
Abstract
The Japanese anchovy (Engraulis japonicus), a finfish with the largest biomass of a single species in the Yellow and East China Seas, plays an important pivotal role in converting zooplanktons into high trophic fish in the food web. As a result, the fish is regard as a key species in its habiting ecosystem. However, the lack of genomic resources hampers our understanding of its genetic diversity and differentiation, as well as the evolutionary dynamics. Here, we firstly report a complex chromosome-level genome assembly of E. japonicus with a large size of 1.4 Gb, with features of high repetitive sequences (54.9%), high heterozygosity (2.3%) and a number of protein-coding genes (24,405). The genome sequence exhibited a remarkable degree of completeness, valued 94.07% of the complete BUSCO. This work firstly reported the genome sequence of E. japonicus, offering the crucial resources for further studies on the genetic diversity and adaptive evolution of this species.
Similar content being viewed by others


Background & Summary
Genomic resources, specifically genome sequences, are of particular importance in various genetic studies. Whole genome sequences are of help in examining the chromosomal evolution through comparative genomics, dissecting the genomic architecture for ecological adaptation, pinpointing the genes responsible for notable phenotypes as well as elucidating the divergence and speciation of organisms1,2,3. The technologies of high-throughput genome sequencing and cost-effective, precise genome assembly algorithms have promoted the assembly and release of numerous genome sequences, meanwhile, have substantially made the progress in genomics, offering comprehensive and novel insights into the fundamental mechanisms behind various biological questions of interest4,5.
The Japanese anchovy (Engraulis japonicus) is a petite marine finfish belonging to the Clupeiformes order, distributing in the northwest Pacific marginal seas, northward from the Sea of Japan and southward to the East China Sea6. This anchovy with a great biomass in the region, plays a pivotal role in the food chain due to being as both a forage and a food fish7. During the late 1990s, its peak annual catch was about one million tons8. However, due to the high capture pressure and adverse effects of global climate change on marine ecosystem, its population size had substantially declining8,9. Unfortunately, the species has recently been classified as overexploited. Like some other migratory fish in the region such as Larimichthys polyactis and L. crocea10, E. japonicus exhibits a migratory behaviour between spawning and overwintering grounds11. So far, the presence of genetic variation among different migratory stocks of E. japonicus remains controversial, primarily due to the use of different genetic markers and variations in the resolution of analytical methods12,13,14,15. Population genetic studies based on sequence variation in mitochondrial cytochrome b (Cyt b) and mitochondrial DNA control region fragments revealed no significant genetic structure across the wide-ranging populations of E. japonicus in the northwestern Pacific12,13. However, another molecular analyse using fragments of the Cyt b gene revealed considerable genetic variation among populations in the southern East China Sea14. Similarly, study utilizing six microsatellite loci detected weak but significant genetic differentiation between populations from the northeastern and southwestern coasts of Taiwan15. Marginally significant genetic differentiation was also observed between regional populations, such as the “Bohai Sea population (BHS)” and the “Japan Sea population (JPS)”, as well as between the “North Yellow Sea population (NYS)” and the “Japan Sea population (JPS)” using restriction-site associated DNA sequencing (RADseq)16. As highlighted above, it should be noted that traditional approaches, which rely on limited genetic data from narrow genomic regions, may not fully capture the population structure of E. japonicus. The discrepancies between these studies may therefore hinder the accuracy and effectiveness of fisheries management and conservation efforts. Recently, genome scans based on the whole genome sequencing data have identified numerous loci under putative natural selection. These genetic loci, with significant genetic differentiation among stocks, can be utilized to assign the different stocks within a given population, which is helpful for management and conservation of fishery resources10,16,17,18. Understandably, these genomic resources are invaluable for those investigations like adaptive evolution, population dynamics, and genetic conservation etc.
Despite the ecological and commercial importance, the genomic features of this species remain unknown. The previous investigations were mostly concerted with the population structure identification by using microsatellite15, and mitochondrial DNA markers12,13, RADseq16. So far, there has not been existed any report about transcriptome or genome sequence datasets of this species. Moreover, genomic data for anchovy fish in general are limited, with genome sequences available for only six species, including Coilia nasus, C. grayii, Encrasicholina punctifer, E. encrasicolus, Setipinna tenuifilis, and Thryssa baelama. This scarcity has greatly hindered our understanding of the evolutionary processes and environmental adaptations within the Engraulidae family and even the broader Clupeiformes order.
To address this, we have utilized the Pacific Biosciences (PacBio) HiFi long-read, Hi-C (chromosome conformation capture), and Illumina short-read sequencing technologies to construct a high-quality chromosome-level genome sequence of the Japanese anchovy. Moreover, we conducted annotation and analysis of the genome in comparison with the related species. The workflow of de novo genome assembly and annotation is shown in the Fig. 1. The highly accurate, chromosome-level reference genome would promote the progress of both population genetics and evolutionary biology of this species, as well as make it possible for the comparative genomics studies among the species of Clupeiformes order.

The overview of the chromosome-level genome assembly and annotation. Chrs: chromosomes. We first used 95.0 Gb short-read sequencing data to predict the assembled genome size was approximately 1,045.1 Mb by K-mer analysis, and the repeat sequences and heterozygosity were approximately 54.0% and 2.3%, respectively. Then, the 51.3 Gb of PacBio ccs data resulted in a 1,467.6 Mb assembly, with contig N50 of 456.3 kb. The contigs were anchored into 24 pseudo-chromosomes covering roughly 95.2% of the genome assembly with the assistance of 109.5 Gb Hi-C reads. The final assembly consisted of 24 pseudo-chromosomes that yielded 1,423.3 Mb of E. japonicus genome, with a scaffold N50 of 55.0 Mb. The genome contained 54.9% repeat sequences and 23,709 genes were functionally annotated from a total of 24,405 (97.15%) predicted protein-coding genes by combination of RNAseq and ISO-Seq annotation, genome sequence, and homolog protein.
Methods
Ethics statement
All experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals in China. All experimental procedures and sample collection methods were approved by the Institutional Animal Care and Use Committee (IACUC) of Yellow Sea Fisheries Research Institute, CAFS under approval No. YSFRI-2022041.
Sample collection and sequencing
A mature female E. japonicus (Fig. 2) was obtained from the coastal waters of the Yellow Sea, close to Qingdao, China. Its dorsal muscle was collected for subsequently DNA extraction using a standard sodium dodecyl sulfate (SDS) extraction method. Subsequently, the concentration and quality of the extracted genomic DNA (gDNA) were quantified and assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) and by running a 0.8% agarose gel, respectively. The high-quality gDNA was initially employed to establish a short-insert library of approximately 350 bp using the TruSeq DNA PCR-Free kit (Illumina, USA). The library was subsequently sequenced on the Illumina NovaSeq 6000 platform (Illumina, USA), and approximately 101 Gb of 2 × 150 bp reads were generated (Table 1). Long-read sequencing was carried out on the same sample using the PacBio HiFi sequencing technology (Pacific Biosciences, USA). A standard PacBio library with an insert size of 20 kb was prepared using the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, USA). Subsequently, the library was sequenced on a PacBio Sequel II system (Pacific Biosciences, USA), yielding a total of 51.3 Gb of PacBio HiFi reads, with an N50 length of 17.4 kb (Table 1). Lastly, a Hi-C library was established according to a previous protocol19 with some modifications20. In summary, muscle samples from the same sequenced individual were cross-linked using 4% formaldehyde. The fixed samples were then homogenized to isolate the nuclei. Following that, the DNA was digested with the MboI restriction enzyme (NEB, USA). The digested products underwent sequential treatments for end repairing, biotin labelling, and ligation of blunt-end fragments. The ligated DNA was subsequently sheared into fragments with a peak size of 400 bp. These fragments were then used to construct a standard DNA library using the TruSeq DNA Sample Prep Kit (Illumina, USA). The Hi-C library was sequenced for 2 × 150 bp reads on the Illumina NovaSeq 6000 platform, generating a total of 109.5 Gb reads (Table 1).

The mature female Japanese anchovy (Engraulis japonicus) obtained from the coastal waters of the Yellow Sea.
For transcriptome sequencing, samples of the brain, ovary, heart, muscle, and liver were obtained from the same sequenced sample for RNA extraction, using TRIzol™ Reagent (Thermo Fisher Scientific, USA). The concentration and quality of the total RNA were quantified and evaluated utilizing a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) and by running a 1.0% agarose gel, respectively. Total RNA from each individual sample was employed to construct mRNA libraries using the TruSeq RNA Library Prep Kit v2 (Illumina, USA). Subsequently, the libraries were sequenced on the Illumina NovaSeq 6000 platform (Illumina, USA), yielding an average of 5.58 Gb of 2 × 150 bp reads for each transcriptome sample (Table 1).
Chromosome-level genome assembly
The Illumina reads were first cleaned using the program NGSQCToolkit v2.321. The cleaned reads were then utilized to estimate genome parameters based on the 17-mer frequency distribution using the program GenomeScope v2.0922. The estimated genome size, heterozygosity, and content of repetitive sequences were found to be 1,045.1 Mb, 2.3%, and 54.0%, respectively. Subsequently, the Pacbio HiFi reads were assembled into contigs using the program Hifiasm v0.19.523, with default parameters. The assembled contigs were then polished using Pilon v1.2224, also with default parameters. The total length and N50 of the assembled contigs were approximately 1,467.6 Mb and 456.3 kb, respectively (Table 2).
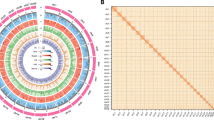
To achieve a chromosome-level assembly, raw Hi-C sequencing reads were first filtered using HiC-Pro v2.8.025. Subsequently, the cleaned reads were employed to anchor the assembled contigs into scaffolds using Juicer26 and 3D-DNA pipelines19. The assembled scaffolds were then manually curated using Juicebox27, with a prior setting of 24 haploid chromosomes28. Consequently, 95.2% of the assembled contigs were anchored to 24 pseudochromosomes (Fig. 3A), with individual chromosome lengths ranging from 47.0 Mb to 69.1 Mb (Fig. 3B and Table 3). The total length of the chromosome-level genome assembly amounted to 1,423.3 Mb, with a scaffold N50 of 55.0 Mb (Table 2). This discrepancy in genome assembly size, as opposed to the previously mentioned prediction, can be attributed to the tendency of short-read sequencing to underestimate the size of highly repetitive and heterozygous genomes29.

Chromosome-level assembly and features of the Engraulis japonicus genome. (A) Genome-wide chromatin interactions in the E. japonicus genome revealed by heatmap. (B) Circos plot of genomic features in the E. japonicus genome in a 100-kb window size. Each circle from outside to inside represents GC content along individual pseudochromosomes with indicated length (a), gene density (b), density of repetitive sequences (c), density of LTR elements (d), density of LINE elements (e) and density of DNA transposable elements (f).
Repetitive sequence annotation
Annotations of repetitive sequences were conducted using Repeatmasker v4.0.630, based on the RepBase database v20210131 and a custom repeat library. The custom repeat library was generated utilizing RepeatModeler v2.0.532, with default parameters. Additionally, the programs LTR_FINDER v1.0633 and Tandem Repeat Finder v4.0734 were independently employed to identify long terminal repeats and tandem repeats, using default parameters. The predictions of these programs were then consolidated to create a nonredundant library of repetitive sequences within the genome, which was subsequently used for annotation within Repeatmasker. A total of 780.9 Mb, constituting 54.9% of the assembled genome, were annotated as repetitive sequences (Table 4). Among these repeats, long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), and long terminal repeats (LTRs) accounted for 6.3%, 0.9%, and 9.1% of the genome, respectively (Table 4).
Gene prediction and functional annotation
Predictions of protein-coding genes were carried out on a repeat-masked genome utilizing homology-, evidence- and ab initio-based prediction methods. For the homology-based gene prediction, protein sequences of Alosa alosa (GCF_017589495.1), A. sapidissima (GCF_018492685.1), S. tenuifilis (v1)35, C. nasus (v1)36, and Danio rerio (NCBI, GCF_000002035.6) were aligned to the E. japonicus genome assembly using BLASTP v2.2.2437 with default parameters. Regarding evidence-based annotation, the mentioned transcriptomes were assembled utilizing Trinity v2.1.138 with default parameters, and then condensed into a nonredundant transcript dataset for utilization as supporting evidence for prediction. The Maker v2.53 pipeline39 was employed to consolidate the predictions from both the homology- and evidence-based approaches. Predicted gene models were iteratively trained using SNAP v2006.07.2840, GeneMark-EP v4.7241, and Augustus v3.3.242 for three iterations. Subsequently, predicted gene models containing transposable element (TE) domains and lacking support from transcripts were filtered out and removed. As a result, a total of 24,405 nonredundant protein-coding genes were predicted. Upon comparing the gene set of E. japonicus with that of A. alosa, A. sapidissima, S. tenuifilis, C. nasus, and D. rerio, a similar distribution pattern in the length of genes (Fig. 4A), exons (Fig. 4B), and coding sequences (CDS) (Fig. 4C) was observed among these studied fish species.

Features of the predicted protein coding genes in the Engraulis japonicus genome. (A) Distribution of the length of genes among six studied species. (B) Distribution of the length of exons. (C) Distribution of the length of coding sequences (CDS). (D) Summary statistics of the number of genes annotated by different databases: NR, InterPro, KEGG, KOG and SwissProt.
Additionally, all predicted genes were functional annotated by mapping to the public databases including SwissProt, Nr, KEGG, and InterPro, COG, KOG, and Pfam. In total, 23,709 genes were classified by at least one of these databases, accounting for 97.1% of all the predicted protein coding genes in the E. japonicus genome (Table 5 and Fig. 4D). Furthermore, genes coding for tRNA were predicted using tRNAscan-SE v1.3.143 with default parameters. Genes for rRNA were predicted by aligning to invertebrate template rRNA sequences using BLASTN v2.2.2437 with an E-value of 1e-5. Genes for both snRNAs and miRNAs were then identified using INFERNAL v1.1.144 against the Rfam database (release 12.0). In total, 23,984 non-coding RNAs (ncRNAs) were predicted, including 19,120 tRNAs, 229 rRNAs, 1,492 miRNAs, and 3,143 snRNAs (Table 6).
Data Records
Technical Validation
Evaluation of the genome assembly
To evaluate the quality of the genome assembly, the completeness of the genome sequence was first assessed by mapping to the Actinopterygii database (actinopterygii_odb10) of Benchmarking Universal Single-Copy Orthologs (BUSCO, v5.7.1). The genome assembly exhibited a high level of completeness, with a complete BUSCO value of 94.07%. Within this value, 88.71% were complete and single-copy while 5.36% were complete and duplicated. Only 1.7% BUSCOs were fragmented, and 4.2% were missing from the genome assembly (Table 7). We retrieved the genome assemblies of Clupeiformes archived in NCBI and found only 23 species with available genome sequences, of which only 10 species had chromosome-level genome assemblies (Table 8). The complete BUSCO value of E. japonicus (94.07%) is comparable to that of the high-quality chromosome-level genome assemblies of Clupeiform species archived in NCBI, which range from 84.5% to 95.6% with a median value of 92% (Table 8). Furthermore, both the PacBio HiFi long reads and Illumina short reads were aligned to the genome assembly using minimap2. The mapping rates for PacBio and Illumina reads were 99.91% and 97.97%, respectively (Table 9). Finally, the consensus quality value (QV), representing per-base consensus accuracy, was estimated using Merqury (v1.3), resulting in a QV of 49.74. Considering these data collectively, it is evident that the genome assembly of E. japonicus is characterized by both high completeness and high quality.
Code availability
No custom codes or scripts were utilized in this study. All bioinformatics programs and pipelines were executed according to the instructions and guidelines provided by the software developers. The specific software versions and corresponding parameters employed have been delineated in the Methods subsection.
References
Wang, L. et al. A chromosome-level reference genome of african oil palm provides insights into its divergence and stress adaptation. Genomics, Proteomics & Bioinformatics 21, 440–454 (2023).
Wang, L. et al. Genomic basis of striking fin shapes and colors in the fighting fish. Molecular Biology and Evolution 38, 3383–3396 (2021).
Yue, G. & Wang, L. Current status of genome sequencing and its applications in aquaculture. Aquaculture 468, 337–347 (2017).
Phillippy, A. M. New advances in sequence assembly. Genome Research 27, xi–xiii (2017).
Jackson, S. A., Iwata, A., Lee, S. H., Schmutz, J. & Shoemaker, R. Sequencing crop genomes: approaches and applications. New Phytologist 191, 915–925 (2011).
Takasuka, A. & Aoki, I. Environmental determinants of growth rates for larval Japanese anchovy Engraulis japonicus in different waters. Fisheries Oceanography 15, 139–149 (2006).
Iversen, S., Zhu, D., Johannessen, A. & Toresen, R. Stock size, distribution and biology of anchovy in the Yellow Sea and East China Sea. Fisheries Research 16, 147–163 (1993).
Yu, H. et al. Potential environmental drivers of Japanese anchovy (Engraulis japonicus) recruitment in the Yellow Sea. Journal of Marine Systems 212, 103431 (2020).
Nakayama, S. I., Takasuka, A., Ichinokawa, M. & Okamura, H. Climate change and interspecific interactions drive species alternations between anchovy and sardine in the western North Pacific: Detection of causality by convergent cross mapping. Fisheries Oceanography 27, 312–322 (2018).
Wang, L., Liu, S., Yang, Y., Meng, Z. & Zhuang, Z. Linked selection, differential introgression and recombination rate variation promote heterogeneous divergence in a pair of yellow croakers. Molecular Ecology 31, 5729–5744 (2022).
Tanaka, H., Ohshimo, S., Takagi, N. & Ichimaru, T. Investigation of the geographical origin and migration of anchovy Engraulis japonicus in Tachibana Bay, Japan: A stable isotope approach. Fisheries Research 102, 217–220 (2010).
Liu, J. X. et al. Late Pleistocene divergence and subsequent population expansion of two closely related fish species, Japanese anchovy (Engraulis japonicus) and Australian anchovy (Engraulis australis). Molecular Phylogenetics and Evolution 40, 712–723 (2006).
Zheng, W., Zou, L. & Han, Z. Genetic analysis of the populations of Japanese anchovy Engraulis japonicus from the Yellow Sea and East China Sea based on mitochondrial cytochrome b sequence. Biochemical Systematics and Ecology 58, 169–177 (2015).
Chen, C. S., Tzeng, C. H. & Chiu, T. S. Morphological and molecular analyses reveal separations among spatiotemporal populations of anchovy (Engraulis japonicus) in the southern East China Sea. Zoological Studies 49, 270–282 (2010).
Yu, H. T., Lee, Y. J., Huang, S. W. & Chiu, T. S. Genetic analysis of the populations of Japanese anchovy (Engraulidae: Engraulis japonicus) using microsatellite DNA. Marine Biotechnology 4, 471–479 (2002).
Zhang, B. D., Li, Y. L., Xue, D. X. & Liu, J. X. Population genomics reveals shallow genetic structure in a connected and ecologically important fish from the Northwestern Pacific Ocean. Frontiers in Marine Science 7, 374 (2020).
Wang, L. et al. Population genetic studies revealed local adaptation in a high gene-flow marine fish, the small yellow croaker (Larimichthys polyactis). PLoS One 8, e83493 (2013a).
Wang, L., Liu, S., Zhuang, Z., Lin, H. & Meng, Z. Mixed-stock analysis of small yellow croaker Larimichthys polyactis providing implications for stock conservation and management. Fisheries Research 161, 86–92 (2015).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Wang, L. et al. A chromosome-level genome assembly of chia provides insights into high omega-3 content and coat color variation of its seeds. Plant Communications 3, 100326 (2022a).
Patel, R. K. & Jain, M. NGSQCToolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7, e30619 (2012).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nature Communications 11, 1432 (2020).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nature Methods 18, 170–175 (2021).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology 16, 259 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 3, 95–98 (2016).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Systems 3, 99–101 (2016).
Jinxing, W., Xiaofan, Z., Xiangmin, W. & Mingcheng, T. Karyotype analysis for seven species of clupeiform and perciform fishes. Zoological Research 15, 76–79 (1994).
Pflug, J. M., Holmes, V. R., Burrus, C., Johnston, J. S. & Maddison, D. R. Measuring genome sizes using read-depth, k-mers, and flow cytometry: methodological comparisons in beetles (Coleoptera). G3: Genes, Genomes, Genetics 10, 3047–3060 (2020).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Current Protocols in Bioinformatics 5, 4.10. 11–14.10. 14 (2004).
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenetic and Genome Research 110, 462–467 (2005).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Liu, B. et al. Chromosome‐level genome assembly and population genomic analysis reveal evolution and local adaptation in common hairfin anchovy (Setipinna tenuifilis). Molecular Ecology 00, 1–18 (2023).
Xu, G. et al. Genome and population sequencing of a chromosome-level genome assembly of the Chinese tapertail anchovy (Coilia nasus) provides novel insights into migratory adaptation. GigaScience 9, giz157 (2020).
McGinnis, S. & Madden, T. L. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Research 32, W20–W25 (2004).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nature Biotechnology 29, 644–652 (2011).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genomics and Bioinformatics 2, lqaa026 (2020).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Research 34, W435–W439 (2006).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods in Molecular Biology 1962, 1–14 (2019).
Nawrocki, E. P., Kolbe, D. L. & Eddy, S. R. Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337 (2009).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP492930 (2024).
Liu, S. et al. Chromosome-level genome assembly and annotation of Japanese anchovy (Engraulis japonicus). figshare https://doi.org/10.6084/m9.figshare.25273354 (2024).
CNGB https://db.cngb.org/search/project/CNP0005377/ (2024).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_040112795.1 (2024).
Acknowledgements
This research was supported by the Project of Laoshan Laboratory (LSK202203802); the National Natural Science Foundation of China (Grant No. 42076132 and 32102768); and the China Agriculture Research System of MOF and MARA (CARS-47).
Author information
Authors and Affiliations
Contributions
S.L. and L.W. conceived and designed this study and drafted the manuscript. S.L., Z.M. and Z.Z. coordinated and supervised the whole study. L.W., R.W. and H.W. conducted the genome assembly and bioinformatics analysis. R.W. and H.W. participated in manuscript improvement. A.L. and C.A. prepared the samples and the figures. S.L. and Z.Z. reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, S., Wang, L., Wang, R. et al. Chromosome-level genome assembly and annotation of Japanese anchovy (Engraulis japonicus). Sci Data 12, 134 (2025). https://doi.org/10.1038/s41597-025-04423-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41597-025-04423-z
