UniProtKB/Swiss-Prot database (Fig. 3a); and (2) text generation (g_AMPs): directly generating non-natural AMPs using AMPGenix (Fig. 3c).

a, Ensembles of NRSPDs. The window size is set to L, with a step size of L/2, where L ranges from 8 to 30. A total of 5 NRSPDs were constructed through a sliding-window technique employed on the complete UniProtKB/Swiss-Prot database, encompassing 410,192,277 short peptide sequences. b, The whole process of SPEL includes modules of AMPSorter, BioToxiPept and wet-lab validation. The antimicrobial activities of the output peptide sequences were validated by wet-lab experiments. c, Ensembles of GNRSPDs. The sequences generated by AMPGenix at the default temperature = 1 parameter were used to construct 10 different GNRSPDs, comprising a total of 7,798 unique short peptide sequences. d,e, Length distribution for 154 m_AMPs (d) and 42 g_AMPs (e). f–h, The prediction probabilities of AMPSorter (f), BioToxiPept (h) and RS predicted by QSAR (g) of 154 m_AMPs and 42 g_AMPs. The boxplots centre on the median and extend to the 25th and 75th percentiles, and the whiskers extend to the furthest point within 1.5× the interquartile range. A two-sided Mann–Whitney U-test was performed for comparison. Exact P values are provided in Source Data Fig. 3. i, UMAP visualization of AMPs, m_AMPs and g_AMPs using k-mer encoding (k = 3). Each point represents a sequence, with the position determined by reducing the high-dimensional k-mer feature space into two dimensions. The schematic in b was created with BioRender.com.
Employing a sliding-window technique, we fragmented all 609,216 non-redundant canonical and isoform sequences into short peptides ranging from 8 to 30 AAs in length (Fig. 3a). Ultimately, multiple non-redundant short-peptide datasets (NRSPDs) were assembled, ready for comprehensive data mining, totalling 410,192,277 peptides. Using SPEL, we identified a total of 121,405,811 peptide candidates (m_AMPs), of which 82,694,928 were recognized as non-cytotoxic (m_AMPs_nontox, Extended Data Table 3) and served as a candidate pool of data mining for further screening different types of AMP.
Using AMPGenix with the default temperature parameter set to 1, 7,798 unique peptide sequences were generated to form the generated non-redundant short-peptide datasets (GNRSPDs), with 76.1% of these sequences identified as AMPs (g_AMPs). After filtering with BioToxiPept, 4,736 (60.7%) sequences labelled as non-toxic formed the candidate pool of text generation (g_AMPs_nontox, Extended Data Table 4).
To independently evaluate the effectiveness of the language models, we selected 20 sequences (PT-series, PT-1–PT-20) with the highest AMPSorter prediction probabilities from the text generation-based candidate pool and conducted pilot test to evaluate their antimicrobial activity in vitro. The strains utilized for minimum inhibitory concentrations (MICs) determination comprised antibiotic-susceptible bacteria (Escherichia coli ATCC25922 and Staphylococcus aureus ATCC25923), clinically isolated superbugs (CRAB and MRSA strains) and Candida albicans ATCC10231. The preliminary test results demonstrated that 90% (18/20) exhibited inhibitory activity against at least one strain (MIC ≤ 512 μg ml−1), with 12 peptides achieving MIC values ≤ 256 μg ml−1 (Extended Data Table 5). Notably, PT-1, PT-4, PT-12, PT-15, PT-18 and PT-20 demonstrated marked broad-spectrum antimicrobial efficacy against both antibiotic-sensitive and antibiotic-resistant bacterial strains, as well as the fungal pathogen, achieving MIC values as low as 1 μg ml−1. Among these, PT-1, PT-4, PT-15, PT-18 and PT-20 demonstrated 2~21.3 μg ml−1 MICs against three CRAB strains, PT-1 and PT-18 showed 4~13.3 μg ml−1 MICs against two MRSA isolates, and five peptides (PT-1, PT-4, PT-12, PT-15 and PT-18) displayed 8~32 μg ml−1 antifungal potency against C. albicans ATCC10231. Strain-specific divergence also emerged, with PT-3 demonstrating potent activity against CRAB strains and PT-8 showing selective efficacy toward MRSA strains. Collectively, these findings underscore AMPSorter’s proficiency in identifying AMP sequences for experimental validation and validate AMPGenix’s capacity to produce a functional AMP candidate repository, specifically tailored to address clinically drug-resistant pathogens.
With the threshold of AMPSorter to 0.9, the numbers of m_AMPs and g_AMPs in the candidate pool were decreased to 26,322,225 (6.4%) and 3,069 (39.4%), respectively (Extended Data Tables 3 and 4). To establish a clear threshold and ensure consistent peptide types (cationic AMPs) for comparison of the efficiency between two strategies, we introduced an algorithm for quantitative structure–activity relationship (QSAR) analysis of antimicrobial activity35,36. This approach functioned as a gatekeeper, ensuring that both strategies met the same criteria during the comparison process and facilitated the selection of an appropriate number of peptides for validation. With the threshold of relative score (RS) set to 0.88, 154 m_AMPs (8–12 AAs; Fig. 3d and Extended Data Table 3) and 42 g_AMPs (11–26 AAs; Fig. 3e and Extended Data Table 4) were selected for experimental validation. Compared with the m_AMPs, these g_AMPs exhibited significantly higher predicted antimicrobial activity (Fig. 3f,g) and lower predicted cytotoxicity compared with m_AMPs (Fig. 3h), which indirectly underscores AMPGenix’s aptitude in learning distinctive features of AMPs.
These 196 peptides selected had overrepresentation of F, I, L, R and W compared with known AMPs present in four public AMP datasets: APD3 (ref. 37), DBAASP38, DRAMP39 and CAMP40 (Extended Data Fig. 2a). The results indicate that the FCD between g_AMPs and AMPs is considerably higher than that between m_AMPs and AMPs (Extended Data Fig. 2b), suggesting that g_AMPs diverge more from known sequences than m_AMPs. In addition, uniform manifold approximation and projection (UMAP) visualization of the sequence space reveals that m_AMPs tend to form tighter clusters, with their distribution closely resembling that of AMPs, while g_AMPs exhibit a more dispersed distribution (Fig. 3i). One possible explanation is that this difference arises because m_AMPs and most AMPs are derived from natural sources, which typically follow conserved sequence patterns, leading to higher sequence similarity. EggNOG mapper revealed that the 154 m_AMPs are predominantly embedded within proteins involved in information storage and processing, cellular processes and signalling (Extended Data Fig. 2c). Consequently, these encrypted AMPs may be constrained by functional requirements, such as the need for conserved regions, ensuring their effective participation in antimicrobial processes or immune regulation35,41. In contrast, g_AMPs, generated using previous knowledge of short peptides from AMPGenix, offer greater flexibility in sequence generation. This flexibility results in a broader distribution in the sequence space.
In vitro evaluation of functional AMPs
The entire workflow of the wet-lab experiments is depicted in Extended Data Fig. 3. A total of 154 m_AMPs and 42 g_AMPs underwent synthesis and in vitro characterization. The Kirby–Bauer disk diffusion assay was used to examine the antibacterial activity against E. coli ATCC25922 and S. aureus ATCC25923 in the presence of 128 μg AMPs initially, revealing that 75.97% (117/154) of m_AMPs and 61.91% (26/42) of g_AMPs exhibited potential bacterial inhibitory properties (Fig. 4a). Subsequently, 53.89% (83/154) of m_AMPs and 40.48% (17/42) of g_AMPs were selected, demonstrating an inhibition zone diameter of ≥10 mm against at least one bacterium. Furthermore, the Kirby–Bauer disk diffusion assays conducted at 64 μg AMPs showed that 66.27% (55/83) of m_AMPs and 100% (17/17) of g_AMPs displayed potent antibacterial activity against E. coli, while 55.42% (46/83) of m_AMPs and 82.3% (14/17) of g_AMPs exhibited strong antibacterial activity against S. aureus. Finally, 44 m_AMPs and 14 g_AMPs with diameter of ≥10 mm against E. coli or S. aureus were selected for further assessment.

a, Proportion of AMPs with bacteriostatic effects on E. coli and S. aureus according to the diameter of the inhibition zone. The number of AMPs is indicated above each bar. b, Antibacterial activity levels of 44 m_AMPs and 14 g_AMPs against E. coli and S. aureus at high inoculum of 108 c.f.u.s ml−1. c, Cytotoxicity and haemolysis ratio of the top 20 candidate AMPs. d,e, Distribution of MIC values (d) and corresponding MIC values (e) of the top 20 candidate AMPs against E. coli, S. aureus, CRAB, MRSA and C. albicans (at standard inoculum of 105 c.f.u.s ml−1). AMPs marked with ‘#’ are the 7 leading AMPs screened for in vivo experiments. CRAB 267, CRAB QLH-2022-267; CRAB 636, CRAB QLH-2022-636; CRAB 637, CRAB QLH-2022-637; MRSA 266, MRSA QLH-2022-266; MRSA 718, MRSA QLH-2022-718.
MICs were determined at 108 colony-forming units (c.f.u.s) ml−1, a high inoculum, revealing the activities of 11 and 17 m_AMPs against E. coli and S. aureus, respectively, with concentrations of ≤160 μg ml−1 (Fig. 4b). Among the g_AMPs, 14 exhibited antimicrobial activities against E. coli and 12 against S. aureus with concentrations of ≤160 μg ml−1, among which 8 reached ≤0.625 μg ml−1 against S. aureus. Combining these results with haemolysis ratios (<8%), we shortlisted the 10 most potent m_AMPs and 10 most potent g_AMPs to evaluate cytotoxicity against human embryonic kidney (HEK293) cells. The results showed that 19 AMPs had negligible cytotoxicity, with cell survival rate of ≥50% (Fig. 4c). Then we measured their MICs at standard inoculum (105 c.f.u.s ml−1) against drug-sensitive or resistant strains (Fig. 4d,e). Five broad-spectrum AMPs (g_AMP14, g_AMP41, g_AMP42, m_AMP46 and m_AMP76) exhibited MIC values <32 μg ml−1 for all bacteria. Among these, m_AMP76 showed the strongest potency with a MIC value of 4 μg ml−1 against CRAB QLH-2022-637, while g_AMP41 reached 8 μg ml−1 against MRSA QLH-2022-718. In addition, g_AMP33 demonstrated a strong inhibitory effect with a specific preference for targeting Gram-negative bacteria such as E. coli and CRAB, while g_AMP35 displayed a more pronounced antibacterial activity against MRSA compared with its effect on CRAB. Interestingly, all these seven AMPs also exhibited strong inhibitory abilities against C. albicans (MIC = 16~64 μg ml−1), a typical pathogenic fungus. On the basis of these in vitro results, we selected these seven leading AMPs for further in vivo experiments.
Therapeutic efficacy evaluation in an in vivo mouse infection model
We established a mouse thigh infection model (Fig. 5a) to assess the efficacy of the seven peptides in treating CRAB QLH-2022-637 or MRSA QLH-2022-718 infections (Extended Data Fig. 4a). As shown in Fig. 5b, five AMP-treated groups exhibited significant reductions of ~70% in CRAB recovered from the thigh tissues compared with the sterile water-treated group (g_AMP14, g_AMP33, g_AMP42, m_AMP46 and m_AMP76). Notably, the elimination rates for g_AMP42 and m_AMP76 reached 83% and 87%, respectively, while the polymyxin B group exhibited an elimination rate of 86%. Four AMPs eliminated ~70% of MRSA c.f.u.s in the thigh tissues (g_AMP14, g_AMP35, m_AMP46 and m_AMP76), and the elimination rate of g_AMP14 reached 85%, with an elimination rate of 89% for the vancomycin group (Fig. 5c). However, despite demonstrating strong in vitro activity against both CRAB and MRSA, g_AMP41 and g_AMP42 were unable to effectively eradicate the pathogens in vivo. To explore this difference, resistance to proteolytic degradation assay was assessed (Fig. 5d). The results indicated that the final concentrations of g_AMP41 and g_AMP42 were below 60%, whereas up to 80% of the initial concentrations of g_AMP14, g_AMP33, g_AMP35, m_AMP46 and m_AMP76 remained after 4 h of continuous exposure to serum proteases (Fig. 5e). Compared with water and antibiotic-treated groups, the composition of the intestinal bacterial community in the AMP-treated groups with significant therapeutic effects (Extended Data Fig. 4b) showed no obvious disturbances at the phylum, class and genus levels (Extended Data Fig. 4c–j). Meanwhile, the Firmicutes/Bacteroidetes (F/B) ratio (widely accepted to have an important influence in maintaining normal intestinal homeostasis42) of AMP-treated groups was similar to that of the water and antibiotic-treated groups (Supplementary Table 3). In addition, g_AMP14, g_AMP33, g_AMP35, g_AMP42, m_AMP46 and m_AMP76 showed anti-inflammatory effects comparable to those of clinical antibiotics commonly used, as evidenced by the absence of obvious inflammatory cell infiltration in colon tissue histology images. A small amount of venous stasis was observed in the kidney tissue histology images of the AMP-treated groups and polymyxin B group. However, in the vancomycin-treated group, the renal tubular epithelial cells of mice exhibited histopathological changes: cytoplasmic rarefaction and pale staining, suggesting protein loss and cellular dysfunction, and pyknosis of the nucleus, indicating apoptosis or necrosis (Extended Data Fig. 4k).

a, Experimental modelling in vivo. b,c, Bacterial counting in thigh tissues of mice infected by CRAB QLH-2022-637 (b) and MRSA QLH-2022-718 (c). For CRAB infection: sterile water (n = 8), polymyxin B (n = 10), g_AMP14 (n = 7), g_AMP33 (n = 9), g_AMP41 (n = 7), g_AMP42 (n = 7), m_AMP46 (n = 7), m_AMP76 (n = 8). For MRSA infection: sterile water (n = 7), vancomycin (n = 8), g_AMP14 (n = 7), g_AMP35 (n = 7), g_AMP41 (n = 7), g_AMP42 (n = 7), m_AMP46 (n = 7), m_AMP76 (n = 6). The lines in each data group represent the first quartile, median and third quartile. Statistical analysis was conducted using two-sided Wilcoxon rank-sum test. Exact P values are provided in Source Data Fig. 5. d, Schematic of the resistance to enzymatic degradation experiment. e, AMPs were exposed for a total period of 4 h to fetal bovine serum that contains several active proteases. Aliquots of the resulting solution were analysed by liquid chromatography–tandem mass spectrometry (LC–MS/MS). Experiments were performed in 2 independent replicates. f,g, Resistance acquisition of CRAB QLH-2022-637 (f) and MRSA QLH-2022-718 (g) under 0.5-fold MIC concentrations of AMPs and antibiotics (n = 3 biologically independent replicates). ‘Water#’ represents DNase/RNase-free and sterile water. The schematics in a and d were created with BioRender.com.
We further evaluated possible drug resistance by culturing CRAB QLH-2022-637 and MRSA QLH-2022-718 with 0.5-fold MIC level of AMPs, using the antibiotic polymyxin B and vancomycin for comparison, respectively. When cultured in the presence of AMPs for 20 passages, the MICs of g_AMP33, g_AMP42 and m_AMP46 were 8 μg ml−1 at the first and 20th passages against CRAB. There were 2-fold and 8-fold increases in the MIC of g_AMP14 (MIC of 16 μg ml−1 at the first passage and 32 μg ml−1 at the last passage) and m_AMP76 (MIC of 4 μg ml−1 at the first passage and 32 μg ml−1 at the last passage) against CRAB, respectively, while exposure to polymyxin B resulted in a 32-fold increase in MIC after 20 passages (Fig. 5f). Serial passaging of MRSA in the presence of subinhibitory concentrations of AMPs did not select for isolates resistant to g_AMP14 (MIC of 16 μg ml−1 at the first and 20th passages), g_AMP35 (MIC of 16 μg ml−1 at the first passage and 32 μg ml−1 at the last passage) and m_AMP76 (MIC of 32 μg ml−1 at the first passage and 64 μg ml−1 at the last passage). There was a 4-fold increase in the MIC of m_AMP46 against MRSA, rising from 32 μg ml−1 at the first passage to 128 μg ml−1 at the last passage. Conversely, exposure to the vancomycin resulted in a rapid increase in MIC after 14 passages, with a ≥128-fold increased MIC after 18 passages (from 0.5 to ≥64 μg ml−1; Fig. 5g).
Functional characterization of identified AMPs
We then delved into elucidating the mechanisms of action (MoA) for the six optimal AMPs with significant therapeutic effects in vivo. Potential morphological changes in CRAB QLH-2022-637 and MRSA QLH-2022-718 cells in the presence of AMPs were monitored using scanning electron microscopy (SEM), revealing wrinkling, deformation or perforation of treated cells (Fig. 6a and Extended Data Fig. 5a,b). AMPs were then investigated with fluorescent probes to determine how they affected the bacterial cytoplasmic membrane (Fig. 6b). After treating CRAB QLH-2022-637 and MRSA QLH-2022-718 cells with AMPs for 2 h, the fluorescence intensity of the membrane-permeable probe propidium iodide (PI) was enhanced with increasing concentrations (1- and 5-fold MIC) compared with non-treated bacteria, indicating disruption of the cytoplasmic membrane (Fig. 6c,d). Next, 3,3’-dipropylthiadicarbocyanine iodide [DiSC3-(5)] was used to assess the depolarization state of the cytoplasmic membrane in CRAB QLH-2022-637 and MRSA QLH-2022-718. The results showed that all AMPs have a certain depolarization effect, with different action speeds. Polymyxin B, a peptide antibiotic with established membrane depolarization activity against A. baumannii35, was employed as the positive control for CRAB, while Triton X-100 served as the positive control for MRSA due to its non-specific membrane disruption capacity. Except for g_AMP42, other AMPs (g_AMP14, g_AMP33, m_AMP46 and m_AMP76) initially demonstrated relatively rapid depolarization kinetics against CRAB QLH-2022-637, with a rapid increase in fluorescence values after 10 min, among which m_AMP76 showed stronger depolarization than polymyxin B (Fig. 6e). Similar to the results with CRAB QLH-2022-637, all AMPs (g_AMP14, g_AMP35, m_AMP46 and m_AMP76) show relatively rapid depolarization kinetics against MRSA QLH-2022-718, with m_AMP76 having the strongest depolarization effect (Fig. 6f). These observations suggest that the MoA of AMPs involves the disruption of the cytoplasmic membrane and the depolarization of the membrane potential. On the basis of the observable deformation using SEM (Extended Data Fig. 5c), we further conducted transcriptomic analysis and found a significant difference in the expression pattern of E. coli ATCC25922 treated with m_AMP76 (renamed Arkwillin, an encrypted peptide mined within the proteins of Clavispora lusitaniae; Supplementary Table 4) compared with the control (Extended Data Fig. 5d,e, P = 0.003). Arkwillin disrupted cell membrane integrity by downregulating the expression of slyB, bamD, bamB and pagP, which encode membrane-associated proteins. It suppressed the expression of protein-encoding genes involved in lipopolysaccharide (LPS) synthesis and transport, biofilm formation and flagellar assembly, while upregulating the expression of membrane protein-encoding genes linked to cell membrane permeability (Fig. 6g).

a, SEM images of CRAB QLH-2022-637 and MRSA QLH-2022-718 treated with m_AMP76. The control groups were untreated corresponding strains. b, Schematic showing the behaviour of PI and DiSC3-(5), the fluorescent probes used to indicate disruption and depolarization of the cytoplasmic membrane caused by the AMPs, respectively. c,d, Changes in cell membrane permeability when CRAB QLH-2022-637 (c) or MRSA QLH-2022-718 (d) were co-cultured with AMPs (at concentrations of 1- and 5-fold MIC, respectively). Experiments were performed in 3 independent replicates. PC group, positive control (high-temperature-treated bacteria, with PI). NC group, negative control (untreated bacteria, with PI). The vertical coordinate is the ratio of FSC-A− and PI-A+ after PI treatment. All AMP-treated groups and the PC group were compared to the NC group. Error bars are the s.d. of triplicate experiments. Statistical analysis was conducted using two-sided t-test. Exact P values are provided in Source Data Fig. 6. e,f, DiSC3-(5) assays show the effect of AMPs on the membrane depolarization of CRAB QLH-2022-637 (e) or MRSA QLH-2022-718 (f). Relative fluorescence was calculated using as baseline the untreated control (buffer + bacteria + fluorescent dye). The graphs show the mean ± s.d. of 3 independent experiments. g, Heat map of differential gene expression related to cell membrane or cell wall synthesis. The colours of the heat map squares represent the magnitude of the ratio, which is the FPKM value of a gene for each sample in the Arkwillin-treated group divided by the mean value of the control group for the same gene. The schematic in b was created with BioRender.com.
Discussion
We present a generative artificial intelligence approach for the discovery of novel antimicrobials, enabling efficient and extensive exploration of the antimicrobial peptide space. Logically, this study can be divided into two major parts: the first part involves the data mining and text generation of AMPs; the second part is a comparison of two development strategies.
Notably, this study underscores the pivotal role of LLMs in the realm of transfer learning: by undergoing pre-training across a broad array of textual data, LLMs demonstrate proficiency in transferring knowledge across diverse datasets and tasks seamlessly. Specifically, ProteoGPT, through meticulous fine-tuning with various peptide datasets, has effectively nurtured transferable submodels capable of generating AMPs (AMPGenix), conducting classifications (AMPSorter) and detecting toxicity (BioToxiPept), harnessing the advantages of both the evolving capabilities of LLMs and domain-specific knowledge. This ‘one for all’ framework exemplifies the adaptability and scalability of LLMs in tackling a multitude of tasks (not only AMPs, but also other bioactive substances), ensuring high accuracy in scenarios with limited sample learning, especially in fields where data availability is restricted or expensive. In contrast to other overlay patterns involving different models, this single-model framework not only simplifies model management and maintenance but also enhances overall processing efficiency and result consistency.
Compared to previous studies where AMP sequence generation31,43 and data mining44,45,46,47 were conducted separately, we have fully leveraged the advantages of LLMs in the field of transfer learning to implement both development strategies within a unified framework. Furthermore, the previous knowledge obtained through transfer learning has made AMP sequence generation more efficient than data mining, a fact that has been thoroughly validated in real-world applications. When comparing g_AMPs and m_AMPs, we observed some intriguing trends: g_AMPs exhibited superior and broader-spectrum antibacterial effects compared with m_AMPs, consistent with the results of AMPSorter and QSAR predictions. G_AMPs were generated through the transfer model AMPGenix, which benefited from transfer learning to acquire knowledge and experience from AMP sequences. This implies that the model could capture a wider range of antibacterial properties and structural features, thus better generating peptides with superior antibacterial effects. In contrast, m_AMPs were identified directly through data mining of existing short peptide sequences, which may be limited by the quality and coverage of the dataset, resulting in limited antibacterial activity and spectrum of the identified AMPs. Notably, the PT-series generated by AMPGenix and selected exclusively through the transfer learning model, achieved notable antimicrobial efficacy, surpassing that of the g_AMPs and m_AMPs tested. These results further emphasize the potential of generative language models in AMP design.
The LLM models developed in this study, such as AMPSorter, function primarily as qualitative classification models for AMPs and are unable to provide quantitative predictions of antimicrobial activity. While the QSAR employed is a quantitative algorithm, it tends to favour cationic peptides with specific physicochemical properties, such as high charge and hydrophobicity, exhibiting an inherent bias that limits its ability to assess a broader spectrum of AMP types. Future research will focus on achieving unbiased quantitative predictions of AMP activity through sequence-based language models.
Methods
Data collection
For model construction, we collected, in total, five kinds of datasets in our study: a large-scale proteome, AMPs, non-AMPs, an external validation dataset, and a toxin and non-toxin dataset.
Proteome
Non-redundant canonical and isoform sequences (609,216) were retrieved from UniProt (http://www.uniprot.org/) (downloaded as of October 2022). The protein library composed of these sequences serves two main purposes: pre-training the ProteoGPT model and constructing different types of NRSPD.
-
(1)
Pre-training the ProteoGPT model
Due to computational constraints and model requirements, all sequences were segmented into subsequences with a maximum length of 1,000 AAs to serve as a training corpus.
-
(2)
Constructing different types of NRSPD
A sliding window with a size of L was employed to scan all protein sequences for constructing multiple NRSPDs. To avoid windows with >50% sequence overlap, we set the sliding step size to L/2. In our study, L was set to 8–30. Five NRSPDs were constructed to be the subject for data mining of AMPs: 8–10 AAs, 11–15 AAs, 16–20 AAs, 21–25 AAs and 26–30 AAs, totalling 410,192,277 non-redundant short peptides.
AMPs
AMP data were primarily sourced from four public AMP datasets: APD3 (ref. 37), DBAASP38, DRAMP39 and CAMP40, which cover most AMP sequences from various origins (downloaded as of November 2022). AMPs exhibiting antibacterial, antiviral and antifungal activities, and having lengths under 50 AAs, were deduplicated and consolidated into an extensive dataset, resulting in 16,062 non-redundant AMPs used for subsequent analysis.
Non-AMPs
We compiled the non-AMP sequences from UniProt, applying the ‘subcellular location’ filter to cytoplasm. Any entries containing keywords such as antimicrobial, antibiotic, antiviral, antifungal, effector or excreted in their functional annotations were excluded (downloaded as of November 2022). We refined the dataset to include only sequences not exceeding 50 AAs in length and removed any duplicates. Concurrently, sequences identical to any known AMPs were eliminated from the non-AMP dataset, resulting in a final tally of 16,549 unique non-AMP sequences. The non-AMP sequence dataset we compiled encompasses a wide range of biological sources, including eukaryotes (for example, humans and other plants and animals), prokaryotes (for example, Escherichia coli and Salmonella enterica) and viruses (for example, HIV-1 and hepatitis B virus) (Supplementary Table 5).
External validation dataset
The AMP sequences in the external validation dataset are distinct from those used in the model construction. These sequences were collected from the APD3 (ref. 37), DBAASP38, DRAMP39, CAMPR4 (ref. 48), LAMP2 (ref. 49) and BaAMPs50 databases. The selection criteria included sequences no longer than 50 AAs and those with antifungal activity stronger than antibacterial activity. In addition, 370 non-AMP sequences (≤50 AAs, without overlap with non-AMPs) were derived from ref. 25. To ensure the independence of the external dataset, any sequences that were part of the model training, validation or testing sets were excluded.
Toxin and non-toxin dataset
We collected 1,932 toxic peptide sequences that were experimentally validated and 1,932 non-toxic peptide sequences with a range of 10–50 residues from ToxIBTL29 (https://server.wei-group.net/ToxIBTL/) (downloaded as of April 2023).
In this study, we collected peptide sequence data generated by five unconstrained generation models (HydrAMP31, Basic31, PepCVAE32, AMP-GAN33, AMP-LM34) to compare their performance in AMP generation with AMPGenix. The following quality control and preprocessing steps were applied to all datasets collected from these models:
Prefix alignment
The first AA of each sequence was restricted to one of the 10 selected residues from the set G, K, F, R, A, L, I, V, S and W.
Quantity alignment
A maximum of 800 sequences per starting AA residue was retained, resulting in a total of 7,000–8,000 sequences for each model.
Length alignment
Sequence lengths were constrained to the range of 5–35 AAs to ensure consistency with the length range of sequences generated by AMPGenix.
After filtering, the final number of valid sequences for each model was as follows: HydrAMP, 8,000; Basic, 7,810; PepCVAE, 7,489; AMP-GAN, 7,906; and AMP-LM, 7,405.
Model building
We built, in total, one pre-trained and three transfer LLMs with different functionalities.
Training dataset, validation dataset and test dataset
The datasets employed for the training of AMPSorter and BioToxiPept were randomly partitioned into training, validation and testing sets at a 6:2:2 ratio (Fig. 2a). These datasets were mutually exclusive. The training set was utilized for model development, the validation set for tuning hyperparameters, and the testing set for assessing the model’s performance. To ensure robust evaluation, a benchmarking set was constructed by applying CD-HIT (v.4.8.1) for sequence filtering. First, sequences with over 90% similarity were removed from the training and validation sets. Then, CD-HIT-2D was used to exclude sequences from the test set that shared >70% identity with any sequence filtered in the first step. In addition, sequences containing UAAs were removed. This resulted in a stringent benchmarking set of 725 AMPs and 1,071 non-AMPs, providing a reliable basis for comparing model performance against existing AMP classification algorithms including AMPlify22, Macrel23, iAMP Pred24, AMP Scanner (v.2)25, Bert-Protein26, AMPir27 and AmPEP28.
Data encoding and decoding
All sequences were encoded and decoded using GPT-2 tokenizer. The tokenizer encoded sequences, converting them into the tokenizer form for model input, and decoded them, transforming model outputs back into AAs. Sequence beginnings/ends were marked with the ‘<|endoftext|>’ label. During the pre-training and fine-tuning of the model, the datasets were divided into multiple pieces to optimize memory usage and balance the training load. This approach is particularly useful when dealing with large datasets, as it helps improve training efficiency and prevent memory overflow.
Model structure
The most fundamental model is ProteoGPT based on GPT-2 architecture. The model structure of AMPGenix is identical to that of ProteoGPT. AMPSorter and BioToxiPept were created by adding an additional classification module at the end of ProteoGPT to reduce the dimension to 2. Comprehensive model structure details can be found in the Supplementary Text.
Model evaluation
A cross-entropy loss function was used to train AMPSorter and BioToxiPept.
where:
N is the number of samples;
yi is the true label of sample i;
p(yi|x) is the predicted probability of sample i by the model, given x.
Accuracy, specificity, sensitivity, precision, F1 score, and MCC, receiver operating characteristic (ROC) curve and PR curve were measured to assess the classification models as follows (TP, true positive; TN, true negative; FP, false positive; FN, false negative; FPR, false positive rate):
The ‘auc’ function in the scikit-learn package (v.1.2.2) was used to compute the AUC and AUPRC values.
Uniqueness (proportion of unique generated sequences to the total number), diversity (average pairwise cosine distance between generated sequences), novelty (average cosine distance between generated sequences and known AMPs) and FCD (Fréchet distance measuring the distributional similarity between generated sequences and known AMPs) were used to evaluate the quality and diversity of sequences generated by generative models.
where N is the total number of sequences, and Xi, Xj are the feature vectors for sequences i and j.
where N is the total number of sequences, M is the number of known AMPs, and Xi, Xj are the feature vectors for the generated and known AMPs, respectively.
where μ1 and Σ1 are the mean and covariance of the generated sequences, and μ2 and Σ2 are the mean and covariance of the known AMPs, respectively.
QSAR analysis
The introduction of the QSAR model has specific background and purpose. To compare data mining and text generation strategies under the same threshold, we incorporated the QSAR model, which offers several advantages:
-
(1)
Clear threshold setting: The QSAR model can provide a clear quantitative threshold, enabling the selection of an appropriate number of peptides for further validation.
-
(2)
Consistent peptide types: The QSAR model, through quantitative structure–activity relationship predictions, predicts the antimicrobial activity of cationic AMPs and ensures that the two groups of peptides being compared are consistent in type, avoiding potential biases from differing peptide types. By introducing QSAR as a third-party tool, we can ensure the selection of consistent types of peptide from the candidate pools, thereby improving the comparability of both strategies in terms of development efficiency and antibacterial efficacy.
Each sequence was scored using the method described in refs. 35,36, which uses peptide charged residues and hydrophobic residues to create a relative score for the propensity of the peptide to present antimicrobial activity:
where C represents the net charge, H represents the total hydrophobicity, and MaxScore is the maximum value of Cm Hn that can be calculated with the given coefficients m = 0.9 and n = 1.1 as described35,36.
Gene function analysis
EggNOG mapper (http://eggnog-mapper.embl.de/) was applied to analyse protein-encoding genes with default parameter settings.
Peptides and chemicals
Peptides with purity >95% were custom synthesized by Synthbio Technology. The powder of each peptide was individually dispensed into several tubes of 2 mg per tube and stored in a refrigerator at −80°C. The powder was prepared on the day of the start of each experiment and used at the appropriate concentration. Dimethylsulfoxide (DMSO), cyclophosphamide, DiSC3-(5) and vancomycin were purchased from Mcklin. HEPES, polymyxin B, MTT Cell Proliferation and Cytotoxicity Assay kit were obtained from Solarbio, and PI was obtained from Yuanye. PBS was obtained from Cytiva. Triton X-100 was purchased from Sigma-Aldrich. DMEM/High glucose medium and penicillin–streptomycin mixed solution (Dual antibody) were purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology. d-glucopyranose and potassium chloride (KCl) were obtained from Hushi. Unless otherwise stated, all chemicals were purchased from Solarbio or the China National Pharmaceutical. Details of the reagents are provided in Supplementary Table 6.
Strains and media
E. coli ATCC25922, S. aureus ATCC25923 and C. albicans ATCC10231 were purchased from the Beijing Microbiological Culture Collection Center (BJMCC). ICU- isolated CRAB QLH-2022-267, CRAB QLH-2022-636, CRAB QLH-2022-637, MRSA QLH-2022-266 and MRSA QLH-2022-718 were provided by the Qilu Hospital Strain Bank, Shandong University. In this study, E. coli ATCC25922, S. aureus ATCC25923, CRAB strains and MRSA strains were cultured in Luria Bertani (LB) broth/agar medium. C. albicans ATCC10231 was cultured in yeast extract peptone dextrose (YPD) broth/agar medium. Mannitol salt agar (MSA), MacConkey agar (MCA), Mueller–Hinton broth (MHB), Mueller–Hinton agar (MHA) and tryptone soybean broth/agar (TSB/TSA) media were used in this study. All media were supplied by Hopebiol. Unless otherwise specified, all strains were incubated at 37 °C.
Mice
All animal experiments were performed according to the ‘Principles of laboratory animal care’ (NIH publication No. 86–23, revised 1985) and approved by the Animal Care and Use Committee of Shandong University (LL20240622). Male BALB/c mice (5-week-old) were purchased from Beijing Vital River Laboratory Animal and kept under a 12 h light/12 h dark cycle, humidity of 50% and temperature of 22 °C in standard specific-pathogen-free (SPF) individually vented cages.
Antimicrobial activity assays
To screen AMPs for better antimicrobial efficacy, a modified Kirby–Bauer disk diffusion assay was used. E. coli ATCC25922 and S. aureus ATCC25923 were cultured to log phase. MHA plates were used as the base medium. A 0.75% agar solution was prepared, sterilized and cooled to ~55 °C. Then, 100 μl of the bacterial suspension was added to 7 ml of the molten agar, mixed thoroughly and poured onto the surface of the pre-solidified MHA base plate to form a double-layer agar plate. The plates were allowed to solidify at room temperature. Sterile filter paper discs (7 mm in diameter) were placed on the surface of the double-layer plates, and the test compound was added to the centre of each disk. All plates were incubated at 37 °C for 16 h. After incubation, the diameter of the inhibition zones was measured in millimetres and recorded to evaluate the antimicrobial activity of the compounds. All 196 peptides were first selected at a content of 128 μg and further selected at a content of 64 μg (Extended Data Fig. 3). To determine MIC values, 58 selected peptides were dissolved into MHB with an initial concentration of 2,560 μg ml−1, followed by 2-fold serial dilutions. Cells of E. coli ATCC25922 and S. aureus ATCC25923 were separately suspended in the assay medium at a density of 1 × 108 c.f.u.s ml−1. Then, 100 μl of each AMP solution was added to 100 μl of the bacterial suspension in a 96-well plate. Plates were incubated at 37 °C for 16 h. Finally, 20 candidate AMPs were selected using the broth microdilution technique46 in MHB. Next, these AMPs were dissolved into MHB with an initial concentration of 1,024 μg ml−1, followed by 2-fold serial dilutions. E. coli ATCC25922, S. aureus ATCC25923, CRAB QLH-2022-267, CRAB QLH-2022-636, CRAB QLH-2022-637, MRSA QLH-2022-266 and MRSA QLH-2022-718 were cultured in LB at 37 °C and 120 r.p.m. to log phase. C. albicans ATCC10231 were cultured in YPD at 30 °C and 120 r.p.m. to log phase. Cells were separately suspended in the assay medium at a density of 105 c.f.u.s ml−1. Then, 100 μl of each AMP solution was added to 100 μl of the bacterial suspension in a 96-well plate. Plates were incubated at 37 °C for 16 h. The optical density (OD) values of 625 nm were measured using a TECAN Spark microplate reader following a previous report47.
Haemolysis effect of candidate AMPs
Freshly collected sheep red blood cells bought from Solarbio were first washed with PBS until the upper phase was clear after centrifugation (491 × g) and allocated onto 96-well flat-bottom plates. Each AMP was diluted and added to the well at a final concentration corresponding to its maximum MIC values against E. coli ATCC25922 and S. aureus ATCC25923 under high inoculum condition of 108 c.f.u.s ml−1. After 1 h at 37 °C, cells were centrifuged at 2,996 × g for 10 min. PBS and Triton X-100 were used as negative and positive controls, respectively. The supernatant was removed and OD450 was measured. All experiments were performed with three independent replicates. The haemolysis rate was calculated using the formula:
Cytotoxicity against mammalian cells
Cytotoxicity of candidate AMPs was determined using the MTT Cell Proliferation and Cytotoxicity Assay kit46. HEK293 cells (FUNHENG BIOLOGY) were inoculated in 96-well flat-bottom plates at 5,000 cells per well in cell culture medium. After 24 h incubation at 37 °C with 5% CO2 in the atmosphere, the medium was replaced with fresh medium, and AMPs (final concentration: 100 μg ml−1) were added, followed by 48 h incubation. Cell viability was monitored by adding MTT solution and measuring OD490 after 4 h. Zero wells and control wells were set during this experimental process. All experiments were performed with three independent replicates. Cell survival rate was calculated using the formula:
Modelling and treatment of neutropenic thigh infections in mice
Mice were injected intraperitoneally with cyclophosphamide 4 d and 1 d before bacterial administration, at concentrations of 150 mg kg−1 and 100 mg kg−1, respectively, to induce neutropenia35,47. MRSA QLH-2022-718 and CRAB QLH-2022-637 were suspended separately in sterile PBS, adjusted to a concentration of ~106 c.f.u.s per infection site and injected into the right thighs of mice in the corresponding experimental groups. Then, AMPs (10-fold MIC, 100 μl; Nmice = 7 for most AMP groups; Nmice = 6 for m_AMP76 group with MRSA infection; Nmice = 9 for g_AMP33 group with CRAB infection; Nmice = 8 for m_AMP76 group with CRAB infection), sterile water (Nmice = 8 with CRAB infection, Nmice = 7 with MRSA infection), polymyxin B (20,000–25,000 U kg−1 day−1, Nmice = 10) or vancomycin (40 mg kg−1, Nmice = 8) were given intraperitoneally at 1, 3, 5 and 7 h after infection. At 24 h after infection, mice were euthanized and thigh wound tissue was collected, weighed, homogenized and serially diluted in sterile PBS. C.f.u.s of MRSA QLH-2022-718 and CRAB QLH-2022-637 were calculated for each thigh wound tissue by diluting the thigh wound homogenate (0.25 g of thigh tissue in 10 ml sterile PBS), inoculating it on MSA plates and MCA plates, respectively, and counting the colonies.
Histologic changes in the kidney and colon of mice
Tissue samples were fixed in 4% paraformaldehyde for at least 24 h, dehydrated in a graded ethanol series, cleared in xylene and embedded in paraffin. Sections (4 μm) were cut, mounted on glass slides and dried at 60 °C. Slides were dewaxed, rehydrated, stained with haematoxylin (3–5 min), differentiated, blued, counterstained with eosin (15 s), dehydrated, cleared and mounted with neutral balsam. Images were acquired using a light microscope.
16S rRNA sequencing and analysis
The DNA of mouse caecum and colon contents were extracted using the EasyPure Stool Genomic DNA kit (EE301-01, TransGene) and sequenced on the Illumina Nova 6000 platform. Shannon index was calculated using QIIME2 (ref. 51). Euclidean distance matrix was computed with the ‘dist’ function and plotted using the ‘hclust’ function (base R package). Principal coordinates analysis (PCoA) and permutational multivariate analysis of variance (PERMANOVA) were conducted using adonis2 (vegan R package).
Resistance to proteolytic degradation assays
AMPs were incubated in fetal bovine serum (FBS) to evaluate resistance to enzymatic degradation45. Peptides were exposed to an aqueous solution of 25% FBS at a concentration of 2 mg ml−1 for 4 h at 37 °C. Aliquots were collected after 0, 0.5, 1, 2 and 4 h, and 200 μl of acetonitrile was added to each sample (100 μl) and incubated for 10 min at 4 °C. Samples were then processed in an AB SCIEX QTRAP 5500 system. The column used was an Agilent ZORBAX Eclipse XDB-C18 (3.5 μm, 2.1 mm × 150 mm). The mobile phases used were A (100% water with 0.1% v/v formic acid) and B (100% acetonitrile with 0.1% v/v formic acid), Fisher optima grades. Measurements were made by multiple reaction monitoring (MRM). The percentage of remaining undamaged peptide was calculated by integrating the AUC related to the peptide at timepoint zero. The time gradient for mobile phase composition is listed in Supplementary Table 7.
Bacterial resistance development assays
In wells of a 96-well polypropylene flat-bottom plate, 5 µl of the overnight bacterial culture was added to 100 μl of AMPs/antibiotic solutions in MHB at 105 c.f.u.s per well. Plates were incubated for 20–24 h at 37 °C. The MIC, the lowest concentration of peptide/antibiotic that caused no visible bacterial growth, was determined for each bacterial species. Thereafter, 5 µl of the growth at the 0.5-fold MIC suspension was added to a fresh medium containing AMPs/antibiotics at 105 c.f.u.s per well, and these mixtures were incubated as described above. This was repeated for 20 passages.
SEM measurement
Strains were grown to the exponential phase. The bacterial suspensions (108 c.f.u.s ml−1) were co-cultured with AMPs at final concentrations of 100 µg ml−1 at 37 °C for 24 h, and untreated cells were used as control. The specimens were observed using the FE-SEM Regulus8100 (Hitachi) scanning electron microscope.
Detection of peptide-induced membrane permeability
Single colonies of strains were inoculated and cultured to the exponential phase, followed by three washes with 10 mM PBS (pH 7.0) and adjustment of OD625 values to 0.08~0.13 with PBS. Subsequently, 150 μl of the bacterial suspension was incubated at 37 °C with 50 μl AMP (dissolved in PBS) for 2 h. PI was added at a final concentration of 50 μg ml−1, and the mixture was incubated in the dark at 37 °C for 30 min. Thereafter, bacterial suspensions were centrifuged (4 °C 1,825 × g, 10 min) and washed with PBS twice. All experiments were performed with three independent replicates. The treated cells were examined by flow cytometry (ThermoFisher Attune NxT). Data were analysed using FlowJo (v.10.8.1).
DiSC3-(5) assay
CRAB QLH-2022-637 and MRSA QLH-2022-718 were grown at 37 °C with agitation until they reached mid-log phase. The cells were then centrifuged and washed twice with washing buffer (20 mmol l−1 glucose, 5 mmol l−1 HEPES, pH 7.2) and resuspended to an OD600 of 0.05 in the same buffer containing 0.1 mol l−1 KCl. The cells (100 μl) were then incubated for 15 min with 20 nmol l−1 of DiSC3-(5) until the reduction of fluorescence stabilized, indicating the incorporation of the dye into the bacterial membrane. Membrane depolarization was then monitored by observing the change in the fluorescence emission intensity of the membrane potential-sensitive dye, DiSC3-(5) (lex = 622 nm, lem = 670 nm), after the addition of the peptides (100 μl solution at MIC values). Relative fluorescence was calculated as:
RNA sequencing and analysis
E. coli ATCC25922 was treated with AMP (1-fold MIC) for 2 h with six replicates, while PBS was used in the control group. RNA sequencing was conducted using the Illumina NovaSeq platform. HTSeq v.0.6.1 was used to count the read numbers mapped to each gene, and then the fragments per kilobase of transcript per million fragments mapped (FPKM) of each gene was calculated. Differential expression analysis was performed using the DESeq R package. PCoA and PERMANOVA were conducted using adonis2 (vegan R package).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Our study contains publicly available proteome, AMP, non-AMP, Toxin and non-toxin, generated sequences, 16S rRNA gene sequencing and RNA-seq data. Protein sequences were retrieved from UniProt (http://www.uniprot.org/) (downloaded as of October 2022). AMP data were mainly collected from six public AMP datasets: APD3 (https://aps.unmc.edu/AP/), DBAASP (https://www.dbaasp.org/home), DRAMP (http://dramp.cpu-bioinfor.org/), CAMP (http://www.camp3.bicnirrh.res.in/), LAMP (http://biotechlab.fudan.edu.cn/database/lamp) and BaAMPs (http://www.baamps.it), which cover most of AMP sequences from various origins (downloaded as of November 2022). The non-AMP dataset was downloaded from UniProt (https://www.uniprot.org) by setting the ‘subcellular location’ filter to cytoplasm and removing any entry that matches the following keywords: antimicrobial, antibiotic, antiviral, antifungal, effector or excreted in their functional annotations (downloaded as of November 2022). Toxin and non-toxin data were collected from https://server.wei-group.net/ToxIBTL/ (downloaded as of April 2023). The relevant data used for model construction, the constructed and generated short-peptide datasets, as well as the prediction results have been uploaded in Zenodo at https://doi.org/10.5281/zenodo.16633186 (ref. 52). Sequence data generated by five unconstrained generation models were collected from https://zenodo.org/records/7420189#.ZBCo4JHMKUk (downloaded as of October 2024). The 16S rRNA gene sequencing and RNA-seq data are available under The National Omics Data Encyclopedia (NODE) accession numbers OEP00006445 and OEP00005095. Source data are provided with this paper.
Code availability
The AMP generation and prediction codes can be found in GitHub at https://github.com/W1V1995/AMP_Project (ref. 53).
References
De Oliveira, D. M. P. et al. antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 33, e00181-19 (2020).
Yang, Q. E. et al. Interphylum dissemination of NDM-5-positive plasmids in hospital wastewater from Fuzhou, China: a single-centre, culture-independent, plasmid transmission study. Lancet Microbe 5, e13–e23 (2024).
Darby, E. M. et al. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 21, 280–295 (2023).
Antimicrobial Resistance Collaborators Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022).
King, A. M. et al. Systematic mining of the human microbiome identifies antimicrobial peptides with diverse activity spectra. Nat. Microbiol. 8, 2420–2434 (2023).
Mhlongo, J. T., Waddad, A. Y., Albericio, F. & de la Torre, B. G. Antimicrobial peptide synergies for fighting infectious diseases. Adv. Sci. 10, 2300472 (2023).
Lazzaro, B. P., Zasloff, M. & Rolff, J. Antimicrobial peptides: application informed by evolution. Science 368, eaau5480 (2020).
Shanahan, M. Talking about large language models. Commun. ACM 67, 68–79 (2024).
Wang, X. et al. UNI-RNA: universal pre-trained models revolutionize RNA research. Preprint at bioRxiv https://doi.org/10.1101/2023.07.11.548588 (2023).
Wang, S., Guo, Y., Wang, Y., Sun, H. & Huang, J. SMILES-BERT: large scale unsupervised pre-training for molecular property prediction. In Proc. 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics 429–436 (ACM, 2019).
Ucak, U. V., Ashyrmamatov, I., Ko, J. & Lee, J. Retrosynthetic reaction pathway prediction through neural machine translation of atomic environments. Nat. Commun. 13, 1186 (2022).
Zhang, Q. et al. Scientific large language models: a survey on biological and chemical domains. ACM Comput. Surv. 57, 161:1–161:38 (2025).
Xue, D. et al. X-MOL: large-scale pre-training for molecular understanding and diverse molecular analysis. Sci. Bull. 67, 899–902 (2022).
Zhang, X.-C. et al. Pushing the boundaries of molecular property prediction for drug discovery with multitask learning BERT enhanced by SMILES enumeration. Research 2022, 0004 (2022).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Flamholz, Z. N., Biller, S. J. & Kelly, L. Large language models improve annotation of prokaryotic viral proteins. Nat. Microbiol. 9, 537–549 (2024).
Nijkamp, E., Ruffolo, J. A., Weinstein, E. N., Naik, N. & Madani, A. ProGen2: exploring the boundaries of protein language models. Cell Syst. 14, 968–978.e3 (2023).
Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203 (2021).
Zhang, Y. et al. Prediction of multiple types of RNA modifications via biological language model. IEEE/ACM Trans. Comput. Biol. Bioinform. 20, 3205–3214 (2023).
Ferruz, N., Schmidt, S. & Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat. Commun. 13, 4348 (2022).
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
Li, C. et al. AMPlify: attentive deep learning model for discovery of novel antimicrobial peptides effective against WHO priority pathogens. BMC Genomics 23, 77 (2022).
Santos-Júnior, C. D., Pan, S., Zhao, X.-M. & Coelho, L. P. Macrel: antimicrobial peptide screening in genomes and metagenomes. PeerJ 8, e10555 (2020).
Meher, P. K., Sahu, T. K., Saini, V. & Rao, A. R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 7, 42362 (2017).
Veltri, D., Kamath, U. & Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 34, 2740–2747 (2018).
Zhang, Y., Lin, J., Zhao, L., Zeng, X. & Liu, X. A novel antibacterial peptide recognition algorithm based on BERT. Brief. Bioinform. 22, bbab200 (2021).
Fingerhut, L. C. H. W., Miller, D.J., Strugnell, J. M., Daly, N. L. & Cooke, I. R. ampir: an R package for fast genome-wide prediction of antimicrobial peptides Bioinformatics 36, 5262–5263 (2021).
Bhadra, P., Yan, J., Li, J., Fong, S. & Siu, S. W. I. AmPEP: sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 8, 1697 (2018).
Wei, L., Ye, X., Sakurai, T., Mu, Z. & Wei, L. ToxIBTL: prediction of peptide toxicity based on information bottleneck and transfer learning. Bioinformatics 38, 1514–1524 (2022).
Sharma, N., Naorem, L. D., Jain, S. & Raghava, G. P. S. ToxinPred2: an improved method for predicting toxicity of proteins. Brief. Bioinform. 23, bbac174 (2022).
Szymczak, P. et al. Discovering highly potent antimicrobial peptides with deep generative model HydrAMP. Nat. Commun. 14, 1453 (2023).
Das, P. et al. PepCVAE: semi-supervised targeted design of antimicrobial peptide sequences. Preprint at https://doi.org/10.48550/arXiv.1810.07743 (2018).
Van Oort, C. M., Ferrell, J. B., Remington, J. M., Wshah, S. & Li, J. AMPGAN v2: machine learning-guided design of antimicrobial peptides. J. Chem. Inf. Model. 61, 2198–2207 (2021).
Nagarajan, D. et al. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem. 293, 3492–3509 (2018).
Torres, M. D. T. et al. Mining for encrypted peptide antibiotics in the human proteome. Nat. Biomed. Eng. 6, 67–75 (2022).
Pane, K. et al. Antimicrobial potency of cationic antimicrobial peptides can be predicted from their amino acid composition: application to the detection of ‘cryptic’ antimicrobial peptides. J. Theor. Biol. 419, 254–265 (2017).
Wang, G., Li, X. & Wang, Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44, D1087–D1093 (2016).
Pirtskhalava, M. et al. DBAASP v3: database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 49, D288–D297 (2021).
Shi, G. et al. DRAMP 3.0: an enhanced comprehensive data repository of antimicrobial peptides. Nucleic Acids Res. 50, D488–D496 (2022).
Waghu, F. H., Barai, R. S., Gurung, P. & Idicula-Thomas, S. CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 44, D1094–D1097 (2016).
Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 415, 389–395 (2002).
Stojanov, S., Berlec, A. & Štrukelj, B. The influence of probiotics on the Firmicutes/Bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms 8, 1715 (2020).
Das, P. et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat. Biomed. Eng. 5, 613–623 (2021).
Santos-Júnior, C. D. et al. Discovery of antimicrobial peptides in the global microbiome with machine learning. Cell 187, 3761–3778.e16 (2024).
Maasch, J. R. M. A., Torres, M. D. T., Melo, M. C. R. & de la Fuente-Nunez, C. Molecular de-extinction of ancient antimicrobial peptides enabled by machine learning. Cell Host Microbe 31, 1260–1274.e6 (2023).
Huang, J. et al. Identification of potent antimicrobial peptides via a machine-learning pipeline that mines the entire space of peptide sequences. Nat. Biomed. Eng. 7, 797–810 (2023).
Ma, Y. et al. Identification of antimicrobial peptides from the human gut microbiome using deep learning. Nat. Biotechnol. 40, 921–931 (2022).
Gawde, U. et al. CAMPR4: a database of natural and synthetic antimicrobial peptides. Nucleic Acids Res. 51, D377–D383 (2023).
Ye, G. et al. LAMP2: a major update of the database linking antimicrobial peptides. Database 2020, baaa061 (2020).
Di Luca, M., Maccari, G., Maisetta, G. & Batoni, G. BaAMPs: the database of biofilm-active antimicrobial peptides. Biofouling 31, 193–199 (2015).
Estaki, M. et al. QIIME 2 enables comprehensive end‐to‐end analysis of diverse microbiome data and comparative studies with publicly available data.Curr. Protoc. Bioinformatics 70, e100 (2020).
Wang, Y. AMP project data [Data set]. Zenodo https://doi.org/10.5281/zenodo.16633186 (2025).
Wang, Y. AMP_Project [Code]. GitHub https://github.com/W1V1995/AMP_Project (2025).
Acknowledgements
Three CRAB strains and two MRSA strains were kindly provided by Y. Zhang and H. Pan (Qilu Hospital, Shandong University). Schematics in Figs. 1a, 3b, 5a,d, 6b, Extended Data Figs. 3 and 5a–c were created using BioRender images as templates, for which the authors possess licences. This study is supported by National Natural Science Foundation of China 82172320, National Natural Science Foundation of China 82370785, Shandong Provincial Natural Science Foundation ZR2024MH220, TaiShan Industrial Experts Program tscy20190612, TaiShan Scholars Program of Shandong Province tshw20120206, Shandong University Outstanding Young Scholars Program to L. Zhang and Shandong Academician Workstation Program 170401 to G.Z.
Author information
Authors and Affiliations
Contributions
L. Zhang and G.Z. conceptualized and managed this study. Y.W. developed all model construction and analysis. L. Zhao, Z.L., Y.X. and Y.P. performed wet-lab experiments and assisted with data interpretation. Y.W. and L. Zhao visualized the results. Y.W. and L. Zhao wrote the original draft, and L. Zhang, G.Z., Y.W. and L. Zhao revised the manuscript. All authors assisted with manuscript editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Yong-Xin Li and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 The performance and evaluation of transfer models on the training, validation, and test set.
a, Performance of the AMPSorter over training and validation datasets across epochs. b, c, AUC (b) and AUPRC (c) of AMPSorter on the test set. d, e, UMAP visualization of training set, validation set, test set (d) and benchmarking set (e) using k-mer encoding (k = 3). Each point represents a sequence, with the position determined by reducing the high-dimensional k-mer feature space into two dimensions. tr-AMP: AMPs in the training set; tr-Non-AMP: Non-AMPs in the training set; v-AMP: AMPs in the validation set; v-Non-AMP: Non-AMPs in the validation set; te-AMP: AMPs in the test set; te-Non-AMP: Non-AMPs in the test set; b-AMP: AMPs in the benchmarking set; b-Non-AMP: Non-AMPs in the benchmarking set. f, Comparison of FCD values between the AMP test set and the benchmarking set. The FCD values were calculated by comparing the test set and benchmarking set against known AMPs using k-mer encoding (k = 3). g, Performance of the BioToxiPept over training and validation datasets across epochs. h, Training process of AMPGenix. Blue solid line indicates the raw loss values, and black solid line depicts the loss values smoothed. i, The ratio of sequences containing UAAs generated by AMPGenix at different temperature parameters, predicted as AMP and Non-AMP by AMPSorter.
Extended Data Fig. 2 The comparison of sequence distributions between m_AMPs and g_AMPs.
a, Amino acids composition of 154 m_AMPs and 42 g_AMPs compared with known AMPs collected. b, Comparison of FCD values between m_AMPs and g_AMPs. The FCD values were calculated by comparing m_AMPs and g_AMPs against known AMPs using k-mer encoding (k = 3). c, The function of genes coding for proteins containing the 154 m_AMPs. The genes exhibit different functional expressions, among which translation, ribosomal structure and biogenesis, signal transduction mechanisms, cell wall/membrane/envelope biogenesis, inorganic ion transport and metabolism and cytoskeleton were the most expressed.
Extended Data Fig. 3 Workflow of AMPs including in vitro evaluation, therapeutic efficiency in vivo, and antimicrobial mechanism.
A total of 154 m_AMPs and 42 g_AMPs were characterized in vitro, and selected for in vivo experiment and mechanism exploration. Figure was created with BioRender.com.
Extended Data Fig. 4 Therapeutic efficacy of mice with seven optimal AMPs.
a, g_AMP14, g_AMP33, g_AMP41, g_AMP42, m_AMP46 and m_AMP76 were used for mice infected with CRAB QLH-2022-637, and g_AMP14, g_AMP35, g_AMP41, g_AMP42, m_AMP46 and m_AMP76 were used for mouse infected with MRSA QLH-2022-718. b, AMPs with significantly therapeutic effects in CRAB and MRSA-infected groups. CRAB 637: CRAB QLH-2022-637; MRSA 718: MRSA QLH-2022-718. c, d, PCoA analysis based on weighted UniFrac distance matrix using asv table. e–j, Clustering analysis with weighted UniFrac distance matrix at the phylum, class, and genus level. k, H&E staining of mouse colon and kidney tissues sections (scale bar, 100 μm). Arrow colors represent different lesions, in colon tissue (black: shedding of intestinal epithelial cells; green: loose arrangement of intestinal glands; blue: karyopyknosis of intestinal epithelial cells; dark blue: focal aggregates of lymphocytes in the submucosa; red: focal erosion of intestinal tissue), and in kidney tissue (black: hydropic degeneration of renal tubular epithelial cells; blue: karyopyknosis of renal tubular epithelial cells; grey: eosinophilic material in renal tubules; orange: cytoplasmic rarefaction and pale staining of renal tubular epithelial cells; yellow: venous stasis; brown: necrosis of renal tubular epithelial cells). Experiments were performed in duplicates with similar results and one representative figure is shown.
Extended Data Fig. 5 Mechanistic studies of optimal AMPs.
a–c, The SEM images of CRAB QLH-2022-637 (a), MRSA QLH-2022-718 (b) and E. coli ATCC25922 (c) treated with AMPs. SEM images of each treatment group are shown at two scale levels: 2 μm (left) and 500 μm (right). d, PCoA represents the Bray-Curtis distance between individuals in the grouping based on the FPKM value and displays the P value and F statistic. Statistical analysis was conducted using PERMANOVA. Corresponding interpreted percentages are indicated in parentheses for each principal axis (PCoA1 and PCoA2). n = 6 in each group. e, Differential gene expression analysis was shown on volcano plot to exhibition the comparison between Arkwillin-treated group and control. The x-axis represents the log2 fold change, and the y-axis shows the -log10 Padj value derived from a two-sided Wald test performed by DESeq2, with multiple testing correction using the Benjamini-Hochberg method. The vertical dashed line depicts the absolute log2foldchange > 0 threshold, and the horizontal dashed line marks the Padj ≤ 0.05 significance threshold. Genes exhibiting significant alterations are highlighted in red dot (up-regulated) or blue dot (down-regulated). Schematic in (a–c) was created with BioRender.com.
Supplementary information
Supplementary Information
Supplementary Text and Tables 1, 2, 6 and 7.
Supplementary Tables
Supplementary Table 3. F/B values of intestinal flora in different treatment groups. Table 4. Blast results of m_AMP76 in NCBI. Table 5. The species distribution of non-AMPs.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Zhao, L., Li, Z. et al. A generative artificial intelligence approach for the discovery of antimicrobial peptides against multidrug-resistant bacteria. Nat Microbiol 10, 2997–3012 (2025). https://doi.org/10.1038/s41564-025-02114-4
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41564-025-02114-4
